

Abstract
Infection of macrophages by bacterial pathogens can trigger Toll-like receptor (TLR) activation as well as Nod-like receptors (NLRs) leading to inflammasome formation and cell death dependent on caspase-1 (pyroptosis). Complicating the study of inflammasome activation is priming. Here, we develop a priming-free NLRC4 inflammasome activation system to address the necessity and role of priming in pyroptotic cell death and damage-associated molecular pattern (DAMP) release. We find pyroptosis is not dependent on priming and when priming is re-introduced pyroptosis is unaffected. Cells undergoing unprimed pyroptosis appear to be independent of mitochondrial involvement and do not produce inflammatory cytokines, nitrous oxide (NO), or reactive oxygen species (ROS). Nevertheless, they undergo an explosive cell death releasing a chemotactic isoform of the DAMP high mobility group protein box 1 (HMGB1). Importantly, priming through surface TLRs but not endosomal TLRs during pyroptosis leads to the release of a new TLR4-agonist cysteine redox isoform of HMGB1. These results show that pyroptosis is dominant to priming signals and indicates that metabolic changes triggered by priming can affect how cell death is perceived by the immune system.
Keywords: HMGB1, inflammasome, Nod-like receptor C4, pyroptosis, Toll-like receptor
Introduction
Activation of innate immune receptors in cells of monocytic lineage promotes anti-microbial defenses. Macrophages in particular are suited to recognize the microbe-associated molecular pattern molecules through germ-line expression of pattern recognition receptors (PRRs). Two primary families of PRRs are the Toll-like receptors (TLRs) and Nod-like receptors (NLRs), which are key in activating innate immune responses to protect the host from infection.
TLRs and NLRs are thought to activate distinct signalling pathways which promote diverse outcomes defending against infection. Activation of TLRs triggers the production of anti-microbial effector molecules nitrous oxide (NO) and mitochondria-dependent reactive oxygen species (mROS) (Kawai and Akira, 2011; West et al, 2011). TLRs also activate the transcription factors NF-κB and IRF3 to induce inflammatory cytokines, the inactive pro-forms of inflammatory cytokines such as IL-1β, and type I interferons and chemokines (Kawai et al, 2001; Kawai and Akira, 2011). Despite this arsenal of anti-microbial effectors, it is often observed that bacterial pathogens activate macrophage cell death (Ashida et al, 2011). Bacterial virulence factors can trigger cell death upon infection and some of these have now been found to be NLR agonists. For example, the mouse NLR Naip5 directly interacts with bacterial flagellin and forms a complex with NLRC4 (Kofoed and Vance, 2011; Zhao et al, 2011). This detection initiates the formation of a multi-protein complex called an inflammasome able to promote caspase-1 and caspase-11 catalytic activity (Schroder and Tschopp, 2010; Akhter et al, 2012). Flagellin activates the NLRC4 inflammasome by the canonical pathway leading to regulated cell death called pyroptosis (defined by a requirement for an ‘activated’ but unprocessed procaspase-1) independently of caspase-11 (Broz et al, 2010; Kayagaki et al, 2011; Motani et al, 2011; Galluzzi et al, 2012). NLRC4 inflammasome activation also induces lysosome exocytosis, phagosome-lysozome fusion, as well as the processing of the pro-form of the inflammatory cytokines IL-1β and IL-18 by a catalytic caspase-1 (Broz et al, 2010; Bergsbaken et al, 2011; Motani et al, 2011; Akhter et al, 2012). In vivo NLRC4 inflammasome activation, and in some cases pyroptosis, leads to anti-microbial activity (Miao et al, 2011) as well as sepsis associated with multi-antibiotic-resistant commensal pathogens in a damaged gut (Ayres et al, 2012).
Confounding the uninfluenced study of inflammasome activation and associated cell death is the presence of ‘signal-1’ or priming. The NLRC4 inflammasome, assisted by Naip5, is activated by the carboxy-terminal tail of flagellin produced by a variety of enteric pathogens (Lightfield et al, 2008, 2011). However, infection of macrophages with flagellated bacteria activates multiple innate immune receptors as well as allows for modulation of NF-κB, inflammasome, and mitochondrial activity via microbe-derived factors (Ashida et al, 2011). This makes the use of infection models unsuited for the study of uninfluenced NLRC4/Naip5 activation. Transduction of macrophages with retroviral vectors expressing flagellin or flagellin COOH tails activates NLRC4/Naip5-dependent pyroptosis (Lightfield et al, 2008; Miao et al, 2010b; Lightfield et al, 2011; Ayres et al, 2012). However, these techniques deliver viral dsRNA activating the viral infection sensor Tank Binding Kinase 1, a key signalling adaptor downstream of RIG-I-like receptors (Sutlu et al, 2012). Transfection of recombinant flagellin protein using cationic lipids activates the primed NLRC4/Naip5 inflammasome and IL-1β processing (Miao et al, 2006; Buzzo et al, 2010; Miao et al, 2010b; Bauernfeind et al, 2011; Lightfield et al, 2011; Zhao et al, 2011) and release of IL-18 without overt priming (Bauernfeind et al, 2011). However, cationic lipids induce autophagy (Man et al, 2010) that affects pro-IL-1β levels, processing, and release (Dupont et al, 2011; Harris et al, 2011). Direct interactions observed between the central autophagy regulator Beclin1 and NLRC4 also suggest that autophagy influences NLRC4/Naip5 responses (Jounai et al, 2011). For these reasons, it is not known if pyroptosis is dependent on, or is affected by, priming and/or activation of other signalling pathways. Furthermore, it is also unknown if NLRC4/Naip5 inflammasome activation leads to the de novo production of NO, ROS, or inflammatory cytokines.
Pyroptosis is associated with anti-microbial activity (Miao et al, 2010a), pathology (Kovarova et al, 2012), and coincides with the release of damage-associated molecular patterns (DAMPs). The DAMP high mobility group protein box 1 (HMGB1) is a highly conserved nuclear protein capable of promoting both sterile and infectious inflammation by numerous mechanisms after release from activated or dying cells (Andersson and Tracey, 2011). HMGB1 contains three cysteine residues (Cys23, Cys45, and Cys106) and the degree of reduction and oxidation (redox), and/or sulphonation of these residues directly affects its structure-to-function relationship. These well-defined, mutually exclusive forms exhibit either chemotactic activity with Cxcl12 (completely reduced ‘all-thiol’), TLR4 agonist activity (oxidized at Cys23-Cys46, reduced Cys106), or an apparently ‘inactive’ form (sulphonated at Cys106) (Yang et al, 2010, 2012; Schiraldi et al, 2012; Venereau et al, 2012). These structural isoforms have a direct relationship to in vivo inflammation where the all thiol form exhibits chemotactic activity and the in vivo oxidation of injected HMGB1 reduces this activity (Venereau et al, 2012). However, it is not known if changes in HMGB1 cysteine redox isoforms are only passive processes in response to environmental oxidation or if specific cellular pathways can effect these changes. The role of cysteine redox isoforms in other functions attributed to HMGB1 is unknown.
Various HMGB1 cysteine redox isoforms have been studied during apoptotic events associated with irradiation (Kazama et al, 2008). Apoptosis generally involves mitochondrial dysfunction and manipulation of mitochondrial ROS production affects HMGB1 cysteine isoforms (Kazama et al, 2008). However, compared to apoptosis, it is not known if pyroptosis involves the mitochondria or ROS production. Priming inflammasomes with LPS induces mitochondrial ROS but it is unknown if these non-lethal changes affect changes in HMGB1 isoforms or if fulminant cell death could suffice.
The release of inflammatory molecules during cell death is likely an important factor in pathology associated with inflammasome activation. Priming, however, complicates our understanding of what responses are specific to inflammasome activation or to the priming event. Understanding these dynamics will help clarify the roles these factors play in the outcomes of cell death and pyroptotic-associated pathology. Here, we develop a priming-free inflammasome activation system to address questions regarding the necessity and role of priming in inflammasome activation, pyroptosis, and DAMP production.
Results
NLRC4/Naip5 agonist expression induces rapid cell death without priming
NLRC4 gene expression is independent of priming (Bauernfeind et al, 2011). To test if NLRC4/Naip5 inflammasome activation is independent of priming, we developed a single-construct retroviral Tet-ON vector to express the C-terminal 34 or 19 amino acids of S. typhimurium flagellin (FliC) fused to Enhanced GFP (EGFP) in macrophages (Figure 1A). Constructs expressing this NLRC4/Naip5 agonist (EGFP-C34) and control (EGFP-C19) were stably transduced into immortalized mouse BMDMs (B10R) expressing macrophage surface markers (Supplementary Figure 1). B10R and inducible-retroviral transductants (called C34 or C19) were tested for the expression of transcripts of NLRC4/Naip5 inflammasome members, associated molecules, and NF-κB and IRF responsive genes. B10R, C34, and C19 cells expressed NLRC4, Naip5, caspase-1, PKR, and ASC, but not the NF-κB-responsive gene IL-1β or the IRF-responsive gene Cxcl10 demonstrating a lack of active TLR and anti-viral signalling (Figure 1B). C34 and C19 cells were then treated with Doxacyclin (Dox) and monitored for inducible gene expression (EGFP signal) as well as cell viability. We observed a rapid increase in fluorescence in C34 and C19 cells, however, only C34 cells underwent cell death (Figure 1C and D). Cell death, as determined by propidium iodide (PI) staining, occurred concurrently with LDH release indicating that PI staining was not likely a function of pore formation in viable cells but stained truly non-viable cells (Figure 1D and E).
Figure 1.
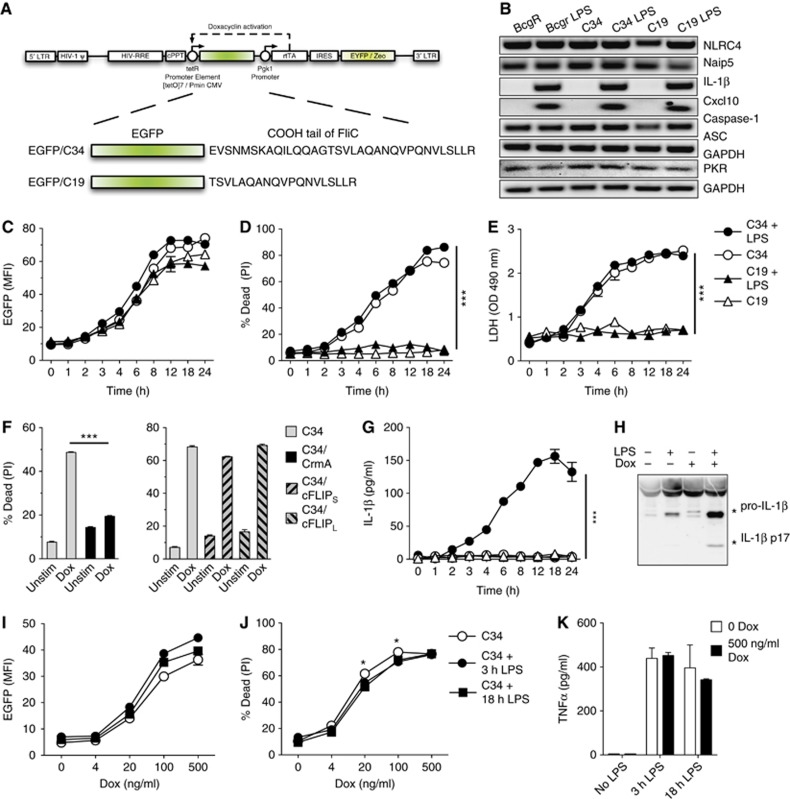
Macrophage pyroptosis by NLRC4/Naip5 agonists expression is priming independent and is largely unaffected by priming. (A) Self-inactivating retroviral vector driving expression of the EGFP-C34 or EGFP-C19 fusions from the tetracycline-regulated promoter (tetR) by the reverse tetracycline-controlled transactivator rtTA2S-M2 (rtTA) by Dox. See Supplementary data for a detailed description of domains. (B) Expression of gene transcripts by B10R, C34, and C19 cells key to NLRC4/Naip5 inflammasome activation and representative of NF-κB, and IRF activation. Transcript expression after 3 h LPS treatment (100 ng/ml) is shown. (C, D) Kinetic analysis of induced C34 and C19 EGFP fusion proteins (C) and cell death (D) determined by flow cytometry±LPS priming (100 ng/ml, 3 h) followed by±Dox (500 ng/ml). (E) Kinetic analysis of cell death in C34 and C19 cells determined by LDH release±LPS priming (100 ng/ml, 3 h) followed by±Dox (500 ng/ml). (F) Determination of cell death by flow cytometry in C34, C34/CrmA, C34/cFLIPS, or C34/cFLIPL cells±Dox (500 ng/ml) for 2 h. (G) Kinetic analysis of IL-1β detection in the cell supernatant as detected by ELISA±LPS priming (100 ng/ml, 3 h) followed by±Dox addition (500 ng/ml) (P-value=0.0023). (H) IL-1β processing in supernatant determined by immunoblot in C34 cells±LPS priming (100 ng/ml, 3 h)±Dox (500 ng/ml) 8 h post Dox addition. (I–K) Induction of C34 and C19 EGFP fusion proteins (I) cell death (J) and TNFα production following±3 or 18 h LPS priming (100 ng/ml) followed by±Dox (0–500 ng/ml) determined by flow cytometry and ELISA 24 h post Dox addition. Data are mean±s.e.m. of triplicate samples representative of two (C, D, E, G), three (I, J), or four (F) independent experiments. (H) Representative of four independent experiments is shown. Significant differences are indicated *P⩽0.05, ***P⩽0.001.
Inflammasome activity and pyroptosis is dependent on caspase-1 (Schroder and Tschopp, 2010). To determine if cell death was dependent on caspase-1, we co-expressed the serpin family caspase-1 inhibitor CrmA in C34 cells. Upon Dox addition, we observed decreased cell death (Figure 1F) but no changes in EGFP expression. CrmA has also been shown to inhibit caspase-8 activity (Smith et al, 1996; Oberst et al, 2011). However, caspase-8 can be specifically inhibited by FLIP-short (cFLIPS) and FLIP-long (cFLIPL) (Weinlich et al, 2011). To determine if cell death involved caspase-8, we overexpressed cFLIPS or cFLIPL in C34 cells. Overexpression of either form of cFLIP was unable to inhibit cell death after Dox addition (Figure 1F) and did not affect EGFP expression despite robust FLIP overexpression (Supplementary Figure 3). Together, these results demonstrate that the presence of a defined inflammasome agonist promotes pyroptotic cell death in the absence of priming and manipulation of signalling pathways.
Priming allows IL-1β production but does not affect pyroptosis after NLRC4/Naip5 agonist expression
Our results indicate that expression of an NLRC4/Naip5 agonist alone induces pyroptosis. However, regulated cell death pathways can be influenced by signalling pathways such as NF-κB supplied by priming (e.g., LPS; Ashida et al, 2011). As inflammasome activation routinely occurs in the presence of priming we assessed the ability of priming to affect cell death. Treatment of B10R, C34, or C19 cells with LPS lead to no discernable changes in NLRC4, Naip5, PKR, or Caspase-1 expression but did upregulate IL-1β and Cxcl10 transcripts demonstrating functional signalling pathways (Figure 1B). However, 3 h pre-treatment with LPS prior to Dox treatment did not lead to changes in the degree of cell death (Figure 1C–E) but did lead to the expression of pro-IL-1β, which underwent processing and release upon Dox treatment in C34 cells but not in C19 cells (Figure 1G and H). In C34 cells, the detection of IL-1β coincided with the degree of death observed (Figure 1G). As the effects of LPS priming may take longer to affect pyroptosis we repeated experiments with both 3 and 18 h LPS priming. Primings were followed by induction of the system with various levels of Dox to allow for variations in NLRC4/Naip5 agonist expression. We observed only a slight trend for priming to effect pyroptosis (∼5–7% inhibition) with 3 or 18 h LPS priming at intermediate levels of agonist expression (Figure 1I and J). Responses to LPS were confirmed by TNFα production (Figure 1K). These results indicate that NLRC4/Naip5 agonist induced pyroptosis is generally refractory to TLR activation.
NLRC4/Naip5 agonist expression induces rapid ‘explosive’ pyroptosis
To understand the physical behaviour of cells undergoing unprimed pyroptosis, we subjected C34 and C19 cells to live-cell confocal microscopy. Cells stained with plasma membrane (PM) dye were imaged after Dox addition and data from PM, EGFP, YFP, and transmission (Trans) images were collected. C34 and C19 cells were observed to be slightly motile and YFP positive at the experimental outset (Figure 2A and B; Supplementary Movies 1 and 2). Induced cells gain EGFP signal that forms a slight, dispersed, punctate staining pattern in both C34 and C19 cells (Figure 2A and B). However, at various time points after induction C34 cells rapidly rupture with little noticeable change in previous behaviour. This rupture leads to an ejection of intracellular contents (Figure 2A, arrow) followed by a collapse in the size of the remaining cell body (defined by PM staining) and loss of EGFP and YFP signals (Figure 2A; Supplementary Movie 1). In contrast, Dox-treated C19 cells remained slightly motile and became progressively EGFP bright without undergoing cell death (Figure 2B; Supplementary Movie 2). Analysis of Dox-treated C34 and C19 cells for the full time course indicated that observations made with individual cells are representative of larger populations. These data demonstrate that pyroptosis is mechanistically different from classic apoptosis and directly suggests a mechanism for inflammatory cytokine and DAMP release.
Figure 2.
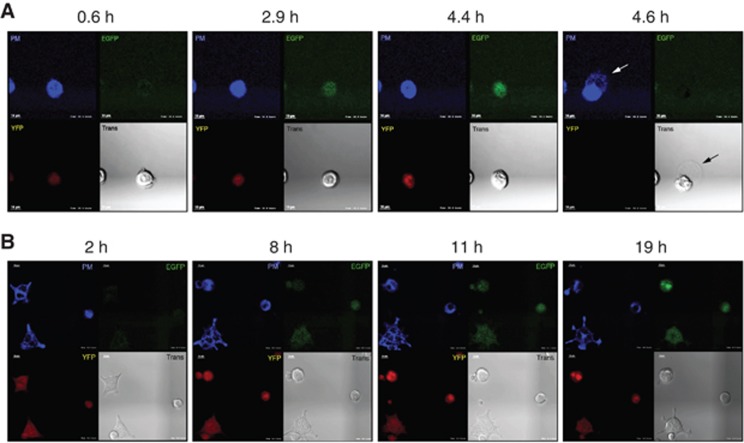
Sudden cell rupture occurs following cytoplasmic NLRC4/Naip5 agonist expression. Upper left: plasma membrane (PM) staining, upper right: EGFP signal, lower left: YFP signal, lower right: transmitted white light (Trans). (A) PM stained C34 cells gain EGFP signal with time then rupture releasing cellular contents. Arrows highlight the release of cellular contents detected by transmission microscopy and PM staining. (B) PM surface stained C19 cells gain EGFP signal with time but fail to rupture and die. Data are representative of ⩾100 independent cell events. Scale bar 10 μm.
NLRC4/Naip5 agonist expression does not lead to inflammatory cytokine expression, NO, or ROS production
Priming of inflammasome systems make it difficult to test whether inflammasome activation leads to the de novo expression of inflammatory cytokines and anti-microbial chemical production typical of innate immune receptor agonism. To study responses typical of TLR agonism during NLRC4/Naip5 activation, we activated C34 and C19 cells for 3 h with Dox or LPS. LPS activation of C34 and C19 cells resulted in the induction of inflammatory transcripts (Figure 3A). However, activation of C34 and C19 cells with Dox revealed no inflammasome-specific increases in gene transcripts for IL-6, Cxcl10, Caspase-11, IL-1β, CSF2, or TNFα despite being activated as indicated by EGFP expression (Figure 3A; Supplementary Figure 4).
Figure 3.
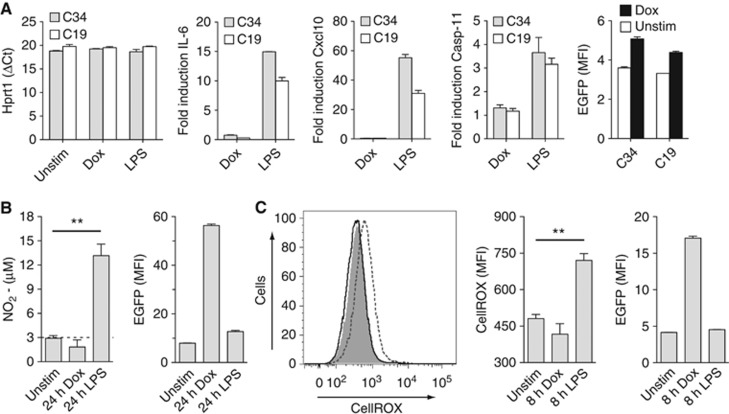
NLRC4/Naip5 agonist expression does not lead to inflammatory cytokine, NO, or ROS production. (A) mRNA and EGFP expression in Dox stimulated (500 ng/ml) or LPS stimulated (100 ng/ml) C34 or C19 cells at 3 h Hprt1 direct transcript values and Hprt1-relative expression data for IL-6, Cxcl10, and Caspse-11. (B) Nitrite levels in culture supernatants of C34 cells 24 h post Dox (500 ng/ml) or LPS (100 ng/ml) addition. (C) ROS levels in living C34 cells 8 h post Dox (500 ng/ml) or LPS (100 ng/ml) addition. First panel: representative histogram of unstimulated (grey fill), LPS-stimulated (dotted line), or Dox-stimulated (solid line) cells. Second panel: quantitative CellROX staining data. Data are mean±s.e.m. of triplicate samples representative of three (A, C) or four (B) independent experiments. Significant differences are indicated **P⩽0.01.
Expression of iNOS is induced by TLR activation leading to the production of the anti-microbial chemical NO that affects inflammatory responses. To see if NLRC4/Naip5-activated cells produce NO, we activated C34 cells with Dox or LPS for 24 h and assayed supernatants for NO (assayed as nitrite). LPS stimulated cells produced NO but activation with Dox did not despite EGFP expression (Figure 3B).
Activation of outer-membrane expressed TLRs in macrophages leads to the production of mitochondrial-derived ROS, which contributes to inflammatory and anti-microbial activity (West et al, 2011). To see if NLRC4/Naip5-activated cells produce ROS, we treated C34 cells with Dox or LPS and assayed living cells 8 h later for the production of whole-cell ROS. LPS stimulated cells produced ROS but activation with Dox did not despite EGFP expression (Figure 3C).
Together these results show that during pyroptosis, activation of the inflammasome does not lead to de novo synthesis of inflammatory cytokines or reactive chemical species associated with inflammation and anti-microbial activity.
NLRC4/Naip5 agonist expression leads to pyroptosis independent of overt mitochondrial dysfunction
Primed apoptosis leading to NLRP3 activation involves a collapse of inner mitochondrial membrane potential and ROS production (Zhou et al, 2011; Shimada et al, 2012). Our results indicating that pyroptosis occurs without ROS production led us to investigate mitochondrial responses in response to NLRP4/Naip5 activation. To do this, we overexpressed the anti-apoptotic Bcl-2-like protein Bcl-XL in C34 cells to inhibit mitochondrial outer membrane permeabilization (MOMP), which occurs during activation of the intrinsic pathway of apoptosis (Kroemer et al, 2007) and inhibits apoptosis associated with NLRP3 activation (Shimada et al, 2012). Overexpression of Bcl-XL in C34 cells inhibited etoposide-induced apoptosis but not pyroptosis (Figure 4A). Bcl-XL expression did not affect EGFP expression after Dox treatment (Figure 4B). These results indicate that standard MOMP is not a response initiated during pyroptosis.
Figure 4.
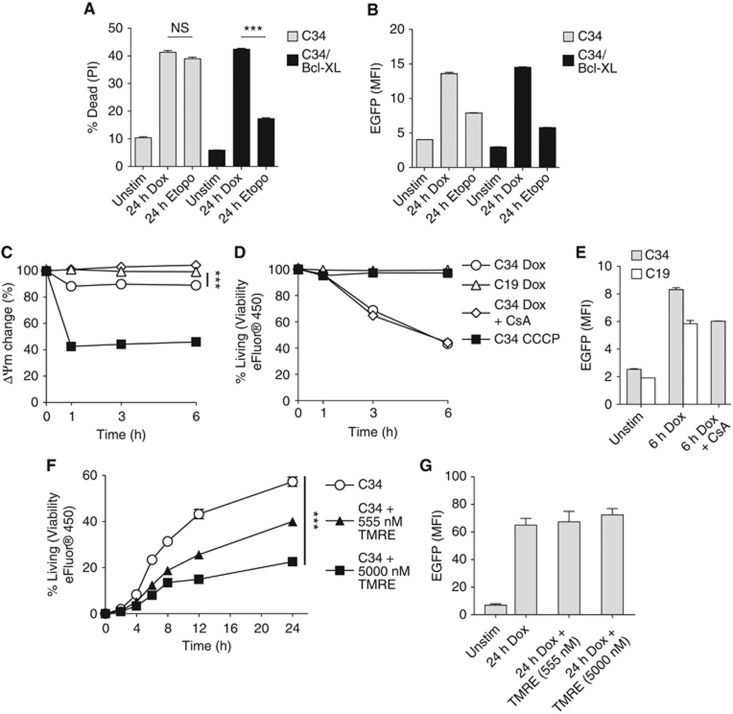
Pyroptosis due to NLRC4/Naip5 agonist expression is unaffected by Bcl-XL and independent of mitochondrial dysfunction. (A, B) Determination of cell death (A) and EGFP expression (B) by flow cytometry in C34 or C34/Bcl-XL cells±Dox (500 ng/ml)±Etoposide (50 μM) after 24 h. (C–E) Changes in ΔΨm (C) viability (D) and EGFP expression (E)±Dox treatment (500 ng/ml)±CsA (10 μM)±CCCP (3 μM) after TMRE loading in C34 or C19 cells measured by flow cytometry. (F, G) Determination of cell death (F) and EGFP expression (G) in C34 cells by flow cytometry during inhibition of state 3 mitochondrial respiration. Data are mean±s.e.m. of triplicate samples representative of three (A–E) or two (F, G) independent experiments. Significant differences are indicated ***P⩽0.001. NS, not significant.
Decreases in the inner mitochondrial transmembrane potential (ΔΨm) can occur following MOMP (Kroemer et al, 2007). Dissipation of ΔΨm leads to reductions in ATP synthesis, increased ROS production, and correlates with the mitochondria release of the NLRP3 agonist oxidized mitochondrial DNA (Shimada et al, 2012). To determine if ΔΨm changes with pyroptosis, we loaded cells with the cationic dye TMRE that accumulates in active mitochondria. Treatment of C34 cells with the mitochondrial decoupler CCCP resulted in a rapid loss of ΔΨm while treatment of cells with Dox only resulted in a slight but significant decrease (Figure 4C). This decrease was dependent on NLRC4/Naip5 agonist expression as it occurred in Dox-treated C34 but not in C19 cells. To determine if this decrease in ΔΨm was functionally correlated with pyroptosis, we treated C34 cells with the mitochondrial permeability transition pore inhibitor Cyclosporin A (CsA) followed by Dox. CsA treatment inhibited the NLRC4/Naip5 inflammasome-dependent ΔΨm decrease (Figure 4C). However, this was unable to prevent pyroptosis (Figure 4D). Neither TMRE loading nor CsA treatment affected EGFP expression after Dox treatment (Figure 4E). These observations show that ΔΨm decreases seen during pyroptosis do not correlate with cell death. This together with observations that C34 cells overexpressing Bcl-XL inhibit MOMP-associated apoptosis but have no effect on pyroptosis suggest that pyroptosis does not involve mitochondria in a manner similar to ‘type II extrinsic’ or ‘intrinsic apoptosis’ demonstrating clear phenotypic differences (Galluzzi et al, 2012).
Programmed and regulated cell death are energy-dependent processes. We reasoned if pyroptosis is a form of regulated cell death it may require a functional mitochondria. To test the energy requirement of pyroptosis, we inhibited the state three respiration step of mitochondria (ADP to ATP phosphorylization) using high-dose TMRE treatment (Scaduto and Grotyohann, 1999). Following inhibition, we treated C34 cells with Dox. Activation led to cell death while pre-treatment with high-dose TMRE inhibited death in a dose-dependent manner without affecting EGFP expression indicating that pyroptosis requires energy (Figure 4E and F). Treatment of C34 cells with the respiration inhibitors 2-deoxyglucose and Antimycin A gave similar results (Supplementary Figure 5). Together, these results indicate that although pyroptotic cell death occurs without overt mitochondrial dysfunction it is dependent on mitochondrial function.
NLRC4/Naip5 agonist expression leads to release of HMGB1
Our data above indicate that pyroptotic cells do not produce inflammatory cytokines, NO, or ROS. However, it may be that other inflammatory and/or anti-microbial factors are produced or released by activated cells. Cellular release of the prototypic DAMP HMGB1 is associated with inflammation and pathology. We analysed culture supernatants of C34 or C19 cells for the presence of the HMGB1. Dox treatment of C34 cells leads to increased HMGB1 release but not Dox-treated C19 cells or unactivated cells (Figure 5A). It has been observed that LPS stimulation of numerous cell types induces HMGB1 release (Andersson and Tracey, 2011). However, after treatment of C34 or C19 cells with highly purified LPS we observed no increase in HMGB1 release (Figure 5B). It may be, however, that LPS priming affects the amount of HMGB1 released during pyroptosis. Priming of C34 or C19 cells for 3 h prior to Dox addition did not affect the total amount of HMGB1 released even though cells responded normally to LPS addition (Figure 5B and C). These results indicate that pyroptosis and not TLR signalling activates HMGB1 release upon innate immune activation of BMDMs. These results are in line with observations using NLRP3 agonists and live bacterial infection (Lamkanfi et al, 2010; Kayagaki et al, 2011; Lu et al, 2012). Additionally, we show that priming does not affect the magnitude of HMGB1 release during pyroptosis.
Figure 5.
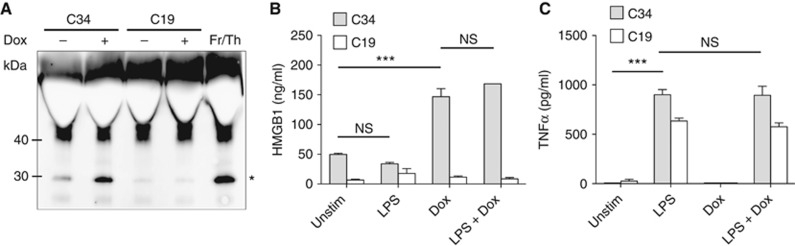
HMGB1 amounts released by pyroptosis are unaffected by priming. (A) Release of HMGB1 into supernatant by Dox-treated C34 or C19 cells determined by immunoblot. Fr/Th indicates supernatants from frozen/thawed C34 cells as positive control. *Indicates observed HMGB1 polypeptide. (B, C) Release of HMGB1 (B) and TNFα production (C) by C34 or C19 cells following±3 h LPS priming (100 ng/ml) followed by±Dox (500 ng/ml) determined by ELISA 24 h post Dox addition. (A) Representative of three experiments is shown. (B, C) Mean±s.e.m. of triplicate samples representative of three independent experiments is shown. Significant differences are indicated ***P⩽0.001. NS, not significant.
Pyroptosis activates the release of an acetylated HMGB1 isoform
LPS treatment of cells triggers the acetylation of multiple lysine residues in two nuclear localization signal (NLS) sequences in HMGB1, whereas HMGB1 released by unstimulated freeze/thaw necrosis is hypo-acetylated (Bonaldi et al, 2003; Lu et al, 2012). This post-translational modification is associated with a cytoplasmic translocation and accumulation of HMGB1 and has been attributed to TLR signalling pathways such as NF-κB, NFATs, JAK/STATs (Kim et al, 2009) and/or MAPKs (Bonaldi et al, 2003). We assessed if unprimed inflammasome activation also induced acetylation of HMGB1 by liquid chromatography tandem mass spectrometric analysis (LC-MS/MS) from extracellular HMGB1 following GluC digestion of extracellular HMGB1. HMGB1 was isolated from supernatants from unactivated C34 and C19 macrophages to assess spontaneous release, or from supernatants from cells treated with Dox or LPS. In C34 and C19 cells, spontaneous release lead to the detection of identical HMGB1 forms with the molecular weights of 1623.8 and 1132.5 Da indicating hypo-acetylated NLS1 and NLS2, respectively (Figure 6A). Analysis of b and y ions generated following MS/MS analysis confirmed the lack of acetylation of each lysine residue within NLS2 (Figure 6C and D). MS/MS analysis of NLS1 also revealed unmodified HMGB1 peptides (Supplementary Figure 6A). Interestingly, Dox treatment of C34 cells resulted in the release of HMGB1-derived peptides with the molecular weights of 1749.8 and 1342.4 Da following MS analysis representing acetylated NLS1 and NLS2, respectively (Figure 6B). Analysis of b and y ions generated following MS/MS analysis confirmed the acetylation of each lysine residue within NLS2 (Figure 6D). MS/MS analysis of NLS1 also revealed an acetylated HMGB1 peptide (Supplementary Figure 6B). The increased mass shift of 126 Da for NLS1 and 210 Da for NLS2 in pyroptotic samples represents the presence of either 3 × acetyl or 5 × acetyl modifications of either NLS1 or NLS2, respectively. HMGB1 from LPS-treated (C34, C19), or Dox-treated (C19) cells was hypo-acetylated (identical to Figure 6A, C and Supplementary Figure 6A). These data indicate that unprimed NLRC4/Naip5 inflammasome activation and co-incident pyroptosis is sufficient to promote the acetylation of HMGB1.
Figure 6.
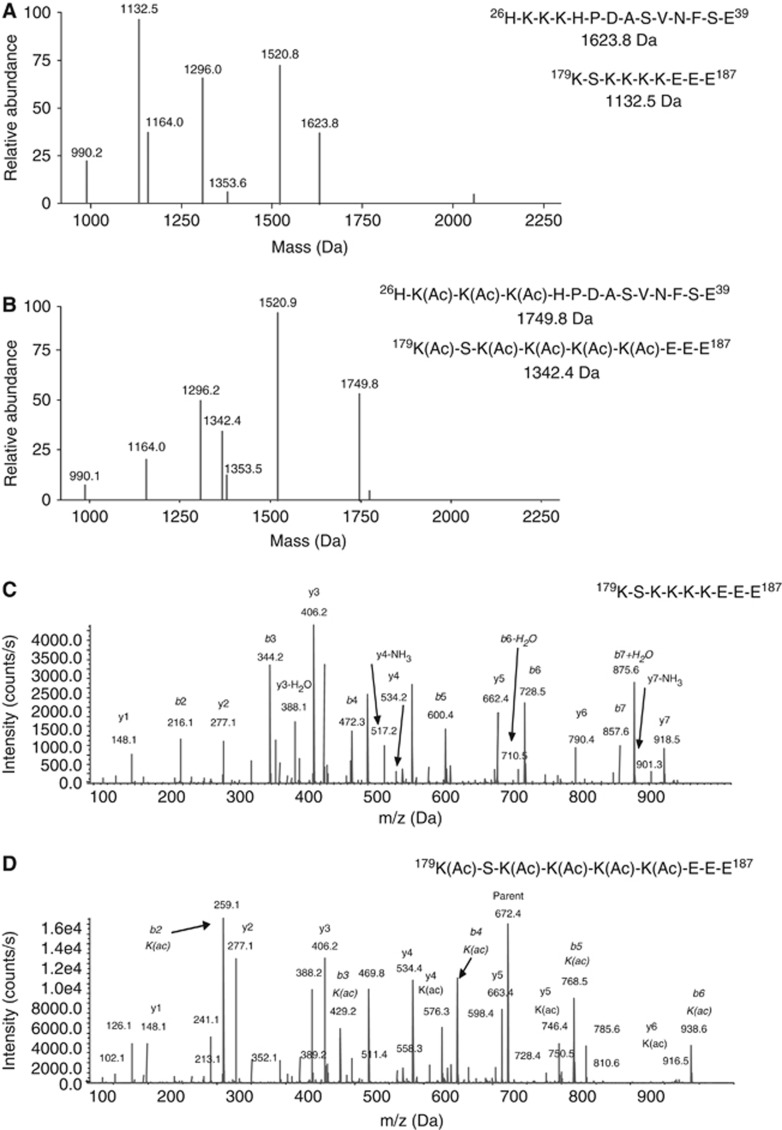
Pyroptosis due to NLRC4/Naip5 agonist expression promotes HMGB1 acetylation. Mass spectrometric characterization of acetyl post-translational modifications of lysine residues of HMGB1 released by C34 cells after 24 h spontaneous release (A) or 24 h Dox treatment (500 ng/ml) (B). Predicted mass of unmodified and modified NLS1 (aa 26–39) and NLS2 (aa 179–187) peptides are indicated. Representative MS/MS spectra of HMGB1 amino acids 179–187 derived from the HMGB1 NLS2 containing non-acetylated (C) or acetylated (D) lysine residues following GluC digestion. Data are representative of two independent experiments.
TLR4 primed pyroptosis regulates HMGB1 cysteine redox isoforms
The redox status of three cysteines in HMGB1 directly alters its functional activity (Venereau et al, 2012; Yang et al, 2012). We observed that induction of unprimed pyroptosis promoted increased release of HMGB1. However, it is unknown what cysteine isoforms of HMGB1 were released. To characterize the redox status of cysteine residues from extracellular HMGB1 derived from unactivated C34 and C19 macrophages to assess spontaneous release, or those treated with Dox or LPS we also employed LC-MS/MS. This was performed using a step-wise HMGB1 immunoprecipitation, iodoacetamide/reduction/N-Ethylmaleimide (NEM) treatment followed by tryptic digestion and peptide characterization. HMGB1 from unactivated samples, or Dox, or LPS treatment of C34 and C19 cells resulted in the detection of peptides with molecular weights that contained Cys23 (1496.7 Da), Cys45 (550.9 Da), and Cys106 (2002.2 Da) (Figure 7A). MS/MS analysis of b and y ions generated from these peptides confirmed the amino-acid sequence of each peptide and also the addition of an iodoacetamide adduct (mass shift 57 Da) on Cys23 (Figure 7C), Cys45 (Figure 7E), and Cys106 (Figure 7G). These data indicate that these cysteines did not participate in disulphide bonds and were present in the thiol form when released from cells. They also indicate that under the conditions of this experiment LPS activation and resulting metabolic changes were unable to modify extracellular HMGB1. Together, these results indicate that although activation of pyroptosis lead to significant increases in the levels of HMGB1 release and the modification of HMGB1 by acetylation, the redox status of the cysteine residues was unchanged compared to spontaneously released HMGB1.
Figure 7.
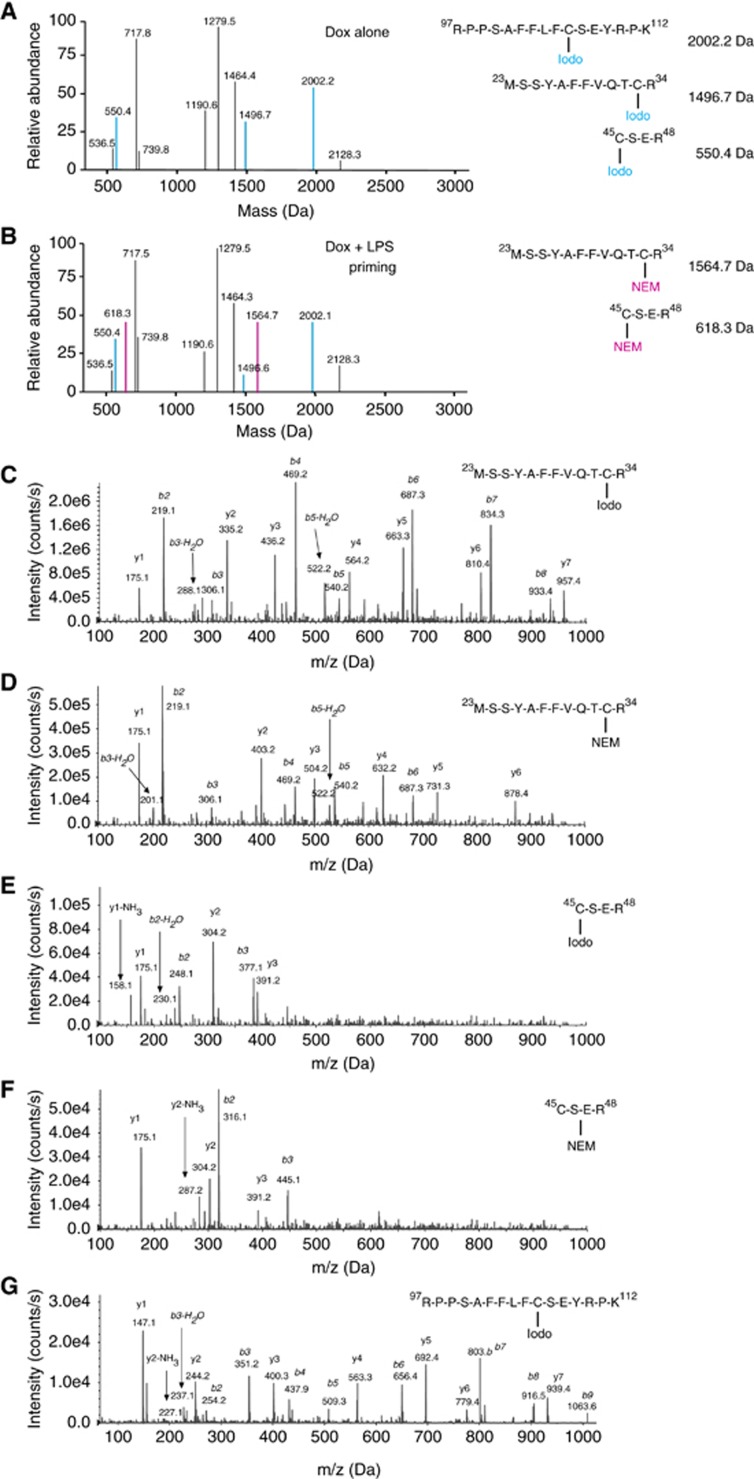
TLR4 primed pyroptosis promotes formation of changes in the cysteine redox isoforms of HMGB1. Mass spectrometric characterization of cysteine redox status of HMGB1 released by C34 and C19 after 24 h Dox treatment (500 ng/ml) (A) or LPS priming (100 ng/ml)+24 h Dox treatment (500 ng/ml) (B). Blue text and mass peaks represent Iodoacetamide (Iodo) capped cysteine residues in HMGB1 peptides, while pink text and mass peaks represent NEM capped cysteine residues in HMGB1 peptides. Representative MS/MS spectra of HMGB1 aa 13–24 peptide from an Iodo (C) or NEM (D) capped cysteine residue following trypsin digestion. Representative MS/MS spectra of HMGB1 aa 45–48 from an Iodo (E) or NEM (F) capped cysteine residue following trypsin digestion. Representative MS/MS spectra of HMGB1 aa 97–112 with an Iodo capped (G) cysteine residue following trypsin digestion. Data are representative of two independent experiments.
Our results show that LPS treatment does not affect the degree of pyroptosis or the amount of HMGB1 release. However, as LPS priming leads to metabolic changes as well as the production of NO and ROS we reasoned that LPS primed pyroptosis could provide signals necessary for modifying the cysteine redox status of HMGB1. To test this, C34 or C19 cells were primed with LPS followed by Dox treatment. HMGB1 from culture supernatants was subjected to step-wise treatments and LS-MS/MS analysis. HMGB1 peptides derived from C34 cells were observed with the molecular weights seen in all thiol form (Figure 7A). However, we also observed the appearance of new peptide masses of Cys23 (1564.7 Da), Cys45 (618.3 Da) and Cys106 (2002.1 Da) (Figure 7B). MS/MS analysis of b and y ions generated from these peptides confirmed the amino-acid sequence of each peptide and also the addition of an NEM (mass shift 125 Da) adduct on Cys23 (Figure 7D) and Cys45 (Figure 7F) and an iodoacetamide adduct (mass shift 57 Da) localized to Cys106 (Figure 7G). Analysis of HMGB1 from LPS+Dox-treated C19 cells revealed that HMGB1 was present in the thiol form (identical to Figure 7A, C). These data indicate that during pyroptosis the TLR4 priming event promotes the joining of Cys23 and Cys45 in a disulphide bond while Cys106 remains in the thiol form. Thus, priming via TLR4 changes the functional isoform of HMGB1 from one with chemotactic ability to one which acts as a TLR4 agonist. No terminal oxidation (sulphonate derivative) was observed on any HMGB1 cysteine residue following any activation (Figure 7C–G).
Cell-surface but not endosomal TLR primed pyroptosis alters HMGB1 cysteine redox isoforms
To determine if TLR activation in general or activation of TLRs endemic to specific cellular locations affect cysteine-redox modifications of HMGB1, we primed C34 cells with media alone or the TLR agonists Pam3CSK4, R848, CpG, or LPS followed by Dox treatment. C34 cells released HMGB1 under all Dox-treated conditions demonstrating that, as with LPS, priming via other TLRs did not affect HMGB1 release (Figure 8A). C34 cells were confirmed to express these TLRs and intact signalling pathways as demonstrated by TNFα production (Figure 8B). HMGB1 from all culture supernatants were subjected to step-wise cysteine labelling as described above and LS-MS/MS analysis was performed. HMGB1 peptides from C34 cells stimulated with Pam3CSK4 were observed with peptide masses indicating the presence of an all-thiol form (Cys23 (1496.3 Da), Cys45 (549.1 Da) and Cys106 (2002.2 Da)) but also peptide masses of Cys23 (1564.7 Da), Cys45 (618.3 Da), indicating joining of Cys23 and Cys45 in a disulphide bond while Cys106 remained in the thiol form (Figure 8C). Interestingly, HMGB1 peptides derived from C34 cells primed via endosomal TLRs with either R848 or CpG were observed with the molecular weights representing only the all-thiol form (Figure 8D and E). MS/MS analysis of b and y ions generated from these peptides from unstimulated, Dox stimulated, and all TLR-primed Dox stimulated C34 cells confirmed the amino-acid sequence of each peptide observed was identical to those seen in Figure 7C–G. These results are summarized in Table I and indicate that activation of cell-surface TLRs can affect changes in HMGB1 cysteine-redox isoforms while activation of endosomal TLRs cannot.
Figure 8.
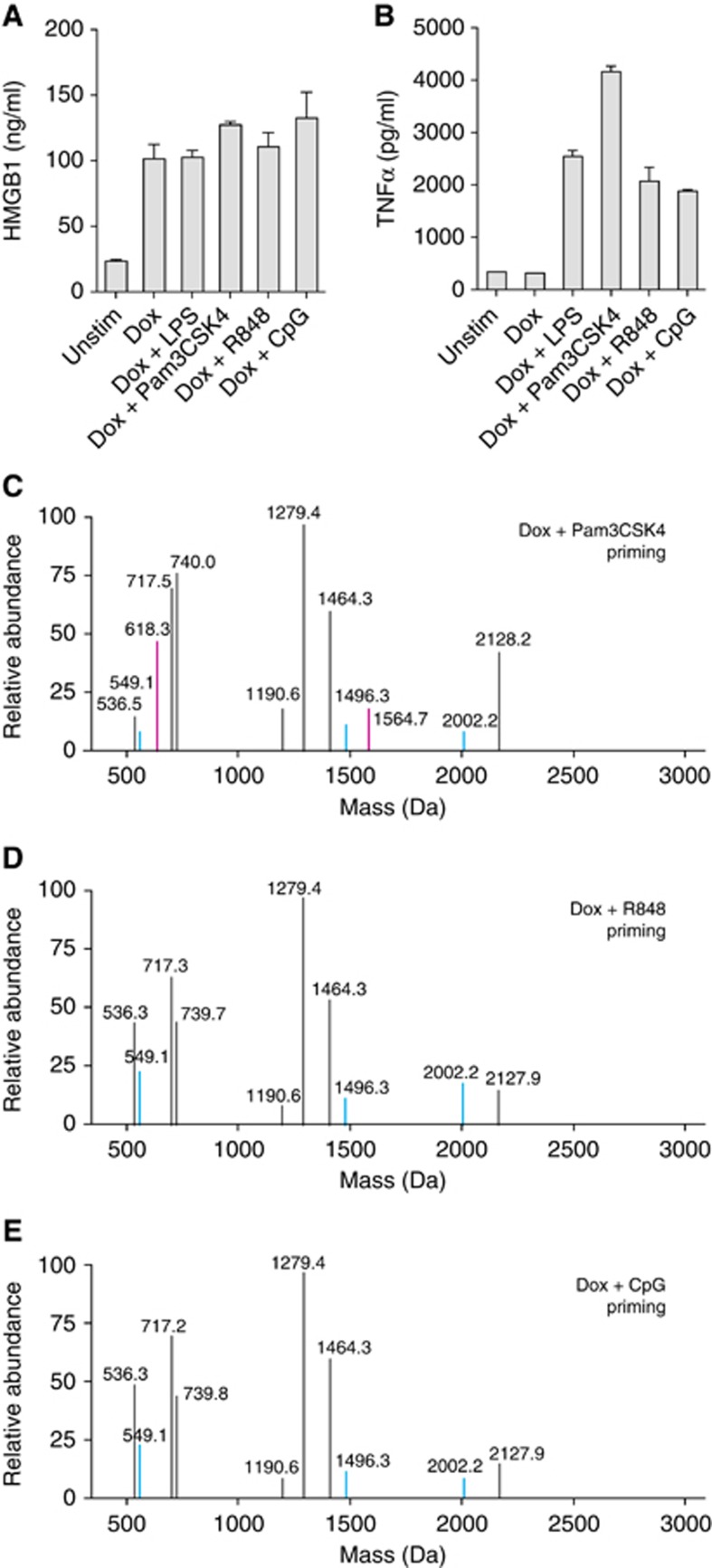
Cell-surface but not endosomal TLR primed pyroptosis promotes formation of changes in the cysteine redox isoforms of HMGB1. Mass spectrometric characterization of cysteine redox status of HMGB1 released by C34 cells. (A, B) Release of HMGB1 (A) and production of TNFα (B) as determined by ELISA from unstimulated or Dox-treated (500 ng/ml)±3 h LPS (100 ng/ml), Pam3CSK4 (200 ng/ml), R848 (4 μg/ml), or CpG (10 μg/ml) priming 24 h post Dox addition. (C–E) Mass spectrometric characterization of cysteine redox status of HMGB1 released by C34 cells primed with Pam3CSK4 (C), R848 (D), or CpG (E) after 24 h Dox treatment. Blue mass peaks represent Iodo capped cysteine residues in HMGB1 peptides while pink mass peaks represent NEM capped cysteine residues in HMGB1 peptides. (A, B) Mean±s.e.m. of triplicate samples is shown. Data are representative of one (A, C–E) or two (B) experiments.
Table I. Effects of pyroptosis and priming on HMGB1 release and isoforms.
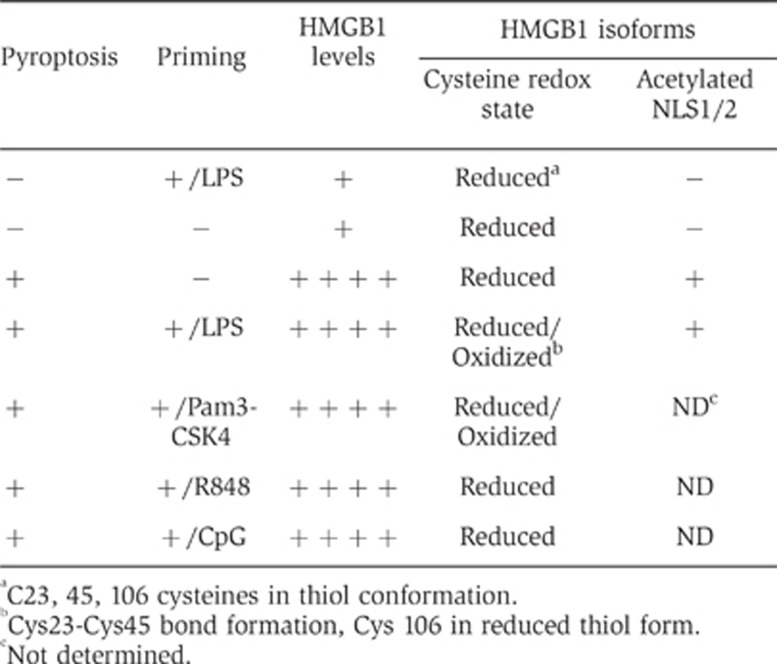
aC23, 45, 106 cysteines in thiol conformation.
bCys23-Cys45 bond formation, Cys 106 in reduced thiol form.
cNot determined.
Discussion
By developing a mimetic NLRC4/Naip5 activation system devoid of other bacterial factors we were able to define the minimum requirements for NLRC4/Naip5 inflammasome activation and downstream outcomes. The re-introduction of bacterial-derived factors and their ability to signal through TLR receptors to this system revealed their ability to promote redox changes to key cysteines altering the functional isoforms of the DAMP HMGB1.
Our data show that the expression of NLRC4/Naip5 agonist alone is sufficient to induce rapid, caspase-1-dependent pyroptotic cell death independent of priming. This death was directly related to the amount of agonist expressed in the cell and occurred with kinetics similar to those seen when the NLRC4/Naip5 inflammasome is activated by flagellin-expressing Salmonella (Fink and Cookson, 2006).
LPS priming prior to NLRC4/Naip5 agonist expression did not lead to great changes in either enhancement or attenuation of pyroptosis. These results suggest a hierarchy of innate immune defense, in which the activation of pathways to prevent macrophages from harbouring cytoplasmic bacteria is dominant over those used to activate classical anti-bacterial effector mechanisms. We suspect this hierarchy plays numerous rolls in host–pathogen interactions. First, this would prevent changes in the presence or form of TLR agonists from affecting NLRC4 immune defense. Second, it would prevent pathogens expressing factors promoting NF-κB-dependent survival signals or preventing NF-κB-dependent innate immune responses from avoiding detection by NLRC4. Third, as priming may ‘mature’ macrophages and promote their migratory potential, pyroptosis functioning independent of priming could inhibit pathogen dissemination. Finally, in intestinal mononuclear phagocytes hypo-responsive to microbial stimulation (e.g., TLR), pyroptotic defense independent of priming would ensure death of infected cells (Smith et al, 2011). Ensuring the potential for pyroptotic success in an environment rife with migratory and/or ‘tolerant’ cells likely to encounter enteric pathogens seems to be a smart evolutionary gambit.
Pyroptotic death has not been studied in response to a defined NLR agonist independent of the activation of other signalling pathways. The rapid release of intracellular constituents from cells undergoing pyroptosis seen here contrasts with the well-defined formation of blebs and apoptotic bodies in apoptotic cells (Kroemer et al, 2007). The ‘explosive’ cell death observed gives evidence for a direct mechanism for the release of inflammatory intracellular constituents. Together with inflammasome-activated lysozomal exocytosis (Bergsbaken et al, 2011), it also demonstrates a direct mechanism for the release of cytoplasmic bacteria. Why pyroptosis exhibits such dramatic structural differences to apoptosis is not known. However, it does suggest that there may be new avenues of cell biology to target, other than the inflammasome, to affect pyroptosis.
In macrophages, TLR signalling activates the transcription factors; NF-κB and IRF3 promoting NLRP3 and pro-IL-1β expression demonstrating the ability of the TLR signalling pathway to ‘cross-talk’ into the inflammasome pathway. Activating the NLRC4/Naip5 inflammasome independently of priming allowed us to study if signals propagated in the reverse direction. NLRC4/Naip5 activation did not lead to a change in general gene dysregulation nor cross-talk into genes regulated by NF-κB or IRF. This indicates that under conditions of inflammasome activation and impending pyroptotic cell death macrophages do not appear to activate transcriptional programs to express known inflammatory factors.
Activating the inflammasome without priming also allowed us to study if cells produced NO or ROS. Interestingly, we did not detect these compounds indicating that their presence is not needed for inflammasome activation or pyroptosis. TLR-induced iNOS expression is controlled transcriptionally and rapid pyroptosis may prevent NO production. Our results contrast other findings where transfected flagellin protein was used to activate the NLRC4/Naip5 inflammasome leading to iNOS expression and NO production (Buzzo et al, 2010). These differences may be due to low-level TLR5 expression by BMDMs allowing flagellin-dependent priming, or contamination of flagellin with other TLR agonists. Our results indicate that the molecular origin of NO does not come from NLRC4/Naip5 inflammasome activation and would, during bacterial infection, likely be dependent on the activation of other innate immune pathways.
The dependence of cell death in C34 cells on caspase-1 indicates that they die by pyroptosis which is unaffected by Bcl-XL overexpression and does not involve ROS production and large decreases in ΔΨm. Although pyroptosis does not involve the proteolytic maturation of caspase-3 (Puri et al, 2012) our results indicate that it does have similarities to extrinsic ‘type I’ apoptosis (Galluzzi et al, 2012). This indicates that pyroptosis appears to occur without standard mitochondrial dysfunction pointing again to the evolution of a system designed to ensure cell death when pathogens employ strategies to affect mitochondrial-associated innate immune responses (Ashida et al, 2011). The exact mechanism of how pyroptosis occurs awaits robust analysis.
Although unprimed pyroptosis does not produce transcription-dependent inflammatory mediators it still leads to the release of HMGB1. Analysis of HMGB1 isoform released by unprimed pyroptosis revealed that activation of the NLRC4/Naip5 inflammasome was able to promote acetylation of NLS sequences (Table I). Acetylation of these domains promote the cytoplasmic accumulation of HMGB1 after LPS treatment (Bonaldi et al, 2003). Our observations that this modification occurs in the absence of NF-κB or IRF activation, indicates that acetylation can also occur by other mechanisms. We favour a model where HMGB1 acetylation is promoted by detection of cell stress and suggests that innate immune stress sensing complexes such as inflammasomes or the initiation of regulated cell death fall into these categories.
Study of the precise structure of HMGB1 revealed that unprimed pyroptosis releases a all-thiol HMGB1 redox isoform (Table I) identical to spontaneously released HMGB1 (data herein) and HMGB1 released by freeze-thaw treatment of cells (Bonaldi et al, 2003; Lu et al, 2012; Venereau et al, 2012), indicating that pyroptosis releases HMGB1 isoform with strong chemotactic behaviour (Schiraldi et al, 2012; Venereau et al, 2012). The release of chemotactic HMGB1 during pyroptotic host defense would ensure recruitment of CXCR4-expressing cell populations such as monocytes, lymphocytes, and neutrophils needed to defend and repair sites of infection. Release of HMGB1 would also recruit these populations regardless of macrophage NF-κB activation and the production of IL-1β which, although chemotactic (Cybulsky et al, 1986), is subject to multiple levels of regulation. Additionally, although not extensively studied, HMGB1 release may also play a role in pyroptotic host defense by providing potent extracellular bactericidal effects (Zetterstrom et al, 2002).
Priming of macrophages via TLR4 promotes numerous signalling and metabolic responses. Nevertheless, we found that pyroptosis and the magnitude of HMGB1 release were not greatly affected by the re-introduction of priming. However, LPS priming as well as priming by other cell-surface TLRs resulted in the oxidative modifications of HMGB1 cysteines functioning as a regulatory switch to convert chemotactic HMGB1 to a TLR4 agonist form (Table I). Interestingly, priming of cells via endosomally located TLR7 or TLR9 was not able to induce this switch. West et al (2011) have shown that agonism of surface TLRs induces mROS production in a TRAF6 and ECSIT-dependent manner while endosomal TLRs are unable to induce mROS. We find a similar functional divergence of these TLR classes in their ability to promote oxidative post-translational modifications of host proteins, indicating that mROS production not only plays a role in host defense but may also affect how cell death, in the context of infections or vaccination, is perceived by the immune system. As activation of different inflammasomes may involve different degrees of mitochondrial and metabolic involvement it will likely not be feasible to eliminate these factors in vivo to address their role in regulating HMGB1 isoforms during pyroptosis. Instead the detailed analysis of HMGB1 isoforms awaits the development of animals lacking cysteines at key locations.
The activation of TLR4 leading to the release of molecules capable of activating TLR4 smacks of a signal amplification loop. Our results suggest that this loop would ensure that pyroptosis is detected as inflammatory should death occur too rapidly to activate inflammatory signals dependent on transcription or if the pathogen evades defenses by manipulating signalling pathways involved in de novo gene transcription. However, these observations also suggest that insufficient redox modification of HMGB1 may prolong inflammatory responses by inhibiting or delaying the formation of ‘inactive’ isoforms.
Evolution has armed the macrophage with an ideal system allowing it to detect the presence of bacterial pathogens in its cytoplasm. Our results reveal a pathway by which activation of two families of innate immune receptors work independently to ensure host defense but also together to alter the functional isoforms of DAMPs released during pyroptosis. As cell death appears to occur frequently during inflammasome activation, an increased understanding of the metabolic changes activated by priming would help us to better understand inflammasome-associated host defense and pathology.
Materials and methods
Cells, culture, reagents, and plasmids
The iBMDM B10R (also called BcgR) has been described previously (Radzioch et al, 1991). Mycoplasma-free B10R and B10R transductants were cultured in High-glucose DMEM with L-glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin (HyClone, Thermo Scientific), and 10% FCS (Sigma-Aldrich). Ultra-pure LPS E. coli 0111:B4, CpG 1826, Pam3CSK4, and R848 from Invivogen. Zeocin, G418 and TMRE from Invitrogen Life Technologies. Doxacyclin from NORDIC Drugs. Polybrene, CCCP, Etoposide, 2-deoxyglucose, and Antimycin A from Sigma-Aldrich. Griess reagent from Merck. CsA from Enzo Life Science.
Single-plasmid Tet-on inducible retroviral transduction vectors with EGFP fusion genes in frame with the c-terminal 34 or 19 aa of FliC are described in Supplementary data. Transduction of B10R cells with these vectors as well as transduction of C34 with CrmA, Bcl-XL, cFLIPS, and cFLIPL retroviral constructs is described in Supplementary data.
ELISA, LDH, and immunoblot assays
TNFα and IL-1β ELISAs (BioLegend). LDH release was determined as suggested by the manufacturer (Promega). Immunoblots were performed using IgG mouse anti-HMGB1 mAb (2G7), rabbit polyclonal anti-IL-1β (Abcam; ab9722), rabbit anti-FLIP mAb (Cell Signaling Technology; D16A8) and mouse anti-actin mAb (MP Biomedicals). Primary antibodies were detected with HRP-goat anti-mouse IgG (Pierce, Thermo Scientific; 31432) or HRP-goat anti-rabbit IgG (Invitrogen Life Technologies; G21234), detected with SuperSignal West Dura Extended Substrate (Pierce, Thermo Scientific), and acquired using a LAS-4000 mini (Fujifilm).
Fluorescent staining and flow cytometry
EGFP, YFP, PI, TMRE, Live/Dead Violet or Aqua viability stain (Invitrogen Life Technologies), Viability Dye eFluor® 450 (eBioscience) and surface staining with fluorophore-conjugated mAb signals were detected using a LSRII-Fortessa (BD Bioscience). See Supplementary Tables I and II for mAb conjugates and filter sets. In all, ⩾25 000 living singlets were analysed for surface stainings. In all, ⩾40,000 non-debris singlets were analysed for EGFP, YFP, and PI viability analysis (gating strategies: Supplementary Figure 2A–C).
Intracellular ROS was detected using CellROX Deep Red as suggested by the manufacturer (Invitrogen Life Technologies). Treated cells were resuspended in PBS (1% FCS) with Live/Dead Aqua for 10 min ⩾25 000 living singlets were analysed for CellROX signals (gating strategies; Supplementary Figure 2D).
Mitochondrial membrane potential was assayed as follows. Cells were incubated with 150 nM of TMRE for 30 min in media, spun down, and plated in full media containing 50 nM TMRE. Cells were activated by CCCP or Dox±CsA for the indicated times. When applicable, CsA was added to cultures 1 h prior to TMRE loading. CCCP or Dox-treated cells were resuspended in media containing Viability Dye eFluor® 450 for 5 min and ⩾25 000 living singlets were analysed for TMRE and EGFP signals (gating strategies; Supplementary Figure 2E and F). FACS data were analysed using FlowJo v9.2 (Tree Star).
Live confocal microscopy
PMs were labelled (Cell Mask Orange; Invitrogen Life Technologies), plated in media with 500 ng/ml Dox, imaged every 10 min for 22.9 h with × 40 air objective (Plan Apo 40X MN2, Numerical Aperture 0.95) in 5% CO2 at 37°C using an A1 spectral detector confocal microscope (Nikon). EGFP and YFP signals were separated based on experimentally recorded control spectra using spectral unmixing software (NIS Elements v3, Nikon). PM signals were pseudo-coloured blue and YFP signals red (Nikon). Signals from four individual channels were compiled into sequential composite images and corrected for brightness and contrast using standard linear settings in Adobe Photoshop CS4 (Adobe). Identical corrections were made to all images.
Quantative RT–PCR analysis
Total RNA was isolated using a RiboPure Kit (Ambion) and cDNA was prepared using High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems). Real-time PCR was performed using RT2; SYBR Green compatible primers (SABiosciences) for Hprt1 (Mm.299381), NLRC4 (Mm.311884), Naip5 (Mm.290476), Caspase-1 (Mm.1051), ASC (Mm.24163), PKR (Mm.378990), IL-6 (Mm.1019), TNFα (Mm.1293), Cxcl10 (Mm.877), IL-1β (Mm.222830), CSF2 (Mm.4922), and Caspase-11 (Mm.1569) all with RT2; qPCR MasterMix (SABiosciences) using a ABI 7500 Real Time system (Applied Biosystems). Expression was normalized to HPRT (ΔΔCt method). For visualization, PCR products were separated, stained with Gel Green Nucleic Acid Stain (Biotium), and acquired using a LAS-4000 mini.
HMGB1 preparation for LC-MS/MS Analysis
All chemicals and solvents were of the highest available grade (Sigma). Culture supernatants were pre-cleared with 50 μl protein G-Sepharose beads for 1 h at 4°C. Supernatant HMGB1 was immunoprecipitated with 5 μg rabbit anti-HMGB1 (Abcam; ab18256) for 16 h at 4°C. Free thiol groups within HMGB1 were alkylated for 90 min with 10 mM iodoacetamide at 4°C. Cysteine residues in disulphide bonds were then reduced with 30 mM dithiothreitol (DTT) at 4°C for 1 h followed by alkylation of newly exposed thiol groups with 90 mM NEM at 4°C for 10 min. Samples were subjected to trypsin (Promega) or GluC (New England Biolabs) digestion according to manufacturer’s instructions and de-salted using ZipTip C18 pipette tips (Millipore). Characterization of cysteine bonds and absolute quantification of acetylated HMGB1 was determined as described (Antoine et al, 2012) using an AB Sciex TripleTOF 5600 (Sciex Inc.).
Statistical analysis
Data were analysed using Prism v5.0d (GraphPad Inc) and is show as mean±standard error of the mean (s.e.m.). A Student’s t-test was performed to compare the differences between treated groups relative to their paired controls. P-values are indicated in the text and figures above the two groups compared with a value of <0.05 (denoted by *) considered as significant (***P<0.001, **P<0.01).
Supplementary Material
Acknowledgments
This work was supported by the Swedish International Development Cooperation Agency (SIDA), as well as the Swedish Research Council. This study was performed in part at the Live Cell Imaging unit, Department of Biosciences and Nutrition, Karolinska Institutet, Huddinge, Sweden, supported by grants from the Knut and Alice Wallenberg Foundation, the Swedish Research Council and the Centre for Biosciences. DJA is supported by the Medical Research Council (MRC) and the Wellcome Trust.
Author contributions: SN, UA, and SEA wrote the manuscript. SN, JL, AG, HE-H, UA, and SEA conceived or designed the experiments. SN, DJA, PL, JL, AFN, AG, KH, and SEA performed the experiments. SN, DJA, PL, JL, AFN, and SEA analysed the data.
The authors declare that they have no conflict of interest.
01/09/2013
Since Advance Online Publication, an extra affiliation has been added.
References
- Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, Voss OH, Doseff AI, Hassan H, Azad AK, Schlesinger LS, Wewers MD, Gavrilin MA, Amer AO (2012) Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37: 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Tracey KJ (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29: 139–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK (2012) Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol 56: 1070–1079 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C (2011) Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Biol 195: 931–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, Trinidad NJ, Vance RE (2012) Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med 18: 799–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187: 613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, den Hartigh AB, Loomis WP, Cookson BT (2011) Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J Immunol 187: 2748–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22: 5551–5560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, von Moltke J, Jones JW, Vance RE, Monack DM (2010) Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8: 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzo CL, Campopiano JC, Massis LM, Lage SL, Cassado AA, Leme-Souza R, Cunha LD, Russo M, Zamboni DS, Amarante-Mendes GP, Bortoluci KR (2010) A novel pathway for inducible nitric-oxide synthase activation through inflammasomes. J Biol Chem 285: 32087–32095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulsky MI, Colditz IG, Movat HZ (1986) The role of interleukin-1 in neutrophil leukocyte emigration induced by endotoxin. Am J Pathol 124: 367–372 [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J 30: 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Cookson BT (2006) Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8: 1812–1825 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W et al. (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19: 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC (2011) Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286: 9587–9597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F (2011) NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol 186: 1646–1655 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650 [DOI] [PubMed] [Google Scholar]
- Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S (2001) Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 167: 5887–5894 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kim SJ, Lee IS, Lee MS, Uematsu S, Akira S, Oh KI (2009) Bacterial endotoxin induces the release of high mobility group box 1 via the IFN-beta signaling pathway. J Immunol 182: 2458–2466 [DOI] [PubMed] [Google Scholar]
- Kofoed EM, Vance RE (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477: 592–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP, Koller BH (2012) NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol 189: 2006–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87: 99–163 [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM (2010) Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185: 4385–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, Monack DM, Tsolis RM, Vance RE (2008) Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 9: 1171–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfield KL, Persson J, Trinidad NJ, Brubaker SW, Kofoed EM, Sauer JD, Dunipace EA, Warren SE, Miao EA, Vance RE (2011) Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun 79: 1606–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, JP-Y Ting, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488: 670–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man N, Chen Y, Zheng F, Zhou W, Wen LP (2010) Induction of genuine autophagy by cationic lipids in mammalian cells. Autophagy 6: 449–454 [DOI] [PubMed] [Google Scholar]
- Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A (2006) Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7: 569–575 [DOI] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010a) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010b) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107: 3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Rajan JV, Aderem A (2011) Caspase-1-induced pyroptotic cell death. Immunol Rev 243: 206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motani K, Kushiyama H, Imamura R, Kinoshita T, Nishiuchi T, Suda T (2011) Caspase-1 protein induces apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC)-mediated necrosis independently of its catalytic activity. J Biol Chem 286: 33963–33972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri AW, Broz P, Shen A, Monack DM, Bogyo M (2012) Caspase-1 activity is required to bypass macrophage apoptosis upon Salmonella infection. Nat Chem Biol 8: 745–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzioch D, Hudson T, Boule M, Barrera L, Urbance JW, Varesio L, Skamene E (1991) Genetic resistance/susceptibility to mycobacteria: phenotypic expression in bone marrow derived macrophage lines. J Leukoc Biol 50: 263–272 [DOI] [PubMed] [Google Scholar]
- Scaduto RC Jr, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76: 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, Thelen M, Varani L, Mellado M, Proudfoot A, Bianchi ME, Uguccioni M (2012) HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med 209: 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010) The inflammasomes. Cell 140: 821–832 [DOI] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KG, Strasser A, Vaux DL (1996) CrmA expression in T lymphocytes of transgenic mice inhibits CD95 (Fas/APO-1)-transduced apoptosis, but does not cause lymphadenopathy or autoimmune disease. EMBO J 15: 5167–5176 [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM (2011) Intestinal macrophages and response to microbial encroachment. Mucosal Immunol 4: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutlu T, Nystrom S, Gilljam M, Stellan B, Applequist SE, Alici E (2012) Inhibition of intracellular anti-viral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther 23: 1090–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venereau E, Casalgrandi M, Schiraldi M, Cattaneo A, De Marchis F, Liu J, Antonelli A, Andersson U, Antoine DJ, Tracey KJ, Uguccioni M, Bachi A, Bianchi ME (2012) Mutually exclusive redox forms of HMGB1 promote cell recuitment or proinflammatory cytokine release. J Exp Med 209: 1519–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R, Dillon CP, Green DR (2011) Ripped to death. Trends Cell Biol 21: 630–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ (2010) A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA 107: 11942–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, Antoine DJ (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 18: 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zetterstrom CK, Bergman T, Rynnel-Dagoo B, Erlandsson Harris H, Soder O, Andersson U, Boman HG (2002) High mobility group box chromosomal protein 1 (HMGB1) is an antibacterial factor produced by the human adenoid. Pediatr Res 52: 148–154 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600 [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.