

Abstract
The antiporter system xc− imports the amino acid cystine, the oxidized form of cysteine, into cells with a 1:1 counter-transport of glutamate. It is composed of a light chain, xCT, and a heavy chain, 4F2 heavy chain (4F2hc), and, thus, belongs to the family of heterodimeric amino acid transporters. Cysteine is the rate-limiting substrate for the important antioxidant glutathione (GSH) and, along with cystine, it also forms a key redox couple on its own. Glutamate is a major neurotransmitter in the central nervous system (CNS). By phylogenetic analysis, we show that system xc− is a rather evolutionarily new amino acid transport system. In addition, we summarize the current knowledge regarding the molecular mechanisms that regulate system xc−, including the transcriptional regulation of the xCT light chain, posttranscriptional mechanisms, and pharmacological inhibitors of system xc−. Moreover, the roles of system xc− in regulating GSH levels, the redox state of the extracellular cystine/cysteine redox couple, and extracellular glutamate levels are discussed. In vitro, glutamate-mediated system xc− inhibition leads to neuronal cell death, a paradigm called oxidative glutamate toxicity, which has successfully been used to identify neuroprotective compounds. In vivo, xCT has a rather restricted expression pattern with the highest levels in the CNS and parts of the immune system. System xc− is also present in the eye. Moreover, an elevated expression of xCT has been reported in cancer. We highlight the diverse roles of system xc− in the regulation of the immune response, in various aspects of cancer and in the eye and the CNS. Antioxid. Redox Signal. 18, 522–555.
-
III. Expression of System xc− In Vitro and In Vivo and Its Functional Consequences
-
IV. The Role of System xc− in Health and Disease
B. The role of system xc− in the immune system and inflammation
-
C. The role of system xc− in cancer and resistance against anti-cancer drugs
1. System xc− is regulated by potentially oncogenic pathways
2. System xc− mediates the infection of cells by oncogenic Kaposi's sarcoma herpesvirus
3. System xc− plays an important role in the multidrug resistance of cancers
4. Inhibition of system xc− reduces cancer cell replication, tissue invasion, and metastasis
5. System xc− expressed in tumor cells may be used as a target for anticancer drug delivery
7. Synopsis of the role of system xc− in cancer and resistance against anti-cancer drugs
I. Introduction
A. Oxidative stress and antioxidant defense
Oxidative stress is defined as an imbalance between the production of free radicals, mostly reactive oxygen species (ROS), and their removal by the antioxidant defense systems present in tissues and body fluids (253) and, thus, results from an increase in ROS production and/or a decrease in antioxidant defense. Oxidative stress leads to the oxidative modification of proteins, lipids, and DNA. Cells contain not only small-molecule antioxidants such as vitamins C and E and the tripeptide glutathione (GSH), which scavenge the ROS produced during the cell's metabolism, but also enzymes whose specific role is the neutralization of ROS [reviewed in (241)]. These include the different isoforms of superoxide dismutase (SOD), which convert superoxide into hydrogen peroxide (H2O2), and catalase, which metabolises H2O2. GSH peroxidases (GPx) GSH-dependently catalyze the decomposition of H2O2 and of organic hydroperoxides while oxidizing GSH to GSH disulfide (GSSG).
Oxidative modification of proteins, lipids, and DNA has been repeatedly shown to be associated with ageing, and it has been frequently demonstrated that GSH levels are decreased in diverse tissues in aged animals or elderly humans (149, 243, 300). Ageing is the major risk factor for many of the most important diseases in the Western World, including diabetes, atherosclerosis, cancer, and neurodegenerative diseases such as Parkinson's disease (PD), Alzheimer's disease (AD), and ischemic stroke. Of note, oxidative stress is thought to play an important role in each of these diseases (88, 274).
B. GSH metabolism
The small-molecule antioxidant GSH is a tripeptide consisting of the amino acids glutamate, glycine, and cysteine. Cells contain approximately millimolar concentrations of GSH. Thus, GSH is one of the most important small-molecule antioxidants in somatic cells.
In most tissues, the rate-limiting amino acid for GSH synthesis is the nonessential amino acid cysteine (160, 179). Cysteine can be imported into cells either directly or in its oxidized form, cystine, via the cystine/glutamate antiporter system xc− (Fig. 1). Within the cell, cystine is immediately reduced to cysteine either by intracellular GSH via the formation of a mixed disulfide intermediate or by thioredoxin reductase 1 (TRR1) (172). Several amino acid transporters that can transport cysteine have been described. System alanine-serine-cysteine (ASC) transports cysteine as well as threonine, asparagine, alanine, serine, and, to some extent, glutamine (40). System A transports glycine, alanine, and proline much more efficiently than cysteine, and system L also transports methionine, valine, phenylalanine, leucine, and isoleucine. In addition, excitatory amino acid transporters (EAATs) have been proposed as playing a role in cysteine import into neurons (36). However, the affinity of EAATs for glutamate is 10 times higher than for cysteine (122).
FIG. 1.
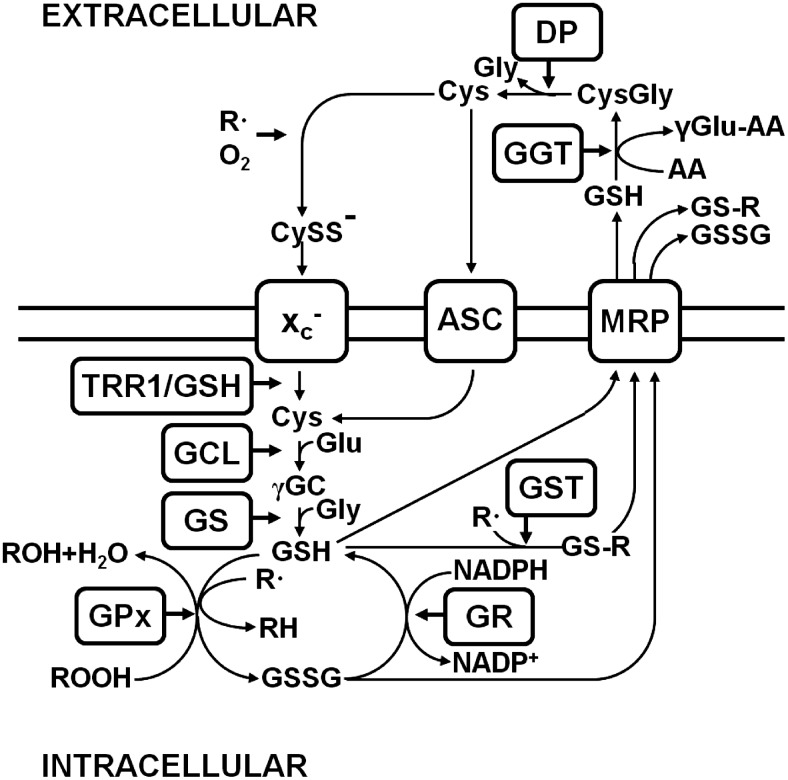
Glutathione (GSH) metabolism. Cystine (CySS−) is taken up by system xc− (xc−). Intracellularly, CySS is reduced to cysteine (Cys) by thioredoxin reductase 1 (TRR1) or GSH. Glutamate cysteine ligase (GCL) catalyzes the synthesis of γ-glutamyl cysteine (γ-GC) from glutamate (Glu) and Cys, and glutathione synthase (GS) generates GSH by adding glycine (Gly). GSH reduces radicals (R•) nonenzymatically and organic hydroperoxides catalyzed by GSH peroxidase (GPx) and is thereby converted to GSH disulfide (GSSG). GSSG is recycled to GSH by GSH reductase (GR), a reaction that uses reduced nicotinamide adenine dinucleotide phosphate (NADPH) as a co-factor. GSH S-transferase (GST) forms GSH adducts (GS-R) from organic molecules (R) and GSH, which along with GSH and GSSG are exported from the cell by multi-drug resistance proteins (MRP). The ecto-enzyme γ-glutamyl transferase (GGT) transfers the γ-glutamyl moiety of GSH to an acceptor amino acid (AA), leading to cysteinyl glycine (CysGly), which is cleaved by a dipeptidase (DP) to Cys and Gly. Both GGT and DP are membrane-bound enzymes. Cys is either taken up by cysteine transporters, among them, system alanine-serine-cysteine (ASC), or extracellularly oxidized to CySS−, which is again taken up by system xc−.
The first step in GSH synthesis, the generation of γ-glutamyl cysteine, is catalyzed by glutamate cysteine ligase (GCL) (182, reviewed in 82). γ-Glutamyl cysteine and glycine then form GSH through the action of GSH synthase. GSH can both nonenzymatically and enzymatically, in a reaction catalyzed by different GPx with distinct substrate specificities, reduce diverse ROS. In scavenging ROS, GSH is oxidized to GSSG, which is either reduced by GSH reductase (GR), in a reaction that requires reduced nicotinamide adenine dinucleotide phosphate (NADPH), or exported from the cell by multi-drug resistance proteins (MRPs). NADPH is generated via the hexose monophosphate shunt, an alternative pathway of glucose metabolism. Moreover, the formation of GSH adducts by GSH S-transferases (GSTs) detoxifies endogenous α,β-unsaturated aldehydes, quinones, epoxides, and hydroperoxides, which are formed as secondary metabolites during cell metabolism and electrophilic xenobiotics, such as chemical carcinogens, environmental pollutants, and antitumor agents (90). The diverse GSH adducts are exported from the cell by MRPs. Both MRPs and/or organic anion transporter proteins (OATPs) release GSH into the extracellular space (65, 181). Extracellular GSH is metabolized by the ectoenzyme γ-glutamyl transferase (GGT), which transfers the γ-glutamyl residue to different acceptor amino acids, leading to the formation of a γ-glutamyl containing dipeptide and the dipeptide cysteinyl glycine, which is either cleaved by extracellular dipeptidases to generate cysteine and glycine or directly taken up by cells (54). The neuroprotective function of GSH and/or GSH-related substrate export via MRP1 in the brain was recently demonstrated in an animal stroke model (200).
Generally, the GSH/GSSG redox couple determines the cell's redox state with a more reducing environment associated with cell proliferation, while a more oxidizing environment is associated with differentiation (237). GSH is not uniformly distributed in cells, but rather different subcellular compartments have distinct GSH levels and GSH/GSSG ratios (42). In the cytosol, the GSH-to-GSSG ratio is high, consistent with a reducing environment, whereas in the endoplasmic reticulum (ER), the GSH redox couple is in a much more oxidized state, in line with the role of this organelle in protein disulfide bond formation (269). The mitochondria contain a separate pool of GSH that plays a key role in maintaining mitochondrial function (173). Other roles for GSH include functioning as a cofactor for a variety of enzymes such as glyoxalase 1, the rate-limiting enzyme for the removal of reactive dicarbonyls (213) and in the glutathionylation of proteins, a potential signaling mechanism somewhat analogous to protein phosphorylation (180) and in the synthesis of the inflammatory mediators cysteinyl leukotrienes (130).
The major transcription factor that regulates GSH metabolism is the ubiquitously expressed protein NF-E2-related factor 2 (Nrf2), a member of the Cap ’n’ Collar family of bZIP proteins [for reviews see (113, 296)]. Under basal conditions, Nrf2-dependent transcription is repressed, because Nrf2 is rapidly degraded by the proteasome, a process mediated by its interaction with Keap1, an Nrf2-specific adaptor protein for the Cul3 ubiquitin ligase complex. On exposure to a variety of different stimuli including oxidative stress, ER stress, nitric oxide (NO), 15d-PGJ2, phenolic compounds, Michael acceptors, isothiocyanates, dithiolethiones, dimercaptans, heavy metals, peroxides and polyenes, Keap1-dependent Nrf2 ubiquitination, and subsequent degradation are blocked, leading to the stabilization and nuclear accumulation of Nrf2, where it induces electrophile response element (EpRE)-dependent gene expression to re-establish cellular redox homeostasis (113). The EpRE is also known as the antioxidant response element (ARE) but since electrophiles rather than antioxidants activate transcription mediated by this element, the term EpRE is preferable. Several key enzymes of GSH metabolism are transcriptionally regulated by Nrf2, including the catalytic and regulatory subunits of GCL, GSH synthase, GPx2, GSTs, and GR (89, 113). Hence, Nrf2 is thought to represent a key transcriptional regulator of GSH metabolism.
C. Glutamate: neurotransmission and neurotoxicity
Glutamate is the most important excitatory neurotransmitter in the brain. When released synaptically, glutamate activates ionotropic glutamate receptors located in the postsynaptic part of the synapse. Ionotropic glutamate receptors are ligand-gated ion channels and include receptors of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainite, and N-methyl-D-aspartic acid (NMDA) types [reviewed in (158)]. While AMPA and kainate receptors primarily mediate sodium influx, NMDA receptors have calcium conductivity. Activation of NMDA receptors plays an important role in synaptic plasticity and learning [reviewed in (183)]. In addition to ionotropic glutamate receptors, eight isoforms of metabotropic glutamate receptors exist, which belong to the family of G protein-coupled receptors and that do not form ion channels but signal via various second-messenger systems [reviewed in (257)].
To ensure adequate neurotransmission, the extracellular glutamate concentration has to be tightly controlled. While the mean glutamate concentration in the brain is 10 mM (127), the extracellular glutamate concentration in the brain is only 2–9 μM (8). Thus, there is a steep concentration gradient with much higher intracellular glutamate concentrations. The rapid removal of released glutamate from the extracellular space is brought about by EAATs, a family of high-affinity Na+/K+-dependent transporters for glutamate and aspartate, of which five different members, EAAT1-5, exist [reviewed in (247)]. EAAT1 and −2 are prominently expressed in astrocytes, especially within their processes surrounding glutamatergic synapses, where they are responsible for the immediate uptake of synaptically released glutamate. However, expression of both EAAT1 and −2 has also been demonstrated in macrophages (218) and EAAT2 expression in microglia (204), indicating other roles for EAATs distinct from neurotransmission. EAAT3 is mainly expressed not only in neurons, but also in the kidney and the intestinal mucosa (109, 203). EAAT4 is most prominently expressed in cerebellar Purkinje cells, and EAAT5 is expressed in rod photoreceptor and bipolar cells of the retina. These transporters co-transport 2 or 3 molecules of Na+ and a proton with each molecule of glutamate (or aspartate) and counter-transport of a K+ ion. Thus, by using the electrochemical gradient of these ions across the plasma membrane as an energy source, they are capable of effectively accumulating glutamate and aspartate in cells against the steep intra- to extracellular concentration gradient of these amino acids. Any change in extracellular glutamate concentrations, within the synaptic cleft or extrasynaptically, can be expected to change the activity of metabotropic or ionotropic glutamate receptors and thereby neuronal activation patterns and, finally, on the highest level, behavior. Overactivation of ionotropic glutamate receptors induces neuronal death, a pathway called excitotoxicity, a term coined by Olney in 1969 (195). Calcium influx via NMDA receptors is especially effective in inducing this cell death pathway [reviewed in (38)]. Thus, any pathophysiological state of the brain that is associated with increased extracellular glutamate concentrations has the propensity to lead to neuronal degeneration. Several cellular processes have been reported as contributing to a pathological rise in extracellular glutamate, including increased exocytotic vesicular release, reduced glutamate uptake via EAATs, and nonvesicular glutamate release via reversal of EAAT-mediated glutamate uptake or opening of astrocytic volume-sensitive organic anion channels (104, 244). Excess glutamate can also be removed by activation of glutamate oxaloacetate transaminase (GOT), which can metabolize glutamate into tricarboxylic acid cycle intermediates, as recently demonstrated by the cerebral glutamate lowering and neuroprotective action of GOT in stroke (219).
Glutamate toxicity has been implicated in the pathogenesis of neuronal injury triggered by many central nervous system (CNS) diseases, including cerebral ischemia, Alzheimer's and Huntington's disease, epilepsy, amyotrophic lateral sclerosis [reviewed in (247)], and multiple sclerosis (MS) [reviewed in (77)].
II. The Cystine/Glutamate Antiporter System xc−
A. Functional and pharmacological characteristics of system xc−
System xc− was first characterized in human fetal lung fibroblasts in culture by Bannai and Kitamura in 1980 (17). Makowske and Christensen described a similar transport system in a transformed rat hepatoma cell line (171). System xc− acts as a sodium-independent and chloride-dependent antiporter of the anionic forms of cystine and glutamate (Fig. 2) (16, 76, 202). Although system xc− can transport both amino acids in both directions (11), since the intracellular pool of cystine is negligibly small because intracellular cystine is rapidly reduced and the intracellular glutamate concentration is generally higher than in the extracellular space, system xc−generally imports cystine while exporting glutamate. This transport appears to be locked in a 1:1 ratio (11). Extracellular glutamate acts as a competitive inhibitor for cystine uptake via system xc−. The reported Ki for glutamate inhibition of cystine uptake by system xc− is 150 μM, while the Ki for cystine inhibition of glutamate uptake is 33 μM (171). The Kms are 78 μM for glutamate and 45 μM for cystine.
FIG. 2.
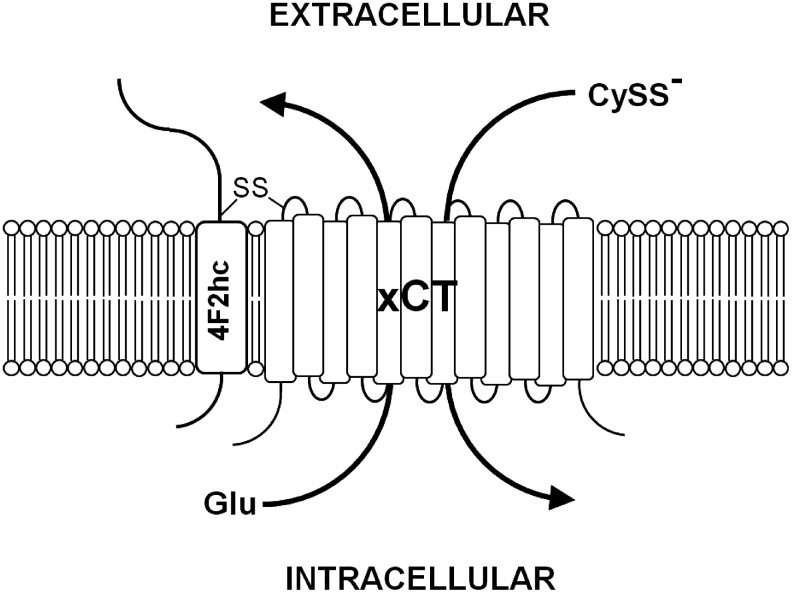
System xc−. System xc− is composed of the 4F2 heavy chain (4F2hc) and the light chain, xCT, which are linked by a disulfide bond (-S-S-). System xc− imports cystine (CySS−) in exchange for glutamate (Glu).
Though all naturally occurring proteinogenic amino acids are neither transported by system xc− nor inhibit glutamate or cystine transport, some structurally related, naturally occurring or artificial molecules do inhibit system xc−. L-α-aminoadipate, an amino acid intermediate in chain length between glutamate and cystine, is an effective substrate inhibitor of system xc−(202). L-α-aminoadipate is a product of lysine metabolism and is present in the brain (32). Other structurally related molecules that inhibit system xc− include β-mercaptolactate-cysteine disulfide, L-homocysteate, L-homocysteine sulfinate, L-α-aminopimelate, and L-serine-O-sulphate (16, 171, 202). β-Mercaptolactate-cysteine disulfide is found in the urine of healthy humans (294). L-homocysteate and L-homocysteine sulfinate are acidic homocysteine derivatives that have been detected in astrocytes (51, 81). L-serine-O-sulfate and L-α-aminopimelate are artificial glutamate analogs. Since L-homocysteate, L-α-aminopimelate, and L-α-aminoadipate activate cystine release from cells under oxidizing conditions, all of these are substrate inhibitors (11). Other inhibitors of system xc− include the cyclic glutamate analogues ibotenate, L-quisqualate, (RS)-4-bromo-homoibotenate, and (S)-4-carboxyphenylglycine [(S)-4-CPG] (202). Ibotenate is a compound that occurs in the mushrooms Amanita muscaria and Amanita pantherina (83), while quisqualate naturally occurs in the seeds of the vine Quisqualis indica. The other compounds do not occur naturally. However, although more potent at inhibiting system xc− than noncyclic glutamate analogs, both cyclic and noncyclic system xc− inhibitors have cross-reactivities, especially with ionotropic and metabotropic glutamate receptors, due to their structural similarity to glutamate. L-homocysteate and L-ibotenate activate NMDA receptors (52, 58) and (RS)-4-bromohomoibotenate and L-quisqualate are AMPA receptor agonists (43, 184). Moreover, L-serine-O-sulfate, L-homocysteate, L-homocysteine sulfinate, L-ibotenate, and L-quisqualate activate metabotropic glutamate receptors (26, 119, 184, 248), and (S)-4-CPG is a Group I metabotropic glutamate receptor antagonist (22). L-serine-O-sulfate also acts as an inhibitor of serine racemase (199) and aspartate aminotransferase (273) as well as an EAAT substrate (278). While L-quisqualate and (S)-4-CPG are the most potent inhibitors of system xc−-mediated glutamate uptake, they are less well transported by system xc− than L-ibotenate and noncyclic glutamate analogs. Recently, AMPA analogues were described as new system xc− inhibitors (201). However, their specificity has not been demonstrated yet. Moreover, the glutamate analogue (4S)-4-(3-[18F]fluoropropyl)-L-glutamate (BAY 94-9392) was shown to be efficiently taken up by system xc− into tumor cells as a possible positron emission tomography (PET) tracer for system xc− activity (123).
Early evidence suggested that nonsteroidal anti-inflammatory drugs also inhibit system xc− (15). On this basis, the Gout lab identified the FDA-approved drug sulfasalazine, commonly used to treat chronic inflammatory diseases such as rheumatoid arthritis, as a potent system xc− inhibitor (79). However, this compound is also a potent inhibitor of nuclear factor kappa B (NF-κB) activation (283). In addition, glutamate uptake via system xc− has been reported to be moderately sensitive to the anion transport inhibitors 4,4′-diisothiocyanatostilbene-2,20-disulfonic acid (DIDS), 4-acetamido-4′-isothiocyanatostilbene-2,2′-disulfonic acid (SITS), and 4,4′-dinitrostilbene-2,2′-disulfonic acid (DNDS) (76).
Taken together, all pharmacological substances commonly used to study system xc− activity have off-target effects. Specific inhibitors of system xc− have not yet been discovered. Moreover, some substrate and nonsubstrate inhibitors of system xc− might have different effects when used as probes to study the role of system xc−, as substrate inhibitors will induce glutamate release in addition to inhibiting cystine uptake. Of the known substrates of system xc− not only cystine and glutamate but also β-mercaptolactate-cysteine disulfide, L-α-aminoadipate, L-homocysteate, and L-homocysteine sulfinate might act as endogenous substrates and/or competitive inhibitors for cystine uptake through system xc−in vivo.
B. The molecular biology of system xc−
Using expression cloning in oocytes and cDNA libraries from peritoneal macrophages where system xc− expression was induced by the electrophilic agent diethyl maleate (DEM) and bacterial lipopolysaccharide (LPS), Sato and co-workers identified the promiscuous 4F2 heavy chain (4F2hc/CD98/SLC3A2) as one subunit and a new 502 amino acid protein named xCT or SLC7A11 as the specific light chain subunit of system xc− (Fig. 2) (232).
The xCT protein shows significant homology with the light chains of heterodimeric amino acid transporters (HATs), a family of amino acid transporters consisting of a light chain and a heavy chain linked by a disulfide bridge [reviewed in (282)]. It was predicted to contain 12 transmembrane domains with the N- and C-termini located inside the cell and a re-entrant loop within intracellular loops 2 and 3, a general structure shared with other HAT light chains (69). As shown for other HAT family members, 4F2hc and xCT are linked by a disulfide bridge.
In mouse macrophages as well as in the murine hippocampal cell line HT22, xCT transcripts of multiple lengths were detected by Northern blotting, of which three of ∼2.5, 3.5 and 12 kb predominate (138, 232). The largest transcript was also shown to be expressed in the cerebral cortex of mice (232) and rats and in cultured astrocytes (76).
Using 5′ rapid amplification of 5′ cDNA ends, Sasaki et al. identified the transcriptional start site in the murine xCT gene (227). This start site predicts that the xCT mRNA contains a 5′ untranslated region (5′UTR) with a length of 329 bp. The longest mouse xCT cDNA (NM_011990.2) published online is 9181 bp, which contains a remarkably long 3′ untranslated region (3′UTR) of 7351 bp (www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=nucleotide&dopt=GenBank&RID=TU0CM91C016&log%24=nucltop&blast_rank=1&list_uids=80861466). Cloning of human xCT from cDNA libraries of W126Va4 cells (233), the human retinal pigment epithelial cell line ARPE-19 by the Ganapathy lab (28), and the human teratoma cell line NT2 (21) yielded putative transcripts of 1885 and 6568 bp (233), 2482 bp (28), and 3144 bp (21), respectively, which all share a 231 bp 5′UTR and a 1506 bp open reading frame (ORF), including the stop codon, but have divergent 3′UTRs. The protein encoded by these cDNAs has 501 amino acids and shows 89% identity and 96% similarity with mouse xCT (28, 233). The human reference cDNA (NM_014331.3) published online (www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=nucleotide&dopt=GenBank&RID=TU4E6EH2014&log%24=nucltop&blast_rank=1&list_uids=80861465) is 9648 bp long with a 280 bp 5′UTR and a 7862 bp 3′UTR. It was demonstrated by Northern blotting that a 12 kb transcript of xCT mRNA predominates in human fibroblasts (233) and in the human brain, which was the tissue with the highest xCT expression among the organs studied (21). Interestingly, the relative expression of the different xCT mRNA transcripts shows a tissue-specific distribution with the 12 kb form much more abundant in brain and meninges as compared with macrophages, where the 2.5 and 3.5 kb forms are more prominently expressed (234). A splice variant (hxCT1b) that results in a 495 amino acid protein with the 20 C-terminal amino acids of the 501 amino acid-long standard form (hxCTa) replaced by 13 divergent amino acids was found to be expressed at high levels in human U87 glioma cells (115). The new sequence for the 3′ end of the ORF and the subsequent 3′UTR is identical with the 3′UTR of the reference sequence (NM_014331.3) starting at bp 9245. When expressed in oocytes, hxCTb cRNA induces system xc− activity when co-injected with 4F2hc cRNA, indicating that the new C-terminus preserves xCT function (115).
In summary, the molecular identity of the 12 kb form of xCT mRNA that seems to predominate in most tissues has not been identified so far. However, several 3′UTRs of different, sometimes remarkable, lengths exist for xCT mRNA, and there is even, at least in humans, a splice variant involving the 3′UTR and part of the xCT ORF. Whether these different xCT mRNAs are involved in the regulation of xCT expression is currently unknown.
C. The phylogeny of xCT, the specific subunit of system xc−
The other members of the HAT family are the amino acid transporters LAT1/SLC7A5, LAT2/SLC7A8, y+LAT1/SLC7A7, y+LAT2/SLC7A6, b0,+AT/SLC7A9, AGT-1/SLC7A13, asc-1/SLC7A10, and the orphan transporter SLC7A15. LAT1 and LAT2 correspond to the functionally defined transporter system L, y+LAT1 and y+LAT2 to system y+L and b0,+AT to system b0,+[reviewed in (282)]. System L mediates sodium-independent exchange of large, neutral amino acids (leucine, histidine, methionine, phenylalanine, and glutamine, LAT2 also alanine), whereas system y+L exchanges extracellular neutral amino acids co-transported with Na+ with cationic amino acids. System b0,+ is a broadly active amino acid transporter that accepts diamino acids, including cationic amino acids and cystine from the outside and, with lower affinity, also large neutral amino acids and exports neutral amino acids (282). AGT-1/SLC7A13 mediates sodium-independent aspartate and glutamate transport (178). Asc-1/SLC7A10 is a sodium-independent transporter for glycine, alanine, L- and D-serine, and cysteine (67). HATs cluster with the members of the cationic transporter family cat-1/SCL7A1, cat-2/SCL7A2, cat-3/SCL7A3, SLC7A4, and the more distantly related SLC7A14 and members of the SLC12 family, transporters for sodium, potassium, and chloride, in the γ-group of the SLC transporter family (66).
The fact that system xc− uses only two proteinogenic amino acids as substrates raises the question of which evolutionary constraints might have required a rather specific cystine transporter in addition to the broadly active system b0,+. Of the nonvertebrate HAT functionally characterized so far, none exhibited system xc−-like activity. When co-expressed with 4F2hc, the SPRM1 protein from Schistosoma mansoni showed an uptake profile that shared similarities with system L and system y+L (177). Both AAT1 and AAT3, two of the three Caenorhabditis elegans putative HAT light chains, AAT1-3, showed a transport activity that shared similarities with system L and asc-1 when expressed in oocytes in parallel with ATG-2, the heavy chain ortholog of C. elegans (281). AAT2 has not been analyzed so far. The HAT light chain encoded by the Drosophila melanogaster gene JhI-21 showed system L activity when co-expressed with the D. melanogaster homolog of 4F2hc/CD98 (216). Reynolds and colleagues also reported that a BLAST search with human LAT1 and LAT2 highlighted five fly amino acid transporters that share a significant degree of homology (homology to human LAT1 and LAT2 in parentheses), CG1607 (47% and 46%), Minidisc (Mnd or CG3297; 49% and 44%), Genderblind (Gb or CG6070; 44% and 40%), JhI-21 (CG12317; 49% and 44%), and CG9413 (38% and 34%). All these molecules share the conserved cysteine that covalently links the light chain to the heavy chain (Cys164 in LAT1). Moreover, they showed that siRNA against CG2791, Mnd, and JhI-21 also down-regulated system L activity in D. melanogaster Schneider cells (216). Surprisingly, Augustin et al. identified the same five Drosophila proteins as putative xCT homologs with 36% to 45% amino acid similarity to murine and human xCT, respectively, and showed that Gb (CG6070) regulates hemolymphe glutamate levels, which was taken as supportive evidence that Gb acts as a system xc−-like transporter in Drosophila (6).
In order to clarify the exact distribution of xCT orthologs in the tree of life, we decided to generate a phylogenetic analysis of xCT proteins from as many vertebrate species as available and also included nonvertebrate representatives of the phylum Chordata and the related phyla Hemichordata and Echinodermata, the five putative D. melanogaster LAT1/2 or xCT homologs, and the three HAT light chain proteins from C. elegans and Schistosoma japonsensis with characterized function. We also included HAT orthologs from four evolutionarily separated vertebrates, the mammal Homo sapiens, the reptile Anolis carolinensis, the frog Xenopus tropicalis, and the fish Danio rerio to avoid species-specific changes or artefacts and to delineate the phylogenetic relationship of questionable xCT orthologs such as Gb within the HAT protein family. The distantly related SLC7A15 transporter was used as an outlier.
The resulting phylogenetic tree nicely separated the eight distinct vertebrate transporter families and is in line with a previous analysis (66). All vertebrates included in the analysis contain an xCT/SLC7A11 ortholog, indicating an important role for xCT in vertebrate amino acid metabolism. Moreover, the tree shows that all analyzed members of the superphylum Deuterostomia (Echinodermata, Hemichordata, and Chordata) also have an xCT/SLC7A11 ortholog. The most phylogenetically primitive animal that seems to have a gene which represents a true xCT ortholog is the sea urchin Strongylocentrotus purpuratus. In addition, our results show that the D. melanogaster HATs cluster with either the LAT/asc-1 or the b0,+AT/SLC7A9 branch and not with xCT/SLC7A11 orthologs (Fig. 3). Based on the data presented here, we conclude that xCT orthologs are only present in Deuterostomia and seem to be absent in the superphylum Protostomia, to which insects belong. Thus, xCT, and presumably also the function of xCT, namely glutamate/cystine antiporter activity, appears to be evolutionarily new. The identification of the specific alterations in amino acid metabolism that are common to all Deuterostomia and might have required system xc− activity requires further investigation.
FIG. 3.
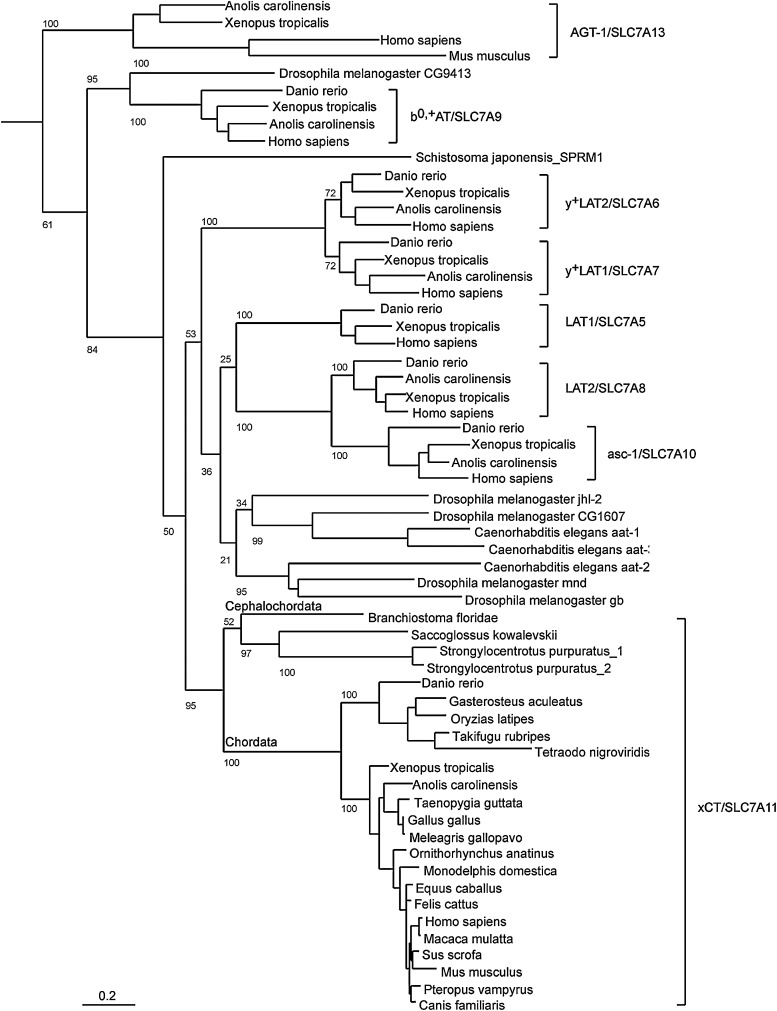
Phylogenetic analysis. Phylogenetic analysis of xCT/SLC7A11 orthologs and related heterodimeric amino acid transporter (HAT) proteins. xCT proteins from vertebrates, cephalochordata, hemichordata, and echinodermata; five putative Drosophila melanogaster LAT1/2 or xCT homologs; the three nonvertebrate HAT light-chain proteins from Caenorhabditis elegans and Schistosoma japonensis with a characterized function; and HAT orthologs from four evolutionarily separated vertebrates were included in the analysis. The distantly related SLC7A14 transporter was used as an outlier. Proteins were identified by BLAST search and used to generate multiple sequence alignments. The output was then used to generate a phylogenetic analysis using Maximum Likelihood and 100 bootstrap steps. The number of bootstrap iterations resulting in the branching shown is given as a number in front of each branch. The scale bar shows 0.2 (or 20%) sequence divergence (for methods see Supplementary Data [available online at www.liebertpub.com/ars]).
D. Regulation of system xc− by transcriptional regulation of its specific subunit xCT
Early functional studies indicated that system xc− represents a highly inducible amino acid transport system. Stimuli that lead to a strong induction of system xc− activity in vitro include not only oxygen (18) and diverse electrophilic agents (12) but also, in certain cell types, bacterial LPS and the inflammatory cytokine tumor necrosis factor α (TNFα) (228). Even regular cell culture conditions with ambient O2 of 21% induce system xc− activity in diverse cell types (226, 261, 286).
Experimental evidence strongly suggests that the transcriptional regulation of xCT expression is by far a much more important determinant for system xc− activity than the expression of the heavy chain, 4F2hc, which is known to form heterodimers with at least five other specific amino acid transporter light chains (282). Overexpression of the xCT light chain alone robustly increases system xc− activity in diverse cell types, including NIH3T3 fibroblasts (284), HEK 293 cells (251), murine hippocampal HT22 cells (142), and astrocytes (250). Moreover, induction of system xc− activity by treatment with the NO donor 3-nitroso-N-acetylpenicillamine in the retinal pigment epithelial cell line ARPE-19 (28) and by tert-butylhydroquinone in HT22 cells (142) was associated with induction of xCT expression alone with no apparent regulation of 4F2hc. Thus, to shed light onto the transcriptional regulation of system xc− activity, most studies focussed on analysis of the xCT promoter.
In 2002, Sasaki and co-workers identified four putative EpREs in the proximal 5′ flanking region of the mouse xCT gene. They showed that the DEM-dependent activation of the xCT promoter in BHK21 hamster kidney cells required the most 5′ EpRE-like element in the xCT promoter. Consistent with this observation, DEM induces system xc− activity and the activity of the xCT promoter, as judged by luciferase reporter analysis, in wild-type but not in Nrf2−/− fibroblasts (227). Moreover, in cultured murine peritoneal macrophages, induction of system xc− activity by oxidative insults, including exposure to glucose oxidase (which generates H2O2), DEM, the superoxide generator paraquat, and the heavy metal cadmium depends on the presence of Nrf2, as induction was absent in macrophages derived from Nrf2−/− mice (101). However, the induction of system xc− either by in vitro culture for 12 h or stimulation by LPS was diminished but not abolished, indicating that Nrf2-independent pathways are also involved (101). Nrf2 also regulates xCT expression in rat primary cortical astrocytes (250). In addition, it was demonstrated that the neuroprotective antibiotic ceftriaxone induces xCT expression and/or system xc− activity in multiple cell types, including hippocampal HT22 cells, cortical and spinal astrocytes, and stem cell-derived motor neurons (136). Ceftriaxone increases nuclear Nrf2 levels and the protective activity of ceftriaxone as well as the effect of ceftriaxone on GSH levels was largely diminished in fibroblasts deficient in Nrf2. Hence, it was concluded that the ceftriaxone-mediated up-regulation of xCT is Nrf2 dependent (136). Thus, xCT and thereby system xc− activity is connected to the network of stress-inducible GSH metabolic enzymes that are coordinately regulated by Nrf2, which on induction by oxidative stress orchestrates the re-establishment of cellular redox homeostasis (see section I.B).
In early works on the regulation of system xc− activity, Shiro Bannai reported that system xc− activity is induced not only by electrophilic agents but also by cystine starvation (12). Of note, cystine starvation causes GSH depletion and thereby also oxidative stress (263). In 2004, Sato et al. reported that the induction of system xc− activity and xCT expression is not only specific for cystine starvation but also occurs when other amino acids are depleted from the medium (230). The common pathway that mediates transcriptional induction by amino acid limitation includes activation of general control non-derepressible-2 (GCN2) protein kinase by free tRNAs and subsequent phosphorylation of the translation initiation factor eIF2α [reviewed in (114)]. eIF2α phosphorylation inhibits cap-dependent translation, while several transcripts, including those of activating transcription factor 4 (ATF4), are preferentially translated. ATF4 heterodimerizes with members of the CCAAT/enhancer-binding protein (C/EBP) and AP1 families and activates transcription on binding to the amino acid response element (AARE). Subsequently, it was shown that the proximal promoter of xCT contains a tandem of two AAREs, the more 5′ of which binds the transcription factor ATF4, and both AAREs cooperatively mediate the activation of the xCT promoter after amino acid starvation (230). The two AAREs, with an intervening sequence of 9 bp oriented in the opposite direction, are completely conserved among mouse, rat, bovine, and human xCT 5′ flanking regions (140), indicating that this mechanism is functionally important across species. While 4F2hc is also induced by amino acid limitation (230) and its expression is suppressed in ATF4-deficient cells (87), the molecular mechanism underlying its regulation remains to be clarified.
At least in cell culture, the phosphorylation status of eIF2α plays a pivotal role in the basal activity of system xc−, as embryonic fibroblasts derived from mice with a homozygous mutation of the eIF2α phosphorylation site, serine 51 to alanine, show very little ATF4 expression, xCT promoter activity, or system xc− activity (140). Moreover, increased eIF2α phosphorylation brought about by the specific eIF2α phosphatase inhibitor salubrinal induced ATF4 protein and system xc− activity in hippocampal HT22 cells and the rat phaeochromocytoma cell line PC12 (140). Through induction of ATF4 and xCT, eIF2α phosphorylation regulates cellular GSH concentrations and cellular sensitivity to oxidative stress (140). Interestingly, PC12 cells selected for resistance against oxidative damage by the amyloid-β peptide, which is involved in the pathogenesis of AD, exhibit a strong activation of the phospho-eIF2α/ATF4/xCT signaling module (140). Since eIF2α phosphorylation and ATF4 expression were also found to be up-regulated in AD brains (140), this pathway might represent an adaptive response to oxidative stress in AD.
GCN2 is only one of four eIF2α kinases. The other three, protein kinase R (PKR), heme-regulated eIF2α kinase (HRI), and PKR-like kinase (PERK), are activated by diverse stimuli [reviewed in (287)]. While GCN2 is activated by not only amino acid deprivation, but also other stresses such as UV radiation and proteasome inhibition, PKR participates in an anti-viral defense mechanism that is mediated by interferon. HRI responds to haem deprivation and oxidative and heat stress in erythroid tissues, and PERK is responsive to ER stress. Thus, in theory, induction of xCT might occur after a variety of cellular insults, which are all relayed through eIF2α and ATF4. However, this hypothesis remains to be experimentally explored.
Inflammatory stimuli that strongly induce system xc− in cells include LPS and TNFα (228). LPS binds to toll-like receptor 4 and thereby activates multiple signaling cascades, including activation of NF-κB, an important mediator of inflammation-induced gene transcription [reviewed in (291)]. A putative binding site for NF-κB was identified in the murine xCT gene 5′ flanking region (229). However, even LPS concentrations too low to activate NF-κB strongly stimulated xCT expression in macrophages (229). Thus, the signaling pathway through which LPS induces xCT expression remains to be clarified. Similarly, how TNFα, a proinflammatory cytokine that binds to two different receptors, which activate multiple intracellular signaling pathways, including NF-κB [reviewed in (84)], activates system xc− has not been clarified. Other extracellular ligands that have been reported to induce xCT expression, also without any detailed information about the intracellular signaling pathways involved, include erythropoietin and interleukin-1β (IL-1β). Erythropoietin has been reported to up-regulate xCT expression in differentiated cortical neural stem cells and B104 neuroblastoma cells (255). The inflammatory cytokine IL-1β specifically up-regulates system xc− activity in astrocytes but not in microglia and neurons by activating the IL1-receptor via induction of xCT but not 4F2hc expression (64, 105). In addition, fibroblast growth factor-2 (FGF-2) induces system xc− activity through induction of xCT expression in astrocytes, but not in neurons or microglia, through activation of fibroblast growth factor receptor 1 (FGFR1), a pathway sensitive to the combined inhibition of the MEK/ERK and phosphoinositide-3 kinase pathways (151). Interestingly, post-transcriptional mechanisms also regulate xCT mRNA levels. The microRNA-26b has been demonstrated as directly targeting and down-regulating xCT transcript expression (153). The pathways that regulate the levels of the xCT transcript are summarized in Figure 4.
FIG. 4.
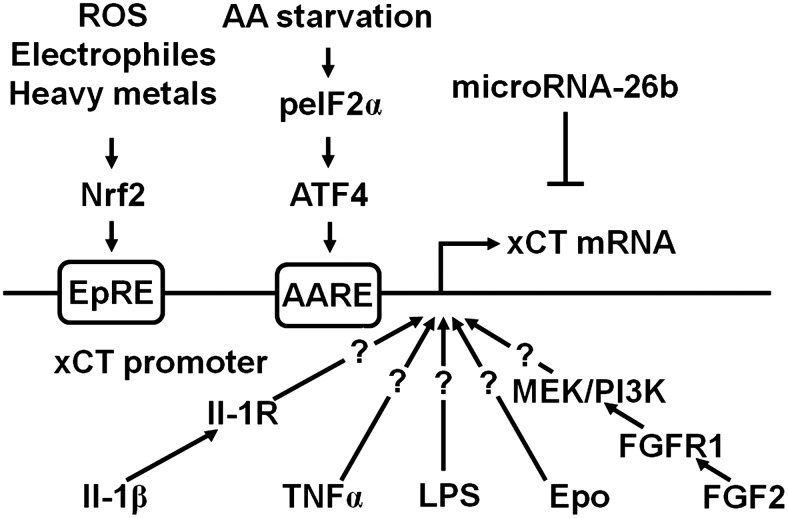
Transcriptional regulation of xCT expression. A variety of stimuli (see section I.B), for example, electrophiles, heavy metals, and reactive oxygen species (ROS), lead to activation of the nuclear factor NF-E2-related factor 2 (Nrf2), which binds to the electrophile response element (EpRE) within the xCT promoter region and activates transcription. Amino acid (AA) starvation leads to phosphorylation of eIF2α (peIF2α), which leads to the translational up-regulation of the transcription factor activating transcription factor 4 (ATF4). ATF4 activates the transcription of xCT by binding to the amino acid response element (AARE) contained in the xCT promoter. Bacterial lipopolysaccharides (LPS), tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), fibroblast growth factor-2 (FGF2), and erythropoietin (EPO) also increase the transcription of xCT through unknown or partially known signaling pathways. IL-1β acts via the IL-1 receptor (IL-1R), while FGF2 activates the FGF receptor 1 (FGFR1) and increases xCT transcription via PI3K and MEK. MicroRNA-26b directly targets xCT mRNA.
E. Regulation of system xc− activity by protein trafficking and protein modification
Although the heavy chain, 4F2hc, is necessary for correct membrane trafficking and insertion of a functionally active system xc−, 4F2hc does not seem to be prominently involved in the regulation of system xc− activity (see above). However, the adhesion molecule CD44v, which is expressed in cancer stem cells, was shown to interact with the xCT-4F2hc heterodimer and regulate its membrane insertion and thereby activity (103). The laboratory of Sylvia Smith demonstrated that a ∼40 kD band immunoreactive for xCT as detected by Western blotting switched from an intracellular localization to the plasma membrane compartment on exposure of retinal Müller glial cells to oxidative stress, whereas the more predominant ∼50 kD band with constitutive membrane insertion did not show any regulation (191). This switch of the ∼40 kD band to the membrane was associated with increased system xc− activity. However, the molecular basis of the different sizes of xCT in Müller cells and whether this pathway is active in other cell types has not been investigated.
Moreover, system xc− activity was found to be down-regulated by signaling through metabotropic group II receptors, which suppress cyclic adenosine 3′,5′ monophosphate (cAMP) synthesis and thereby protein kinase A (PKA) activation, in a cAMP-dependent manner in striatal slices (9). Human xCT contains two putative PKA phosphorylation sites (9). However, direct evidence for the regulation of transport activity or membrane insertion by phosphorylation of xCT has not been published.
F. Regulation of system xc− activity by substrate availability
Since extracellular glutamate is a competitive inhibitor of cystine import via system xc− while intracellular glutamate drives cystine import, pathways that regulate the intra- as well as the extracellular glutamate concentrations might be potent, indirect regulators of cystine import via system xc−. In fibroblasts, glutamine was shown to be taken up via system ASC and converted to glutamate to activate cystine import via system xc−(13). In 2000, Rimaniol et al. reported that system xc− as well as EAAT1 and EAAT2 are expressed in human monocyte-derived macrophages (217). Glutamate amplified the cystine-induced increase in GSH levels, an effect that was sensitive to EAAT inhibitors. This indicated that glutamate uptake via EAATs trans-stimulates cystine import via system xc−. Moreover, the same group reported that not only glutamate but also glutamine and aspartate stimulated GSH synthesis in these cells. These observations led to the hypothesis that while glutamate uptake via EAATs directly transactivates system xc−, the uptake of glutamine and L-aspartate, which are then converted to glutamate by glutaminase and aspartate aminotransferase, respectively, also contribute to the intracellular glutamate pool that drives cystine import (218). In addition, some of the glutamate released by system xc− is the subject of immediate re-uptake by EAATs (138, 202). In hippocampal HT22 cells, transient overexpression of the neuronal EAAT, EAAT3, increased intracellular GSH in the presence of high glutamate concentrations and protected HT22 cells from oxidative glutamate toxicity. These effects were especially pronounced when EAAT3 was co-overexpressed with xCT (138). In summary, cystine uptake via system xc− can be supported by multiple pathways that increase the intracellular concentration of the exported substrate glutamate. Glutamate uptake via EAATs, in theory, might be especially effective, because it causes a concomitant decrease in the extracellular glutamate that inhibits cystine uptake (Fig. 5).
FIG. 5.
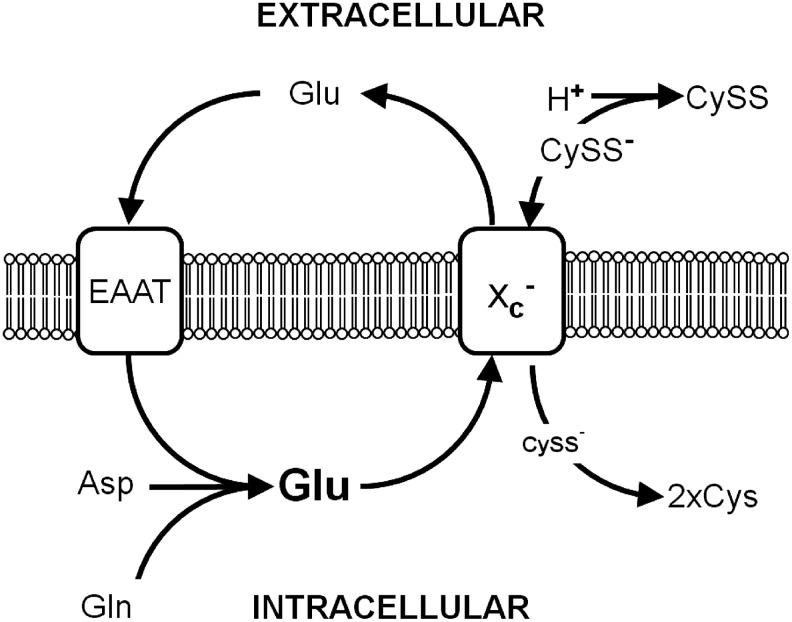
Regulation of system xc− by substrate availability. Glutamate released by system xc− is taken up by excitatory amino acid transporters (EAAT). Intracellular glutamate, which also can be synthesized from aspartate (Asp) and glutamine (Gln), fuels import of the anionic form of cystine (CySS−) by system xc−. Extracellular protons (H+) lead to the formation of neutral cystine (CySS), which is not accepted as a substrate by system xc−.
Early reports about the substrate requirements of system xc− indicated that the exchanger only accepts the anionic form of cystine (16) (Fig. 5). The two amino groups of cystine have pK values of 7.48 and 9.02. As a result, at physiological pH, the neutral (cystine I) and the anionic (cystine II) forms of cystine predominate, while at alkaline pH, cystine is predicted to exhibit two negative charges (cystine III). A decrease in pH reduces the concentration of cystine II, the substrate of system xc−. As a consequence, a shift to a more acidic pH decreased cystine uptake, whereas the uptake of glutamate, which does not change its ionic state within this pH range, was hardly affected (16, 21, 137). The regulation of system xc− by pH might be of pathophysiogical relevance, as many disease states, including critical illness (4, 44) and diabetes (279), are associated with both acidosis and oxidative stress. Inhibition of system xc− by lactate in rat cortical astrocytes has been reported (124). However, Lewerenz and co-workers could demonstrate that in the hippocampal cell line HT22, system xc− is resistant to extracellular lactate in concentrations of approximately 20 mM (137). The reason for this discrepancy is not known.
III. Expression of System xc− In Vitro and In Vivo and Its Functional Consequences
A. In the absence of disease, system xc− shows a rather restricted expression pattern in vivo
Northern blot analysis revealed that xCT mRNA is prominently expressed in the brain in mice (232). Even higher expression levels of xCT were found in the thymus and spleen, tissues that belong to the immune system (260). No xCT expression or very low levels were found in lung, heart, liver, and kidney. In human tissues, Kim et al. also detected high levels of xCT expression in the brain and spinal cord. However, very low or no expression of xCT was detected in peripheral leucocytes, spleen, thymus, and lymph nodes (115). xCT mRNA was also demonstrated to be present in the pancreas (115).
The expression of xCT in the brain was confirmed in the protein level by Western blotting and immunohistochemistry (30, 176, 250). Especially high levels were found in the meninges (250). Functionally, as judged by the uptake of L-aminoadipate in brain slices and the subsequent detection of L-aminoadipate-like immunoreactivity, astrocytes seem to show the highest system xc− activity (209). Along with high expression levels in the meninges, compatible with the results of Shih et al. (250), nonradioactive in situ hybridization showed expression of xCT only in restricted areas of the mouse brain, including the area postrema, subfornical organ, habenular nucleus, hypothalamic area, and ependymal cells of the lateral wall of the third ventricle (234). These results are in contrast to data that indicate the presence of xCT protein in the cortex, hippocampus, cerebellum, and striatum (250) and functional system xc− activity in diverse areas of the brain, including the nucleus accumbens, the striatum, and the hippocampus (8, 9, 48). Most probably, these differences are explained by the limited sensitivity of nonradioactive in situ hybridization. No or very low system xc− activity has been reported in freshly prepared hepatocytes (261), macrophages (286), and granulocytes (226).
Taken together, the expression of xCT and system xc− seems to be rather restricted in vivo. Lymphoid organs, although the data in mice and humans are conflicting, and the CNS are the primary tissues of constitutive xCT expression. The detailed expression patterns of xCT in the eye, also a part of the CNS, are discussed next (see section IV.D). Clearly, more work needs to be done to fully characterize xCT expression in vivo.
B. System xc− is induced in most cultured cells
As described in section II.D, system xc− is readily induced in primary cells of diverse origin on culture in vitro (18, 101, 226, 261, 286), most probably due to the high partial pressure of oxygen (18). In contrast, prolonged culture of fibroblasts under conditions of reduced oxygen partial pressure decreases the activity of system xc− almost fivefold (18). Further research is needed to determine whether culture conditions such as these help to retain a pattern of system xc− expression that more closely resembles the expression pattern in vivo.
The dependence of cultured cells on system xc−-mediated cystine import is illustrated by the fact that cells derived from xCT knock-out (−/−) mice or mice with a naturally occurring deletion in the xCT gene fail to grow and finally die in vitro unless thiol-containing antioxidants are added to the cell culture medium (192, 231, 250). Thus, it is not surprising that system xc− was detected in numerous cell lines and cultured primary cells, even though no xCT expression had been demonstrated either in the tissue of origin or in freshly prepared cells (226, 261, 286). However, there are some notable exceptions. First, lymphocytes and T cell lines seem to be incapable of inducing system xc− in culture (73, 100, 102). In contrast, the cysteine transporter system ASC is up-regulated on activation (73, 102). As a consequence, these cells require either β-mercaptoethanol in the culture medium or a feeder layer of fibroblasts that can provide the immune cells with cysteine (60, 100). Second, some reports indicate that mature neurons in culture cannot use cystine as a GSH precursor in vitro (125, 221). This is surprising, because robust system xc− activity was demonstrated in freshly dissociated brain cells from rat E17 embryos (220), a developmental stage where astrocytes have not been differentiated yet, and immature neuronal cultures take up cystine and die on inhibition of system xc− (190). The existence of cystine/cysteine cycling between astrocytes and neurons was hypothesized with astrocytes taking up cystine via system xc− and releasing cysteine or GSH, which is then metabolized to cysteine, for neuronal GSH synthesis (55, 221). Thus, astrocytes in mixed neuronal/astrocytic cultures may play a similar role as the fibroblast feeder layer in lymphocyte cultures and compensate for insufficient system xc− expression.
C. The role of system xc− in the regulation of GSH synthesis, the extracellular redox milieu, and extracellular glutamate levels
The most obvious function of system xc− activity in vitro is the delivery of cystine, which is intracellularly reduced to cysteine, for GSH synthesis. This is demonstrated by the fact that in many cells, including neuronal cell lines, in vitro inhibition of system xc− activity induces GSH depletion and cell death by oxidative stress, a cell death pathway termed oxidative glutamate toxicity [reviewed in (2)] (see section III.D). Furthermore, induction of system xc− activity by eIF2α phosphorylation and subsequent ATF4 up-regulation prominently increased cellular GSH levels in PC12 cells (140). However, plasma levels of reduced GSH are lowered by more than 50% in xCT−/− mice (231), whereas deletion of xCT does not decrease striatal or hippocampal GSH levels (48, 175), indicating that in vivo alternative mechanisms can compensate for system xc− deficiency to maintain the intracellular GSH pool.
System xc− has also been implicated in the cellular regulation of the extracellular cystine/cysteine redox couple via several different mechanisms. GSH synthesized from cyst(e)ine taken up by system xc− is, in part, exported from cells and thereby can regulate extracellular cysteine levels by disulfide exchange with cystine, leading to the formation of GSH-cysteinyl disulfide and cysteine or by catabolism of GSH through GGT and dipeptidases (55, 285). However, early evidence in fibroblasts suggested that cystine taken up by cells via system xc− and immediately reduced to cysteine might be directly exported by system ASC, leading to an increase in extracellular cysteine concentrations without the intermediate step of GSH synthesis (14). In line with this observation, Anderson et al. demonstrated that the regulation of the extracellular cystine/cysteine redox couple occurs independently of GSH synthesis, export, and extracellular GSH redox regulation (3). Interestingly, Banjac et al. showed that overexpression of xCT in the Burkitt lymphoma cell line HH514 BL does not change the intracellular GSH pool but rather increases extracellular cysteine concentrations. These findings suggest that the import of cystine via system xc−, its intracellular reduction, and the subsequent release of cysteine might be the mechanism through which cells modify the redox state of the extracellular cystine/cysteine redox couple (10). Taken together, these data suggest that system xc− might not only provide intracellular cysteine for GSH synthesis but may also comprise, along with TRR1 or intracellular GSH, which reduce imported cystine, and system ASC, which can act as a cysteine exporter, the third critical constituent of a molecular machinery through which cells adjust their extracellular environment to a more reduced state (Fig. 6). The importance of this system is supported by the observation that xCT−/− mice show a more oxidized state of the cysteine/cystine redox couple in their plasma (231). In human plasma, a more oxidized state of the cysteine/cystine redox couple is associated with risk factors for cardiovascular disease; for example, aging, smoking, and obesity and rodent and vascular cell studies show that the extracellular cysteine/cystine redox state can play a vital role in controlling cardiovascular disease through proinflammatory signaling [for review see (75)].
FIG. 6.
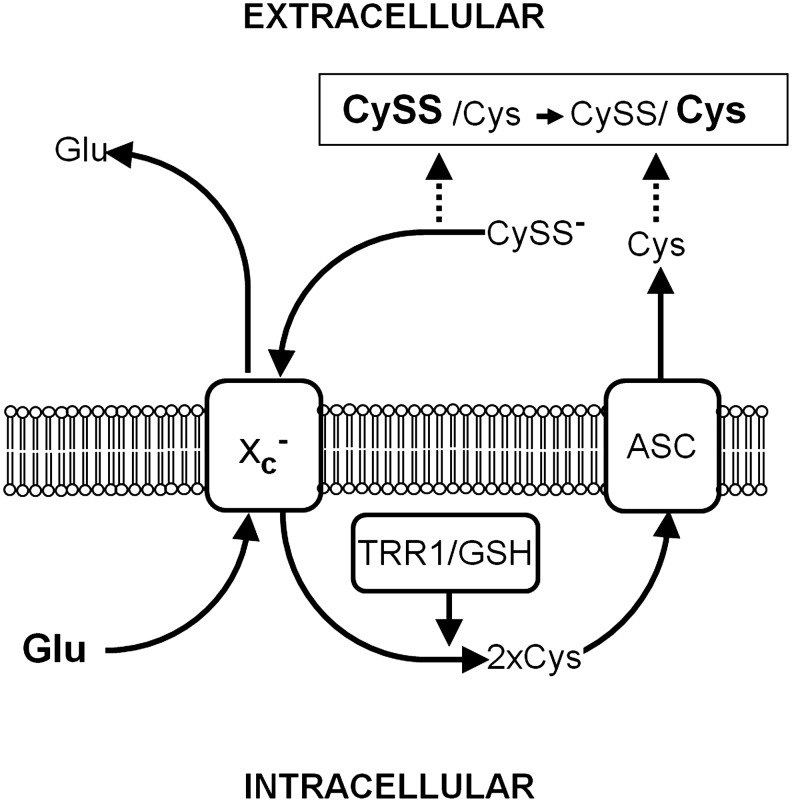
System xc− regulates the extracellular cystine/cysteine redox couple. Cystine (CySS−) imported by system xc− is intracellularly reduced to cysteine (Cys) by thioredoxin reductase 1 and/or GSH (TRR1/GSH). Cys can be directly exported by system ASC; therefore, system xc− changes the ratio of the extracellular CySS/Cys redox couple in favor of Cys.
Since system xc− exports one molecule of glutamate for each molecule of cystine imported into the cell, it should also impact extracellular glutamate concentrations with a plethora of consequences (see section I.C). Indeed, cystine concentrations in the physiological range increased extracellular glutamate levels in hippocampal slices (185). Moreover, several groups demonstrated in microdialysis experiments in vivo that system xc− inhibitors reduced extrasynaptic glutamate concentrations in the striatum and hippocampus (9, 48). The role of system xc− in regulating extracellular glutamate levels in the brain was independently substantiated by microdialysis experiments in xCT−/−mice, which showed reduced extracellular glutamate concentrations in the striatum and hippocampus (48, 175).
In summary, cystine import via system xc− is involved in (i) GSH synthesis and thereby the enhancement of the intracellular defenses against oxidative stress; (ii) the modification of the extracellular redox milieu; and (iii) the regulation of extracellular glutamate levels.
D. Oxidative glutamate toxicity—an in vitro paradigm for neuronal death induced by system xc− inhibition
1. The cell death pathway in oxidative glutamate toxicity
In 1989, Murphy and colleagues reported that in the N18-RE-105 neuroblastoma X retina cell line, glutamate induced calcium-dependent cell death by inhibition of cystine import via system xc−, resulting in GSH depletion and oxidative stress (189). This type of cytotoxicity has been named oxidative glutamate toxicity or oxytosis (264) and has been extensively studied by the Maher lab in the hippocampal cell line HT22, a glutamate-sensitive subclone of the hippocampal cell line HT4 (47). Oxidative glutamate toxicity is distinct from excitotoxicity where increased extracellular glutamate over-stimulates ionotropic glutamate receptors, thereby leading to a massive calcium influx and rather rapid nerve cell death (38). Most notably, the sensitivity to oxidative glutamate toxicity is dependent on cell density, with higher densities rendering cells more resistant to system xc− inhibition (Fig. 7A). GSH, which is cell impermeable, dose-dependently protects HT22 cells against glutamate toxicity but not cystine-free medium (Fig. 7B). Since HT22 cells release GSH (137) and do not express GGT (222), some of the protection by high cell density might be a consequence of GSH release followed by disulfide exchange reactions between GSH and cystine, leading to the generation of cysteine, which can then be taken up by transporters other than system xc−.
FIG. 7.
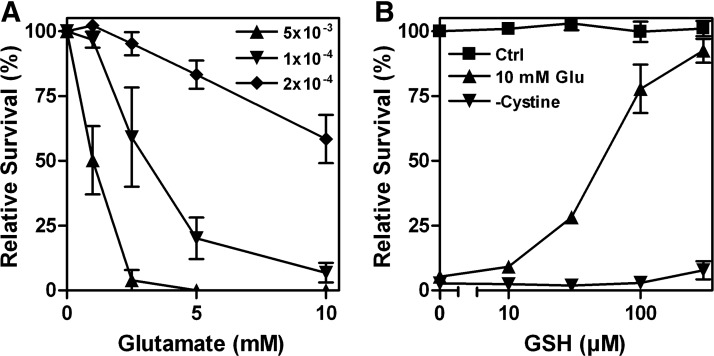
Cell density and disulfide exchange of cystine with extracellular GSH regulate sensitivity of HT22 cells to system xc− inhibition by glutamate. (A) HT22 cells were seeded at the indicated densities per well in 96-well plates, and glutamate was added after 24 h. (B) After 24 h in culture, HT22 cells were treated with the indicated concentrations of GSH in the absence of glutamate in a normal medium (Ctrl), along with 10 mM glutamate (10 mM Glu) or with a medium exchanged for a cystine-free medium (-Cystine). (A/B) Survival was measured by the MTT assay after 24 h and normalized to cells not treated with glutamate (A) or GSH (B). Graphs represent the means of three independent experiments.
The series of events leading to cell death by oxytosis have been quite well characterized, although some questions and controversies remain. Following the inhibition of system xc− by glutamate, GSH levels drop in a time-dependent manner. When the GSH levels fall below ∼20% (about 6 h after glutamate treatment), ROS start to increase exponentially (263). It is important to note that the ROS themselves do not kill the cells but rather give rise to the activation of signaling pathways which culminate in cell death. Thus, the accumulation of large amounts of intracellular ROS is not sufficient to cause death, but it is a necessary step in the cell death process. Consistent with this idea, compounds that block signaling pathways downstream of ROS accumulation can be protective even in the presence of elevated levels of ROS (e.g., 170, 245). The major source of these ROS appears to be complex I of the mitochondrial electron transport chain (263). The importance of mitochondrial ROS production is supported by the observation that the mitochondrial uncoupler cyanide p-trifluoromethoxyphenylhydrazone (FCCP) and other mitochondrial inhibitors protect nerve cells from oxidative glutamate toxicity (263). However, other sources of ROS, including the NADPH oxidase Nox4 (85) and lysosomes (126), may also contribute to the increase in ROS. GSH depletion also results in the activation of 12/15 lipoxygenase (12/15-LOX) (144) most probably because of the resulting inhibition of GSH peroxidase 4 (GPx4) (242), which depends on an adequate supply of GSH for activity. GPx4 is unique in its ability to reduce lipid hydroperoxides embedded in membranes (97). Activation of 12/15-LOX generates 12- and 15-hydroxyeicosatetraenoic acid. These eicosanoids activate soluble guanylate cyclases, which then generate cGMP (145). Elevated cGMP eventually opens an uncharacterized calcium channel, resulting in a detrimental influx of calcium (145). Activated 12/15-LOX may also have direct effects on mitochondria, thereby further increasing ROS production (197). About 10–12 h after the induction of oxidative glutamate toxicity, when both ROS and intracellular calcium levels have reached their maximum, the pro-apoptotic Bcl-2 family member BH3-interacting domain death agonist (Bid) translocates to the mitochondria, and Bid-loaded mitochondria accumulate around the nucleus and lose their membrane integrity (131). At this time, apoptosis-inducing factor (AIF) translocates from the mitochondria to the nucleus, where it rapidly induces caspase-independent cell death (131). The Maher laboratory observed a transient hyperpolarization of the mitochondria during the exponential increases in ROS and intracellular calcium (264), whereas others reported depolarization on the release of AIF from the mitochondria in the final phase of oxidative glutamate toxicity (131). However, the question of whether these divergent observations result from distinct properties of the HT22 cells in the different laboratories or whether hyperpolarization actually precedes the final depolarization has not been answered yet. Although caspase-3 is activated during oxidative glutamate toxicity in HT22 cells (131), caspase inhibitors do not inhibit cell death (59, 131). However, several protease inhibitors protect against oxidative glutamate toxicity, indicating a role for serine proteases and calpains in the cell death pathway (59, 265). The series of events taking place after the induction of oxytosis is depicted in Figure 8.
FIG. 8.
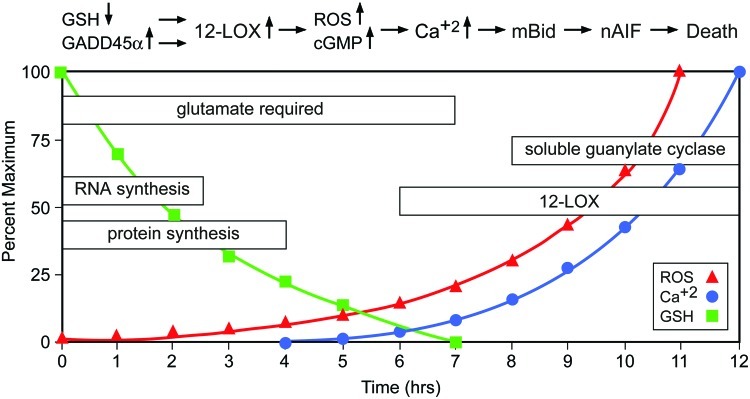
Oxidative glutamate toxicity—neuronal cell death induced by system xc− inhibition in vitro. In hippocampal HT22 cells, glutamate-mediated inhibition of cystine uptake via system xc− causes a decrease of intracellular GSH. GSH levels below 20% lead to an increase of ROS. In the early phase, de novo synthesis of RNA and proteins is necessary. One of the proteins induced is GADD45α. 12-lipoxygenase (12-LOX) and soluble guanylate cyclase are activated and mediate an accumulation of ROS and cGMP, and, subsequently, Ca++ influx. Ca++ induces cell death mediated by truncated BH3-interacting domain death agonist (Bid) and nuclear translocation of apoptosis-inducing factor (nAIF) (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Since macromolecular synthesis inhibitors block oxidative glutamate toxicity in HT22 cells (265), synthesis of RNA and protein seems to be required for the execution of the cell death pathway. A recent study provided further insight into that requirement by demonstrating a critical role for p53-mediated GADD45α synthesis in glutamate-mediated cell death in HT22 cells (39), but the precise relationships between GSH depletion, ROS production, and p53 activation still remain to be determined. However, others have argued that inhibition of protein synthesis simply reduces the consumption of cysteine, which is then used for GSH synthesis (215). This argument is contradicted by the observation that macromolecular synthesis inhibitors also protect HT22 cells from cell death induced by treatment with buthionine sulfoximine, an inhibitor of GSH synthesis (265).
While oxidative glutamate toxicity has some of the features of apoptosis such as the requirement for protein synthesis, it is missing many others such as nuclear and DNA fragmentation and chromatin condensation (265). HT22 cells exposed to glutamate for 10 h show severe damage to their ER, Golgi apparatus, and mitochondria (265). These observations as well as many of the other features of oxytosis are consistent with a recently described form of cell death called necroptosis [for review see (277)]. Necroptosis is a regulated form of necrosis that is dependent on ROS production from mitochondrial complex I and induction of caspase-independent, AIF-mediated cell death (23). Importantly, a highly specific inhibitor of necroptosis, necrostatin 1 (Nec-1), was identified (49) and can be used to determine the contribution of necroptosis in various cell death paradigms. Nec-1 was shown to inhibit oxidative glutamate toxicity in HT22 cells, at least in part, by preventing the nuclear translocation of AIF (289). Together, these observations suggest that oxytosis is a form of necroptosis. In vitro, oxytosis has been studied using neuronal cell lines (47, 189) as well as primary cultures of cells originating from the CNS, including oligodendrocytes (193), astrocytes (33), and immature cortical primary neurons (190, 240). In immature primary neurons, glutamate excitotoxicity does not occur, as ionotropic glutamate receptors are not yet fully expressed (240). However, even in maturing primary neuronal cultures, which become increasingly susceptible to excitotoxicity, it was observed that after a short exposure to glutamate, cell death in a subset of the neurons occurs by oxidative glutamate toxicity (240).
Two of the major practical advantages of the HT22 model for oxytosis are its reproducibility and the simplicity of the experimental procedure. Moreover, the oxidative stress induced in this model is not due to externally applied oxidants, as for example in H2O2 toxicity, but rather the ROS are generated endogenously within the cell and, thus, this kind of oxidative stress might, in theory, be more closely related to oxidative stress occurring in vivo. Of note, in immature primary neurons, oxidative glutamate toxicity generally yields the same results as in HT22 cells. Other lines of evidence suggesting the general importance of this model is that HT22 cells selected for resistance to oxidative glutamate toxicity were shown to be also less vulnerable to amyloid-β toxicity, an in vitro model for AD (46), ER stress brought about by the glycosylation inhibitor tunicamycin, and over-expression of the pro-apoptotic protein Bax (50). Although, at least in part, the sensitivity of cells to system xc− inhibition is an artifact of the increased dependence of cells in culture on cystine import for GSH synthesis, together these data highlight the pathophysiological importance of oxytosis and the potential therapeutic use of compounds that protect against this particular kind of cell death.
2. Using oxidative glutamate toxicity to identify neuroprotective pathways
Due to its simplicity and reproducibility, the HT22 model of oxidative glutamate toxicity is an excellent tool to screen for and analyze pathways involved in both neuroprotection and GSH metabolism. In particular, the role of different G-protein coupled receptors (GPCRs) in oxidative glutamate toxicity has been extensively studied. Dopamine D4 receptor activation protects by inhibition of ROS production without affecting GSH depletion (98). Activation of group I mGluRs protects by increasing GSH levels (224). Activation of stimulatory G proteins attenuated the glutamate-induced accumulation of ROS and calcium influx, at least in part, by causing an increase in GSH due to improved uptake of cystine mediated by the induction of xCT or, additionally, by the up-regulation of the anti-apoptotic protein Bcl-2 (139).
Protein kinase C (PKC) is activated by Gq-coupled receptors via phospholipase C and diacylglycerol. Phorbol esters also activate PKC and in a series of studies (47, 164), the Maher lab demonstrated that the phorbol ester-mediated activation of PKCα and PKCɛ combined with the phorbol ester-mediated down-regulation of PKCδ, activation of JNK, and inhibition of p38 MAP kinase activation protected HT22 cells from oxidative glutamate toxicity by preventing the glutamate-induced increase in ROS without any effect on GSH levels (164). In a complementary study, Aharoni-Simon et al. demonstrated that the phorbol ester TPA protects by increasing glutamate-induced AP-1 activity, which is downstream of JNK activation. ROS increased AP-1 activity and AP-1 contraintuitively stimulated ROS production, indicating that, at least in this context, ROS possibly serve protective functions (1).
To identify potentially neuroprotective GPCRs, the Methner lab screened for the transcriptional up-regulation of a large group of well-characterized GPCRs by quantitative real-time PCR in HT22 cells selected for resistance against glutamate and identified the receptor VPAC2, which is activated by the vasoactive intestinal peptide VIP, as up-regulated in glutamate resistance. VPAC2 activation or overexpression protected from oxidative glutamate toxicity by increasing anti-apoptotic Bcl-2 (225). In a similar screen comparing the expression of all known orphan GPCRs, GPCRs with no known ligands, the constitutively active orphan GPCR GPR39 was found most prominently up-regulated in glutamate-resistant cells (50). GPR39 protected against oxidative glutamate toxicity through coupling to Gα13. In a broader screen to identify genes up-regulated in the same glutamate resistant HT22 cells, a new SOD motif-containing peroxisomal protein that has neuroprotective properties was identified (268). We conclude that glutamate-resistant HT22 cells can serve as a screening tool to identify novel neuroprotective genes.
The Maher lab (165) as well as another laboratory (276) found that inhibition of the proteasome using low doses of several structurally distinct proteasome inhibitors could prevent oxidative glutamate toxicity. While the proteasome inhibitors had no effect on GSH levels, they did prevent the increase in ROS (165). Surprisingly, the neuroprotective effects of the proteasome inhibitors appeared to be at least partially mediated by the induction of the transcription factor NF-κB, as protection was significantly reduced in HT22 cells expressing a specific NF-κB repressor (165). This observation is consistent with the majority of the data on NF-κB which suggest that it is important for both normal nerve cell survival and the survival of nerve cells exposed to oxidative stress (108). Moreover, the neuromodulator hydrogen sulfide (H2S) protects cortical neurons mainly by increasing GSH levels (118) and HT22 cells by both increasing GSH and activating plasma membrane KATP and CFTR Cl− channels (116). H2S also increases both cystine and cysteine uptake and enhances GSH import into mitochondria (117).
3. Using oxidative glutamate toxicity to screen for neuroprotective drugs
We and others have used the model of oxidative glutamate toxicity to identify a number of compounds that might be useful in the treatment of neurological disorders. For example, the Maher lab identified tyrphostins, known inhibitors of protein tyrosine kinases, as compounds that block oxidative glutamate toxicity at different steps in the cell death pathway independent of their effects on tyrosine kinase activity (223). Some tyrphostins were inducers of GCL, whereas others acted by stabilizing the mitochondrial membrane potential or were direct antioxidants (223).
In a related study, flavonoids, natural plant compounds, were investigated (99). Different flavonoids also acted at distinct steps in the cell death pathway, including maintenance of GSH levels, prevention of ROS accumulation, and inhibition of calcium influx. In addition, since many flavonoids are inducers of Nrf2 (section I.B) (95), it is likely that at least some of their neuroprotective effects are mediated via this transcription factor. Interestingly, very small structural changes, such as the shifting of the placement of a single hydroxyl group, resulted in very different activities for this group of compounds. Together, these results are in agreement with the emerging idea that the protective effects of flavonoids result from their ability to modulate multiple signaling pathways rather than acting as direct antioxidants (239, 254).
Among the flavonoids tested, the flavonol fisetin, which is found in strawberries, proved to be highly effective. Fisetin not only has antioxidant activity, but is also able to maintain GSH levels. Consistent with this observation, fisetin was shown to increase Nrf2 levels (163). The value of oxidative glutamate toxicity for the identification of possible therapeutics is highlighted by the observation that fisetin proved to be protective in rabbit and mouse models of stroke (70, 169) and a mouse model of Huntington's disease (166) and type 1 diabetes (167). In another screen, cystine conjugates of catechin were tested for their ability to protect against oxidative glutamate toxicity (267). This led to the identification of a novel mechanism by which one of these compounds, cysteamine epicatechin, protects against oxidative glutamate toxicity and increases GSH synthesis, which involves the generation of extracellular cysteine via a disulfide exchange mechanism (168).
Moreover, the HT22 cell model of oxidative glutamate toxicity was used to identify novel neuroprotective derivatives of the curry spice turmeric (154). A derivative with a 15-fold decrease in EC50 in this assay, CNB-001, was found to have enhanced neuroprotective activity in a variety of other assays, including protection against Aβ toxicity (154). Further studies showed that this novel curcumin derivative was protective in a rabbit model of stroke (133) and a rat model of traumatic brain injury (288). Along with other in vitro paradigms of neuronal cell death, trophic factor withdrawal, and amyloid-β toxicity, oxidative glutamate toxicity was used to identify another novel, curcumin-based, broadly neuroprotective compound, J147, which prevented the loss of synaptic proteins and cognitive decline in a transgenic AD mouse model (34).
4. Oxidative glutamate toxicity in vivo
As indicated earlier, the susceptibility of cultured cells to glutamate-mediated system xc− inhibition might result from the enhanced reliance of cells in culture on cystine import for GSH synthesis. However, necroptosis and oxytosis have many features in common (23), and the necroptosis inhibitor Nec-1 can protect HT-22 cells from oxidative glutamate toxicity (289). Nec-1 can also reduce lesion volume in mice after transient middle cerebral artery occlusion, a model of ischemic stroke (49, 290), and in a model of traumatic brain injury (293). Very recently, Nec-1 was shown to exhibit protective activity in cell culture and mouse models of Huntington's disease (299). Together, these data suggest that oxytosis, or at least forms of cell death that share a very similar mechanism, may play an important role in a variety of neurological disorders.
IV. The Role of System xc− in Health and Disease
A. System xc− in vivo–lessons from xCT-deficient mice
With the exception of a redox imbalance with the cystine/cysteine redox couple in the plasma shifted to −89 mV compared with −100 mV in wild-type control animals and reduced plasma GSH, xCT−/− mice show no obvious phenotype (231). Even in the brain, where xCT is constitutively expressed, no accelerated hippocampal atrophy up to the age of 18 months could be demonstrated by Massie and co-workers (48). These findings indicate that loss of xCT in these mice is largely compensated for by unknown mechanisms. However, sut/sut mice, which harbor a spontaneous 481,280 bp deletion that includes parts of the xCT gene, show changes in fur color due to a deficiency in the cysteine-dependent yellow/red pigment, pheomelanin (37), and prominent brain atrophy, including in the hippocampus (250). Whether these divergent results can be explained by the different genetic backgrounds, C57BL/6 for xCT−/− and C3H/HeSnJ for sut/sut mice, or are due to the different molecular bases for xCT deficiency remains to be determined.
B. The role of system xc− in the immune system and inflammation
The immune system comprises cells of myeloid origin (monocytes, macrophages, dendritic cells, microglia, and granulocytes) and lymphocytes. From a functional point of view, two different systems can be distinguished. The innate immune system responds to a limited set of conserved danger signals such as double-stranded RNA or LPS that indicate the presence of infectious agents or other harmful conditions. The adaptive immune system generates a more specific response. Here, antigen-presenting cells (macrophages, dendritic cells, or stromal cells of the thymus) present specific antigens to the different types of lymphocytes that specifically respond to this type of antigen. These lymphocytes then proliferate, thereby generating an antigen-specific immune response against a virtually unlimited number of target structures. Both systems show extensive interactions at all levels and are interdependent.
Several lines of evidence suggest that system xc− might play a role in the regulation of the innate immune response. First, activation of macrophages and granulocytes is associated with excessive production of pathogen-toxic ROS, the so-called respiratory burst, leading to oxidative stress. Macrophages prominently up-regulate system xc− expression on activation with bacterial LPS or the pro-inflammatory cytokine TNFα (228). Similarly, in bleomycin-induced pneumonitis, infiltrating leukocytes prominently express xCT (192). Thus, induction of system xc− might be an auto-protective response during activation of these cells within the innate immune response. In line with this assumption, macrophages from xCT-deficient mice die on activation with LPS (192).
System xc− may also modulate the adaptive immune response. xCT is robustly expressed in vivo in the spleen, the organ of B lymphocyte maturation, and the thymus, where the selection and maturation of T lymphocytes takes place. On systemic treatment with LPS, the expression of xCT is heavily up-regulated in the cortex of the thymus and the white pulp of the spleen (260). However, lymphocytes are the only cell type where it has been consistently reported that system xc− expression cannot be induced (73, 100, 102). Nevertheless, spleen lymphocytes increase intracellular GSH levels on activation with LPS by up-regulation of cysteine uptake via system ASC (102). Cellular GSH levels have been reported to modulate intracellular signaling after stimulation of T-cell receptors or treatment with TNFα (259). Moreover, the availability of cysteine for GSH synthesis during maturation of stimulated lymphocytes in vitro has been shown to strongly influence the generation of cytotoxic T-cell activity (72). In contrast to lymphocytes, macrophages prominently up-regulate system xc− activity and, as a consequence, release cysteine into the extracellular space (72), probably through a system L-like transporter (235). Therefore, it has been postulated that, in addition to the exposure to the specific antigen and cytokines, lymphocyte activation is modulated by cysteine released from antigen-presenting cells (Fig. 9).
FIG. 9.
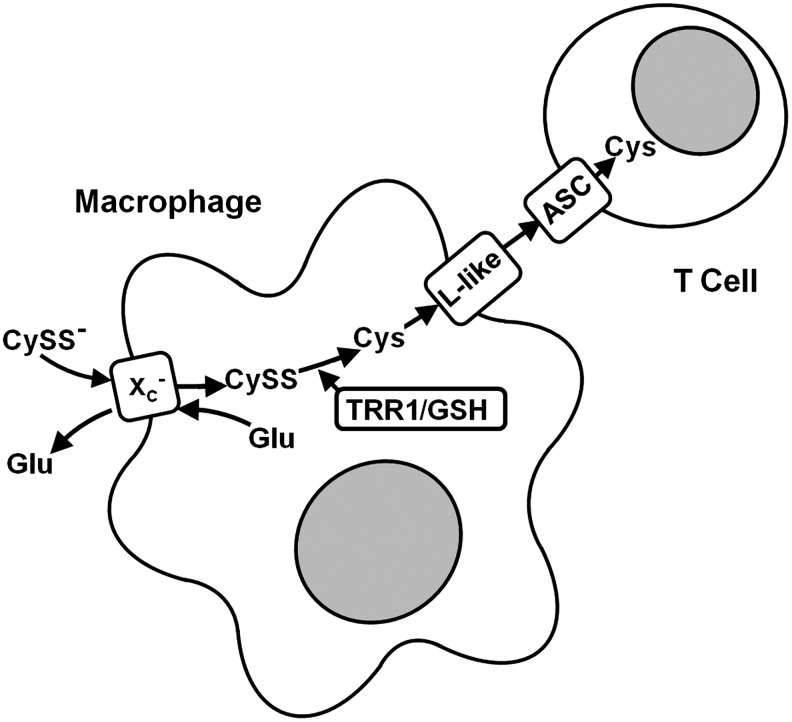
System xc− expressed in macrophages regulates T-cell proliferation and activity through cysteine release. Macrophages import cystine (CySS−) via system xc−. Intracellularly, cystine is reduced to cysteine (Cys) by GSH or thioredoxin reductase 1 (TRR1/GSH), which is subsequently released to the extracellular space and taken up by T lymphocytes for GSH synthesis.
Evidence from inflammatory bowel disease supports this concept (252). In the normal intestinal mucosa, the T-cell response is suppressed by low T-cell GSH. However, this does not result from altered properties of the mucosal T cells themselves but most probably is a consequence of very low system xc− activity and subsequently low cysteine release from the resident macrophages. In contrast, in the inflamed gut, where the natural inhibition of the T cell response is known to be abolished, the presence of cysteine-releasing macrophages with high system xc− activity was demonstrated. These results suggest that xCT expression in antigen-presenting cells might be a critical regulator of autoimmunity.
Further data indicate that xCT might also play a role in the termination of an inflammatory response. In a model of focal chronic inflammation induced in mice by a subcutaneous injection of 3-methylcholanthrene (3-MCA), where normally prominent expression of xCT is observed in the infiltrating immune cells, it was shown that a deficiency of xCT increases the expression of the pro-inflammatory cytokines IL-1β and TNFα within the lesion (192).
In summary, system xc− seems to play a dual role in the immune system. First, by fostering GSH synthesis, it serves as a mechanism for the auto-protection of activated macrophages and granulocytes from the high levels of released ROS and, second, it indirectly regulates the cysteine supply to lymphocytes and thereby their proliferation and activation, which might be a mechanism for fine-tuning the antigen-specific immune response. The role of xCT and system xc− in the specific setting of neuroinflammation will be discussed in section IV.E.
C. The role of system xc− in cancer and resistance against anti-cancer drugs
1. System xc− is regulated by potentially oncogenic pathways
xCT up-regulation has been demonstrated at the mRNA or protein level in lymphomas (80), gliomas (236, 292), and pancreatic cancers (156). As a corollary, the system xc− substrate and PET tracer (4S)-4-(3-[18F]fluoropropyl)-L-glutamate (BAY 94-9392) has been successfully used to visualize tumors in animal models (123). Here, we will summarize which of the pathways that regulate xCT expression are potentially oncogenic or were observed in cancers of diverse origin. Recently, somatic loss of function mutations in KEAP1 leading to increased nuclear Nrf2 protein levels and thereby to constitutive activation of Nrf2-mediated gene expression have been found with a high frequency in primary cancer samples and cell lines of lung, breast, gall bladder, and prostate cancer (196, 256, 297). The constitutive activation of Nrf2-mediated signaling may result in xCT up-regulation, although this has not been experimentally demonstrated. The other candidate nuclear factor that regulates xCT expression is ATF4, which has been reported to be up-regulated in various cancers (25), and its expression level is associated with chemoresistance in cancer cell lines (96). Enhanced resistance to chemo- or radiotherapy of cancer stem cells has been linked to a high level of CD44 expression. As shown by Ishimoto et al. (103), expression of the variant isoform of CD44, CD44v, contributes to the defense of cancer cells against ROS by interacting with and stabilizing xCT and increasing system xc−activity. In contrast, the microRNA miR-26b, which targets xCT mRNA, was found to be down-regulated in human breast cancer specimens and cell lines, and transfection of miR-26b into breast cancer cell lines led to inhibition of xCT expression (153). Thus, system xc− is under the control of a variety of potentially oncogenic pathways.
2. System xc− mediates the infection of cells by oncogenic Kaposi's sarcoma herpesvirus
Kaposi's sarcoma–associated herpesvirus (KSHV, human herpesvirus 8) is the causative agent of Kaposi's sarcoma (KS) and other lymphoproliferative diseases associated with HIV/AIDS. In a cDNA screen, Kaleeba and Berger identified xCT as a factor required for KSHV-mediated cell fusion (106) and postulated that xCT acts as a co-receptor for KSHV entry. However, using radiolabeled KSHV particles, Veetil et al demonstrated that the xCT/4F2hc heterodimer is involved in neither the binding and entry of KSHV nor the delivery of KSHV DNA to the nucleus (280). In contrast, they demonstrated that system xc− is required for KHSV gene expression in the postentry stage of KSHV infection via an unknown mechanism, possibly by mediating signal transduction (280). Of note, elevated expression of xCT protein was found in KS tissues (295). Consistent with the up-regulation of xCT expression in KS, Qin et al. demonstrated that KSHV-encoded microRNAs up-regulate xCT expression largely through suppression of BACH-1, a negative regulator of EpRE-mediated gene transcription, a process that eventually facilitates the dissemination of the KSHV via a positive feed-back loop (212).
3. System xc− plays an important role in the multidrug resistance of cancers
The ability of cancer cells to become concurrently resistant to different drugs, a trait known as multidrug resistance, remains a significant impediment to successful chemotherapy. Cancer cells can become resistant to anticancer drugs via several mechanisms, including (i) augmentation of the activity of efflux pumps (e.g., ATP-dependent transporters) that enhances extrusion of the drugs; (ii) reduction of drug influx; and (iii) activation of detoxifying pathways such as those mediated by GSH, GSTs, and MRPs (78).
Using a pharmacogenomic approach (92, 94), it was demonstrated that increased xCT expression is associated with the chemoresistance of tumor cells, as its expression level was negatively correlated with drug potency across the NCI-60 cancer cell lines (93). The number of significant target-drug correlations was much greater for xCT than for known resistance genes such as ABCB1 (encoding for P-glycoprotein or MDR1) and GSTs (45), suggesting that system xc− plays a key role in anticancer drug resistance. Anti-cancer compounds whose activity was negatively correlated with xCT expression tended to contain structural features amenable to GSH reactivity, such as Mannich bases (45), indicating GSH-dependent inactivation as a possible pathway through which xCT expression induces chemoresistance (Fig. 10). In support of this idea, system xc− inhibitors can lead to intracellular GSH depletion and reverse the multidrug resistance of cells to certain anticancer drugs in vitro (45, 93, 150, 206). In another study, it was demonstrated that system xc− specifically mediates resistance to anticancer drugs that produce high amounts of ROS, such as geldanamycin and celastrol (150, 206).
FIG. 10.
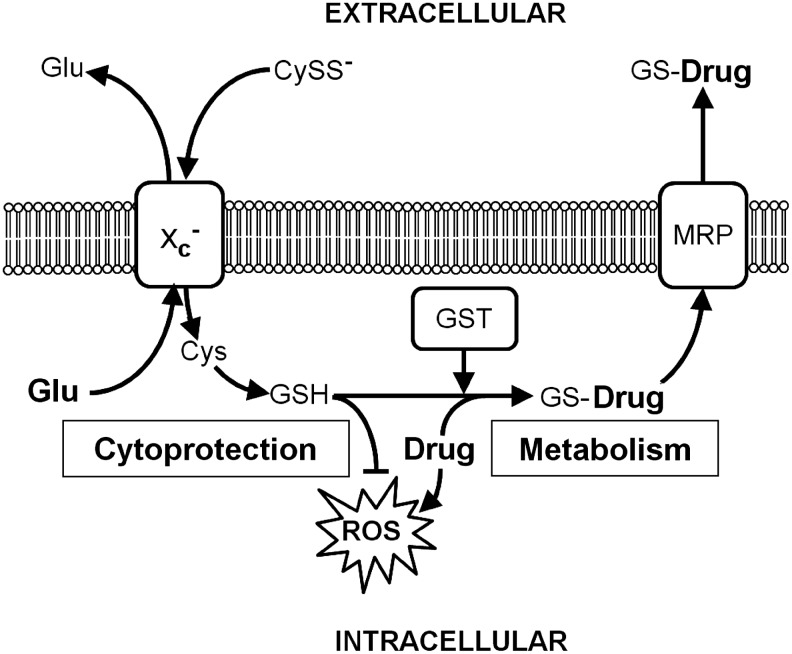
GSH-mediated chemoresistance in tumor cells though system xc−. In cancer cells, GSH is synthesized from cystine (CySS−) taken up via system xc−. GSH protects cells from ROS induced by anticancer drugs (Drug) and/or is used as a co-substrate for GSTs, which form GSH-drug adducts (GS-Drug). GSH adducts are exported from the cell by MRP.
4. Inhibition of system xc− reduces cancer cell replication, tissue invasion, and metastasis
Since system xc− has been proposed as a potential target for cancer therapy, various pharmacological and genetic approaches have been applied to inhibit its function. Several system xc− inhibitors are available (see section II.A). The system xc− substrate and competitive cystine uptake inhibitor glutamate, which as a proteinogenic amino acid cannot be used as a therapeutic drug, can severely inhibit or completely arrest in vitro proliferation of malignant cells that depend for growth on system xc−-mediated uptake of cystine (80, 156). The cyclic glutamate analog (S)-4-CPG has antiproliferative and cytotoxic effects on certain glioma cell lines in the micromolar range (41). Moreover, the growth of cultures of system xc−-dependent lymphoma cells and certain breast, prostate, lung, and pancreatic cancer cells can be completely inhibited by the system xc− inhibitor sulfasalazine (79) at patient-tolerated levels (range 0.05–0.3 mM) via specific inhibition of system xc− (156, 157). In vivo, sulfasalazine acts primarily as a cytostatic agent, without leading to major toxicity in the host (41, 79). Although sulfasalazine also inhibits NF-κB activation, its growth-inhibitory activity was found to be specifically based on system xc− inhibition (leading to cysteine and GSH depletion) but not on NF-κB inhibition (155). System xc− has been reported as playing a role not only in tumor growth but also in the ability of tumors to metastasize, a process that is regulated by cell adhesion. xCT-deficient melanocytes derived from sut/sut mice showed a higher cell-cell as well as cell-matrix adhesion compared with wild-type melanocytes (35). In addition, in an esophageal cancer cell line, inhibition of system xc− by siRNA specific for xCT or sulfasalazine increased adhesion and inhibited cell invasion in vitro, and sulfasalzine decreased tumor metastasis in vivo (35). The role of xCT in cancers of the CNS will be discussed in section IV.E.
5. System xc− expressed in tumor cells may be used as a target for anticancer drug delivery
Although xCT expression in most cases induces resistance against chemotherapeutic drugs, xCT expression can also sensitize tumor cells to some substances. Recently, it was demonstrated that system xc− is involved in the cellular uptake and cytotoxicity of the selenium salt, selenite (SeO32−) (194). The uptake of selenite into cancer cells apparently requires its extracellular reduction, probably to selenide, by cysteine released from cells in a system xc−-dependent manner (see section III.C) (Fig. 11).
FIG. 11.
System xc− sensitizes tumor cells to selenite toxicity. Cystine taken up by tumor cells is intracellularly reduced to cysteine, which is subsequently exported from the cell. Extracellular cysteine reduces selenite, probably to selenide, which is subsequently imported into the cell by an unknown transport mechanism, where it induces cytotoxicity.
The pharmacogenomic studies conducted by the Huang lab suggest that several other compounds have enhanced efficacy against cancer cells, exhibiting increased xCT expression (93). One of these drugs was the amino acid analog L-alanosine whose toxicity in various tumor cell lines was reduced by inhibition of system xc− (93). L-alanosine is an L-α-amino acid analog with a structure that is distantly related to glutamate. Thus, it is a potential substrate for system xc−. However, this hypothesis has not been experimentally verified.
Thus, unknown, probably xCT-dependent, mechanisms exist through which some chemotherapeutic drugs gain enhanced activity against otherwise drug-resistant tumors with high xCT expression. These observations hint at the possibility of using these drugs as potential therapeutics for chemoresistant cancers.
6. Up-regulation of system xc− in normal cells provides protection against carcinogenesis—a possible role in cancer prevention
System xc− may also be a target for cancer chemoprevention. Common features of human carcinogenesis amendable to chemoprevention include mutagenesis, oxidative stress, and inflammation (258). The key molecular target for the chemopreventive agents sulforaphane and oltipraz is the transcription factor Nrf2 (128), which can regulate xCT expression (see section II.D). Indeed, Nrf2−/− mice have been found to be more susceptible to carcinogenic challenges (214). System xc− itself might be responsible for some of the chemopreventive effects of Nrf2 inducers, as xCT-mutant mice showed accelerated generation of fibrosarcomas induced by 3-MCA (192).
7. Synopsis of the role of system xc− in cancer and resistance against anti-cancer drugs
Recent studies indicate that system xc− plays an important role in various aspects of cancer, including (1) the growth and malignant progression of cancer cells; (2) GSH-mediated drug resistance to anticancer drugs; (3) infection with KSHV, a causative agent for KS; (4) anticancer drug delivery; and (5) protection of normal cells against oxidative damage induced by carcinogens and (6) might serve as a target to monitor cancer progression using PET imaging. While further studies are needed, the results just cited illustrate the impressive potential of system xc− to act as a target for cancer therapy and prevention.
D. System xc− and diseases of the eye
The eye transmits visual information through the transparent cornea and lens to the retina, from where the photic stimuli trigger a cis-to-trans isomerization within the visual pigment rhodopsin activating the neurochemical stimulation of retinal neurons for transmission of the signal via the optic nerve to the brain (Fig. 12). Excessive exposure of the eye to light can damage various ocular structures, including the cornea, lens, and retina. In addition to radiation, oxidative stress is associated with ocular diseases such as diabetic retinopathy, glaucoma, macular degeneration, and cataract. Production of GSH is a major mechanism used by the eye to cope with these stressors. Thus, it is not surprising that the expression and function of system xc− have been investigated in the eye.
FIG. 12.
The anatomy of the eye.
1. Studies of system xc− in the retina
The retina is comprised of multiple cellular and synaptic layers, which are shown in Figure 13a. The outermost layer is a single cuboidal epithelial cell layer, the retinal pigment epithelium (RPE) that provides nutritive support for adjacent photoreceptor cells, phagocytoses that shed photoreceptor outer segment (OS) discs, and regulate the transport of vitamin A to photoreceptors. The microvillous processes of RPE cells interdigitate with the OS of adjacent photoreceptor cells. The cell bodies of the photoreceptor cells, known as rods and cones, constitute the outer nuclear layer and are the first-order neurons of the visual pathway. Rods and cones synapse in the outer plexiform layer (OPL) with bipolar cells, the second-order neurons of the visual pathway. The neuronal signals are modulated by horizontal cells and amacrine cells. These interneurons have their cell bodies in the inner nuclear layer. Axons of the bipolar cells synapse in the inner plexiform layer with dendrites of the ganglion cells (gcl), the third-order neurons of the pathway whose axons form the optic nerve. Spanning the entire retinal thickness are retinal Müller glial cells (Fig. 13b). System xc− has been studied in RPE, Müller cells, gcl, and the OPL. An early work by Kato et al. demonstrated that system xc− is active in the retina and that an intravitreal injection of the known substrate inhibitor of system xc−, DL-α-aminoadipate (see section II.A), can decrease retinal GSH concentrations and induce retinal dysfunction in vivo (111), indicating that system xc− is functionally important in the retina.
FIG. 13.
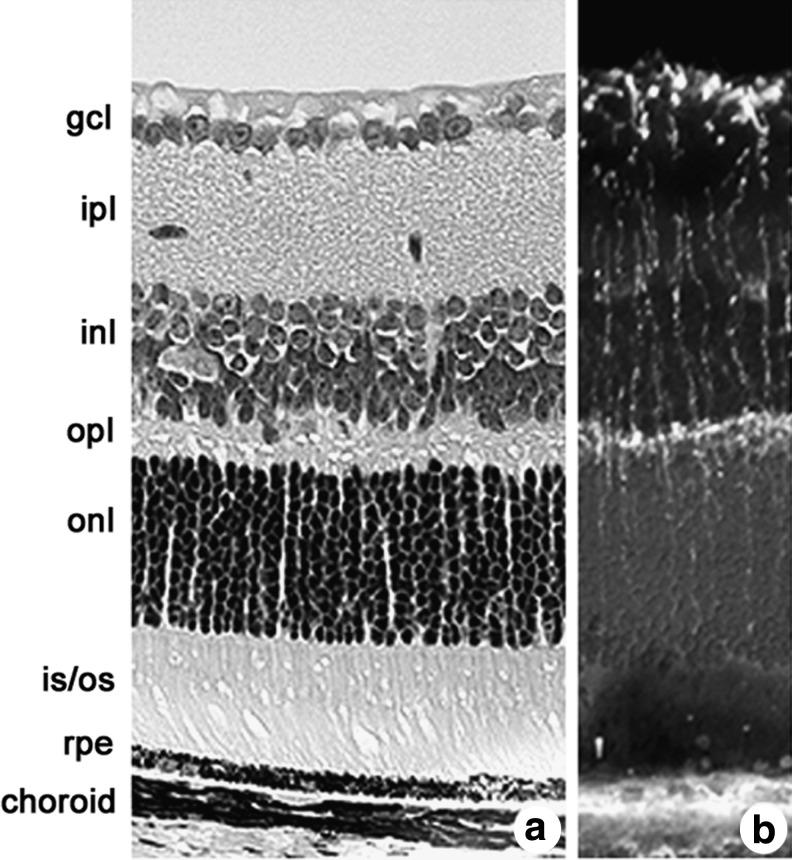
The anatomy of the retina. (a) Hematoxylin-eosin stained section of the retina. The outermost layer is the retinal pigment epithelium (RPE). The microvillous processes of RPE cells interdigitate with the outer segments (OS) of adjacent photoreceptor cells. The cell bodies of the photoreceptor cells, known as rods and cones, constitute the outer nuclear layer (ONL). Photoreceptor cells synapse in the outer plexiform layer (OPL) with bipolar cells. Bipolar cells, horizontal cells, and amacrine cells have their cell bodies in the inner nuclear layer (INL). Axons of the bipolar cells synapse in the inner plexiform layer (IPL) with dendrites of the ganglion cells (gcl). (b) Immunohistochemical labeling with antibodies against vimentin of the radially oriented fibers of Müller cells that span the entire retina.
Oxidative stress to the RPE is implicated in the devastating disease age-related macular degeneration (AMD), and GSH protects these cells from oxidant-induced apoptosis [reviewed in (31)]. The Ganapathy lab demonstrated system xc− activity in the ARPE-19 human RPE cell line, from which human xCT was subsequently cloned (28). Infection with HIV-1 in humans is associated with decreased levels of GSH, which is mediated by Tat (56). The Ganapathy lab demonstrated that HIV-Tat decreased GSH levels in ARPE-19 cells (27) and, most possibly as a consequence, induced system xc− activity and xCT and 4F2hc expression at the mRNA and protein levels through an unknown mechanism (27).
Excess levels of iron have been reported in patients with AMD (86). Investigators in the McGahan lab showed that intracellular iron regulates the activity of system xc− in RPE cells via cytosolic aconitase and possibly synthesis of glutamate (129). The fact that iron plays a critical role in the regulation of xCT in RPE was supported by studies in HFE knock-out mice (HFE−/−), a model for the human iron overload disorder haemochromatosis. In the retina, HFE is predominantly expressed in RPE (174). HFE−/− mice undergo RPE hypertrophy and hyperplasia (74). An analysis of cultured HFE−/− primary RPE cells revealed increased expression of xCT protein and mRNA and system xc− activity (74). This finding is important given the documented critical role that xCT plays in cell proliferation (157). Thus, in RPE, the cell that nourishes and maintains photoreceptor cells, a number of factors that increase oxidative stress (NO, iron overload, and HIV-Tat), leads to up-regulation of system xc−.
A second retinal cell type in which system xc− has been investigated is the Müller glial cell. The GSH concentration in the retina is relatively high (1.2 μmol/g wet weight in freshly isolated rat retina) and, as demonstrated by immunohistochemistry with antibodies against GSH, much of the GSH is present in Müller cells (210). Indeed, Müller cells protect adjacent retinal neurons by providing GSH to neurons under ischemic conditions. In early studies looking at uptake of radiolabeled cystine in freshly dissected retinae, Kato et al. reported a pattern of cystine accumulation that is consistent with high activity of system xc− in Müller cells (111). Pow obtained similar results after loading retinas with the system xc− substrate L-α-aminoadipate and visualizing intracellular L-α-aminoadipate accumulation by immunocytochemistry with a specific antiserum (209). Taken together, these studies indicate that Müller glia show system xc− activity in situ.
In vitro studies from the Hosoya lab showed that the conditionally immortalized rat Müller cell line (TR-MUL) exhibits system xc− activity and expresses xCT and 4F2hc. System xc−-mediated L-cystine uptake and xCT expression could be induced by the GSH-depleting agent DEM (266). The laboratory of Sylvia Smith obtained similar results in primary cultured Müller cells subjected to H2O2-mediated oxidative stress brought about by xanthine/xanthine oxidase, which also increased xCT mRNA expression and system xc− activity (191). Moreover, as mentioned earlier (see sectionII.E), on oxidative stress, a shift of a ∼40 kD isoform of xCT from the intracellular compartment to the plasma membrane was observed, whereas for the predominant form of xCT, with a molecular weight of ∼50 kD, no detectable change could be demonstrated (191).
In another disease of the retina, diabetic retinopathy, increased glutamate levels were found in the vitreous humor, indicating disturbed glutamate metabolism (159). However, 8 days of hyperglycemia neither altered the uptake of glutamate by EAAT1 or system xc−, nor altered the expression of xCT mRNA or protein in primary cultured Müller cells (191). The regulation of system xc− activity in the Müller glia by oxidative stress may be relevant, because, in theory, glutamate release by system xc− might induce excitotoxic damage to retinal neurons.
Although earlier studies highlighted the prominent system xc− activity in Müller glia (111, 209), initial immunohistochemical studies using an antibody against xCT suggested that it was actually present in several neuronal layers in the retina (27). Studies from two laboratories working independently examined this comprehensively and concluded that system xc− was indeed present in retinal neurons (57, 91). Dun and co-workers showed that primary gcl from neonatal and adult mouse retina were positive for xCT and 4F2hc expression and detected both xCT and 4F2hc as well as oxidative and nitrosative stress-inducible system xc− activity in the retinal neuronal cell line RGC-5 (57). Electron microscopy immunolocalization studies showed that xCT was present in the ribbon synapses of rod and cone photoreceptor cells and the dendrites of rod bipolar cells in diverse species (91). It was demonstrated that developmentally the onset of xCT expression in rat retina occurred at postnatal day 10, the time when photoreceptor cells make functional contact with bipolar cells. Taken together, molecular, biochemical, and functional analyses indicate that system xc− is present in several retinal cell types (RPE, Müller, ganglion, and photoreceptor/bipolar) and may play a key role in providing the rate-limiting precursor, cyst(e)ine for the synthesis of GSH.
2. Studies of system xc− in the lens and cornea
The lens is a transparent convex disk positioned between the iris and the vitreous body; it is suspended within the globe by zonular fibers that extend from the ciliary body (Fig. 12). The lens is comprised of (1) a capsule, which is a thickened basement membrane that envelops the lens; (2) a simple, cuboidal epithelium, which lines the anterior lens surface; and (3) lens fibers, comprising the inner and outer cortex of the lens, which are elongated cells that eventually lose their nuclei and are specialized to allow passage of light. An opacity of the lens is termed a cataract. Excessive oxidative stress, which can lead to protein modification, is implicated in the formation of cataracts [for review see (24)]. While young healthy eyes benefit from an active ROS scavenging system largely comprised of GSH, aging eyes have diminished capacity to cope with oxidative stress. Members of the Donaldson laboratory found that xCT is present at the mRNA and protein levels in lens fiber cells (143, 146, 148). Immunocytochemical studies of the lens showed a cytoplasmic distribution of xCT-like immunoreactivity in outer cortical fibers and a membranous distribution in the inner fiber cells (146). This team combined immunocytochemistry for cystine using antisera against glutaraldehyde-cystine conjugates to map the distribution of free cystine and to determine its relationship to the expression of xCT (143). In our opinion, the cystine immunoreactivity represents the distribution of both cysteine and cystine in vivo as cysteine should be oxidized to cystine during the process of preparation and fixation. They reported a bimodal distribution of cystine immunoreactivity (intense in outer cortex, diminished in inner cortex, and intense in lens core), which correlated well with xCT immunoreactivity. It has been proposed that in the lens cortex, xCT works with EAAT4 to accumulate cystine for the synthesis of GSH; whereas in the lens nucleus (a site where GSH is not synthesized), xCT may work with ASCT2 to accumulate cystine, which when reduced to cysteine may itself act as a low-molecular-mass antioxidant (146–148). Interestingly, Lall and co-workers performed immunohistochemical localization studies of xCT in the canine lens using the same antiserum and reported that the highest levels of xCT were in lens cortical fibers, but found no xCT in the nuclear region of the lens (129). In this same study, it was shown that iron, which is known to induce oxidative damage, regulates the activity of system xc− in the lens.
The cornea represents the anterior 1/6th of the eyeball and is responsible for most of the refraction of light entering the eye (Fig. 12). It is comprised of five layers, including (from outermost to innermost) a multi-layered epithelium, an anterior limiting lamina (Bowman's membrane), the substantia propria (stroma), the posterior limiting lamina (Descemet's membrane), and the endothelium (Fig. 14a). The corneal epithelial layer is composed of two to three layers of flattened superficial cells, two to three layers of wing cells, and a single layer of columnar cells that are attached to the underlying Bowman's membrane. In the cornea, GSH maintains the normal hydration level and protects cellular membrane integrity (68). In the only published investigation of system xc−in cornea to date, Langford and colleagues localized xCT immunoreactivity in the superficial epithelial layer and the columnar cells of human corneal epithelium from nondiabetic, nonglaucomatous donor eyes (132). The authors speculated that this localization facilitates the transport of cystine from stroma and tears. As shown in Figure 14b, a similar pattern of distribution is observed in mouse cornea with labeling of the superficial cells and also robust xCT expression in the columnar cells. We also observed intense xCT immunoreactivity in the epithelium of the ciliary body (Fig. 14c). Along with the iris, the ciliary body (depicted in Fig. 14c) is the anterior-most extension of the uvea (vascular tunic of the eye) and plays a key role in the synthesis of the aqueous humor, which nourishes the cornea and lens. xCT was also expressed in the iridial epithelium (data not shown). Whether xCT expression in the ciliary epithelium is involved in the redox homeostasis of the aqueous humor remains to be explored.
FIG. 14.
Distribution of xCT immunoreactivity in the corneal epithelium and the ciliary body. Hematoxylin-eosin stained sections of (a) the cornea and (c) the ciliary body (cb). (a) The corneal epithelium (EP) is comprised of a layer of columnar cells (c) attached to the Bowman's membrane (BM), wing cells (w), and superficial cells (s). The other layers of the cornea, from outside to inside, are the stroma (ST), the Descemet's membrane (DM), and the endothelial layer (EN). (B/D) The distribution of xCT immunoreactivity in (b) the corneal epithelium and (d) the ciliary body (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars.)
3. Synopsis and future directions for system xc− and diseases of the eye
Given the considerable oxidative and irradiative stress to which the eye is regularly subjected, it is not surprising that myriad ocular cell types express xCT and 4F2hc in situ. The fact that inhibition of system xc− by an intravitreal injection of the inhibitor DL-α-aminoadipate decreases GSH in vivo indicates an important role for system xc− in the GSH homeostasis of the retina. Future studies on the function of system xc− in the cornea and lens would be extremely enlightening, especially as it relates to corneal injury or cataract, respectively. Another area that is ripe for investigation is to systematically examine alterations in the expression or function of system xc− in animal models of visual diseases, including cataract, corneal disease, diabetic retinopathy, macular degeneration, and glaucoma. Finally, an important area of investigation will be to determine the effects of xCT loss on the lens, corneal, and retinal function in xCT−/− mice (231).
E. The role of system xc− in diseases of the CNS
Oxidative stress, inflammation, mitochondrial dysfunction, and excitotoxicity are key players in the pathogenesis of many neurological diseases/disorders, and a vast body of evidence indicates that these processes are not independent but inextricably linked. Two important players in these numerous pathological conditions are GSH and glutamate. In many diseases of the CNS, including AD, Huntington's disease, epilepsy, and cerebral ischemia, perturbed function of EAATs, which clear the glutamate released by system xc−, has been reported [for review see (247)]. The subsequent increase in extracellular glutamate as an excitotoxic insult has the propensity to induce the formation of ROS, which contributes to mitochondrial dysfunction, thereby further increasing the formation of ROS and oxidative stress. ROS also increase/induce the formation of inflammatory factors, which can enhance excitotoxicity by affecting glial glutamate transport (105). GSH depletion, which can result from and in oxidative stress, can also induce the release of inflammatory mediators and excitotoxic substances from astrocytes and microglia (135), as well as the expression of xCT (see section II.D). Likewise, the neurodegenerative consequences of inflammation could result from the conversion of oxidative stress to excitotoxic stress (20). Finally, increased extracellular glutamate levels, which induce excitotoxic damage, can also compromise the proper functioning of system xc−, resulting in GSH depletion and cell death (see section III.D). Hence, cellular perturbations occurring under neuropathological states can induce several, nonmutually exclusive feed-forward loops that ultimately lead to neuronal cell death.
The in vitro study by Piani and Fontana (207), which demonstrated that neuronal killing by macrophage-mediated glutamate release was dependent on system xc− activity, was perhaps the first to show the neurotoxic potential for this transporter (207). It was subsequently shown that the export of glutamate via system xc− from glioma cells produces an excitotoxic necrosis that aids in tumor growth, migration, and invasion (161, 292) both in vitro and in vivo. Significantly, pharmacological inhibition of system xc− activity suppressed the growth of primary brain tumors in vitro and in vivo (41), and siRNA-induced silencing or knockdown of xCT in gliomas ameliorated tumor-induced necrosis (236). Moreover, in mice, the system xc− inhibitor sulfasalazine reduced peritumoral epileptic activity induced by glutamate released through system xc− from implanted human glioma cells (29).
The suggestion that system xc− activity could contribute to pathology in AD soon followed when Barger and Basile demonstrated in vitro that secreted amyloid precursor protein–treated microglia killed neurons via system xc− mediated excitotoxicity (19). Similar results were found using aggregated β-amyloid (Aβ) (211). Significantly, increased xCT gene expression is seen in adult mouse hippocampus after an injection of Aβ and in activated microglia in the cerebral cortex of Thy1-APP751 transgenic mice (211). In the cortex of aged APP23 mice, xCT overexpression is accompanied by reduced glutamate uptake and increased extracellular glutamate levels (238). With regard to models of PD, an ipsilateral increase in striatal xCT protein levels occurred in 6-hydroxydopamine (6-OHDA)-lesioned hemi-Parkinson rats 3 weeks after an injection (176). Although the functional significance was not determined in the former study, follow-up studies using xCT−/− mice showed that dopaminergic neurons in the substantia nigra of system xc−-deficient mice were highly protected against 6-OHDA-induced neurodegeneration (175).
It is well known that alterations in glutamate homeostasis contribute to oligodendrocyte cell death and neurological deficits in animal models of MS [reviewed in (77)]. However, the molecular source of the glutamate has not been well defined. A recent work by Pampliega and colleagues (198) demonstrated increased expression of xCT (mRNA and protein) in microglia, meningeal, and dendritic cells in the spinal cord of Lewis rats subjected to experimental autoimmune encephalomyelitis (EAE), as well as in chronic EAE mice, animal models for MS. Most importantly, xCT expression was similarly altered in the CNS (postmortem optic nerve) and in peripheral blood cells (isolated leukocytes) from human MS patients, suggesting that immune-activated up-regulation of system xc− levels may result in higher glutamate release and contribute to excitotoxic damage to oligodendrocytes (198). In vitro experiments support this supposition (53). In addition, the oxidative stress and excitotoxic neuronal injury induced by β-N-methylamino-l-alanine (BMAA), a neurotoxin associated with amyotrophic lateral sclerosis/Parkinson-dementia complex in Guam, has been linked to system xc−, with BMAA acting as a transportable inhibitor simultaneously blocking cystine uptake while facilitating glutamate release (152).
Adopting both a pharmacological and a genetic approach, recent in vitro data demonstrated that astrocytic system xc− is responsible for the deleterious, excitotoxic effects of IL-1β, a cytokine known to be up-regulated and to contribute to cerebral ischemic injury [for review see (62)], in hypoxia of astrocyte/neuron co-cultures (63, 105). While the role of system xc− activity during in vivo ischemia remains to be confirmed, it should be noted that the same system xc−/mGluR1a antagonist (LY367385), used in vitro (63, 105), reduced infarct volume in a rat model of ischemia (186), a protective effect originally ascribed to inhibition of mGluR1. However, curiously, compared with wild-type controls, neuronal injury after experimental ischemia is not reduced in mGluR1α-deficient mice (61). Thus, the idea that inhibition of system xc−might underlie the protection by this so-called mGluR1 antagonist after cerebral ischemia deserves further attention.
With regard to epilepsy, alterations in xCT expression levels in the hippocampus of the EL mouse, used as a model of generalized epilepsy with complex partial seizures, as well as in the cortex of the WAG/Rij rat, a genetic model for absence seizures, were noted (262) (A. Massie and I. Smolders, unpublished observations). Indeed, xCT protein levels differed when comparing rodents displaying seizures with younger presymptomatic animals. How or whether these changes causally relate to alterations in seizure susceptibility remains to be definitely determined, because other factors might contribute to the complex process of epileptogenesis in these models. Interestingly, xCT−/− mice, however, showed an elevated seizure threshold to the chemoconvulsants pilocarpine and kainic acid as compared with xCT+/+ mice, indicating that in these models for limbic seizures, the deletion of glutamate/cystine exchange is anticonvulsant (48). Consistent with the observations just described, N-acetylcysteine (NAC), a cyst(e)ine prodrug that activates system xc−, increased hippocampal glutamate levels in rats and had proconvulsant properties in models of limbic seizures (48).
In summary, it is clear from the studies just mentioned that in most animal models for neurological disorders analyzed so far, xCT expression is up-regulated. Intuitively, one might think that system xc− activity should be enhanced in order to protect the CNS against oxidative stress associated with most CNS diseases. This idea is strengthened by reports that describe increased xCT expression along with rescued GSH levels after treatment with levetiracetam and zonisamide, both anticonvulsant drugs with neuroprotective properties (5, 71, 271). However, as in animal models for epilepsy, up-regulation of xCT in the PD model might paradoxically contribute to injury. Most likely, the increased nonsynaptic glutamate release via system xc− overrides a putative protective effect through excitotoxic side effects via activation of ionotropic glutamate receptors. The deleterious excitotoxic side effects of increased system xc− activity might be amplified in many diseases of the CNS as down-regulation of EAATs, which, usually balance system xc− mediated glutamate release, is commonly observed [for review see (247)] (Fig. 15). In line with this hypothesis, deletion of xCT has been shown to exacerbate ischemic kidney injury (249), supporting a protective role for system xc− in tissues where excitotoxicity is not involved.
FIG. 15.
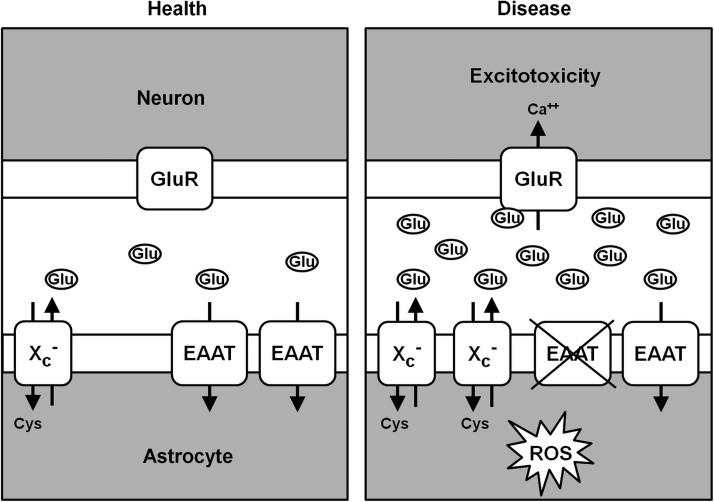
Hypothetical roles of system xc−in diseases of the central nervous system. (Left panel) In the healthy brain, glutamate release by system xc− (here only depicted in astrocytes, as conflicting data about neuronal expression have been published) is balanced by glutamate uptake by EAATs, which leads to negligible activation of ionotropic glutamate receptors (GluR). (Right panel) In many disease states of the brain, oxidative stress is present (ROS), which might lead to the up-regulation of system xc− and subsequent increased glutamate release. Activated microglia may also contribute to the increase in glutamate release via system xc− (not shown). Simultaneously, EAATs are down-regulated. The increased extracellular glutamate concentration activates ionotropic glutamate receptors and induces excitotoxicity.
Hence, it might be concluded that inhibition of system xc−could be a therapeutic approach for CNS disorders. However, it has to be kept in mind that the absence of decreased GSH levels in xCT−/− mice might be the result of long-term, adaptive changes in GSH metabolism in these animals. While inhibition of system xc− in neurological diseases might be effective at re-establishing the balance of glutamate release via system xc− and uptake by down-regulated EAATs at a lower level and thereby decrease excitotoxicity, this goal might be achieved at the expense of increased oxidative stress through impaired GSH synthesis.
Thus, in theory, drugs that induce both xCT and EAATs might both decrease the excitotoxic side effects of system xc−induction and strengthen antioxidant defense. Examples of this group of substances are the xCT-inducing anticonvulsants levetiracetam and zonisamide, which were shown to also induce EAAT3 expression (270, 272) and the EAAT2-inducing neuroprotective antibiotic ceftriaxone, which has been shown to attenuate damage in models of both acute and chronic neurodegenerative disorders [for review see (247)] and increases xCT expression in vitro (136) as well as in the brain in vivo (121).
F. The role of system xc− activity in memory and behavior
As described in section III.C, system xc− is not only involved in the maintenance of cellular redox homeostasis but also regulates extracellular glutamate levels. Altered redox homeostasis can have an impact on numerous intracellular signaling pathways [reviewed in (141)], and glutamate is the major neurotransmitter in the brain. Both altered neurotransmission and intracellular signaling might finally lead to changes in the level of higher brain function such as memory and behavior.
Using mice with a genetic deletion of xCT (231), Massie and co-workers investigated the impact of system xc− activity on memory by comparing their behavior to that of their wild-type littermates in the water maze and Y-maze assays (48). In the water maze, young xCT−/− mice showed a tendency to learn the hidden platform task less efficiently, but this tendency remained below the level of significance. In addition, young xCT−/− mice, but not aged xCT−/− mice, showed a spatial working memory failure in the Y-maze. The fact that the genotype differences were no longer apparent in aged mice was due to a less pronounced trend in working memory decline with ageing in xCT−/− compared with wild-type mice. Whether this observation results from long-term adaptive changes to xCT deficiency or the reduction of chronic excitotoxicity due to lower extracellular glutamate levels remains to be determined.
Schizophrenia is a psychiatric disease that is thought to involve disturbed glutamate signaling. In a rodent model of schizophrenia, where the NMDA antagonist phencyclidine (PCP) is administered, deficits in working memory, as evaluated using a t-maze, are linked to increased synaptic glutamate release in prefrontal cortex. Baker et al. showed that these deficits can be restored by increasing the activity of system xc− and, as such, inducing a negative feedback loop via group II metabotropic glutamate receptor activation, which subsequently leads to reduced presynaptic glutamate release (7).
Addictive disorders are among the most important psychiatric diseases. Over the last 10 years, a role for system xc−in the vulnerability to relapse has been characterized by the Kalivas laboratory as well as other groups. In 1996, it was observed using microdialysis that chronic treatment with cocaine reduced the extracellular levels of glutamate in the nucleus accumbens of rats by ∼50% (208). Since the glutamate measured by microdialysis is not derived from synaptic activity (275), other sources of extracellular glutamate were examined. It was found that the reduction in extracellular glutamate after chronic treatment with either cocaine or nicotine resulted from reduced system xc− activity and levels of xCT expression in the accumbens (8, 120). The reduced activity of system xc− was linked to the drug-induced reductions in extracellular glutamate by showing that stimulating system xc− with either the cyst(e)ine prodrug NAC or by intra-accumbens infusion of cystine restores extracellular glutamate levels to normal (8, 162), and this effect was reversed by infusing the system xc− antagonist (S)-4-CPG into the accumbens (112). The importance of down-regulated system xc− in the reinstatement of cocaine-seeking behavior, an animal model of relapse (246), was revealed by finding that NAC-mediated restoration of system xc− activity prevents reinstated cocaine seeking (8, 112). Similarly, NAC prevents relapse to heroin seeking (298). When NAC is chronically given, an enduring restoration of xCT levels in the accumbens is measured in rats withdrawn from self-administered cocaine (121). Importantly, the restoration of xCT by chronic NAC administration is associated with an enduring inhibition of reinstated cocaine seeking and a restoration of basal levels of glutamate (162, 188). These behavioural findings have led to moderate success using NAC to inhibit craving and drug use in cocaine addicts and cigarette smokers (120, 134).
The mechanism by which activating system xc− inhibits reinstated drug seeking has been linked to stimulation of group II metabotropic glutamate receptors (mGluR2/3). Thus, increasing cystine/glutamate exchange via system xc−elevates basal levels of extracellular extrasynaptic glutamate that, in turn, stimulate presynaptic mGluR2/3 (188), and stimulating mGluR2/3 decreases synaptic glutamate release probability in prefrontal projections into the accumbens (107, 185). In addition, the reduction in reinstated cocaine seeking elicited by activating system xc− with NAC is prevented by pretreatment with an mGluR2/3 antagonist (185). Given this interaction between system xc− and mGluR2/3 to reduce synaptic glutamate release probability and reinstated cocaine seeking, it is not surprising that directly stimulating mGluR2/3 with a pharmacological agonist also inhibits reinstated cocaine seeking (205). The fact that activating system xc−indirectly regulates synaptic glutamate release raises the possibility that system xc− may modulate synaptic plasticity. This possibility was tested in cocaine-withdrawn animals that show a deficit in the capacity to induce long-term potentiation (LTP) or long-term depression (LTD) in glutamatergic prefrontal afferents into the nucleus accumbens (110, 187). Treatment with NAC restored both LTP and LTD in prefrontal synapses in the accumbens, and this was accomplished by increasing stimulation of mGluR2/3 and mGluR5 with glutamate derived from the activation of system xc−(187). The role of system xc− derived glutamate in the modification of synaptic function, including the effect of NAC, is summarized in Figure 16.
FIG. 16.
Glutamate released by system xc− modulates synaptic activity. Glutamate released into the synaptic cleft is taken up by astrocytic glutamate transporters (EAAT) Extrasynaptically, glutamate released by system xc− activates presynaptic metabotropic glutamate receptors 2 and 3 (mGluR2/3) and thereby reduces the release probability of vesicular glutamate into the synaptic cleft. Postsynaptically, glutamate released by system xc− activates metabotropic glutamate receptor 5 (mGluR5). As a result, long-term depression (LTD) is permitted. The cyst(e)ine pro-drug N-acetyl cysteine can increase extracellular cystine, thereby activating glutamate release by system xc− and subsequently increasing signaling through pre- and postsynaptic mGluRs.
In summary, system xc− age-dependently modifies hippocampal-dependent memory. Moreover, dysregulation of system xc− activity plays a role in addictive behavior in rodents. These findings indicate that system xc− activity in the CNS affects higher brain functions.
V. Conclusion
System xc− plays a role in the physiology and pathophysiology of many cells and tissues. Although its specific subunit, xCT, shows a rather restricted expression pattern under normal conditions in vivo, it is induced in disease states where oxidative stress is present. Inflammatory stimuli also induce system xc−. Cystine imported by system xc− increases GSH synthesis, thereby strengthens the antioxidant defense, and, thus, should have a protective function. In addition, system xc− regulates the extracellular redox milieu by shifting the cystine/cysteine redox couple to a more reduced state.
Transcriptional regulation through the nuclear factors Nrf2 and ATF4 is the best characterized molecular mechanism by which xCT expression is induced. Other pathways, including its induction in certain cell types through inflammatory stimuli such as LPS, TNFα, and IL-1β, remain to be characterized in detail. On the functional level system, xc− activity is regulated by substrate availability and pH. Most importantly, glutamate transporters, EAATs, might regulate system xc−activity by increasing intracellular glutamate levels as the driving force for cystine uptake and reducing the inhibitory action of extracellular glutamate.
We show that the specific subunit of system xc−, xCT, and thus most probably system xc−-like activity, is an evolutionarily rather new member of the HAT light chain family, as orthologs of xCT are restricted to Deuterostomia and are absent in the Protostomia. Which evolutionary contraints required system xc− activity remain to be defined. However, since xCT-deficient mice are viable and show an inconsistent phenotype, loss of system xc− can be compensated for by unknown mechanisms.
In vivo xCT is most prominently expressed in the CNS and in parts of the immune system. Experimental evidence suggests that system xc− activity plays a role in regulating the immune response. In the CNS, the fact that the amino acid glutamate, which is exported by system xc−, also acts as a neurotransmitter explains why system xc− has the propensity to modulate memory and behavior. However, since higher levels of glutamate induce excitotoxic neuronal death, up-regulation of system xc− in disease states of the brain might represent a double-edged sword by inducing excitotoxicity. This could explain why xCT deficiency is protective in animal models of PD and epilepsy. Whether induction of system xc−activity is beneficial or detrimental might depend on the pathway of induction and whether increased glutamate release can be compensated for by glutamate uptake through EAATs, which are frequently down-regulated in diseases of the brain. Whether inhibition of system xc− or dual induction of xCT and EAAT is the more promising neuroprotective strategy remains to be explored. In addition, similar to the brain, system xc− activity might also play a role in both antioxidant defense and neurotransmission in the eye. Up-regulation of xCT also has been frequently observed in cancers. Here, xCT expression correlates with resistance against many chemotherapeutic drugs, most likely through GSH-mediated drug resistance. However, xCT-overexpressing resistant cancer cells are uniquely sensitive to some specific compounds, for example, selenite.
In contrast to the in vivo situation, system xc− activity is indispensable to the growth and survival of most cell types in cell culture. Nerve cell death induced by glutamate-mediated inhibition of system xc− activity, a pathway called oxidative glutamate toxicity, has been successfully used to characterize neuroprotective signaling pathways, genes, and putative therapeutic substances.
Future research designed to (i) characterize the functional relevance of system xc− activity in disease states with oxidative stress and inflammation; (ii) understand the regulation of xCT in diverse cells types in greater detail; and (iii) develop more specific pharmacological tools that inhibit and induce system xc− might pave the way for treating disorders as diverse as autoimmunity, neurodegeneration, and addiction. With regard to clinical trials, in our opinion, the most important question to be addressed is whether xCT expression in tumours correlates with disease progression and resistance to chemotherapy in humans in vivo. Moreover, larger clinical trials are warranted to substantiate the efficacy of the indirect system xc− activator NAC to treat addiction.
Supplementary Material
Abbreviations Used
- 3′UTR
3′ untranslated region
- 3-MCA
3-methylcholanthrene
- 4F2hc
4F2 heavy chain
- 5′UTR
5′ untranslated region
- 6-OHDA
6-hydroxydopamine
- 12/15-LOX
12/15 lipoxygenase
- AARE
amino acid response element
- AD
Alzheimer's disease
- AIF
apoptosis-inducing factor
- AMD
age-related macular degeneration
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ARE
antioxidant response element
- ASC
alanine-serine-cysteine
- ATF4
activating transcription factor 4
- Aβ
β-amyloid
- Bid
BH3-interacting domain death agonist
- BMAA
β-N-methylamino-l-alanine
- C/EBP
CCAAT/enhancer-binding protein
- cAMP
cyclic adenosine 3′,5′ monophosphate
- CNS
central nervous system
- DEM
diethyl maleate
- DIDS
4,4′-diisothiocyanatostilbene-2,20-disulfonic acid
- DNDS
4,4′-dinitrostilbene-2,2′-disulfonic acid
- EAAT
excitatory amino acid transporter
- EAE
experimental autoimmune encephalomyelitis
- EpRE
electrophile response element
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- FCCP
cyanide p-trifluoromethoxyphenylhydrazone
- FGF-2
fibroblast growth factor-2
- FGFR1
fibroblast growth factor receptor 1
- Gb
genderblind
- gcl
ganglion cells
- GCL
glutamate cysteine ligase
- GCN2
general control non-derepressible-2
- GGT
γ-glutamyl transferase
- GOT
glutamate oxaloacetate transaminase
- GPCRs
G-protein coupled receptors
- GPx
glutathione peroxidase
- GPx4
glutathione peroxidase 4
- GR
glutathione reductase
- GS
glutathione synthase
- GSH
glutathione
- GSSG
glutathione disulfide
- GST
glutathione S-transferase
- H2O2
hydrogen peroxide
- H2S
hydrogen sulfide
- HATs
heterodimeric amino acid transporters
- HRI
heme-regulated eIF2α kinase
- IL-1β
interleukin-1β
- IPL
inner plexiform layer
- KS
Kaposi's sarcoma
- KSHV
Kaposi's sarcoma–associated herpesvirus
- LPS
lipopolysaccharide
- LTD
long-term depression
- LTP
long-term potentiation
- MEK
mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- Mnd
minidisc
- MRP
multi-drug resistance protein
- MS
multiple sclerosis
- NAC
N-acetylcysteine
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- Nec-1
necrostatin 1
- NF-κB
nuclear factor kappa B (transcription factor)
- NMDA
N-methyl-D-aspartic acid
- NO
nitric oxide
- Nrf2
NF-E2-related factor 2
- OATPs
organic anion transporter proteins
- ONL
outer nuclear layer
- OPL
outer plexiform layer
- ORF
open reading frame
- OS
outer segments
- PD
Parkinson's disease
- PERK
PKR-like kinase
- PET
positron emission tomography
- PKA
protein kinase A
- PKC
protein kinase C
- PKR
protein kinase R
- ROS
reactive oxygen species
- RPE
retinal pigment epithelium
- (S)-4-CPG
(S)-4-carboxyphenylglycine
- SITS
4-acetamido-4′-isothiocyanatostilbene-2,2′-disulfonic acid
- SOD
superoxide dismutase
- TNFα
tumor necrosis factor α
- TRR
thioredoxin reductase
Acknowledgments
J. Lewerenz. and A. Methner were supported by the the Fritz-Thyssen-Stiftung (Az10.08.1.174); S.B. Smith and V. Ganapathy, by the National Institutes of Health NEI (2R01 EY012830 and 2R01 EY014560); S. Hewett, by the National Institutes of Health NINDS (NS051445); A. Massie and I. Smolders, by the Medical Foundation Queen Elisabeth and the Charcot Foundation; and P. Maher, by the Alzheimer's Association (IRG-07-58874). S.B.S and V.G. thank Mrs. L. McKie for preparing Figure 12 and Dr. A. Tawfik for immunolocalization data shown in Figure 14.
References
- 1.Aharoni-Simon M. Reifen R. Tirosh O. ROS-production-mediated activation of AP-1 but not NFkappaB inhibits glutamate-induced HT4 neuronal cell death. Antioxid Redox Signal. 2006;8:1339–1349. doi: 10.1089/ars.2006.8.1339. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht P. Lewerenz J. Dittmer S. Noack R. Maher P. Methner A. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc− as a neuroprotective drug target. CNS Neurol Disord Drug Targets. 2010;9:373–382. doi: 10.2174/187152710791292567. [DOI] [PubMed] [Google Scholar]
- 3.Anderson CL. Iyer SS. Ziegler TR. Jones DP. Control of extracellular cysteine/cystine redox state by HT-29 cells is independent of cellular glutathione. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1069–R1075. doi: 10.1152/ajpregu.00195.2007. [DOI] [PubMed] [Google Scholar]
- 4.Antonini B. Piva S. Paltenghi M. Candiani A. Latronico N. The early phase of critical illness is a progressive acidic state due to unmeasured anions. Eur J Anaesthesiol. 2008;25:566–571. doi: 10.1017/S0265021508003669. [DOI] [PubMed] [Google Scholar]
- 5.Asanuma M. Miyazaki I. Diaz-Corrales FJ. Kimoto N. Kikkawa Y. Takeshima M. Miyoshi K. Murata M. Neuroprotective effects of zonisamide target astrocyte. Ann Neurol. 2010;67:239–249. doi: 10.1002/ana.21885. [DOI] [PubMed] [Google Scholar]
- 6.Augustin H. Grosjean Y. Chen K. Sheng Q. Featherstone DE. Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J Neurosci. 2007;27:111–123. doi: 10.1523/JNEUROSCI.4770-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker DA. Madayag A. Kristiansen LV. Meador-Woodruff JH. Haroutunian V. Raju I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology. 2008;33:1760–1772. doi: 10.1038/sj.npp.1301532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker DA. McFarland K. Lake RW. Shen H. Tang XC. Toda S. Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- 9.Baker DA. Xi ZX. Shen H. Swanson CJ. Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banjac A. Perisic T. Sato H. Seiler A. Bannai S. Weiss N. Kolle P. Tschoep K. Issels RD. Daniel PT. Conrad M. Bornkamm GW. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27:1618–1628. doi: 10.1038/sj.onc.1210796. [DOI] [PubMed] [Google Scholar]
- 11.Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261:2256–2263. [PubMed] [Google Scholar]
- 12.Bannai S. Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J Biol Chem. 1984;259:2435–2440. [PubMed] [Google Scholar]
- 13.Bannai S. Ishii T. A novel function of glutamine in cell culture: utilization of glutamine for the uptake of cystine in human fibroblasts. J Cell Physiol. 1988;137:360–366. doi: 10.1002/jcp.1041370221. [DOI] [PubMed] [Google Scholar]
- 14.Bannai S. Ishii T. Transport of cystine and cysteine and cell growth in cultured human diploid fibroblasts: effect of glutamate and homocysteate. J Cell Physiol. 1982;112:265–272. doi: 10.1002/jcp.1041120216. [DOI] [PubMed] [Google Scholar]
- 15.Bannai S. Kasuga H. Anti-inflammatory drug inhibition of transport of cystine and glutamate in cultured human fibroblasts. Biochem Pharmacol. 1985;34:1852–1854. doi: 10.1016/0006-2952(85)90663-x. [DOI] [PubMed] [Google Scholar]
- 16.Bannai S. Kitamura E. Role of proton dissociation in the transport of cystine and glutamate in human diploid fibroblasts in culture. J Biol Chem. 1981;256:5770–5772. [PubMed] [Google Scholar]
- 17.Bannai S. Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. 1980;255:2372–2376. [PubMed] [Google Scholar]
- 18.Bannai S. Sato H. Ishii T. Sugita Y. Induction of cystine transport activity in human fibroblasts by oxygen. J Biol Chem. 1989;264:18480–18484. [PubMed] [Google Scholar]
- 19.Barger SW. Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- 20.Barger SW. Goodwin ME. Porter MM. Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassi MT. Gasol E. Manzoni M. Pineda M. Riboni M. Martin R. Zorzano A. Borsani G. Palacin M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc−. Pflugers Arch. 2001;442:286–296. doi: 10.1007/s004240100537. [DOI] [PubMed] [Google Scholar]
- 22.Bedingfield JS. Kemp MC. Jane DE. Tse HW. Roberts PJ. Watkins JC. Structure-activity relationships for a series of phenylglycine derivatives acting at metabotropic glutamate receptors (mGluRs) Br J Pharmacol. 1995;116:3323–3329. doi: 10.1111/j.1476-5381.1995.tb15142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berghe TV. Vanlangenakker N. Parthoens E. Deckers W. Devos M. Festjens N. Guerin CJ. Brunk UT. Declercq W. Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 24.Berthoud VM. Beyer EC. Oxidative stress, lens gap junctions, and cataracts. Antioxid Redox Signal. 2009;11:339–353. doi: 10.1089/ars.2008.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bi M. Naczki C. Koritzinsky M. Fels D. Blais J. Hu N. Harding H. Novoa I. Varia M. Raleigh J. Scheuner D. Kaufman RJ. Bell J. Ron D. Wouters BG. Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–3481. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brauner-Osborne H. Nielsen B. Krogsgaard-Larsen P. Molecular pharmacology of homologues of ibotenic acid at cloned metabotropic glutamic acid receptors. Eur J Pharmacol. 1998;350:311–316. doi: 10.1016/s0014-2999(98)00246-5. [DOI] [PubMed] [Google Scholar]
- 27.Bridges CC. Hu H. Miyauchi S. Siddaramappa UN. Ganapathy ME. Ignatowicz L. Maddox DM. Smith SB. Ganapathy V. Induction of cystine-glutamate transporter xc− by human immunodeficiency virus type 1 transactivator protein tat in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2004;45:2906–2914. doi: 10.1167/iovs.03-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bridges CC. Kekuda R. Wang H. Prasad PD. Mehta P. Huang W. Smith SB. Ganapathy V. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42:47–54. [PubMed] [Google Scholar]
- 29.Buckingham SC. Campbell SL. Haas BR. Montana V. Robel S. Ogunrinu T. Sontheimer H. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17:1269–1274. doi: 10.1038/nm.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burdo J. Dargusch R. Schubert D. Distribution of the cystine/glutamate antiporter system xc− in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54:549–557. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- 31.Cai J. Nelson KC. Wu M. Sternberg P., Jr. Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 32.Chang YF. Lysine metabolism in the human and the monkey: demonstration of pipecolic acid formation in the brain and other organs. Neurochem Res. 1982;7:577–588. doi: 10.1007/BF00965124. [DOI] [PubMed] [Google Scholar]
- 33.Chen CJ. Liao SL. Kuo JS. Gliotoxic action of glutamate on cultured astrocytes. J Neurochem. 2000;75:1557–1565. doi: 10.1046/j.1471-4159.2000.0751557.x. [DOI] [PubMed] [Google Scholar]
- 34.Chen Q. Prior M. Dargusch R. Roberts A. Riek R. Eichmann C. Chiruta C. Akaishi T. Abe K. Maher P. Schubert D. A novel neurotrophic drug for cognitive enhancement and Alzheimer's disease. PLoS One. 2011;6:e27865. doi: 10.1371/journal.pone.0027865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen RS. Song YM. Zhou ZY. Tong T. Li Y. Fu M. Guo XL. Dong LJ. He X. Qiao HX. Zhan QM. Li W. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene. 2009;28:599–609. doi: 10.1038/onc.2008.414. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y. Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem. 2003;84:1332–1339. doi: 10.1046/j.1471-4159.2003.01630.x. [DOI] [PubMed] [Google Scholar]
- 37.Chintala S. Li W. Lamoreux ML. Ito S. Wakamatsu K. Sviderskaya EV. Bennett DC. Park YM. Gahl WA. Huizing M. Spritz RA. Ben S. Novak EK. Tan J. Swank RT. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc Natl Acad Sci U S A. 2005;102:10964–10969. doi: 10.1073/pnas.0502856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 39.Choi HJ. Kang KS. Fukui M. Zhu BT. Critical role of the JNK-p53-GADD45alpha apoptotic cascade in mediating oxidative cytotoxicity in hippocampal neurons. Br J Pharmacol. 2011;162:175–192. doi: 10.1111/j.1476-5381.2010.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christensen HN. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol Rev. 1990;70:43–77. doi: 10.1152/physrev.1990.70.1.43. [DOI] [PubMed] [Google Scholar]
- 41.Chung WJ. Lyons SA. Nelson GM. Hamza H. Gladson CL. Gillespie GY. Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Circu ML. Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coquelle T. Christensen JK. Banke TG. Madsen U. Schousboe A. Pickering DS. Agonist discrimination between AMPA receptor subtypes. Neuroreport. 2000;11:2643–2648. doi: 10.1097/00001756-200008210-00008. [DOI] [PubMed] [Google Scholar]
- 44.Crimi E. Sica V. Williams-Ignarro S. Zhang H. Slutsky AS. Ignarro LJ. Napoli C. The role of oxidative stress in adult critical care. Free Radic Biol Med. 2006;40:398–406. doi: 10.1016/j.freeradbiomed.2005.10.054. [DOI] [PubMed] [Google Scholar]
- 45.Dai Z. Huang Y. Sadee W. Blower P. Chemoinformatics analysis identifies cytotoxic compounds susceptible to chemoresistance mediated by glutathione and cystine/glutamate transport system xc−. J Med Chem. 2007;50:1896–1906. doi: 10.1021/jm060960h. [DOI] [PubMed] [Google Scholar]
- 46.Dargusch R. Schubert D. Specificity of resistance to oxidative stress. J Neurochem. 2002;81:1394–1400. doi: 10.1046/j.1471-4159.2002.00950.x. [DOI] [PubMed] [Google Scholar]
- 47.Davis JB. Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 48.De Bundel D. Schallier A. Loyens E. Fernando R. Miyashita H. Van Liefferinge J. Vermoesen K. Bannai S. Sato H. Michotte Y. Smolders I. Massie A. Loss of system xc−formula does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci. 2011;31:5792–5803. doi: 10.1523/JNEUROSCI.5465-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Degterev A. Huang Z. Boyce M. Li Y. Jagtap P. Mizushima N. Cuny GD. Mitchison TJ. Moskowitz MA. Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 50.Dittmer S. Sahin M. Pantlen A. Saxena A. Toutzaris D. Pina AL. Geerts A. Golz S. Methner A. The constitutively active orphan G-protein-coupled receptor GPR39 protects from cell death by increasing secretion of pigment epithelium-derived growth factor. J Biol Chem. 2008;283:7074–7081. doi: 10.1074/jbc.M704323200. [DOI] [PubMed] [Google Scholar]
- 51.Do KQ. Benz B. Sorg O. Pellerin L. Magistretti PJ. beta-Adrenergic stimulation promotes homocysteic acid release from astrocyte cultures: evidence for a role of astrocytes in the modulation of synaptic transmission. J Neurochem. 1997;68:2386–2394. doi: 10.1046/j.1471-4159.1997.68062386.x. [DOI] [PubMed] [Google Scholar]
- 52.Do KQ. Herrling PL. Streit P. Cuenod M. Release of neuroactive substances: homocysteic acid as an endogenous agonist of the NMDA receptor. J Neural Transm. 1988;72:185–190. doi: 10.1007/BF01243418. [DOI] [PubMed] [Google Scholar]
- 53.Domercq M. Sanchez-Gomez MV. Sherwin C. Etxebarria E. Fern R. Matute C. System xc− and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178:6549–6556. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- 54.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 55.Dringen R. Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 56.Droge W. Cysteine and glutathione deficiency in AIDS patients: a rationale for the treatment with N-acetyl-cysteine. Pharmacology. 1993;46:61–65. doi: 10.1159/000139029. [DOI] [PubMed] [Google Scholar]
- 57.Dun Y. Mysona B. Van Ells T. Amarnath L. Ola MS. Ganapathy V. Smith SB. Expression of the cystine-glutamate exchanger xc− in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324:189–202. doi: 10.1007/s00441-005-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ebert B. Madsen U. Johansen TN. Krogsgaard-Larsen P. NMDA receptor agonists: relationships between structure and biological activity. Adv Exp Med Biol. 1991;287:483–487. doi: 10.1007/978-1-4684-5907-4_42. [DOI] [PubMed] [Google Scholar]
- 59.Elphick LM. Hawat M. Toms NJ. Meinander A. Mikhailov A. Eriksson JE. Kass GE. Opposing roles for caspase and calpain death proteases in L-glutamate-induced oxidative neurotoxicity. Toxicol Appl Pharmacol. 2008;232:258–267. doi: 10.1016/j.taap.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 60.Falk MH. Hultner L. Milner A. Gregory CD. Bornkamm GW. Irradiated fibroblasts protect Burkitt lymphoma cells from apoptosis by a mechanism independent of bcl-2. Int J Cancer. 1993;55:485–491. doi: 10.1002/ijc.2910550327. [DOI] [PubMed] [Google Scholar]
- 61.Ferraguti F. Pietra C. Valerio E. Corti C. Chiamulera C. Conquet F. Evidence against a permissive role of the metabotropic glutamate receptor 1 in acute excitotoxicity. Neuroscience. 1997;79:1–5. doi: 10.1016/s0306-4522(97)00074-2. [DOI] [PubMed] [Google Scholar]
- 62.Fogal B. Hewett SJ. Interleukin-1beta: a bridge between inflammation and excitotoxicity? J Neurochem. 2008;106:1–23. doi: 10.1111/j.1471-4159.2008.05315.x. [DOI] [PubMed] [Google Scholar]
- 63.Fogal B. Li J. Lobner D. McCullough LD. Hewett SJ. System xc− activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J Neurosci. 2007;27:10094–10105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fogal B. Trettel J. Uliasz TF. Levine ES. Hewett SJ. Changes in secondary glutamate release underlie the developmental regulation of excitotoxic neuronal cell death. Neuroscience. 2005;132:929–942. doi: 10.1016/j.neuroscience.2005.01.036. [DOI] [PubMed] [Google Scholar]
- 65.Franco R. Cidlowski JA. SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J Biol Chem. 2006;281:29542–29557. doi: 10.1074/jbc.M602500200. [DOI] [PubMed] [Google Scholar]
- 66.Fredriksson R. Nordstrom KJ. Stephansson O. Hagglund MG. Schioth HB. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett. 2008;582:3811–3816. doi: 10.1016/j.febslet.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 67.Fukasawa Y. Segawa H. Kim JY. Chairoungdua A. Kim DK. Matsuo H. Cha SH. Endou H. Kanai Y. Identification and characterization of a Na+-independent neutral amino acid transporter that associates with the 4F2 heavy chain and exhibits substrate selectivity for small neutral D- and L-amino acids. J Biol Chem. 2000;275:9690–9698. doi: 10.1074/jbc.275.13.9690. [DOI] [PubMed] [Google Scholar]
- 68.Ganea E. Harding JJ. Glutathione-related enzymes and the eye. Curr Eye Res. 2006;31:1–11. doi: 10.1080/02713680500477347. [DOI] [PubMed] [Google Scholar]
- 69.Gasol E. Jimenez-Vidal M. Chillaron J. Zorzano A. Palacin M. Membrane topology of system xc− light subunit reveals a re-entrant loop with substrate-restricted accessibility. J Biol Chem. 2004;279:31228–31236. doi: 10.1074/jbc.M402428200. [DOI] [PubMed] [Google Scholar]
- 70.Gelderblom M. Leypoldt F. Lewerenz J. Birkenmayer G. Orozco D. Ludewig P. Thundyil J. Arumugam TV. Gerloff C. Tolosa E. Maher P. Magnus T. The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J Cereb Blood Flow Metab. 2012;32:835–843. doi: 10.1038/jcbfm.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gibbs JE. Walker MC. Cock HR. Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia. 2006;47:469–478. doi: 10.1111/j.1528-1167.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 72.Gmunder H. Eck HP. Benninghoff B. Roth S. Droge W. Macrophages regulate intracellular glutathione levels of lymphocytes. Evidence for an immunoregulatory role of cysteine. Cell Immunol. 1990;129:32–46. doi: 10.1016/0008-8749(90)90184-s. [DOI] [PubMed] [Google Scholar]
- 73.Gmunder H. Eck HP. Droge W. Low membrane transport activity for cystine in resting and mitogenically stimulated human lymphocyte preparations and human T cell clones. Eur J Biochem. 1991;201:113–117. doi: 10.1111/j.1432-1033.1991.tb16263.x. [DOI] [PubMed] [Google Scholar]
- 74.Gnana-Prakasam JP. Thangaraju M. Liu K. Ha Y. Martin PM. Smith SB. Ganapathy V. Absence of iron-regulatory protein Hfe results in hyperproliferation of retinal pigment epithelium: role of cystine/glutamate exchanger. Biochem J. 2009;424:243–252. doi: 10.1042/BJ20090424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Go YM. Jones DP. Cysteine/cystine redox signaling in cardiovascular disease. Free Radic Biol Med. 2011;50:495–509. doi: 10.1016/j.freeradbiomed.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gochenauer GE. Robinson MB. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent L-[3H]glutamate transport and expression of both system xc−subunits. J Neurochem. 2001;78:276–286. doi: 10.1046/j.1471-4159.2001.00385.x. [DOI] [PubMed] [Google Scholar]
- 77.Gonsette RE. Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J Neurol Sci. 2008;274:48–53. doi: 10.1016/j.jns.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 78.Gottesman MM. Fojo T. Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 79.Gout PW. Buckley AR. Simms CR. Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc− cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 80.Gout PW. Kang YJ. Buckley DJ. Bruchovsky N. Buckley AR. Increased cystine uptake capability associated with malignant progression of Nb2 lymphoma cells. Leukemia. 1997;11:1329–1337. doi: 10.1038/sj.leu.2400739. [DOI] [PubMed] [Google Scholar]
- 81.Grieve A. Griffiths R. Simultaneous measurement by HPLC of the excitatory amino acid transmitter candidates homocysteate and homocysteine sulphinate supports a predominant astrocytic localisation. Neurosci Lett. 1992;145:1–5. doi: 10.1016/0304-3940(92)90189-e. [DOI] [PubMed] [Google Scholar]
- 82.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 83.Guldin WO. Markowitsch HJ. Epidural kainate, but not ibotenate, produces lesions in local and distant regions of the brain. A comparison of the intracerebral actions of kainic acid and ibotenic acid. J Neurosci Methods. 1982;5:83–93. doi: 10.1016/0165-0270(82)90055-3. [DOI] [PubMed] [Google Scholar]
- 84.Gupta S. A decision between life and death during TNF-alpha-induced signaling. J Clin Immunol. 2002;22:185–194. doi: 10.1023/a:1016089607548. [DOI] [PubMed] [Google Scholar]
- 85.Ha JS. Lim HM. Park SS. Extracellular hydrogen peroxide contributes to oxidative glutamate toxicity. Brain Res. 2010;1359:291–297. doi: 10.1016/j.brainres.2010.08.086. [DOI] [PubMed] [Google Scholar]
- 86.Hahn P. Milam AH. Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch's membrane. Arch Ophthalmol. 2003;121:1099–1105. doi: 10.1001/archopht.121.8.1099. [DOI] [PubMed] [Google Scholar]
- 87.Harding HP. Zhang Y. Zeng H. Novoa I. Lu PD. Calfon M. Sadri N. Yun C. Popko B. Paules R. Stojdl DF. Bell JC. Hettmann T. Leiden JM. Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 88.Harris RA. Amor S. Sweet and sour—oxidative and carbonyl stress in neurological disorders. CNS Neurol Disord Drug Targets. 2011;10:82–107. doi: 10.2174/187152711794488656. [DOI] [PubMed] [Google Scholar]
- 89.Harvey CJ. Thimmulappa RK. Singh A. Blake DJ. Ling G. Wakabayashi N. Fujii J. Myers A. Biswal S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med. 2009;46:443–453. doi: 10.1016/j.freeradbiomed.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hayes JD. Flanagan JU. Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 91.Hu RG. Lim J. Donaldson PJ. Kalloniatis M. Characterization of the cystine/glutamate transporter in the outer plexiform layer of the vertebrate retina. Eur J Neurosci. 2008;28:1491–1502. doi: 10.1111/j.1460-9568.2008.06435.x. [DOI] [PubMed] [Google Scholar]
- 92.Huang Y. Pharmacogenetics/genomics of membrane transporters in cancer chemotherapy. Cancer Metastasis Rev. 2007;26:183–201. doi: 10.1007/s10555-007-9050-6. [DOI] [PubMed] [Google Scholar]
- 93.Huang Y. Dai Z. Barbacioru C. Sadee W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65:7446–7454. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- 94.Huang Y. Penchala S. Pham AN. Wang J. Genetic variations and gene expression of transporters in drug disposition and response. Expert Opin Drug Metab Toxicol. 2008;4:237–254. doi: 10.1517/17425255.4.3.237. [DOI] [PubMed] [Google Scholar]
- 95.Hur W. Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15:162–173. doi: 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 96.Igarashi T. Izumi H. Uchiumi T. Nishio K. Arao T. Tanabe M. Uramoto H. Sugio K. Yasumoto K. Sasaguri Y. Wang KY. Otsuji Y. Kohno K. Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene. 2007;26:4749–4760. doi: 10.1038/sj.onc.1210289. [DOI] [PubMed] [Google Scholar]
- 97.Imai H. Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 98.Ishige K. Chen Q. Sagara Y. Schubert D. The activation of dopamine D4 receptors inhibits oxidative stress-induced nerve cell death. J Neurosci. 2001;21:6069–6076. doi: 10.1523/JNEUROSCI.21-16-06069.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ishige K. Schubert D. Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med. 2001;30:433–446. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]
- 100.Ishii T. Hishinuma I. Bannai S. Sugita Y. Mechanism of growth promotion of mouse lymphoma L1210 cells in vitro by feeder layer or 2-mercaptoethanol. J Cell Physiol. 1981;107:283–293. doi: 10.1002/jcp.1041070215. [DOI] [PubMed] [Google Scholar]
- 101.Ishii T. Itoh K. Takahashi S. Sato H. Yanagawa T. Katoh Y. Bannai S. Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 102.Ishii T. Sugita Y. Bannai S. Regulation of glutathione levels in mouse spleen lymphocytes by transport of cysteine. J Cell Physiol. 1987;133:330–336. doi: 10.1002/jcp.1041330217. [DOI] [PubMed] [Google Scholar]
- 103.Ishimoto T. Nagano O. Yae T. Tamada M. Motohara T. Oshima H. Oshima M. Ikeda T. Asaba R. Yagi H. Masuko T. Shimizu T. Ishikawa T. Kai K. Takahashi E. Imamura Y. Baba Y. Ohmura M. Suematsu M. Baba H. Saya H. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc− and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 104.Jabaudon D. Scanziani M. Gahwiler BH. Gerber U. Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci U S A. 2000;97:5610–5615. doi: 10.1073/pnas.97.10.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jackman NA. Uliasz TF. Hewett JA. Hewett SJ. Regulation of system xc− activity and expression in astrocytes by interleukin-1beta: implications for hypoxic neuronal injury. Glia. 2010;58:1806–1815. doi: 10.1002/glia.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaleeba JA. Berger EA. Kaposi's sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science. 2006;311:1921–1924. doi: 10.1126/science.1120878. [DOI] [PubMed] [Google Scholar]
- 107.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 108.Kaltschmidt B. Widera D. Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta. 2005;1745:287–299. doi: 10.1016/j.bbamcr.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 109.Kanai Y. Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 110.Kasanetz F. Deroche-Gamonet V. Berson N. Balado E. Lafourcade M. Manzoni O. Piazza PV. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science. 2010;328:1709–1712. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- 111.Kato S. Ishita S. Sugawara K. Mawatari K. Cystine/glutamate antiporter expression in retinal Muller glial cells: implications for DL-alpha-aminoadipate toxicity. Neuroscience. 1993;57:473–482. doi: 10.1016/0306-4522(93)90080-y. [DOI] [PubMed] [Google Scholar]
- 112.Kau KS. Madayag A. Mantsch JR. Grier MD. Abdulhameed O. Baker DA. Blunted cystine-glutamate antiporter function in the nucleus accumbens promotes cocaine-induced drug seeking. Neuroscience. 2008;155:530–537. doi: 10.1016/j.neuroscience.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kensler TW. Wakabayashi N. Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 114.Kilberg MS. Shan J. Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim JY. Kanai Y. Chairoungdua A. Cha SH. Matsuo H. Kim DK. Inatomi J. Sawa H. Ida Y. Endou H. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim Biophys Acta. 2001;1512:335–344. doi: 10.1016/s0005-2736(01)00338-8. [DOI] [PubMed] [Google Scholar]
- 116.Kimura Y. Dargusch R. Schubert D. Kimura H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid Redox Signal. 2006;8:661–670. doi: 10.1089/ars.2006.8.661. [DOI] [PubMed] [Google Scholar]
- 117.Kimura Y. Goto Y. Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- 118.Kimura Y. Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 119.Kingston AE. Lowndes J. Evans N. Clark B. Tomlinson R. Burnett JP. Mayne NG. Cockerham SL. Lodge D. Sulphur-containing amino acids are agonists for group 1 metabotropic receptors expressed in clonal RGT cell lines. Neuropharmacology. 1998;37:277–287. doi: 10.1016/s0028-3908(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 120.Knackstedt LA. LaRowe S. Mardikian P. Malcolm R. Upadhyaya H. Hedden S. Markou A. Kalivas PW. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65:841–845. doi: 10.1016/j.biopsych.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Knackstedt LA. Melendez RI. Kalivas PW. Ceftriaxone Restores Glutamate Homeostasis and Prevents Relapse to Cocaine Seeking. Biol Psychiatry. 2009;67:81–84. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knickelbein RG. Seres T. Lam G. Johnston RB., Jr. Warshaw JB. Characterization of multiple cysteine and cystine transporters in rat alveolar type II cells. Am J Physiol. 1997;273:L1147–L1155. doi: 10.1152/ajplung.1997.273.6.L1147. [DOI] [PubMed] [Google Scholar]
- 123.Koglin N. Mueller A. Berndt M. Schmitt-Willich H. Toschi L. Stephens AW. Gekeler V. Friebe M. Dinkelborg LM. Specific PET imaging of xc− transporter activity using a 18F-labeled glutamate derivative reveals a dominant pathway in tumor metabolism. Clin Cancer Res. 2011;17:6000–6011. doi: 10.1158/1078-0432.CCR-11-0687. [DOI] [PubMed] [Google Scholar]
- 124.Koyama Y. Kimura Y. Hashimoto H. Matsuda T. Baba A. L-lactate inhibits L-cystine/L-glutamate exchange transport and decreases glutathione content in rat cultured astrocytes. J Neurosci Res. 2000;59:685–691. doi: 10.1002/(SICI)1097-4547(20000301)59:5<685::AID-JNR12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 125.Kranich O. Hamprecht B. Dringen R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci Lett. 1996;219:211–214. doi: 10.1016/s0304-3940(96)13217-1. [DOI] [PubMed] [Google Scholar]
- 126.Kubota C. Torii S. Hou N. Saito N. Yoshimoto Y. Imai H. Takeuchi T. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem. 2010;285:667–674. doi: 10.1074/jbc.M109.053058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kvamme E. Schousboe A. Hertz L. Torgner IA. Svenneby G. Developmental change of endogenous glutamate and gamma-glutamyl transferase in cultured cerebral cortical interneurons and cerebellar granule cells, and in mouse cerebral cortex and cerebellum in vivo. Neurochem Res. 1985;10:993–1008. doi: 10.1007/BF00964635. [DOI] [PubMed] [Google Scholar]
- 128.Kwak MK. Kensler TW. Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol. 2010;244:66–76. doi: 10.1016/j.taap.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lall MM. Ferrell J. Nagar S. Fleisher LN. McGahan MC. Iron regulates L-cystine uptake and glutathione levels in lens epithelial and retinal pigment epithelial cells by its effect on cytosolic aconitase. Invest Ophthalmol Vis Sci. 2008;49:310–319. doi: 10.1167/iovs.07-1041. [DOI] [PubMed] [Google Scholar]
- 130.Lam BK. Leukotriene C4 synthase: a critical enzyme for the biosynthesis of SRS-A. Front Biosci. 1997;2:d380–d386. doi: 10.2741/a198. [DOI] [PubMed] [Google Scholar]
- 131.Landshamer S. Hoehn M. Barth N. Duvezin-Caubet S. Schwake G. Tobaben S. Kazhdan I. Becattini B. Zahler S. Vollmar A. Pellecchia M. Reichert A. Plesnila N. Wagner E. Culmsee C. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 2008;15:1553–1563. doi: 10.1038/cdd.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Langford MP. Redmond P. Chanis R. Misra RP. Redens TB. Glutamate, excitatory amino acid transporters, xc− antiporter, glutamine synthetase, and gamma-glutamyltranspeptidase in human corneal epithelium. Curr Eye Res. 2010;35:202–211. doi: 10.3109/02713680903461489. [DOI] [PubMed] [Google Scholar]
- 133.Lapchak PA. Schubert DR. Maher PA. Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke. J Neurochem. 2011;116:122–131. doi: 10.1111/j.1471-4159.2010.07090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.LaRowe SD. Myrick H. Hedden S. Mardikian P. Saladin M. McRae A. Brady K. Kalivas PW. Malcolm R. Is cocaine desire reduced by N-acetylcysteine? Am J Psychiatry. 2007;164:1115–1117. doi: 10.1176/ajp.2007.164.7.1115. [DOI] [PubMed] [Google Scholar]
- 135.Lee M. Cho T. Jantaratnotai N. Wang YT. McGeer E. McGeer PL. Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degenerative neurological diseases. FASEB J. 2010;24:2533–2545. doi: 10.1096/fj.09-149997. [DOI] [PubMed] [Google Scholar]
- 136.Lewerenz J. Albrecht P. Tien ML. Henke N. Karumbayaram S. Kornblum HI. Wiedau-Pazos M. Schubert D. Maher P. Methner A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- 137.Lewerenz J. Dargusch R. Maher P. Lactacidosis Modulates Glutathione Metabolism and Oxidative Glutamate Toxicity. J Neurochem. 2010;113:502–514. doi: 10.1111/j.1471-4159.2010.06621.x. [DOI] [PubMed] [Google Scholar]
- 138.Lewerenz J. Klein M. Methner A. Cooperative action of glutamate transporters and cystine/glutamate antiporter system xc− protects from oxidative glutamate toxicity. J Neurochem. 2006;98:916–925. doi: 10.1111/j.1471-4159.2006.03921.x. [DOI] [PubMed] [Google Scholar]
- 139.Lewerenz J. Letz J. Methner A. Activation of stimulatory heterotrimeric G proteins increases glutathione and protects neuronal cells against oxidative stress. J Neurochem. 2003;87:522–531. doi: 10.1046/j.1471-4159.2003.02019.x. [DOI] [PubMed] [Google Scholar]
- 140.Lewerenz J. Maher P. Basal levels of eIF2alpha phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem. 2009;284:1106–1115. doi: 10.1074/jbc.M807325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lewerenz J. Maher P. Control of redox state and redox signaling by neural antioxidant systems. Antioxid Redox Signal. 2011;14:1449–1465. doi: 10.1089/ars.2010.3600. [DOI] [PubMed] [Google Scholar]
- 142.Lewerenz J. Maher P. Methner A. Regulation of xCT expression and system xc− function in neuronal cells. Amino Acids. 2012;42:171–179. doi: 10.1007/s00726-011-0862-x. [DOI] [PubMed] [Google Scholar]
- 143.Li L. Lim J. Jacobs MD. Kistler J. Donaldson PJ. Regional differences in cystine accumulation point to a sutural delivery pathway to the lens core. Invest Ophthalmol Vis Sci. 2007;48:1253–1260. doi: 10.1167/iovs.06-0861. [DOI] [PubMed] [Google Scholar]
- 144.Li Y. Maher P. Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 145.Li Y. Maher P. Schubert D. Requirement for cGMP in nerve cell death caused by glutathione depletion. J Cell Biol. 1997;139:1317–1324. doi: 10.1083/jcb.139.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lim J. Lam YC. Kistler J. Donaldson PJ. Molecular characterization of the cystine/glutamate exchanger and the excitatory amino acid transporters in the rat lens. Invest Ophthalmol Vis Sci. 2005;46:2869–2877. doi: 10.1167/iovs.05-0156. [DOI] [PubMed] [Google Scholar]
- 147.Lim J. Lorentzen KA. Kistler J. Donaldson PJ. Molecular identification and characterisation of the glycine transporter (GLYT1) and the glutamine/glutamate transporter (ASCT2) in the rat lens. Exp Eye Res. 2006;83:447–455. doi: 10.1016/j.exer.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 148.Lim JC. Donaldson PJ. Focus on molecules: the cystine/glutamate exchanger (System xc−) Exp Eye Res. 2011;92:162–163. doi: 10.1016/j.exer.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 149.Liu H. Wang H. Shenvi S. Hagen TM. Liu RM. Glutathione metabolism during aging and in Alzheimer disease. Ann N Y Acad Sci. 2004;1019:346–349. doi: 10.1196/annals.1297.059. [DOI] [PubMed] [Google Scholar]
- 150.Liu R. Blower PE. Pham AN. Fang J. Dai Z. Wise C. Green B. Teitel CH. Ning B. Ling W. Lyn-Cook BD. Kadlubar FF. Sadee W. Huang Y. Cystine-glutamate transporter SLC7A11 mediates resistance to geldanamycin but not to 17-(allylamino)-17-demethoxygeldanamycin. Mol Pharmacol. 2007;72:1637–1646. doi: 10.1124/mol.107.039644. [DOI] [PubMed] [Google Scholar]
- 151.Liu X. Resch J. Rush T. Lobner D. Functional upregulation of system xc− by fibroblast growth factor-2. Neuropharmacology. 2012;62:901–906. doi: 10.1016/j.neuropharm.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 152.Liu X. Rush T. Zapata J. Lobner D. beta-N-methylamino-l-alanine induces oxidative stress and glutamate release through action on system xc−. Exp Neurol. 2009;217:429–433. doi: 10.1016/j.expneurol.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 153.Liu XX. Li XJ. Zhang B. Liang YJ. Zhou CX. Cao DX. He M. Chen GQ. He JR. Zhao Q. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 2011;585:1363–1367. doi: 10.1016/j.febslet.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 154.Liu Y. Dargusch R. Maher P. Schubert D. A broadly neuroprotective derivative of curcumin. J Neurochem. 2008;105:1336–1345. doi: 10.1111/j.1471-4159.2008.05236.x. [DOI] [PubMed] [Google Scholar]
- 155.Lo M. Ling V. Low C. Wang YZ. Gout PW. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol. 2010;17:9–16. doi: 10.3747/co.v17i3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lo M. Ling V. Wang YZ. Gout PW. The xc− cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer. 2008;99:464–472. doi: 10.1038/sj.bjc.6604485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lo M. Wang YZ. Gout PW. The xc− cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215:593–602. doi: 10.1002/jcp.21366. [DOI] [PubMed] [Google Scholar]
- 158.Lodge D. The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclature. Neuropharmacology. 2009;56:6–21. doi: 10.1016/j.neuropharm.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 159.Lu MJ. Pulido JS. McCannel CA. Pulido JE. Hatfield RM. Dundervill RF., 3rd Shippy SA. Detection of elevated signaling amino acids in human diabetic vitreous by rapid capillary electrophoresis. Exp Diabetes Res. 2007;2007:39765. doi: 10.1155/2007/39765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lyons SA. Chung WJ. Weaver AK. Ogunrinu T. Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Madayag A. Lobner D. Kau KS. Mantsch JR. Abdulhameed O. Hearing M. Grier MD. Baker DA. Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci. 2007;27:13968–13976. doi: 10.1523/JNEUROSCI.2808-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Maher P. A comparison of the neurotrophic activities of the flavonoid fisetin and some of its derivatives. Free Radic Res. 2006;40:1105–1111. doi: 10.1080/10715760600672509. [DOI] [PubMed] [Google Scholar]
- 164.Maher P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J Neurosci. 2001;21:2929–2938. doi: 10.1523/JNEUROSCI.21-09-02929.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Maher P. Proteasome inhibitors prevent oxidative stress-induced nerve cell death by a novel mechanism. Biochem Pharmacol. 2008;75:1994–2006. doi: 10.1016/j.bcp.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Maher P. Dargusch R. Bodai L. Gerard PE. Purcell JM. Marsh JL. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington's disease. Hum Mol Genet. 2011;20:261–270. doi: 10.1093/hmg/ddq460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Maher P. Dargusch R. Ehren JL. Okada S. Sharma K. Schubert D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One. 2011;6:e21226. doi: 10.1371/journal.pone.0021226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Maher P. Lewerenz J. Lozano C. Torres JL. A novel approach to enhancing cellular glutathione levels. J Neurochem. 2008;107:690–700. doi: 10.1111/j.1471-4159.2008.05620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Maher P. Salgado KF. Zivin JA. Lapchak PA. A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res. 2007;1173:117–125. doi: 10.1016/j.brainres.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Maher P. Schubert D. Signaling by reactive oxygen species in the nervous system. Cell Mol Life Sci. 2000;57:1287–1305. doi: 10.1007/PL00000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Makowske M. Christensen HN. Contrasts in transport systems for anionic amino acids in hepatocytes and a hepatoma cell line HTC. J Biol Chem. 1982;257:5663–5670. [PubMed] [Google Scholar]
- 172.Mandal PK. Seiler A. Perisic T. Kolle P. Banjac Canak A. Forster H. Weiss N. Kremmer E. Lieberman MW. Bannai S. Kuhlencordt P. Sato H. Bornkamm GW. Conrad M. System xc− and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285:22244–22253. doi: 10.1074/jbc.M110.121327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mari M. Morales A. Colell A. Garcia-Ruiz C. Fernandez-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal. 2009;11:2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Martin PM. Gnana-Prakasam JP. Roon P. Smith RG. Smith SB. Ganapathy V. Expression and polarized localization of the hemochromatosis gene product HFE in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2006;47:4238–4244. doi: 10.1167/iovs.06-0026. [DOI] [PubMed] [Google Scholar]
- 175.Massie A. Schallier A. Kim SW. Fernando R. Kobayashi S. Beck H. De Bundel D. Vermoesen K. Bannai S. Smolders I. Conrad M. Plesnila N. Sato H. Michotte Y. Dopaminergic neurons of system xc−deficient mice are highly protected against 6-hydroxydopamine-induced toxicity. FASEB J. 2011;25:1359–1369. doi: 10.1096/fj.10-177212. [DOI] [PubMed] [Google Scholar]
- 176.Massie A. Schallier A. Mertens B. Vermoesen K. Bannai S. Sato H. Smolders I. Michotte Y. Time-dependent changes in striatal xCT protein expression in hemi-Parkinson rats. Neuroreport. 2008;19:1589–1592. doi: 10.1097/WNR.0b013e328312181c. [DOI] [PubMed] [Google Scholar]
- 177.Mastroberardino L. Spindler B. Pfeiffer R. Skelly PJ. Loffing J. Shoemaker CB. Verrey F. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature. 1998;395:288–291. doi: 10.1038/26246. [DOI] [PubMed] [Google Scholar]
- 178.Matsuo H. Kanai Y. Kim JY. Chairoungdua A. Kim DK. Inatomi J. Shigeta Y. Ishimine H. Chaekuntode S. Tachampa K. Choi HW. Babu E. Fukuda J. Endou H. Identification of a novel Na+-independent acidic amino acid transporter with structural similarity to the member of a heterodimeric amino acid transporter family associated with unknown heavy chains. J Biol Chem. 2002;277:21017–21026. doi: 10.1074/jbc.M200019200. [DOI] [PubMed] [Google Scholar]
- 179.Meister A. Metabolism and function of glutathione. In: Dolphin D, editor; Poulsen R, editor; Avramovic O, editor. Glutathione: Chemical, Biochemical and Medical Aspects. New York: John Wiley & Sons; 1989. pp. 367–474. [Google Scholar]
- 180.Mieyal JJ. Chock PB. Posttranslational modification of cysteine in redox signaling and oxidative stress: focus on s-glutathionylation. Antioxid Redox Signal. 2012;16:471–475. doi: 10.1089/ars.2011.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Minich T. Riemer J. Schulz JB. Wielinga P. Wijnholds J. Dringen R. The multidrug resistance protein 1 (Mrp1), but not Mrp5, mediates export of glutathione and glutathione disulfide from brain astrocytes. J Neurochem. 2006;97:373–384. doi: 10.1111/j.1471-4159.2006.03737.x. [DOI] [PubMed] [Google Scholar]
- 182.Misra I. Griffith OW. Expression and purification of human gamma-glutamylcysteine synthetase. Protein Expr Purif. 1998;13:268–276. doi: 10.1006/prep.1998.0897. [DOI] [PubMed] [Google Scholar]
- 183.Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- 184.Monaghan DT. Howson PA. Jane DE. Bridges RJ. The excitatory amino acid system. In: D'haenen H, editor; den Boer JA, editor; Westenberg H, editor; Willner P, editor. Biological Psychiatry. Chichester: John Wiley and Sons Ltd.; 2002. pp. 76–84. [Google Scholar]
- 185.Moran MM. McFarland K. Melendez RI. Kalivas PW. Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Moroni F. Attucci S. Cozzi A. Meli E. Picca R. Scheideler MA. Pellicciari R. Noe C. Sarichelou I. Pellegrini-Giampietro DE. The novel and systemically active metabotropic glutamate 1 (mGlu1) receptor antagonist 3-MATIDA reduces post-ischemic neuronal death. Neuropharmacology. 2002;42:741–751. doi: 10.1016/s0028-3908(02)00033-3. [DOI] [PubMed] [Google Scholar]
- 187.Moussawi K. Pacchioni A. Moran M. Olive MF. Gass JT. Lavin A. Kalivas PW. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Moussawi K. Zhou W. Shen H. Reichel CM. See RE. Carr DB. Kalivas PW. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci U S A. 2011;108:385–390. doi: 10.1073/pnas.1011265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Murphy TH. Miyamoto M. Sastre A. Schnaar RL. Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 190.Murphy TH. Schnaar RL. Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- 191.Mysona B. Dun Y. Duplantier J. Ganapathy V. Smith SB. Effects of hyperglycemia and oxidative stress on the glutamate transporters GLAST and system xc− in mouse retinal Muller glial cells. Cell Tissue Res. 2009;335:477–488. doi: 10.1007/s00441-008-0742-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nabeyama A. Kurita A. Asano K. Miyake Y. Yasuda T. Miura I. Nishitai G. Arakawa S. Shimizu S. Wakana S. Yoshida H. Tanaka M. xCT deficiency accelerates chemically induced tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:6436–6441. doi: 10.1073/pnas.0912827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Oka A. Belliveau MJ. Rosenberg PA. Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Olm E. Fernandes AP. Hebert C. Rundlof AK. Larsen EH. Danielsson O. Bjornstedt M. Extracellular thiol-assisted selenium uptake dependent on the xc− cystine transporter explains the cancer-specific cytotoxicity of selenite. Proc Natl Acad Sci U S A. 2009;106:11400–11405. doi: 10.1073/pnas.0902204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–721. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- 196.Padmanabhan B. Tong KI. Ohta T. Nakamura Y. Scharlock M. Ohtsuji M. Kang MI. Kobayashi A. Yokoyama S. Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 197.Pallast S. Arai K. Wang X. Lo EH. van Leyen K. 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J Neurochem. 2009;111:882–889. doi: 10.1111/j.1471-4159.2009.06379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pampliega O. Domercq M. Soria FN. Villoslada P. Rodriguez-Antiguedad A. Matute C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J Neuroinflammation. 2011;8:63. doi: 10.1186/1742-2094-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Panizzutti R. De Miranda J. Ribeiro CS. Engelender S. Wolosker H. A new strategy to decrease N-methyl-D-aspartate (NMDA) receptor coactivation: inhibition of D-serine synthesis by converting serine racemase into an eliminase. Proc Natl Acad Sci U S A. 2001;98:5294–5299. doi: 10.1073/pnas.091002298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Park HA. Kubicki N. Gnyawali S. Chan YC. Roy S. Khanna S. Sen CK. Natural vitamin E alpha-tocotrienol protects against ischemic stroke by induction of multidrug resistance-associated protein 1. Stroke; J Cereb Circ. 2011;42:2308–2314. doi: 10.1161/STROKEAHA.110.608547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Patel SA. Rajale T. O'Brien E. Burkhart DJ. Nelson JK. Twamley B. Blumenfeld A. Szabon-Watola MI. Gerdes JM. Bridges RJ. Natale NR. Isoxazole analogues bind the system xc− transporter: structure-activity relationship and pharmacophore model. Bioorg Med Chem. 2010;18:202–213. doi: 10.1016/j.bmc.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Patel SA. Warren BA. Rhoderick JF. Bridges RJ. Differentiation of substrate and non-substrate inhibitors of transport system xc−: an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology. 2004;46:273–284. doi: 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 203.Peghini P. Janzen J. Stoffel W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997;16:3822–3832. doi: 10.1093/emboj/16.13.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Persson M. Brantefjord M. Hansson E. Ronnback L. Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-alpha. Glia. 2005;51:111–120. doi: 10.1002/glia.20191. [DOI] [PubMed] [Google Scholar]
- 205.Peters J. Kalivas PW. The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology (Berl) 2006;186:143–149. doi: 10.1007/s00213-006-0372-9. [DOI] [PubMed] [Google Scholar]
- 206.Pham AN. Blower PE. Alvarado O. Ravula R. Gout PW. Huang Y. Pharmacogenomic approach reveals a role for the xc− cystine/glutamate antiporter in growth and celastrol resistance of glioma cell lines. J Pharmacol Exp Ther. 2010;332:949–958. doi: 10.1124/jpet.109.162248. [DOI] [PubMed] [Google Scholar]
- 207.Piani D. Fontana A. Involvement of the cystine transport system xc− in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152:3578–3585. [PubMed] [Google Scholar]
- 208.Pierce RC. Bell K. Duffy P. Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pow DV. Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia. 2001;34:27–38. doi: 10.1002/glia.1037. [DOI] [PubMed] [Google Scholar]
- 210.Pow DV. Crook DK. Immunocytochemical evidence for the presence of high levels of reduced glutathione in radial glial cells and horizontal cells in the rabbit retina. Neurosci Lett. 1995;193:25–28. doi: 10.1016/0304-3940(95)11657-i. [DOI] [PubMed] [Google Scholar]
- 211.Qin S. Colin C. Hinners I. Gervais A. Cheret C. Mallat M. System xc− and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1–40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Qin Z. Freitas E. Sullivan R. Mohan S. Bacelieri R. Branch D. Romano M. Kearney P. Oates J. Plaisance K. Renne R. Kaleeba J. Parsons C. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 2010;6:e1000742. doi: 10.1371/journal.ppat.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Rabbani N. Thornalley PJ. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2012;42:1133–1142. doi: 10.1007/s00726-010-0783-0. [DOI] [PubMed] [Google Scholar]
- 214.Ramos-Gomez M. Kwak MK. Dolan PM. Itoh K. Yamamoto M. Talalay P. Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ratan RR. Murphy TH. Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Reynolds B. Roversi P. Laynes R. Kazi S. Boyd CA. Goberdhan DC. Drosophila expresses a CD98 transporter with an evolutionarily conserved structure and amino acid-transport properties. Biochem J. 2009;420:363–372. doi: 10.1042/BJ20082198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rimaniol AC. Haik S. Martin M. Le Grand R. Boussin FD. Dereuddre-Bosquet N. Gras G. Dormont D. Na+-dependent high-affinity glutamate transport in macrophages. J Immunol. 2000;164:5430–5438. doi: 10.4049/jimmunol.164.10.5430. [DOI] [PubMed] [Google Scholar]
- 218.Rimaniol AC. Mialocq P. Clayette P. Dormont D. Gras G. Role of glutamate transporters in the regulation of glutathione levels in human macrophages. Am J Physiol Cell Physiol. 2001;281:C1964–C1970. doi: 10.1152/ajpcell.2001.281.6.C1964. [DOI] [PubMed] [Google Scholar]
- 219.Rink C. Gnyawali S. Peterson L. Khanna S. Oxygen-inducible glutamate oxaloacetate transaminase as protective switch transforming neurotoxic glutamate to metabolic fuel during acute ischemic stroke. Antioxid Redox Signal. 2011;14:1777–1785. doi: 10.1089/ars.2011.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sagara J. Miura K. Bannai S. Cystine uptake and glutathione level in fetal brain cells in primary culture and in suspension. J Neurochem. 1993;61:1667–1671. doi: 10.1111/j.1471-4159.1993.tb09801.x. [DOI] [PubMed] [Google Scholar]
- 221.Sagara JI. Miura K. Bannai S. Maintenance of neuronal glutathione by glial cells. J Neurochem. 1993;61:1672–1676. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- 222.Sagara Y. Dargusch R. Chambers D. Davis J. Schubert D. Maher P. Cellular mechanisms of resistance to chronic oxidative stress. Free Radic Biol Med. 1998;24:1375–1389. doi: 10.1016/s0891-5849(97)00457-7. [DOI] [PubMed] [Google Scholar]
- 223.Sagara Y. Ishige K. Tsai C. Maher P. Tyrphostins protect neuronal cells from oxidative stress. J Biol Chem. 2002;277:36204–36215. doi: 10.1074/jbc.M203895200. [DOI] [PubMed] [Google Scholar]
- 224.Sagara Y. Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J Neurosci. 1998;18:6662–6671. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sahin M. Saxena A. Joost P. Lewerenz J. Methner A. Induction of Bcl-2 by functional regulation of G-protein coupled receptors protects from oxidative glutamate toxicity by increasing glutathione. Free Radic Res. 2006;40:1113–1123. doi: 10.1080/10715760600838191. [DOI] [PubMed] [Google Scholar]
- 226.Sakakura Y. Sato H. Shiiya A. Tamba M. Sagara J. Matsuda M. Okamura N. Makino N. Bannai S. Expression and function of cystine/glutamate transporter in neutrophils. J Leukoc Biol. 2007;81:974–982. doi: 10.1189/jlb.0606385. [DOI] [PubMed] [Google Scholar]
- 227.Sasaki H. Sato H. Kuriyama-Matsumura K. Sato K. Maebara K. Wang H. Tamba M. Itoh K. Yamamoto M. Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 228.Sato H. Fujiwara K. Sagara J. Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem J. 1995;310(Pt 2):547–551. doi: 10.1042/bj3100547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sato H. Kuriyama-Matsumura K. Hashimoto T. Sasaki H. Wang H. Ishii T. Mann GE. Bannai S. Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages. J Biol Chem. 2001;276:10407–10412. doi: 10.1074/jbc.M007216200. [DOI] [PubMed] [Google Scholar]
- 230.Sato H. Nomura S. Maebara K. Sato K. Tamba M. Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun. 2004;325:109–116. doi: 10.1016/j.bbrc.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 231.Sato H. Shiiya A. Kimata M. Maebara K. Tamba M. Sakakura Y. Makino N. Sugiyama F. Yagami K. Moriguchi T. Takahashi S. Bannai S. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280:37423–37429. doi: 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- 232.Sato H. Tamba M. Ishii T. Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 233.Sato H. Tamba M. Kuriyama-Matsumura K. Okuno S. Bannai S. Molecular cloning and expression of human xCT, the light chain of amino acid transport system xc−. Antioxid Redox Signal. 2000;2:665–671. doi: 10.1089/ars.2000.2.4-665. [DOI] [PubMed] [Google Scholar]
- 234.Sato H. Tamba M. Okuno S. Sato K. Keino-Masu K. Masu M. Bannai S. Distribution of cystine/glutamate exchange transporter, system xc−, in the mouse brain. J Neurosci. 2002;22:8028–8033. doi: 10.1523/JNEUROSCI.22-18-08028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sato H. Watanabe H. Ishii T. Bannai S. Neutral amino acid transport in mouse peritoneal macrophages. J Biol Chem. 1987;262:13015–13019. [PubMed] [Google Scholar]
- 236.Savaskan NE. Heckel A. Hahnen E. Engelhorn T. Doerfler A. Ganslandt O. Nimsky C. Buchfelder M. Eyupoglu IY. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat Med. 2008;14:629–632. doi: 10.1038/nm1772. [DOI] [PubMed] [Google Scholar]
- 237.Schafer FQ. Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 238.Schallier A. Smolders I. Van Dam D. Loyens E. De Deyn PP. Michotte A. Michotte Y. Massie A. Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer's disease. J Alzheimers Dis. 2011;24:287–300. doi: 10.3233/JAD-2011-101005. [DOI] [PubMed] [Google Scholar]
- 239.Schewe T. Steffen Y. Sies H. How do dietary flavanols improve vascular function? A position paper. Arch Biochem Biophys. 2008;476:102–106. doi: 10.1016/j.abb.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 240.Schubert D. Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21:7455–7462. doi: 10.1523/JNEUROSCI.21-19-07455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Schulz JB. Lindenau J. Seyfried J. Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 242.Seiler A. Schneider M. Forster H. Roth S. Wirth EK. Culmsee C. Plesnila N. Kremmer E. Radmark O. Wurst W. Bornkamm GW. Schweizer U. Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 243.Sekhar RV. Patel SG. Guthikonda AP. Reid M. Balasubramanyam A. Taffet GE. Jahoor F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am J Clin Nutr. 2011;94:847–853. doi: 10.3945/ajcn.110.003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Seki Y. Feustel PJ. Keller RW., Jr. Tranmer BI. Kimelberg HK. Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke; J Cereb Circ. 1999;30:433–440. doi: 10.1161/01.str.30.2.433. [DOI] [PubMed] [Google Scholar]
- 245.Sen CK. Khanna S. Roy S. Packer L. Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60c−Src kinase activation and death of HT4 neuronal cells. J Biol Chem. 2000;275:13049–13055. doi: 10.1074/jbc.275.17.13049. [DOI] [PubMed] [Google Scholar]
- 246.Shalev U. Grimm JW. Shaham Y. Neurobiology of relapse to heroin and cocaine seeking: a review. Pharmacol Rev. 2002;54:1–42. doi: 10.1124/pr.54.1.1. [DOI] [PubMed] [Google Scholar]
- 247.Sheldon AL. Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Shi Q. Savage JE. Hufeisen SJ. Rauser L. Grajkowska E. Ernsberger P. Wroblewski JT. Nadeau JH. Roth BL. L-homocysteine sulfinic acid and other acidic homocysteine derivatives are potent and selective metabotropic glutamate receptor agonists. J Pharmacol Exp Ther. 2003;305:131–142. doi: 10.1124/jpet.102.047092. [DOI] [PubMed] [Google Scholar]
- 249.Shibasaki T. Iuchi Y. Okada F. Kuwata K. Yamanobe T. Bannai S. Tomita Y. Sato H. Fujii J. Aggravation of ischemia-reperfusion-triggered acute renal failure in xCT-deficient mice. Arch Biochem Biophys. 2009;490:63–69. doi: 10.1016/j.abb.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 250.Shih AY. Erb H. Sun X. Toda S. Kalivas PW. Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Shih AY. Murphy TH. xCt cystine transporter expression in HEK293 cells: pharmacology and localization. Biochem Biophys Res Commun. 2001;282:1132–1137. doi: 10.1006/bbrc.2001.4703. [DOI] [PubMed] [Google Scholar]
- 252.Sido B. Lasitschka F. Giese T. Gassler N. Funke B. Schroder-Braunstein J. Brunnemer U. Meuer SC. Autschbach F. A prominent role for mucosal cystine/cysteine metabolism in intestinal immunoregulation. Gastroenterology. 2008;134:179–191. doi: 10.1053/j.gastro.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 253.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 254.Sies H. Polyphenols and health: update and perspectives. Arch Biochem Biophys. 2010;501:2–5. doi: 10.1016/j.abb.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 255.Sims B. Clarke M. Njah W. Hopkins ES. Sontheimer H. Erythropoietin-induced neuroprotection requires cystine glutamate exchanger activity. Brain Res. 2010;1321:88–95. doi: 10.1016/j.brainres.2010.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Singh A. Misra V. Thimmulappa RK. Lee H. Ames S. Hoque MO. Herman JG. Baylin SB. Sidransky D. Gabrielson E. Brock MV. Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Spooren W. Lesage A. Lavreysen H. Gasparini F. Steckler T. Metabotropic glutamate receptors: their therapeutic potential in anxiety. Curr Top Behav Neurosci. 2010;2:391–413. doi: 10.1007/7854_2010_36. [DOI] [PubMed] [Google Scholar]
- 258.Sporn MB. Liby KT. Cancer chemoprevention: scientific promise, clinical uncertainty. Nat Clin Pract Oncol. 2005;2:518–525. doi: 10.1038/ncponc0319. [DOI] [PubMed] [Google Scholar]
- 259.Staal FJ. Anderson MT. Staal GE. Herzenberg LA. Gitler C. Redox regulation of signal transduction: tyrosine phosphorylation and calcium influx. Proc Natl Acad Sci U S A. 1994;91:3619–3622. doi: 10.1073/pnas.91.9.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Taguchi K. Tamba M. Bannai S. Sato H. Induction of cystine/glutamate transporter in bacterial lipopolysaccharide induced endotoxemia in mice. J Inflamm (Lond) 2007;4:20. doi: 10.1186/1476-9255-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Takada A. Bannai S. Transport of cystine in isolated rat hepatocytes in primary culture. J Biol Chem. 1984;259:2441–2445. [PubMed] [Google Scholar]
- 262.Takaki M. Ueda Y. Doi T. Nagatomo K. Murashima YL. Kannan H. Molecular regulation of antioxidant ability in the hippocampus of EL mice. Brain Res. 2008;1228:1–5. doi: 10.1016/j.brainres.2008.06.041. [DOI] [PubMed] [Google Scholar]
- 263.Tan S. Sagara Y. Liu Y. Maher P. Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Tan S. Schubert D. Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- 265.Tan S. Wood M. Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J Neurochem. 1998;71:95–105. doi: 10.1046/j.1471-4159.1998.71010095.x. [DOI] [PubMed] [Google Scholar]
- 266.Tomi M. Funaki T. Abukawa H. Katayama K. Kondo T. Ohtsuki S. Ueda M. Obinata M. Terasaki T. Hosoya K. Expression and regulation of L-cystine transporter, system xc−, in the newly developed rat retinal Muller cell line (TR-MUL) Glia. 2003;43:208–217. doi: 10.1002/glia.10253. [DOI] [PubMed] [Google Scholar]
- 267.Torres JL. Lozano C. Maher P. Conjugation of catechins with cysteine generates antioxidant compounds with enhanced neuroprotective activity. Phytochemistry. 2005;66:2032–2037. doi: 10.1016/j.phytochem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 268.Toutzaris D. Lewerenz J. Albrecht P. Jensen LT. Letz J. Geerts A. Golz S. Methner A. A novel giant peroxisomal superoxide dismutase motif-containing protein. Free Radic Biol Med. 2010;48:811–820. doi: 10.1016/j.freeradbiomed.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 269.Tu BP. Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ueda Y. Doi T. Nagatomo K. Tokumaru J. Takaki M. Willmore LJ. Effect of levetiracetam on molecular regulation of hippocampal glutamate and GABA transporters in rats with chronic seizures induced by amygdalar FeCl3 injection. Brain Res. 2007;1151:55–61. doi: 10.1016/j.brainres.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 271.Ueda Y. Doi T. Takaki M. Nagatomo K. Nakajima A. Willmore LJ. Levetiracetam enhances endogenous antioxidant in the hippocampus of rats: in vivo evaluation by brain microdialysis combined with ESR spectroscopy. Brain Res. 2009;1266:1–7. doi: 10.1016/j.brainres.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 272.Ueda Y. Doi T. Tokumaru J. Willmore LJ. Effect of zonisamide on molecular regulation of glutamate and GABA transporter proteins during epileptogenesis in rats with hippocampal seizures. Brain Res. 2003;116:1–6. doi: 10.1016/s0169-328x(03)00183-9. [DOI] [PubMed] [Google Scholar]
- 273.Ueno H. Likos JJ. Metzler DE. Chemistry of the inactivation of cytosolic aspartate aminotransferase by serine O-sulfate. Biochemistry. 1982;21:4387–4393. doi: 10.1021/bi00261a030. [DOI] [PubMed] [Google Scholar]
- 274.Valko M. Leibfritz D. Moncol J. Cronin MT. Mazur M. Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 275.van der Zeyden M. Oldenziel WH. Rea K. Cremers TI. Westerink BH. Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav. 2008;90:135–147. doi: 10.1016/j.pbb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 276.van Leyen K. Siddiq A. Ratan RR. Lo EH. Proteasome inhibition protects HT22 neuronal cells from oxidative glutamate toxicity. J Neurochem. 2005;92:824–830. doi: 10.1111/j.1471-4159.2004.02915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Vandenabeele P. Galluzzi L. Vanden Berghe T. Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 278.Vandenberg RJ. Mitrovic AD. Johnston GA. Serine-O-sulphate transport by the human glutamate transporter, EAAT2. Br J Pharmacol. 1998;123:1593–1600. doi: 10.1038/sj.bjp.0701776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Vantyghem MC. Balduyck M. Zerimech F. Martin A. Douillard C. Bans S. Degand PM. Lefebvre J. Oxidative markers in diabetic ketoacidosis. J Endocrinol Invest. 2000;23:732–736. doi: 10.1007/BF03345062. [DOI] [PubMed] [Google Scholar]
- 280.Veettil MV. Sadagopan S. Sharma-Walia N. Wang FZ. Raghu H. Varga L. Chandran B. Kaposi's sarcoma-associated herpesvirus forms a multimolecular complex of integrins (alphaVbeta5, alphaVbeta3, and alpha3beta1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J Virol. 2008;82:12126–12144. doi: 10.1128/JVI.01146-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Veljkovic E. Stasiuk S. Skelly PJ. Shoemaker CB. Verrey F. Functional characterization of Caenorhabditis elegans heteromeric amino acid transporters. J Biol Chem. 2004;279:7655–7662. doi: 10.1074/jbc.M309528200. [DOI] [PubMed] [Google Scholar]
- 282.Verrey F. Closs EI. Wagner CA. Palacin M. Endou H. Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 2004;447:532–542. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- 283.Wahl C. Liptay S. Adler G. Schmid RM. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J Clin Invest. 1998;101:1163–1174. doi: 10.1172/JCI992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wang H. Tamba M. Kimata M. Sakamoto K. Bannai S. Sato H. Expression of the activity of cystine/glutamate exchange transporter, system xc−, by xCT and rBAT. Biochem Biophys Res Commun. 2003;305:611–618. doi: 10.1016/s0006-291x(03)00808-8. [DOI] [PubMed] [Google Scholar]
- 285.Wang XF. Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- 286.Watanabe H. Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages. J Exp Med. 1987;165:628–640. doi: 10.1084/jem.165.3.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Wek RC. Jiang HY. Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 288.Wu A. Ying Z. Schubert D. Gomez-Pinilla F. Brain and spinal cord interaction: a dietary curcumin derivative counteracts locomotor and cognitive deficits after brain trauma. Neurorehabil Neural Repair. 2011;25:332–342. doi: 10.1177/1545968310397706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Xu X. Chua CC. Kong J. Kostrzewa RM. Kumaraguru U. Hamdy RC. Chua BH. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007;103:2004–2014. doi: 10.1111/j.1471-4159.2007.04884.x. [DOI] [PubMed] [Google Scholar]
- 290.Xu X. Chua KW. Chua CC. Liu CF. Hamdy RC. Chua BH. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010;1355:189–194. doi: 10.1016/j.brainres.2010.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Yamamoto M. Takeda K. Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. 2010;2010:240365. doi: 10.1155/2010/240365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ye ZC. Rothstein JD. Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.You Z. Savitz SI. Yang J. Degterev A. Yuan J. Cuny GD. Moskowitz MA. Whalen MJ. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Yuasa S. Akagi R. Ubuka T. Masuoka N. Yao K. Excretion of 3-mercaptolactate-cysteine mixed disulfide, sulfate and taurine in human urine before and after oral administration of sulfur-containing amino acids. Acta Med Okayama. 1990;44:117–122. doi: 10.18926/AMO/30459. [DOI] [PubMed] [Google Scholar]
- 295.Zeng Y. Li Y. Chen RS. He X. Yang L. Li W. Overexpression of xCT induces up-regulation of 14–3-3beta in Kaposi's sarcoma. Biosci Rep. 2010;30:277–283. doi: 10.1042/BSR20090163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 297.Zhang P. Singh A. Yegnasubramanian S. Esopi D. Kombairaju P. Bodas M. Wu H. Bova SG. Biswal S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010;9:336–346. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Zhou W. Kalivas PW. N-acetylcysteine reduces extinction responding and induces enduring reductions in cue- and heroin-induced drug-seeking. Biol Psychiatry. 2008;63:338–340. doi: 10.1016/j.biopsych.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zhu S. Zhang Y. Bai G. Li H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington's disease. Cell Death Dis. 2011;2:e115. doi: 10.1038/cddis.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Zhu Y. Carvey PM. Ling Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006;1090:35–44. doi: 10.1016/j.brainres.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.