

Abstract
Non-homologous end joining (NHEJ) is critical for the maintenance of genetic integrity and DNA double-strand break (DSB) repair. NHEJ is regulated by a series of interactions between core components of the pathway, including Ku heterodimer, XLF/Cernunnos, and XRCC4/DNA Ligase 4 (Lig4). However, the mechanisms by which these proteins assemble into functional protein–DNA complexes are not fully understood. Here, we show that the von Willebrand (vWA) domain of Ku80 fulfills a critical role in this process by recruiting Aprataxin-and-PNK-Like Factor (APLF) into Ku-DNA complexes. APLF, in turn, functions as a scaffold protein and promotes the recruitment and/or retention of XRCC4-Lig4 and XLF, thereby assembling multi-protein Ku complexes capable of efficient DNA ligation in vitro and in cells. Disruption of the interactions between APLF and either Ku80 or XRCC4-Lig4 disrupts the assembly and activity of Ku complexes, and confers cellular hypersensitivity and reduced rates of chromosomal DSB repair in avian and human cells, respectively. Collectively, these data identify a role for the vWA domain of Ku80 and a molecular mechanism by which DNA ligase proficient complexes are assembled during NHEJ in mammalian cells, and reveal APLF to be a structural component of this critical DSB repair pathway.
Keywords: DNA repair, DNA strand break, end joining
Introduction
DNA double-strand breaks (DSBs) are a major threat to genetic stability and hereditary defects in DSB repair result in a variety of disease pathologies (McKinnon and Caldecott, 2007). To date, two cellular pathways have been identified that repair DSBs; a mechanism in which undamaged sister chromatids and homologous recombination are employed and a mechanism denoted non-homologous end joining (NHEJ) in which the DNA ends are spliced together (Jackson and Bartek, 2009). Loss of homologous recombination-mediated repair is associated with increased predisposition to cancer, including hereditary breast cancer, whereas loss of NHEJ is typically associated with immunodeficiency and neurological dysfunction (McKinnon and Caldecott, 2007). During the classical and most commonly employed pathway for NHEJ, DSBs are bound by the Ku heterodimer, which comprises 70 kDa (Ku70) and 80 kDa (Ku80) subunits (Mahaney et al, 2009; Lieber, 2010). Ku then recruits a number of proteins to the DSB including DNA-PKcs, XLF/Cernunnos (from now on termed as XLF), and XRCC4-Lig4 complex. DNA-PKcs is a DNA-dependent protein kinase that is activated at DNA ends and promotes DNA end processing by Artemis (Ma et al, 2002; Goodarzi et al, 2006), whereas XLF and XRCC4-Lig4 facilitate the final step of DNA ligation (Ahnesorg et al, 2006; Buck et al, 2006; Gu et al, 2007; Lu et al, 2007; Tsai et al, 2007). With the exception of DNA-PKcs, which evolutionarily is a relatively recent addition, these proteins comprise the core components of NHEJ. Importantly, while NHEJ involves multiple interactions between the core components of this process, the mechanisms by which these interactions are regulated and/or organised is unclear.
Recently, Aprataxin-and-PNK-Like Factor (APLF; also denoted as PALF/C2orf13/XIP1) was identified as a novel component of NHEJ that promotes intracellular re-joining of transfected linear plasmid DNA molecules and accelerates the repair of chromosomal DSBs following γ-irradiation (Bekker-Jensen et al, 2007; Iles et al, 2007; Kanno et al, 2007; Macrae et al, 2008; Rulten et al, 2011). APLF is recruited to sites of DNA strand breakage via interaction of a C-terminal tandem zinc finger domain with poly (ADP-ribose); a nucleic acid-like structure synthesised at sites of DNA breakage by poly (ADP-ribose) polymerases (Ahel et al, 2008; Rulten et al, 2008; Eustermann et al, 2010; Li et al, 2010). Importantly, APLF appears to accelerate the rate of chromosomal NHEJ during the first few hours following γ-irradiation, suggesting that it is an accessory factor for NHEJ (Iles et al, 2007; Rulten et al, 2011). The molecular mechanism by which APLF accelerates NHEJ is unclear. One possibility is that APLF is an end processing factor, because it has been reported to possess nuclease activity in vitro, and thus may process incompatible DNA termini (Kanno et al, 2007; Li et al, 2011). However, we recently demonstrated that APLF-depleted cells exhibit reduced levels of XRCC4 in chromatin, and that overexpression of XRCC4-Lig4 complex can circumvent the requirement for APLF for rapid rates of NHEJ, suggesting that APLF accelerates NHEJ primarily by promoting DNA ligase activity (Rulten et al, 2011). It is possible that APLF promotes DNA ligation by modifying chromatin structure directly, since this protein interacts with histone H3/H4 and exhibits histone chaperone activity, in vitro (Mehrotra et al, 2011). However, APLF also possesses an amino-terminal fork-head associated (FHA) domain that interacts directly with the XRCC4 (Iles et al, 2007; Kanno et al, 2007; Macrae et al, 2008) and an MID domain that interacts directly with Ku heterodimer (Iles et al, 2007; Kanno et al, 2007; Macrae et al, 2008). The role and importance of these interactions remains unknown, but it seems likely that they will underpin the molecular mechanism by which APLF accelerates DNA ligation during NHEJ. Indeed, we show here that APLF promotes the assembly and activity of multi-protein Ku-DNA complexes containing all of the NHEJ factors required for DNA ligation, and describe the mechanism by which this is achieved. Collectively, our data identify APLF as a novel structural component of the NHEJ pathway, in mammalian cells.
Results
Identification of a conserved peptide motif in APLF sufficient for binding Ku80
In a yeast 2-hybrid (Y2H) screen employing full-length human APLF as bait we recovered multiple clones encoding XRCC1 and XRCC4, both of which interact with the amino-terminal FHA domain of APLF (Figure 1A). In addition, we recovered seven clones encoding the 80-kDa subunit of Ku (Ku80), but none encoding the 70-kDa subunit (Ku70). Additional Y2H experiments confirmed that the MID domain of APLF spanning residues 94–358 is sufficient for interaction with Ku80, consistent with a previously reported interaction of this region with Ku heterodimer (Macrae et al, 2008) (Figure 1B). Close inspection of the MID domain revealed a conserved peptide motif between residues 182 and 191, comprising a short patch of 3 or 4 basic amino acids followed by a hydrophobic patch of 6–9 amino acids that includes an invariant tryptophan (Figure 1C; Supplementary Figure 1). Mutation of the three basic residues RKR (182–184) resulted in an APLF protein that constitutively transactivated Y2H reporter genes, and so was uninformative (data not shown). However, mutation of the invariant tryptophan (W189) ablated Ku80-dependent activation of Y2H reporter genes, suggesting that W189 is critical for the interaction of APLF with Ku80 (Figure 1B; Supplementary Figure 2A).
Figure 1.
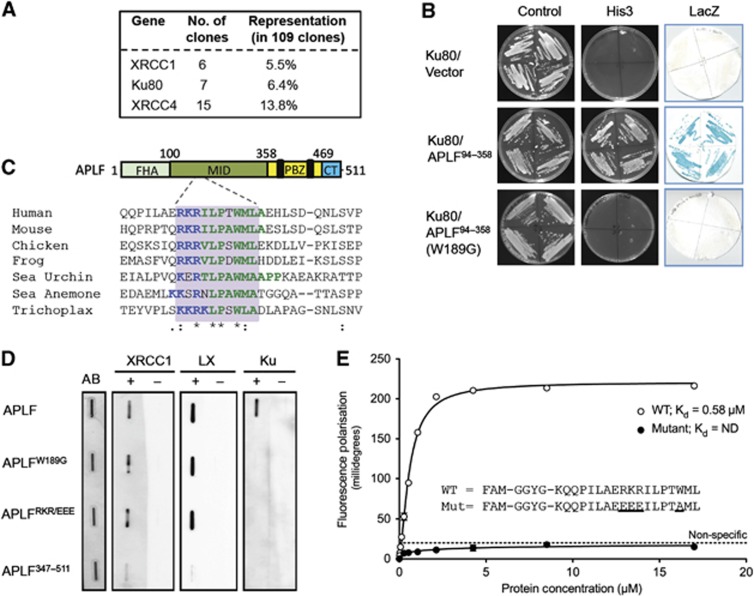
Identification of a novel peptide motif in APLF that binds Ku80. (A) Recovery of human cDNA clones encoding XRCC1, XRCC4, or Ku80 in a yeast 2-hybrid (Y2H) screen employing APLF as bait. The number and relative representation of each clone type is indicated. (B) Ku80 interacts with APLF residues 94–358, and requires W189 for this interaction. Y190 budding yeast harbouring pACT-Ku80 (library clone 5) and empty pGBKT7, pGBKT7-APLF94–358, or pGBKT7-APLFW189G was examined for activation of the His3 and LacZ reporter genes by Y2H analysis. (C) Schematic of APLF depicting a conserved Ku80-binding peptide motif in the MID domain. Top, APLF cartoon depicting the interaction domains; FHA domain that interacts with XRCC1 and XRCC4 (FHA), MID domain that interacts with Ku80, PBZ domain that interacts with poly (ADP-ribose), and C-terminal (CT) domain that interacts with histone H3/H4. Bottom, ClustalW alignment of the putative Ku80-binding peptide motif (grey box) from the indicated organisms. The NCBI accession numbers employed are NP_775816, NP_001163960, XP_419335, NM_001113131, XP_788055, XP_001635068, and XP_002117261. (D) Mutation of the conserved peptide motif in APLF prevents Ku binding, in vitro. One microgram of wild-type APLF, APLFW189G, APLFRKR(182–184)EEE, or APLF347–511 was slot blotted onto nitrocellulose and individual membrane strips stained with Amido black as a loading control (AB) or mock incubated (−) or incubated (+) with 100 nM of recombinant XRCC1, XRCC4-Lig4 heterodimer (LX), or Ku70/80 as indicated. (E) The APLF conserved peptide motif is sufficient to bind Ku. The indicated fluorescein-labelled peptides spanning the wild-type (WT) or mutant APLF conserved peptide motif were examined for binding to Ku70/80ΔC by fluorescence polarisation. Data points are the average of three independent experiments, with error bars representing one standard deviation.
To confirm the importance of the conserved peptide motif for Ku80 binding, we slot-blotted wild-type and mutant recombinant APLF proteins onto nitrocellulose and measured Ku binding in solution. Whereas Ku heterodimer (Ku70/80) bound wild-type APLF in these experiments, it failed to bind a C-terminal fragment of APLF in which the MID domain was missing (APLF347–511) or full-length APLF in which RKR (182–184) or W189 was mutated (Figure 1D). Neither of the point mutations disrupted binding of APLF to XRCC1 or XRCC4-Lig4 (LX), which are known to interact with the amino-terminal FHA domain of APLF (Bekker-Jensen et al, 2007; Iles et al, 2007; Kanno et al, 2007; Macrae et al, 2008). Next, we examined whether the conserved peptide motif is sufficient for interaction with Ku heterodimer, using fluorescence polarisation. We employed Ku heterodimer containing Ku80ΔC for these experiments, and for some of the experiments described later, because this protein lacks the highly flexible C-terminal 141 residues and appears to be more homogenous in structural conformation (Walker et al, 2001). Notably, whereas an 18-mer synthetic peptide spanning the conserved peptide motif bound Ku with a Kd of ∼0.6 μM, Ku binding by a mutant peptide in which RKR (182–184) and W189 were mutated was too low to quantify (Figure 1E). Together, these data identify a conserved peptide motif in APLF that is both required and sufficient for interaction with Ku80.
The Ku80-binding motif is an autonomous module that recruits proteins into Ku-DNA complexes
To examine whether the Ku-binding motif is required to recruit APLF into DNA complexes containing Ku heterodimer (Ku70/80), we employed electrophoretic mobility shift assays (EMSAs). Whereas full-length APLF and a truncated derivative harbouring the MID domain (APLF1–469) super-shifted DNA complexes containing Ku (Figure 2A, lanes 7–12), N-terminal (APLF1–164) and C-terminal (APLF360–511) fragments lacking the MID domain did not (Figure 2A, lanes 13–18). None of the APLF proteins bound DNA substrate in the absence of Ku, confirming that APLF did not bind DNA independently of Ku under the conditions employed (Figure 2A, lanes 1–5). More importantly, mutation of the Ku-binding motif ablated the ability of APLF to super-shift Ku-DNA complexes, confirming that this motif is required for recruitment of APLF into DNA complexes containing Ku (Figure 2B).
Figure 2.
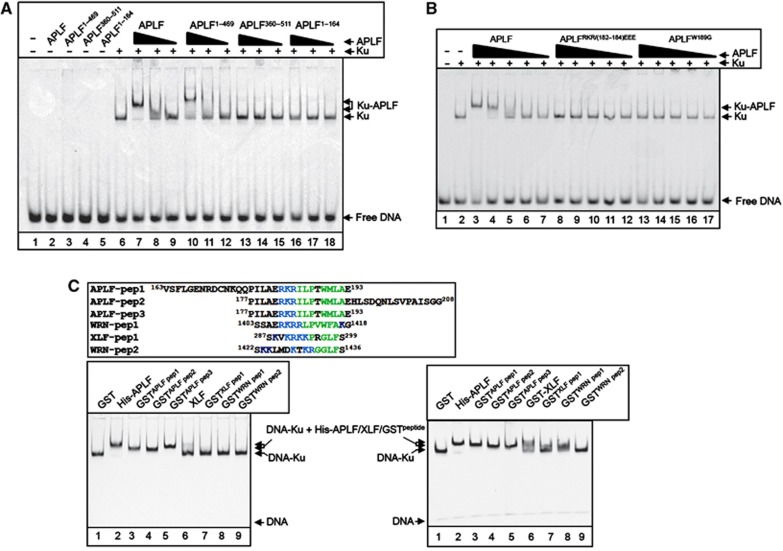
The Ku-binding motif is a novel autonomous module for recruiting proteins into Ku-DNA complexes. (A) The MID domain is required for APLF recruitment into DNA complexes containing Ku. A Cy3-labelled 30-bp duplex oligonucleotide (10 nM) harbouring blunt-ended termini was incubated with (+) or without (−) recombinant Ku70/80 (20 nM) in the absence (−) or presence of 150, 50, or 17 nM of the indicated recombinant APLF protein and employed in EMSA. Where a single concentration of APLF is indicated, the protein was employed at 150 nM. The composition of the protein DNA complexes is indicated on the right (arrows). (B) The Ku-binding motif is required for APLF recruitment into DNA complexes containing Ku. EMSAs were conducted as above. Where indicated, Ku70/80 was present at 20 nM and the indicated wild-type and mutant APLF proteins were employed at 150, 50, 17, 6 and 2 nM. (C) The Ku-binding motif is an autonomous module for protein recruitment into Ku-DNA complexes. Top left, alignment of the peptide sequences from APLF, XLF, and WRN that were fused to GST and analysed in the bottom panels, by EMSA. The conserved basic and hydrophobic patches that characterise the Ku-binding motif are indicated in blue and green, respectively. Bottom left, Cy3-labelled 30-bp duplex oligonucleotide (10 nM) was incubated with Ku70/80 (10 nM) in the presence of 0.5 μM of GST, the indicated GST peptides, full-length His-APLF (lane 2), or full-length XLF (lane 6), and employed in EMSAs as described above. Bottom right, Cy3-labelled 30-bp duplex oligonucleotide was incubated as above in the presence of GST (3 μM), His-APLF (0.5 μM), GST-XLF (1.6 μM), or the indicated GST-APLF (1.3 μM), GST-XLF (1.6 μM), or GST-WRN (3 μM) peptides.
We next examined whether the Ku-binding motif can function autonomously, and thus target an unrelated protein into Ku-DNA complexes in vitro. Indeed, GST fusion proteins harbouring short peptides that span the Ku-binding motif in APLF were efficiently recruited into Ku complexes on a 30-bp duplex DNA substrate, whereas GST lacking the motif was not (Figure 2C, bottom left, lanes 1–5). Similar results were observed on a shorter, 19-bp, duplex substrate that is approximately the same size as the footprint of a single Ku heterodimer, suggesting that the GST peptides assemble into Ku-DNA complex by direct interaction Ku (Supplementary Figure 2). Indeed, none of the GST peptides bound the 19-bp duplex in the absence of Ku (Supplementary Figure 2). Intriguingly, sequence analyses revealed a similar motif at the C-terminus of XLF and two similar motifs in tandem at the C-terminus of Werner (WRN) protein (Figure 2C, top left). Both XLF and WRN are involved in NHEJ and have been reported to interact with Ku via their C-termini, suggesting that the APLF-like motifs similarly might function as Ku binding modules in these proteins (Cooper et al, 2000; Karmakar et al, 2002; Yano et al, 2011). Consistent with this idea, GST fusion proteins harbouring the putative Ku-binding motif from XLF or the first of the two motifs from WRN (residues 1403–1418) were recruited into DNA complexes containing Ku, albeit less efficiently than the APLF motif (Figure 2C bottom panels, lanes 7–9).
Ku80 binding promotes APLF accumulation at chromosome damage
APLF is recruited and/or retained at cellular DNA strand breaks via PBZ and FHA domain-mediated interactions with poly (ADP-ribose) and XRCC1/XRCC4, respectively (Bekker-Jensen et al, 2007; Iles et al, 2007; Kanno et al, 2007; Ahel et al, 2008; Macrae et al, 2008; Rulten et al, 2008). To address the contribution of the Ku-binding motif in this process, we compared YFP-tagged wild-type and mutant derivatives of APLF for ability to accumulate at sites of UVA laser damage. As expected, YFP-APLFR27A and YFP-APLFZFD proteins harbouring mutated FHA and PBZ domains, respectively, were recruited and/or retained at sites of UVA laser damage to a lesser extent than was wild-type YFP-APLF (Figure 3A, bottom; ‘R27A’ and ‘ZFD’). However, YFP-APLFW189G (‘WG’) harbouring a mutated Ku-binding motif also exhibited reduced retention at such sites, and the accumulation of YFP-APLFZFD/W189G or YFP-APLFZFD/R27A/W189G harbouring mutations in both the PBZ and Ku80-binding domains or in all three domains was reduced to ∼15 and 5% of wild type, respectively (Figure 3A, bottom). We noted during these experiments that mutation of the Ku-binding motif affected the subcellular distribution of YFP- or mRFP-tagged APLF (Supplementary Figure 3A). However this did not account for the impact of W189G on APLF accumulation at sites of UVA laser damage, because only cells harbouring similar levels of nuclear YFP-APLF or YFP-APLFW189G were scored in these experiments. Moreover, YFP-nls-APLFW189G protein, which harboured an engineered SV40 nuclear localisation signal and was entirely nuclear, similarly exhibited reduced retention at sites of UVA laser damage (Supplementary Figure 3B).
Figure 3.
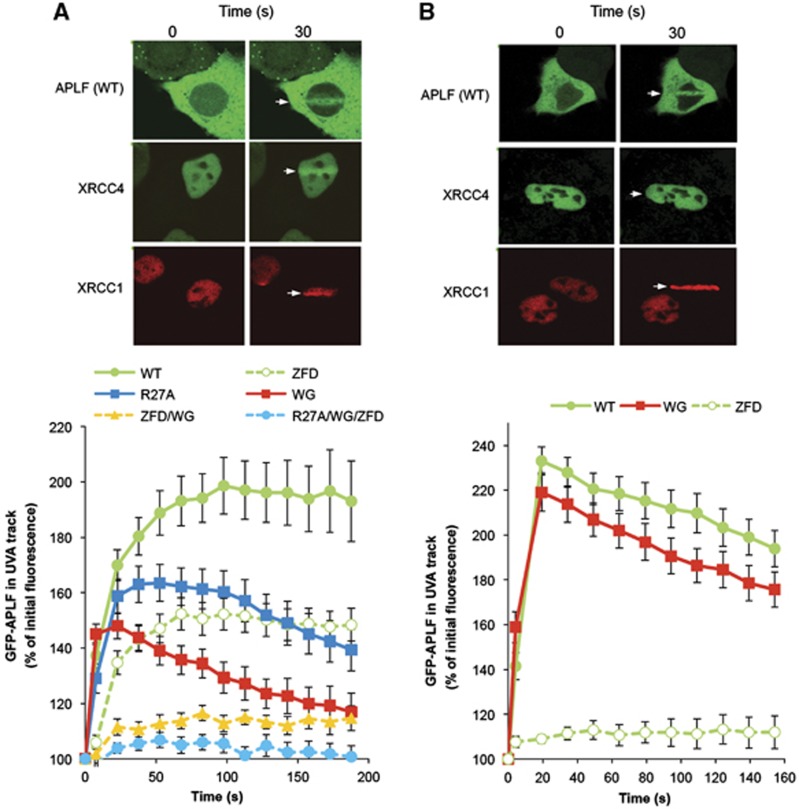
The Ku80-binding motif promotes APLF accumulation at chromosome damage. A549 cells were transiently transfected with constructs encoding wild-type YFP-APLF (WT) or YFP-APLF harbouring point mutations in the PBZ domain (YFP-APLFZFD; ‘ZFD’), FHA domain (YFP-APLFR27A; ‘R27A’), Ku-binding motif (YFP-APLFW189G; ‘WG’), or in combinations of these (‘ZFD/WG’, ‘R27A/WG/ZFD’). mRFP-XRCC1 and GFP-XRCC4 were used as markers of recruitment to single- and double-strand breaks, respectively. Cells were irradiated with 4.36 J/m2 (A) or 0.22 J/m2 (B) using a UVA laser (arrow). Images were captured at 15 s intervals after laser irradiation. For each data point, data are normalised to the YFP fluorescence intensity prior to irradiation (set to 100%). Data are the mean (±s.e.m.) of 10 or more individual cells per data point. Representative examples of YFP-APLF, GFP-XRCC4, and mRFP-XRCC1 accumulation at sites of UVA laser damage are shown (top).
The amount of UVA damage introduced in the above experiments was sufficient to induce both single-strand breaks (SSBs) and DSBs, as indicated by the accumulation of both the SSB repair protein XRCC1 and the DSB repair protein, XRCC4 (Figure 3A, top). Consequently, since APLF interacts with both of these proteins and is involved in the repair of both SSBs and DSBs, we wished to confirm that the Ku80-binding motif was required specifically for accumulation of APLF at the latter. To do this, we employed a 20-fold lower level of UVA damage; an experimental condition in which the level of SSB damage was still sufficient to trigger XRCC1 accumulation but in which the level of DSB damage was too low to trigger significant recruitment of XRCC4 or Ku (Figure 3B, top, and data not shown). The accumulation of APLF at sites of UVA laser damage remained dependent on the PBZ domain under these conditions, consistent with APLF recruitment at chromosomal SSBs, but was largely independent of W189 (Figure 3B, bottom). Together, these data suggest that the Ku-binding motif is required to promote the retention of APLF at sites of chromosomal DSBs.
APLF interacts with the von Willebrand-like domain of Ku80
The seven Ku80 cDNA clones recovered by APLF in our Y2H screen encoded only the amino-terminal 258–278 amino acids, and thus just the von-Willebrand Factor A (WA)-like domain (Aravind and Koonin, 2001) (Figure 4A). This observation is significant, because while this putative protein–protein interaction domain is highly conserved in eukaryotic Ku80, its protein partner/s have not yet been identified and its molecular function is unknown. Consequently, we attempted to map the site of APLF interaction in more detail. Given the partially hydrophobic character of the Ku-binding motif in APLF, we considered it likely that the corresponding interface in Ku80 is also hydrophobic. We therefore identified and mutated nine highly conserved hydrophobic residues predicted to be present on the surface of the vWA domain (Supplementary Figure 4), and examined the impact of these mutations on the interaction of Ku80 with the APLF MID domain in Y2H experiments. Whereas six of the nine mutations did not measurably impact on the interaction with the APLF MID domain, three of the mutations (Ku80L68R, Ku80Y74R, and KuI112R) greatly reduced or ablated it (Figure 4B). Strikingly, these three residues co-localise in Ku80 on the surface of the vWA domain (Walker et al, 2001), consistent with them forming part of a hydrophobic interface for the Ku-binding motif (Figure 4C). Indeed, in contrast to wild-type Ku (Ku70/80ΔC), KuL68R harbouring a mutant Ku80 vWA domain (Ku70/80ΔCL68R) failed to recruit APLF into DNA complexes in vitro (Figure 4D, compare lanes 3–5 with 7–9). This was not a non-specific impact of L68R on Ku folding, because the mutant heterodimer bound DNA in vitro (Figure 4D, lane 6) and was recruited to sites of chromosome damage in cells (Figure 4E). To confirm the impact of mutating the vWA domain on APLF behaviour in cells, we exploited our observation that the nuclear retention of mRFP-APLF is increased by co-expression with recombinant Ku80 (see Supplementary Figure 3A). Whereas co-transfection with GFP-Ku70/GFP-Ku80 increased the fraction of A549 cells harbouring predominantly nuclear mRFP-APLF, co-transfection with GFP-Ku70/GFP-Ku80L68R failed to do so (Figure 4F). Importantly, neither GFP-Ku70/GFP-Ku80 nor GFP-Ku70/GFP-Ku80L68R promoted the nuclear retention of mRFP-APLFW189G, confirming that the vWA domain promoted mRFP-APLF nuclear retention only if the Ku-binding motif was intact. Collectively, these results reveal that APLF interacts directly with the vWA domain of Ku80, thereby identifying a protein partner and molecular role for this domain.
Figure 4.
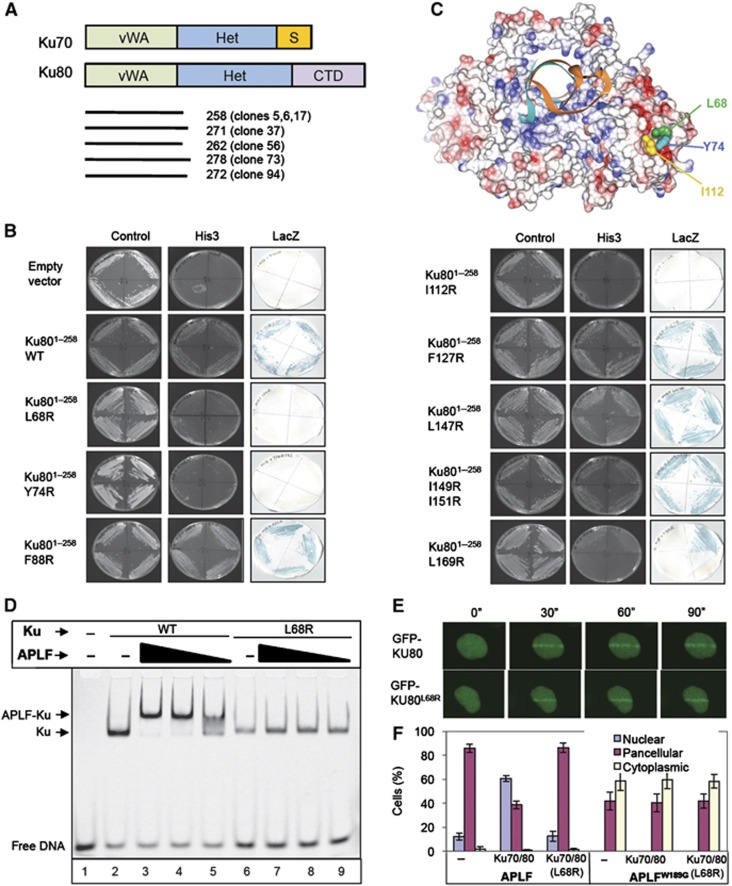
APLF interacts with the vWA domain of Ku80. (A) Cartoon of Ku70 and Ku80, depicting the von Willebrand-like (‘vWA’) domains, heterodimerisation domains (‘Het’), Ku70 SAP domain (‘S’), and Ku80 C-terminal domain (‘CTD’). The regions of Ku80 recovered by APLF in the Y2H screen depicted in Figure 1A are shown (bottom). (B) Mutation of the Ku80 vWA domain disrupts interaction with APLF. Y190 cells harbouring empty pGBKT7 or pGBKT7-APLF94–358 (encoding the MID domain) and either empty pACT2 or the indicated wild-type or mutant derivative of pACT-Ku801–258 (clone 5) were examined for His3 and LacZ reporter gene expression. (C) Residues required for APLF interaction co-localise in a hydrophobic interface on the surface of the Ku80 vWA domain. The location of L68, Y74, and I112 within the Ku heterodimer (RCSB PDB entry; 1JEY) (Walker et al, 2001) is shown. Blue and red denote basic and acidic regions, respectively. (D) Mutation of the Ku80 vWA domain prevents recruitment of APLF into DNA complexes containing Ku. A Cy3-labelled 30-bp duplex (10 nM) was incubated with (+) or without (−) 10 nM wild-type Ku (Ku70/80Δ) or mutant Ku (Ku70/80ΔL68R) in the absence (−) or presence of 700, 350, or 175 nM of the indicated recombinant APLF protein and employed in EMSA. (E) Normal accumulation of Ku70/GFP-Ku80L68R at sites of chromosome damage. A549 cells were transiently co-transfected with His-Ku70 and either GFP-Ku80 or GFP-Ku80L68R prior to UVA laser irradiation. Images were captured at 30 s intervals after laser irradiation. (F) Impact of mutations in the vWA domain on the subcellular localisation of APLF. mRFP localisation following co-transfection with mRFP-APLF or mRFP-APLFW189G and GFP vector, GFP-Ku70/GFP-Ku80, or GFP-Ku70/GFP-Ku80L68R. Data are the mean of three independent experiments (±s.e.m.).
APLF promotes the stability of NHEJ protein complexes, in vitro
The data described above define the mechanism by which APLF and Ku interact, and demonstrate that this interaction is important for APLF recruitment into Ku complexes and for retention at chromosome damage. However, these results do not explain how APLF accelerates DNA ligation during NHEJ. To address this question, we examined the impact of APLF on the stable assembly of the DNA ligase heterodimer, XRCC4-Lig4, into DNA complexes containing Ku. Surprisingly, despite the established ability of XRCC4-Lig4 to interact directly with Ku (Nick McElhinny et al, 2000; Hsu et al, 2002; Costantini et al, 2007), we observed little if any incorporation of XRCC4-Lig4 into DNA complexes containing Ku heterodimer (Ku70/80ΔC) in the absence of APLF, under the conditions employed (Figure 5A, lanes 1 and 2). In contrast, in the presence of APLF, all of the DNA complexes containing Ku were super-shifted by XRCC4-Lig4 into a single protein complex of lower mobility (Figure 5A, lanes 3 and 4). Importantly, both anti-XRCC4 and anti-APLF antibodies were able to super-shift these Ku–DNA complexes, confirming the presence of both APLF and XRCC4-Lig4 (Figure 5B, lanes 4–6). In contrast, neither antibody super-shifted Ku-DNA complexes in the absence of APLF, confirming their specificity (Figure 5B, lanes 7–9). Note that while Ku heterodimer harbouring Ku80ΔC was employed for the above experiments, similar results were observed for Ku heterodimer harbouring full-length Ku80 (Supplementary Figure 6A).
Figure 5.

APLF promotes NHEJ complex assembly. (A) Co-assembly of APLF and XRCC4-Lig4 into 19-bp DNA complexes containing Ku. A Cy3-labelled 19-bp duplex (10 nM) was incubated with Ku70/80ΔC (20 nM), in the absence or presence of APLF (0.4 μM) and/or XRCC4-Lig4 (LX; 0.4 μM) and then employed in EMSA. (B) NHEJ protein complexes assembled by APLF are super-shifted by anti-APLF and anti-XRCC4 antibodies. A Cy3-labelled 19-bp duplex was incubated with Ku70/80ΔC as described above in the presence or absence of APLF (0.4 μM) and/or XRCC4-Lig4 (LX; 0.55 μM) and/or the indicated anti-XRCC4 (Santa Cruz; sc-8285) or anti-APLF (Abmart 2G11) antibody and employed in EMSA. (C) Co-assembly of APLF and XRCC4-Lig4 into 60-bp DNA complexes containing Ku. A Cy3-labelled 60-bp duplex (10 nM) was incubated with full-length Ku70/80 (20 nM) in the presence and absence of APLF (0.4 μM) and 0–140 nM XRCC4-Lig4 (LX), as indicated. (D) APLF promotes assembly of both XRCC4-Lig4 and XLF into Ku-DNA complexes and functions as a molecular scaffold. A Cy3-labelled 19-bp duplex (10 nM) was incubated in the absence or presence of full-length Ku70/80 (20 nM), XRCC4-Lig4 (LX; 0.2 μM), XLF (1 μM), and the indicated wild-type or mutant APLF (0.2 μM), and then employed in EMSA.
It is worth noting that we employed a short DNA duplex probe of 19 bp in the above experiments, which is similar to the footprint of a single Ku heterodimer, to prevent DNA binding by XRCC4-Lig4 and so specifically address the role of protein–protein interactions on XRCC4-Lig4 assembly into Ku-DNA complexes (Kysela et al, 2003; Lu et al, 2007). However, similar results were observed with a longer duplex probe (60 bp) that allows DNA binding by multiple Ku heterodimers and direct DNA binding by XRCC4-Lig4 (Figure 5C). As reported previously (Nick McElhinny et al, 2000), XRCC4-Lig4 was able to super-shift Ku complexes in the absence of APLF on the longer substrate, at the highest concentration of XRCC4-Lig4 employed at least (Figure 5C, lane 2, orange asterisk). Nevertheless, APLF promoted incorporation of XRCC4-Lig4 into Ku complexes even on this substrate, with almost all Ku complexes super-shifted by XRCC4-Lig4 in the presence of APLF even at the lowest concentration of ligase employed (Figure 5C, lane 10, blue asterisk).
Next, we expanded EMSAs to include XLF, since this protein is another core component of DNA ligation reactions during NHEJ (Ahnesorg et al, 2006; Buck et al, 2006; Lu et al, 2007; Tsai et al, 2007; Riballo et al, 2009). In addition, since XRCC4-Lig4 and XLF most likely function together during DNA ligation, we examined whether APLF also promotes the assembly of larger Ku complexes containing both of these proteins. Indeed, whereas we failed to detect assembly of XLF and/or XRCC4-Lig4 into DNA complexes containing Ku heterodimer (Ku70/80) in the absence of APLF (Figure 5D, lanes 2–5), in the presence of APLF more than half of the Ku-DNA complexes were super-shifted by XLF and/or XRCC4-Lig4 into complexes of lower mobility (Figure 5D, lanes 7–9, green, blue, and red asterisks). Importantly, the ability of APLF to stimulate assembly of NHEJ complexes containing XLF and/or XRCC4-Lig4 was completely dependent on the presence of Ku, because neither XLF nor XRCC4-Lig4 super-shifted DNA in the absence of Ku, irrespective of whether or not APLF was present (Supplementary Figure 6B).
To understand how APLF promotes assembly of NHEJ complexes, we employed mutant derivatives of this protein. APLFW189G failed to promote the incorporation and/or retention of either XLF or XRCC4-Lig4 into Ku-DNA complexes, indicating that direct interaction with Ku80 is essential for APLF-mediated assembly of NHEJ complexes (Figure 5D, compare lanes 6–9 with lanes 14–17) and thus that APLF functions as a molecular scaffold. Consistent with this idea, while APLFR27A protein harbouring a mutated FHA domain was still recruited into DNA complexes containing Ku (Figure 5D, lane 10, black asterisk), and was able to weakly promote incorporation of XLF into Ku-DNA complexes (Figure 5D, lanes 11 and 13, green asterisks), it was unable to promote the incorporation and/or retention of XRCC4-Lig4 (Figure 5D, compare lanes 8 and 12, blue asterisk). Consistent with the dependence on protein phosphorylation for interaction of the APLF FHA domain with XRCC4, pre-treatment of XRCC4-Lig4 complex with lambda protein phosphatase prevented wild-type APLF from promoting the assembly XRCC4-Lig4 into Ku-DNA complexes (Supplementary Figure 6C). Collectively, these results reveal that APLF is a molecular scaffold protein that promotes the assembly and/or stability of Ku complexes containing the core components required for DNA ligation.
APLF promotes the retention of NHEJ DNA ligase proteins at sites of chromosome damage
To address the relevance of the above observations to the situation in cells, we examined the recruitment and retention of Ku, XLF, and XRCC4-Lig4 at sites of UVA laser-induced chromosomal damage in wild-type and APLF-depleted A549 cells. In agreement with our EMSAs, GFP-tagged Ku heterodimer accumulated at sites of UVA laser damage to a similar extent in wild-type and APLF-depleted A549 cells, supporting the idea that APLF functions downstream of Ku binding (Figure 6A). In contrast, as reported previously (Rulten et al, 2011), the accumulation of GFP-tagged XRCC4 at sites of UVA laser-induced chromosomal damage was reduced in APLF-depleted cells, again consistent with our EMSA data (Figure 6B). More importantly, whereas co-transfection of expression construct encoding shRNA-resistant mRFP-APLF restored normal levels of GFP-XRCC4 accumulation at sites of UVA damage, co-transfection with empty vector or with expression constructs encoding shRNA-resistant mRFP-APLF that cannot bind XRCC4 (mRFP-APLFR27A) or Ku80 (mRFP-APLFW189G) failed to do so (Figure 6B). Similar results were observed for GFP-tagged XLF, which also accumulated at sites of UVA damage to a lesser extent in APLF-depleted cells than in wild-type cells; a phenotype which again was complemented by co-expression of mRFP-APLF but not mRFP-APLFR27A or mRFP-APLFW189G (Figure 6C). Note that the inability of mRFP-APLFW189G to support normal levels of GFP-XRCC4 or GFP-XLF accumulation at UVA damage did not reflect the reduced nuclear localisation of the mutant protein, because mRFP-nls-APLFW189G harbouring an SV40 nuclear localisation signal was similarly unable to support normal GFP-XRCC4 or GFP-XLF accumulation (Supplementary Figure 7). Based on these experiments and our biochemical assays, we conclude that APLF is recruited into Ku complexes by interaction with Ku80 and, in turn, promotes the recruitment and/or retention of the key NHEJ factors required for DNA ligation.
Figure 6.
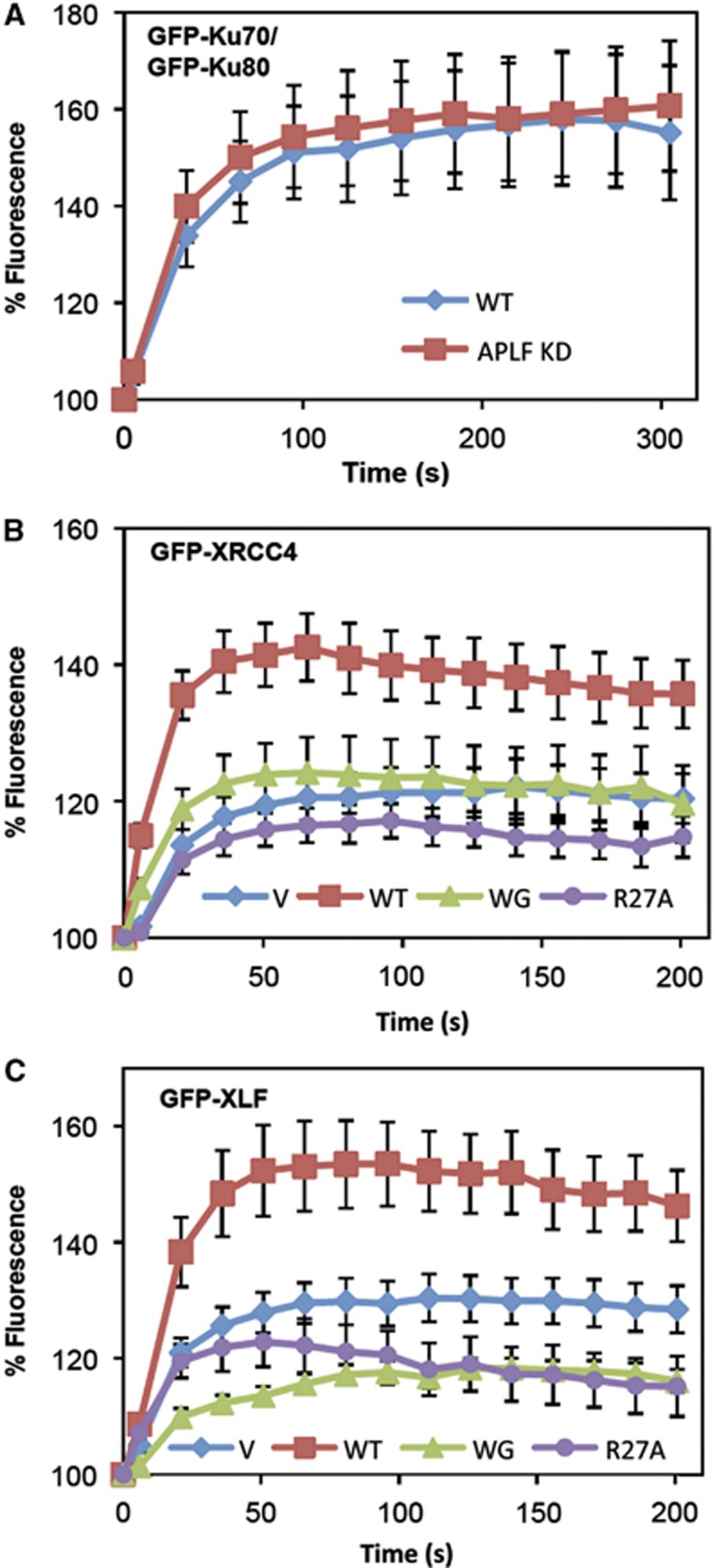
APLF promotes the recruitment and/or retention of XRCC4-Lig4 and XLF at sites of chromosome damage. (A) APLF is not required for recruitment/retention of Ku at sites of UVA damage. A549 cells mock depleted (WT) or stably depleted of APLF (APLF KD) were transiently transfected with expression constructs encoding GFP-Ku70 and GFP-Ku80 and images were captured at the indicated times after laser UVA micro-irradiation. For each data point, data are normalised to the GFP fluorescence intensity prior to irradiation (set to 100%). Data are the mean (±s.e.m.) of 10 or more individual cells. (B) APLF promotes recruitment/retention of GFP-XRCC4 at sites of UVA damage. APLF KD cells were transiently transfected with GFP-XRCC4 and either mRFP vector (V) or shRNA-resistant mRFP-APLF (WT), mRFP-APLFW189G (WG), or mRFP-APLFR27A (R27A), and analysed as described above. (C) APLF promotes recruitment/retention of GFP-XLF at sites of UVA damage. APLF KD cells were co-transfected with GFP-XLF and mRFP vector (V), mRFP-APLF (WT), mRFP-APLFW189G (WG), or mRFP-APLFR27A (R27A), and analysed as described above.
APLF scaffold activity promotes NHEJ in human cells and resistance to γ irradiation in DT40
We have shown previously that APLF accelerates the rate of NHEJ in cells, most likely by promoting DNA ligation (Rulten et al, 2011). The latter was suggested by the ability of XRCC4-Lig4 overexpression to restore normal rates of DSB repair in APLF-depleted cells. We therefore examined whether the novel role for APLF, described here, can influence DNA ligation by XRCC4-Lig4 in vitro. Indeed, in experiments employing a 247-bp DNA duplex with a single ligatable blunt DSB terminus, APLF stimulated ligation by XRCC4-Lig4 above that observed in the presence of Ku alone (Figure 7A, lanes 1–3). PCR amplification of the head-to-tail junctions of these reactions confirmed that at least 9 out 10 of the junctions were the product of direct ligation (Supplementary Figure 8A). Mutation of either the FHA domain or Ku-binding motif reduced the impact of APLF, and mutation of both ablated it, confirming the collective importance of the Ku80 and XRCC4-Lig4 interactions for stimulation of DNA ligation by APLF, in vitro (Figure 7A, lanes 4–6).
Figure 7.
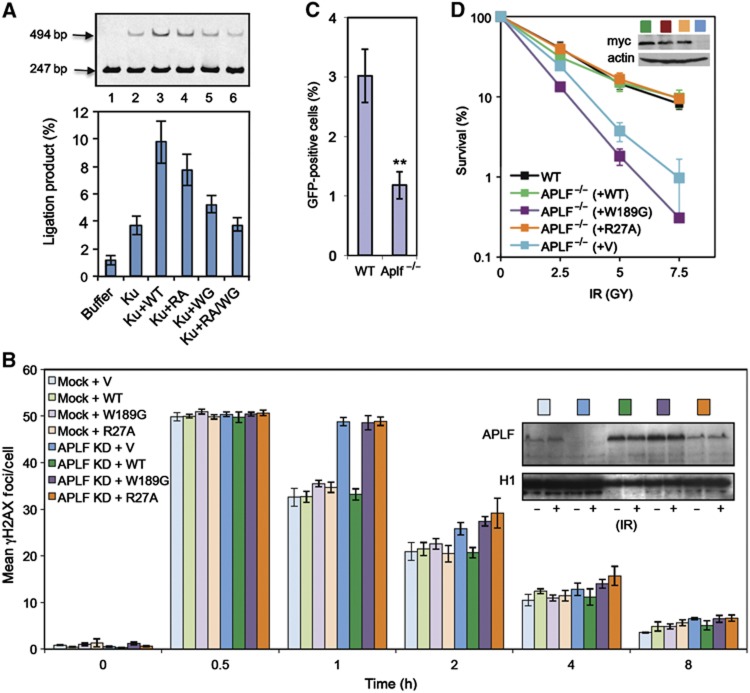
APLF scaffold activity stimulates DNA ligation in vitro and is required for rapid repair of, and resistance to, chromosomal DSBs. (A) APLF stimulates XRCC4-Lig4 activity in vitro. A Cy3-labelled 247-bp DNA duplex with one ligatable end was incubated for 30 min with recombinant XRCC4-Lig4 (10 nM) alone (‘buffer’) or in the presence or absence as indicated of recombinant Ku70/80 (‘Ku’; 20 nM), wild-type APLF (‘WT’), APLFW189G (‘WG’), APLFR27A (‘RA’), or APLFW189G/R27A (‘RA/WG’). Reaction products were fractionated by denaturing PAGE. A representative image from one experiment is presented (top), along with quantitative data from four independent experiments showing the mean (±s.e.m.) fraction of total DNA converted to a 494-bp product (bottom). (B) APLF scaffold activity is required to accelerate DSB repair. A549 cells were transiently co-transfected with empty pSUPER (‘Mock’) or pSUPER-APLF (‘APLF KD’) and either empty pcD2E expression vector (‘V’) or pcD2E encoding shRNA-resistant wild-type APLF (‘WT’), APLFR27A (‘R27A’), or APLFW189G (‘W189G’). Transfected cells were mock irradiated or γ irradiated (2 Gy) and allowed to recover for the times indicated prior to immunostaining. Data are the mean number (±s.e.m.) of γH2AX foci scored in G1 cells from three independent experiments. Inset, immunoblotting for levels of APLF and histone H1 (loading control) in untreated cells and γ-irradiated cells (30 min after irradiation). (C) Reduced plasmid re-joining in APLF−/− DT40 cells. Wild-type (Clone 18) and APLF−/− DT40 cells were co-transfected with circular RFP and linear GFP vector, 18 h before analysis by FACS. Data are the percentage of RFP-positive cells expressing GFP and are the mean (±s.e.m.) of five independent experiments. **P<0.01 by Student’s t-test. (D) Hypersensitivity of APLF−/− DT40 cells to γ radiation. Wild-type DT40 cells (clone 18), APLF−/− DT40 cells, and APLF−/− DT40 cells stably transfected with empty expression vector or with expression construct encoding myc-tagged wild-type (WT) human APLF or the indicated myc-tagged mutant human APLF were γ irradiated with the indicated dose and cell colonies counted 10 days after treatment. Inset, anti-myc Western blot depicting expression levels of the indicated recombinant APLF protein. Note that the APLF proteins expressed in these experiments were additionally tagged with an SV40 nuclear localisation signal.
To confirm the importance of the APLF scaffolding activity in cells, we examined the importance of the FHA domain and Ku-binding motif for chromosomal NHEJ and cellular resistance to DSBs. Consistent with our previous data (Rulten et al, 2011), shRNA-mediated depletion of APLF reduced the rate of NHEJ in A549 cells at early times following γ irradiation, as measured by γH2AX immunostaining (Figure 7B, compare light blue and dark blue bars). γH2AX immunostaining is a reliable and sensitive marker for DSBs following γ irradiation, allowing accurate quantification of DSB repair rates at clinically relevant levels of DSB breakage (Löbrich et al, 2010). Importantly, co-transfection of pSUPER-APLF with an expression construct encoding shRNA-resistant wild-type APLF prevented this reduction in DSB repair rate, confirming that reduced DSB repair was the result of APLF depletion (‘APLF KD+WT’; dark green bars). In contrast, co-transfection of pSUPER-APLF with expression constructs encoding either shRNA-resistant APLFR27A (‘APLF KD+R27A’; orange bars) or shRNA-resistant APLFW189G (‘APLF KD+W189G’; purple bars) failed to prevent the reduction in DSB repair rate. This was not due to differences in the level of expression of the mutant APLF proteins, because these were similar or greater than that of endogenous APLF (Figure 7B, inset). Nor did this reflect the impact of W189G on nuclear localisation, because shRNA-resistant APLFW189G harbouring an SV40 NLS was similarly unable to support normal rates of DSB repair (Supplementary Figure 8B). Together, these data demonstrate that the APLF FHA and Ku binding domains that promote assembly of NHEJ complexes in vitro are required to accelerate NHEJ, in cells.
As further support for the role of APLF in cellular NHEJ, we deleted this gene in avian DT40 cells (Supplementary Figure 9). NHEJ efficiency was reduced 2- to 3-fold in APLF−/− cells in plasmid re-joining assays, as measured by the level of GFP expression in cells transiently transfected with a linearised GFP reporter plasmid (Figure 7C). In addition, APLF−/− cells were hypersensitive to a range of DSB inducing agents, including etoposide and γ irradiation (Figure 7D; Supplementary Figure 9). Importantly, whereas wild-type human APLF restored the sensitivity of APLF−/− cells to γ rays to normal, APLFW189G mutant protein that cannot assemble NHEJ complexes was unable to do so.
Discussion
A role for APLF in the NHEJ pathway for DSB repair is indicated by several observations. First, APLF-depleted or deleted cells exhibit a reduced ability to ligate linear plasmid DNA in human (Macrae et al, 2008) and avian DT40 cells (this work), similar to cells harbouring defects in other components of NHEJ. Second, the rate of repair of γ ray-induced chromosomal DSBs is reduced in APLF-depleted human cells in G0/G1 phase of the cell cycle (Iles et al, 2007; Rulten et al, 2011), conditions in which NHEJ is the primary mechanism for DSB repair. Third, DNA products of class switch recombination in APLF−/− murine B cells exhibit mildly increased levels of microhomology at switched junctions in vitro, a phenotype consistent with a reduction in efficiency of the ‘classical’ pathway for NHEJ (Rulten et al, 2011). Fourth, APLF physically associates with Ku heterodimer and with XRCC4-Lig4 (Bekker-Jensen et al, 2007; Iles et al, 2007; Kanno et al, 2007; Macrae et al, 2008; Rulten et al, 2011), both of which are core components of NHEJ, and overexpression of XRCC4-Lig4 circumvents the requirement for APLF for normal rates of DSB repair following γ irradiation (Rulten et al, 2011). Collectively, these observations provide compelling evidence that APLF accelerates DSB repair, most likely by promoting DNA ligation during NHEJ.
While APLF clearly influences DSB repair it is important to note that this protein is an accessory factor that accelerates this process, rather than an essential component. This is suggested by the observation that APLF depletion slows DSB repair only at early times (first 1–2 h) after γ irradiation, such that after longer periods (>4 h) the residual level of DSBs is similar irrespective of whether or not APLF is present. Consistent with this, neither APLF-depleted human cells nor APLF−/− MEFs exhibit significant hypersensitivity to DNA damage (unpublished observations). Surprisingly, APLF−/− DT40 cells are hypersensitive to ionising radiation, suggesting that APLF is required for cellular resistance to DNA damage in some cellular contexts. However, while the requirement for APLF for resistance to ionising radiation in DT40 required the Ku-binding motif, it did not require the FHA domain. It is thus likely that the hypersensitivity of APLF−/− DT40 cells to ionising radiation reflects a role for the interaction between Ku and APLF other than that involving XRCC4-Lig4 recruitment.
In contrast to its accessory nature in cells, APLF was essential for the assembly of XRCC4-Lig4 into Ku complexes on a 19-bp DNA duplex, in vitro. This apparent contradiction is resolved by the observation that XRCC4-Lig4 can assemble into Ku complexes in the absence of APLF, albeit at reduced efficiency, if a longer DNA duplex (60 bp) is employed that enables direct DNA binding by XRCC4-Lig4 (this work and Nick McElhinny et al, 2000). Collectively, these results suggest that DNA binding by XRCC4-Lig4 can enable incorporation of the ligase into Ku complexes, presumably in conjunction with the established interaction between XRCC4-Lig4 and Ku (Nick McElhinny et al, 2000), but that this incorporation is promoted or stabilised by the interaction of APLF with both Ku and XRCC4-Lig4. In addition to XRCC4-Lig4, APLF also promoted the incorporation of XLF into Ku-DNA complexes, resulting in assembly of a complex putatively containing six polypeptides. To our knowledge, this is the first description of such a complex, and suggests that APLF promotes the assembly of a DNA ligase ‘holocomplex’, during NHEJ.
Our finding that APLF interacts with the vWA domain of Ku80 is intriguing, because the role of this domain has remained elusive in human cells. To our knowledge, APLF is the first identified partner of this domain in human Ku80, thereby identifying a molecular function. Another notable finding is that APLF binds Ku80 via a conserved motif of 10–15 amino acids (spanning residues 177–193), and that this motif can function as an autonomous module that can target GST fusion proteins into DNA complexes containing Ku. Interestingly, the Ku80-binding motif is also present at the C-termini of XLF and WRN; two additional NHEJ proteins that bind Ku (Li and Comai, 2000, 2001; Karmakar et al, 2002; Yano et al, 2008, 2011). The C-terminus of WRN contains two such motifs, only one of which (spanning residues 1403–1418) bound Ku-DNA complexes in EMSA. Of the two WRN motifs, WRN1403–1418 is the most similar to the APLF motif and contains the highly conserved tryptophan (W1414 in WRN). The XLF motif (XLF287–299) lacks this residue but nevertheless still binds Ku-DNA complexes, albeit with lower affinity than that of either APLF177–193 or WRN1403–1418. Despite this apparent lower affinity, deletion of C-terminal amino acids encoding the Ku-binding motif prevents XLF accumulation at sites of laser-induced chromosome damage, suggesting that the putative Ku-binding motif is important for XLF recruitment/retention in cells (Yano et al, 2011). Given that both APLF and XLF bound a single Ku heterodimer in our EMSA experiments it seems unlikely that both proteins interact with the same region of Ku80. Perhaps the putative Ku-binding motif in XLF interacts with a different region of Ku80, or with the vWA domain in Ku70.
Both the Ku-binding motif and FHA domain were required for APLF to promote the assembly and/or stability of Ku complexes, consistent with APLF operating as a molecular scaffold protein. Our data support a model in which Ku80 recruits APLF into NHEJ complexes, which in turn promotes the recruitment/retention of XLF and XRCC4-Lig4. While it is clear that the FHA domain is critical for APLF-mediated recruitment/retention of XRCC4-Lig4 into Ku complexes, it is not clear how APLF promotes recruitment/retention of XLF. Perhaps, APLF interacts directly with XLF or, alternatively, exerts an allosteric impact on the interaction between XLF and Ku. Consistent with our EMSAs, APLF also stimulated the accumulation and/or retention of XLF and XRCC4-Lig4 at sites of chromosome damage. Moreover, this role required an intact Ku-binding motif in APLF, consistent with it reflecting the role of APLF in promoting the assembly/stability of NHEJ complexes. Notably, APLF also stimulated the enzymatic activity of XRCC4-Lig4 in vitro and this again was greatly reduced by mutation of the Ku-binding motif, and was ablated by additional mutation of the FHA domain. Interestingly, mutating only the FHA domain reduced XRCC4-Lig4 activity only slightly, despite the severe impact of this mutation on APLF-mediated assembly of NHEJ complexes and APLF-mediated acceleration of NHEJ. The FHA domain-mediated interaction with XRCC4 is thus more important for DNA ligase activity in a cellular context; a notion consistent with our observation that depletion of APLF results in reduced concentrations of XRCC4 in chromatin (Rulten et al, 2011). It is worth noting that the acidic C-terminal tail of APLF is also likely to be important for NHEJ in chromatin, since this domain interacts with histone H3/H4 and possesses histone chaperone activity in vitro (Mehrotra et al, 2011). It is also worth noting that APLF has been reported to possess nuclease activity, raising the possibility that this protein also functions during the end-processing step of NHEJ, upstream of DNA ligation (Kanno et al, 2007; Li et al, 2011). However, as discussed above, overexpression of XRCC4-Lig4 complements the defect in DSB repair in APLF-depleted cells, suggesting that APLF accelerates NHEJ largely or solely by promoting the DNA ligase step, at γ ray-induced DSBs at least (Rulten et al, 2011).
Based on these observations, we propose the following model for APLF function (Figure 8). We suggest that APLF is initially recruited to chromosomal DSBs in part via interaction of its C-terminal tandem zinc finger (PBZ) domain with poly (ADP-ribose) (Ahel et al, 2008; Rulten et al, 2008; Eustermann et al, 2010; Li et al, 2010). The ribosylated target of the APLF PBZ domain has not yet been identified, but is most likely auto-ribosylated poly (ADP-ribose) polymerase or a trans-ribosylated component of chromatin (Rulten et al, 2008; 2011, 36). It is worth noting that even in some organisms that lack APLF, such as Dictyostelium discoideum, for example, NHEJ components such as Ku can contain their own PBZ domain, suggesting that the use of poly (ADP-ribose) to stimulate NHEJ is evolutionarily widespread (Iles et al, 2007; Couto et al, 2011). We suggest that APLF is then incorporated and retained in Ku complexes via interactions with Ku80, and to a lesser extent XRCC4, and in turn then promotes the recruitment and/or retention of XLF and XRCC4-Lig4. We propose that while XLF and XRCC4-Lig4 still bind Ku in the absence of APLF they do so less efficiently, resulting in reduced retention time at chromosomal DSBs and reduced rates of DNA ligation. The role of APLF interaction with Ku80 and XRCC4-Lig4 in stabilising assembly of multi-protein complexes is in agreement with recent models of NHEJ, in which interactions between Ku, XLF, and XRCC4-Lig4 are proposed to fulfill a similar function (Wu et al, 2007; Yano and Chen, 2008; Yano et al, 2008). Our data demonstrate that APLF is a novel component in this respect; promoting the assembly of Ku complexes containing both XLF and XRCC4-Lig4 and thus all of the components required for DNA ligation.
Figure 8.
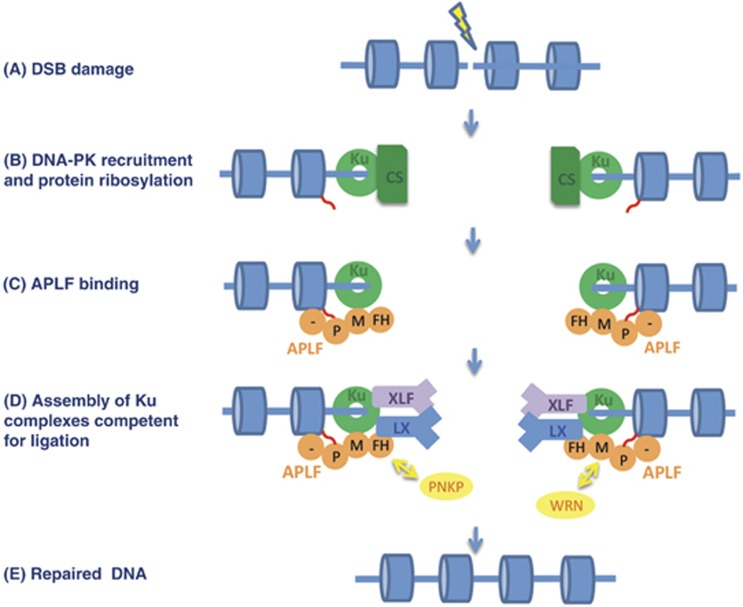
A model for APLF scaffold activity during NHEJ. (A) DSB induction. (B) Ku heterodimer binds the DSB and recruits DNA-PKcs, and PARP3 ribosylates (red wavy lines) nucleosomes (blue cylinders) near to the break. (C) APLF (orange) binds ribosylated nucleosomes via its C-terminal poly (ADP-ribose)-binding domain (PBZ domain; ‘P’) and histone-binding acidic tail, and binds Ku heterodimer via its Ku80-binding MID domain (‘M’). (D) Binding and retention of the DNA ligase co-factors XLF and XRCC4-Lig4 is promoted by their interaction with both Ku and APLF, forming a ‘holocomplex’ competent for ligation. We suggest that this complex is highly dynamic, allowing exchange between different NHEJ proteins and individual APLF domains as-and-when required. For example, APLF might be replaced by PNKP during DNA end processing, should the latter detect the presence of its DNA substrate, via interchange between their respective XRCC4-binding FHA domains. Similarly, APLF may exchange with WRN, via interchange between their respective Ku80-binding motifs. (E) Following end processing (and gap filling if required), XLF and XRCC4-Lig4 ligate the DSB.
Materials and methods
Constructs
A full description and table of constructs is included in Supplementary data.
Cell lines
Human A549 and DT40 cells were maintained as described (Rulten et al, 2011; Zeng et al, 2012).
Recombinant proteins
Recombinant His-APLF proteins were expressed from appropriate pET16b constructs in BL21(DE3) and purified by metal chelate affinity chromatography (IMAC) and gel filtration, as described previously (Iles et al, 2007; Rulten et al, 2008). Baculoviral co-expression of full length his-tagged Ku70 and Ku80 and co-expression of full-length XRCC4 and His-Lig4 were as previously described (Riballo et al, 2001; Arosio et al, 2002). Ku heterodimer (Ku70/80) and XRCC4-Lig4 were purified by IMAC followed by gel filtration in 20 mM Tris–HCl pH 7.5, 0.3 M NaCl, 5% glycerol, 1 mM DTT. Ku heterodimers composed of full-length Ku70 and wild-type or mutant (L68R) Ku80 with a C-terminal deletion (deleted amino acids 591–732) were expressed from pFBDM-His-Ku70/Ku80ΔC or pFBDM-His-Ku70/Ku80ΔCL68R using a baculovirus system and purified by IMAC and gel filtration (described in Supplementary data). GST-XLF was expressed in BL21(DE3) from pGEX3-XLF and XLF released from glutathione beads by digestion with thrombin (Hentges et al, 2006; Riballo et al, 2009). Alternatively, where indicated, non-digested GST-XLF (or other GST-fusions) was eluted with 20 mM glutathione. Note that SDS–PAGE gels of the proteins used in this work are shown in Supplementary Figure 5.
Antibodies
The antibodies employed in this study were anti-Xrcc1 (mAb 33-2-5), anti-Lig4 polyclonal (Abcam; Ab80514), anti-Ku80 Mab (Ab80592; Abcam), anti-APLF polyclonal (SK3595; Iles et al, 2007), anti-histone H1.2 polyclonal (Ab17677, Abcam), anti-CENPF (Abcam, ab5), anti-phospho-γH2AX (Ser 139, clone JBW301; Millipore) and anti-myc (Cell Signaling, 9B11).
Yeast 2-hybrid screen and analysis
For library screening, competent cells were generated from a single Saccharomyces cerevisiae Y190 clone harbouring pGBKT7-APLF (full length) and transformed with a pACT human cDNA library (a kind gift from S Elledge/J Diffley). Transformants were selected for histidine prototrophy (to detect activation of the His3 reporter gene) on minimal medium plates supplemented with 40 μg/ml adenine and 50 mM 3-amino triazole (3-AT), and screened for β-galactosidase expression by colony lift assays (to detect activation of the LacZ reporter gene) (Iles et al, 2007). For interaction analysis, Y190 cells were co-transformed with the indicated plasmids and screened for activation of the His3 and LacZ reporter genes by growth on media lacking histidine and containing 3-AT and by β-galactosidase filter lift assays, respectively.
Slot-blots
1 μg aliquots of APLF were applied to nitrocellulose membrane by vacuum and the membrane then cut into strips. One strip was stained with Amido black (Sigma) as a loading control and the others were blocked in non-fat dried milk (5% in PBS) for 2 h. The blocked strips were washed in binding buffer (BB; 20 mM Tris–HCl pH 7.5, 100 mM NaCl) (3 times, 5 min) and incubated with 100 nM of the indicated proteins in BB containing 5% non-fat dried milk (MBB) for 30 min at room temperature. Filters were rinsed three times for 5 min in BB, and incubated in MBB containing anti-XRCC1, anti-Lig4, or anti-Ku80 antibody as indicated, followed by an appropriate secondary antibody-HRP conjugate (DAKO).
Fluorescence polarisation
Fluorescein-labelled peptides at a concentration of 100 nM were incubated at room temperature, for 10 min with increasing concentrations of Ku70/80ΔC in 20 mM HEPES pH 7.5, 200 mM NaCl, and 0.5 mM TCEP. The samples (50 μl) were then transferred to a black 96-well polypropylene plate for measurement of fluorescence polarisation in a POLARstar Omega microplate reader (BMG Labtech GmbH, Ortenberg, Germany). Fifty flashes were recorded for each well with an excitation wavelength of 485 nm, and simultaneous detection of emission at 520 nm with parallel and perpendicular polarisers in-line. Background fluorescence in wells containing only buffer was subtracted from all values obtained for the samples. Polarisation data were analysed using GraphPad Prism 5.0 by non-linear fitting with a one-site total binding model. All data represent the mean of three separate experiments, and error bars represent one standard deviation.
Electrophoretic mobility shift assays
5′ Cy3-labelled oligonucleotides (Cy3-rDNAF, Cy3-30F, and cy3-60F) were annealed to their complementary oligonucleotides (19B, 30B, and 60B) to generate 19, 30, and 60 bp blunt-ended duplex substrates, respectively (Supplementary Table 3). The indicated recombinant proteins were incubated at the concentrations indicated in 10 μl binding buffer (20 mM Tris–HCl pH 7.5, 50 mM NaCl, 0.5% glycerol, 0.1 mg/ml BSA) containing 10 nM Cy3-labelled DNA substrate for 15 min at room temperature. Glycerol was then added to 5% total volume and the mixtures fractionated on 4.5% native PAGE gels (37:1 acrylamide:bis-acrylamide) in 0.4 × TBE at room temperature at 10 mA or less. Gels were scanned on a Fujifilm FLA-500 instrument using a 532-nm laser and Cy3 filter.
Ligation assay
To generate the 247-bp blunt-ended duplex for DNA ligation assays, rDNA (a kind gift from Jessica Downs, Sussex University) was employed as a template in PCRs using the Cy3-labelled rDNAF primer (see above), a 5′ phosphorylated reverse primer (rDNARP, 5′-GACAAGCGTGTCAGTACCTATCT-3′), and KOD polymerase (Merck). The Cy3-labelled 247-bp product was then purified by gel extraction (4.5% PAGE). The 247-bp DNA substrate (10 nM) was incubated in ligation buffer (50 mM triethanolamine pH 7.5, 2 mM Mg-acetate, 2 mM DTT, 0.1 mg/ml BSA, 12% polyethylene glycol MW 6000) (Kysela et al, 2003) in the absence or presence of 20 nM Ku, 150 nM wild-type or mutant APLF, or 150 nM XLF as indicated. After equilibration at 30°C for 5 min, ligation reactions were initiated by the addition of 10 nM XRCC4-Lig4. Reactions were stopped when indicated with 50 mM EDTA, 1% SDS, and 2 mg/ml Proteinase K. Following digestion for 1 h at 37°C, reactions were fractionated on 4.5% polyacrylamide gels and reaction products detected by scanning as in EMSA. The percentage of substrate converted to the ligation product was quantified using ImageQuant TL (GE).
Laser micro-irradiation
Wild-type A549 cells, or A549 cells stably transfected with either empty pSUPER or pSUPER-APLF, were subjected to UVA laser micro-irradiation as previously described (Iles et al, 2007; Rulten et al, 2011). A UVA dose of 4.36 J/m2 was used unless otherwise stated. For complementation studies, stable APLF knockdown cells were co-transfected with equal amounts of pEGFP-XRCC4 or pEGFP-XLF and pmRFP-C1, shRNA-resistant pmRFP-C1-APLF or site-directed mutants thereof.
RNA interference and γH2AX assays
Transient APLF knockdown and complementation was carried out in A549 cells as described previously (Rulten et al, 2011) using pSUPER-APLF (Iles et al, 2007) and either pcD2E or shRNA targeting-resistant versions of pcD2E-APLFWT, pcD2E-APLFW189G, or pcD2E-APLFR27A at a ratio of 1 μg pSUPER-APLF:2 μg pcD2E construct. Transfected cells were γ irradiated (2 Gy), allowed to recover for the indicated repair periods and immunolabelled as previously described (Rulten et al, 2011). γH2AX foci were counted in CENPF-negative (G1) cells only.
Generation of APLF mutant DT40 cells
The description of the generation of APLF−/− DT40 cells is described in Supplementary data.
DT40 survival assays
To measure survival, DT40 cells were treated where indicated with γ irradiation (in complete medium) or UVC (in PBS) and plated in medium containing 1.5% methylcellulose as described previously (Zeng et al, 2012). Where indicated, media contained the indicated concentration of methyl methanesulfonate (MMS), cisplatin, or etoposide, throughout the growth period. Cells were incubated for 7–11 days and visible colonies counted.
Plasmid religation assay
5 μg NheI-linearised plasmid pEGFP-C1 was co-transfected with 5 μg circular pRFP-C1 into 5 × 106 wild-type (clone 18) or APLF−/− DT40 cells by nucleofection using Amaxa (Lonza). Eighteen hours later, cells were harvested and analysed by FACS Canto (BD Biosciences). RFP-positive cells were counted, and plasmid religation efficiency was calculated as the percentage of GFP-positive cells within the RFP-positive population.
Supplementary Material
Acknowledgments
This work was funded by MRC and CR-UK grants to KWC. We thank Penny Jeggo and Aidan Doherty for provision of expression plasmids and baculovirus.
Author contributions: GG, SR, and RAB/AO designed, conducted, and interpreted the biochemical, cellular, and fluorescence polarisation experiments, respectively. GG and RAB expressed and purified recombinant proteins. NI conducted the yeast-2-hybrid library screen and KM carried out initial 2-hybrid analyses. ZZ carried out DT40 knockout work. KWC designed and interpreted experiments, coordinated the project, and wrote the manuscript. All authors edited the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, West SC (2008) Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451: 81–85 [DOI] [PubMed] [Google Scholar]
- Ahnesorg P, Smith P, Jackson SP (2006) XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124: 301–313 [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV (2001) Prokaryotic homologs of the eukaryotic DNA-end-binding protein Ku, novel domains in the Ku protein and prediction of a prokaryotic double-strand break repair system. Genome Res 11: 1365–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio D, Cui S, Ortega C, Chovanec M, Di Marco S, Baldini G, Falaschi A, Vindigni A (2002) Studies on the mode of Ku interaction with DNA. J Biol Chem 277: 9741–9748 [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Fugger K, Danielsen JR, Gromova I, Sehested M, Celis J, Bartek J, Lukas J, Mailand N (2007) Human Xip1 (C2orf13) is a novel regulator of cellular responses to DNA strand breaks. J Biol Chem 282: 19638–19643 [DOI] [PubMed] [Google Scholar]
- Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche M-C, Sanal O, Plebani A, Stéphan J-L, Hufnagel M, le Deist F, Fischer A, Durandy A, de Villartay J-P, Revy P (2006) Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124: 287–299 [DOI] [PubMed] [Google Scholar]
- Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA (2000) Ku complex interacts with and stimulates the Werner protein. Genes Dev 14: 907–912 [PMC free article] [PubMed] [Google Scholar]
- Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A (2007) Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst) 6: 712–722 [DOI] [PubMed] [Google Scholar]
- Couto CA-M, Wang H-Y, Green JCA, Kiely R, Siddaway R, Borer C, Pears CJ, Lakin ND (2011) PARP regulates nonhomologous end joining through retention of Ku at double-strand breaks. J Cell Biol 194(3): 367–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustermann S, Brockmann C, Mehrotra PV, Yang J-C, Loakes D, West SC, Ahel I, Neuhaus D (2010) Solution structures of the two PBZ domains from human APLF and their interaction with poly(ADP-ribose). Nat Struct Mol Biol 17: 241–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Härer C, Marchetti C, Morrice N, Jeggo PA, Lees-Miller SP (2006) DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J 25: 3880–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lu H, Tsai AG, Schwarz K, Lieber MR (2007) Single-stranded DNA ligation and XLF-stimulated incompatible DNA end ligation by the XRCC4-DNA ligase IV complex: influence of terminal DNA sequence. Nucleic Acids Res 35: 5755–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges P, Ahnesorg P, Pitcher RS, Bruce CK, Kysela B, Green AJ, Bianchi J, Wilson TE, Jackson SP, Doherty AJ (2006) Evolutionary and functional conservation of the DNA non-homologous end-joining protein, XLF/Cernunnos. J Biol Chem 281: 37517–37526 [DOI] [PubMed] [Google Scholar]
- Hsu HL, Yannone SM, Chen DJ (2002) Defining interactions between DNA-PK and ligase IV/XRCC4. DNA Repair (Amst) 1: 225–235 [DOI] [PubMed] [Google Scholar]
- Iles N, Rulten S, El-Khamisy SF, Caldecott KW (2007) APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol Cell Biol 27: 3793–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno S-I, Kuzuoka H, Sasao S, Hong Z, Lan L, Nakajima S, Yasui A (2007) A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J 26: 2094–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar P, Snowden CM, Ramsden DA, Bohr VA (2002) Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res 30: 3583–3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kysela B, Doherty AJ, Chovanec M, Stiff T, Ameer-Beg SM, Vojnovic B, Girard P-M, Jeggo PA (2003) Ku stimulation of DNA ligase IV-dependent ligation requires inward movement along the DNA molecule. J Biol Chem 278: 22466–22474 [DOI] [PubMed] [Google Scholar]
- Li B, Comai L (2000) Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J Biol Chem 275: 28349–28352 [DOI] [PubMed] [Google Scholar]
- Li B, Comai L (2001) Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-Ku70/80 complex. J Biol Chem 276: 9896–9902 [DOI] [PubMed] [Google Scholar]
- Li G-Y, McCulloch RD, Fenton AL, Cheung M, Meng L, Ikura M, Koch CA (2010) Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc Natl Acad Sci USA 107: 9129–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kanno S-I, Watanabe R, Ogiwara H, Kohno T, Watanabe G, Yasui A, Lieber MR (2011) Polynucleotide kinase and aprataxin-like forkhead-associated protein (PALF) acts as both a single-stranded DNA endonuclease and a single-stranded DNA 3' exonuclease and can participate in DNA end joining in a biochemical system. J Biol Chem 286: 36368–36377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79: 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löbrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA (2010) gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle 9: 662–669 [DOI] [PubMed] [Google Scholar]
- Lu H, Pannicke U, Schwarz K, Lieber MR (2007) Length-dependent binding of human XLF to DNA and stimulation of XRCC4.DNA ligase IV activity. J Biol Chem 282: 11155–11162 [DOI] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, Lieber MR (2002) Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108: 781–794 [DOI] [PubMed] [Google Scholar]
- Macrae CJ, McCulloch RD, Ylanko J, Durocher D, Koch CA (2008) APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair (Amst) 7: 292–302 [DOI] [PubMed] [Google Scholar]
- Mahaney BL, Meek K, Lees-Miller SP (2009) Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J 417: 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ, Caldecott KW (2007) DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet 8: 37–55 [DOI] [PubMed] [Google Scholar]
- Mehrotra PV, Ahel D, Ryan DP, Weston R, Wiechens N, Kraehenbuehl R, Owen-Hughes T, Ahel I (2011) DNA repair factor APLF is a histone chaperone. Mol Cell 41: 46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA (2000) Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol Cell Biol 20: 2996–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riballo E, Doherty AJ, Dai Y, Stiff T, Oettinger MA, Jeggo PA, Kysela B (2001) Cellular and biochemical impact of a mutation in DNA ligase IV conferring clinical radiosensitivity. J Biol Chem 276: 31124–31132 [DOI] [PubMed] [Google Scholar]
- Riballo E, Woodbine L, Stiff T, Walker SA, Goodarzi AA, Jeggo PA (2009) XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Res 37: 482–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Cortes Ledesma F, Guo L, Iles NJ, Caldecott KW (2008) APLF (C2orf13) is a novel component of poly(ADP-ribose) signaling in mammalian cells. Mol Cell Biol 28: 4620–4628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Fisher AEO, Robert I, Zuma MC, Rouleau M, Ju L, Poirier G, Reina-San-Martin B, Caldecott KW (2011) PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell 41: 33–45 [DOI] [PubMed] [Google Scholar]
- Tsai CJ, Kim SA, Chu G (2007) Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc Natl Acad Sci USA 104: 7851–7856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412: 607–614 [DOI] [PubMed] [Google Scholar]
- Wu P-Y, Frit P, Malivert L, Revy P, Biard D, Salles B, Calsou P (2007) Interplay between Cernunnos-XLF and nonhomologous end-joining proteins at DNA ends in the cell. J Biol Chem 282: 31937–31943 [DOI] [PubMed] [Google Scholar]
- Yano K-I, Chen DJ (2008) Live cell imaging of XLF and XRCC4 reveals a novel view of protein assembly in the non-homologous end-joining pathway. Cell Cycle 7: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Yano K-I, Morotomi-Yano K, Lee K-J, Chen DJ (2011) Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF. FEBS Lett 585: 841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano K-I, Morotomi-Yano K, Wang S-Y, Uematsu N, Lee K-J, Asaithamby A, Weterings E, Chen DJ (2008) Ku recruits XLF to DNA double-strand breaks. EMBO Rep 9: 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Sharma A, Ju L, Murai J, Umans L, Vermeire L, Pommier Y, Takeda S, Huylebroeck D, Caldecott KW, El-Khamisy SF (2012) TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Res 40(17): 8371–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.