

Abstract
Aims: Hydrogen peroxide (H2O2), a nonradical oxidant, is employed to ascertain the role of redox mechanisms in regulation of vascular tone. Where both dilation and constriction have been reported, we examined the hypothesis that the ability of H2O2 to effect vasoconstriction or dilation is conditioned by redox mechanisms and may be modulated by antioxidants. Results: Exogenous H2O2 (0.1–10.0 μM), dose-dependently reduced the internal diameter of rat renal interlobular and 3rd-order mesenteric arteries (p<0.05). This response was obliterated in arteries pretreated with antioxidants, including tempol, pegylated superoxide dismutase (PEG-SOD), butylated hydroxytoluene (BHT), and biliverdin (BV). However, as opposed to tempol or PEG-SOD, BHT & BV, antioxidants targeting radicals downstream of H2O2, also uncovered vasodilation. Innovations: Redox-dependent vasoconstriction to H2O2 was blocked by inhibitors of cyclooxygenase (COX) (indomethacin-10 μM), thromboxane (TP) synthase (CGS13080-10 μM), and TP receptor antagonist (SQ29548-1 μM). However, H2O2 did not increase vascular thromboxane B2 release; instead, it sensitized the vasculature to a TP agonist, U46619, an effect reversed by PEG-SOD. Antioxidant-conditioned dilatory response to H2O2 was accompanied by enhanced vascular heme oxygenase (HO)-dependent carbon monoxide generation and was abolished by HO inhibitors or by HO-1 & 2 antisense oligodeoxynucleotides treatment of SD rats. Conclusion: These results demonstrate that H2O2 has antioxidant-modifiable pleiotropic vascular effects, where constriction and dilation are brought about in the same vascular segment. H2O2-induced oxidative stress increases vascular TP sensitivity and predisposes these arterial segments to constrictor prostanoids. Conversely, vasodilation is reliant upon HO-derived products whose synthesis is stimulated only in the presence of antioxidants targeting radicals downstream of H2O2. Antioxid. Redox Signal. 18, 471–480.
Introduction
The tone of resistance arterial vessels is regulated by the interplay of mechanisms promoting vasoconstriction and dilation via complex networks of interacting signaling pathways. Reactive oxygen species (ROS) contribute to regulation of vasomotor tone in physiological and pathophysiological settings (26, 46), with superoxide anion (O2•−), hydrogen peroxide (H2O2), and other ROS displaying constrictor and/or dilator activities (20). H2O2 is particularly interesting, because it affects vasoconstriction as well as dilation (1), sometimes in the same vascular preparation (10, 28). These outcomes are determined by the concentration of H2O2, vessel type, and experimental conditions (11, 12).
H2O2-induced vasoconstriction has been related to stimulation of vascular smooth muscle thromboxane A2 (TxA2)/prostaglandin endoperoxide receptors (thromboxane [TP] receptors) by a product of arachidonic acid metabolism via COX (13, 27, 32). It has also been linked to elevation of cytosolic calcium (49) and/or activation of protein kinases (18, 41). Conversely, H2O2-induced vasodilation has been associated with activation of guanylate cyclase (33), increasing cellular cAMP levels (16), and stimulation of vascular smooth muscle K+ channels (4, 15). Vasodilation has also been linked to augmented synthesis of vasodilator mediators, including prostaglandins (16) and endothelium-derived nitric oxide (NO) (50). According to previous studies, whether H2O2-promotes vasoconstriction or dilation depends on the functional status of K+ channels in the target vessels, viz., constriction occurs when vascular smooth muscle K+ channels are functionally impaired, and dilation happens when they are not (28). The redox status of the vessels influences functionality of vascular K+ channels (40, 48), as well as of other signaling proteins important for Ca2+-dependent regulation of vasomotor tone (26, 37). Redox mechanisms also modulate the expression of TP receptors (42, 44), the activity heme oxygenase (HO) (21, 23, 29), and the vascular actions of its vasodilatory product—carbon monoxide (CO) (25). Hence, it is plausible that one or more of these redox-controlled vasoregulatory systems condition vasoconstriction or dilation to this nonradical oxidant, H2O2.
Innovation.
Our study examines the vascular effects of exogenous hydrogen peroxide (H2O2) in light of its oxidant properties. This radical invokes a constrictive response in resistance arteries, where an antioxidant milieu is not provided. Both constriction and dilation are observed in the same arterial preparation, depending upon the use of antioxidants targeting radicals derived-from or leading-to H2O2. Where thromboxane sensitization underlies vasoconstriction, increased heme oxygenase (HO)-dependent carbon monoxide (CO) generation causes vasodilation. Notably, radicals downstream to H2O2 interfere with its ability to stimulate HO-dependent CO release; targeting these oxidants increases vascular CO and leads to H2O2-induced vasodilation. This study lays the foundation to explore vasoreactivity of endogenous H2O2, in models of chronic oxidative stress where application of broad-spectrum antioxidants should provide further evidence of pleiotropic vascular effects of H2O2.
We undertook the present study to test the hypothesis that the response of resistance arteries to physiologically relevant concentrations of H2O2 is dictated by the redox status of the preparations. This was achieved via activation and/or suppression of redox-modulated vasoactive systems that promote constriction or dilation. First, we contrasted the effect of H2O2 on internal diameter (ID) of pressurized rat renal interlobular (RIA) and 3rd order mesenteric artery (MA), in the absence and presence of antioxidants. Second, we connected the redox-dependent constrictor action of H2O2 to an associated increase in responsiveness to TP receptor stimulation. Third, we linked the redox-dependent dilator action of H2O2 to stimulation of HO-derived CO.
Results
Effect of H2O2 on the ID of pressurized arterial vessels: comparison in preparations pretreated and not pretreated with antioxidants
The notion that redox mechanisms influence the response of resistance arteries to H2O2 was addressed by contrasting the effects of this oxidant on vascular diameter in preparations exposed and not exposed to agents selected for their ability to create an antioxidant setting. As illustrated in Figure 1, RIA and MA not pretreated with an antioxidant demonstrated a dose-dependent decrease in ID to exogenous H2O2 (p<0.05). In contrast, H2O2 challenge in RIA and MA pretreated with 1 mM butylated hydroxytoluene (BHT), a general antioxidant with an ability to quench free radicals downstream of H2O2 (24), resulted in increment (p<0.05) rather than reduction of ID. H2O2 also increased the ID of MA and/or RIA pretreated with desferroxamine (DES), an iron chelator that by disrupting the Fenton's reaction decreases generation of hydroxyl radical (OH·), and subsequent lipid peroxidation (2, 45), dimethyl thiourea (DMTU), OH· scavenger (22), or biliverdin (BV), an antioxidant effective against radicals downstream of H2O2 and a known inhibitor of lipid peroxidation (17, 39) (Fig. 2).
FIG. 1.
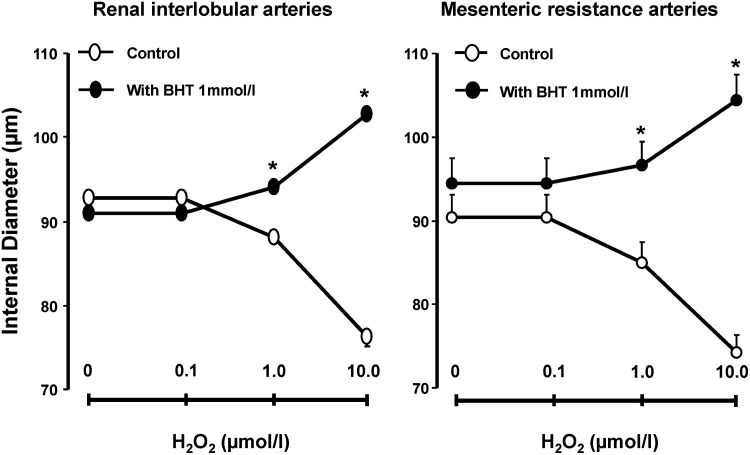
Effect of H2O2 on the ID of renal interlobular and mesenteric resistance arteries, in arteries not exposed and exposed to BHT. Results are means±SE, n=6/group.*p<0.05 versus control. BHT, butylated hydroxytoluene; H2O2, hydrogen peroxide; ID, internal diameter; SE, standard error.
FIG. 2.
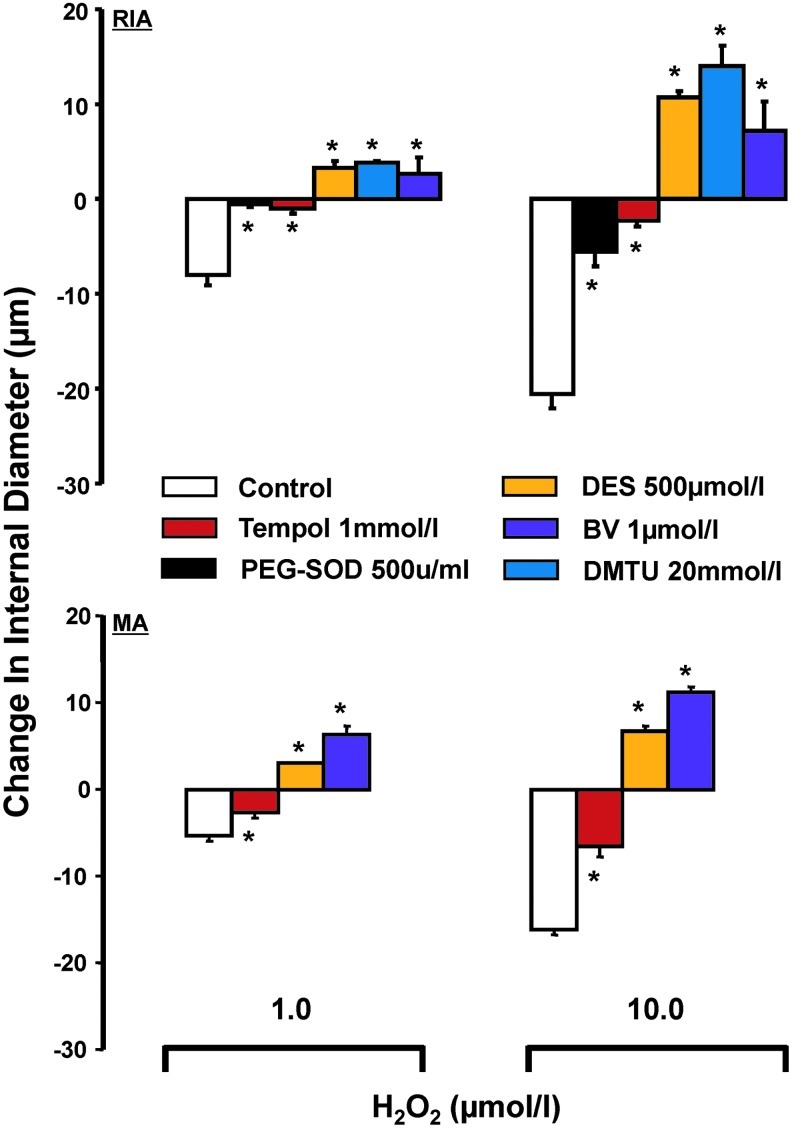
Effect of H2O2 on change in ID, over baseline, of renal interlobular (n: Control-5, PEG-SOD-4, tempol-4, Des-4, DMTU-5, and BV-6) and mesenteric arteries (n=4), in arteries not exposed and exposed to antioxidants (as shown). Results are means±SE. *p<0.05 versus control. BV, biliverdin; DES, desferroxamine; DMTU, dimethyl thiourea; PEG-SOD, pegylated superoxide dismutase.
Notably, arteries pretreated with pegylated superoxide dismutase (PEG-SOD), an antioxidant that dismutates O2− anion to H2O2, or tempol, a SOD mimetic (34), did not respond to exogenous H2O2 with an increase of ID. This was observed, even though these pretreatments greatly attenuated the ability of H2O2 to effect decrease of vascular diameter, as observed in vehicle-pretreated controls (Fig. 2). Accordingly, the response of resistance arteries to exogenous H2O2 appears conditioned by their redox status: constriction versus blunted constriction or dilation, respectively, in vascular preparations not afforded and afforded protection from oxidative stress.
Relative to this point, complementary experiments documented that H2O2 (10 μM) increase O2− levels as revealed by lucigenin chemiluminescence (from 66.9±10.9 CPM/μg protein to 87.5±7.7 CPM/μg protein, n=7, p<0.05). Vessels exposed to H2O2 also displayed increased levels of TBARS (from 433.9±168.8 to 1892.3±498.0 nmoles/mg protein, n=4, p<0.05), an index of lipid peroxidation and oxidative stress (35). This increase was blunted in vessels concurrently pretreated with BHT (234.3±76.2 vs. 138.4±15.6 nmoles/mg protein, n=4), BV (185.7±56.1 vs. 214.9±89.1 nmoles/mg protein, n=4), tempol (103.5±7.52 vs. 190.2±26.1 nmoles/mg protein, n=4), PEG-SOD (96.6±4.1 vs. 144.7±15.0 nmoles/mg protein, n=3), or DES (321.8±162.3 vs. 207.5±42.3 nmoles/mg protein, n=3), whereas arterial vessels exposed to H2O2 did not display any significant change in endogenous SOD activity (control: 331.3±68.6 vs. H2O2: 281.7±71.4 U/mg protein).
Effect of H2O2 on the ID of pressurized arterial vessels: comparison in preparations with and without endothelium
To investigate contribution of the vascular endothelium to the redox-dependent vascular actions of H2O2, RIAs, denuded and not denuded of endothelium were contrasted in terms of responsiveness to H2O2, in the absence and presence of BHT. As shown in Figure 3A, endothelium removal did not affect H2O2-induced vasoconstriction in vessels without antioxidant pretreatment. On the other hand, as depicted in Figure 3B, H2O2-induced increase of vascular ID was blunted by endothelium removal in arteries pretreated with BHT. Accordingly, antioxidant-conditioned H2O2-induced vasodilation is endothelium dependent, whereas constriction of vessels without antioxidant pretreatment is not.
FIG. 3.
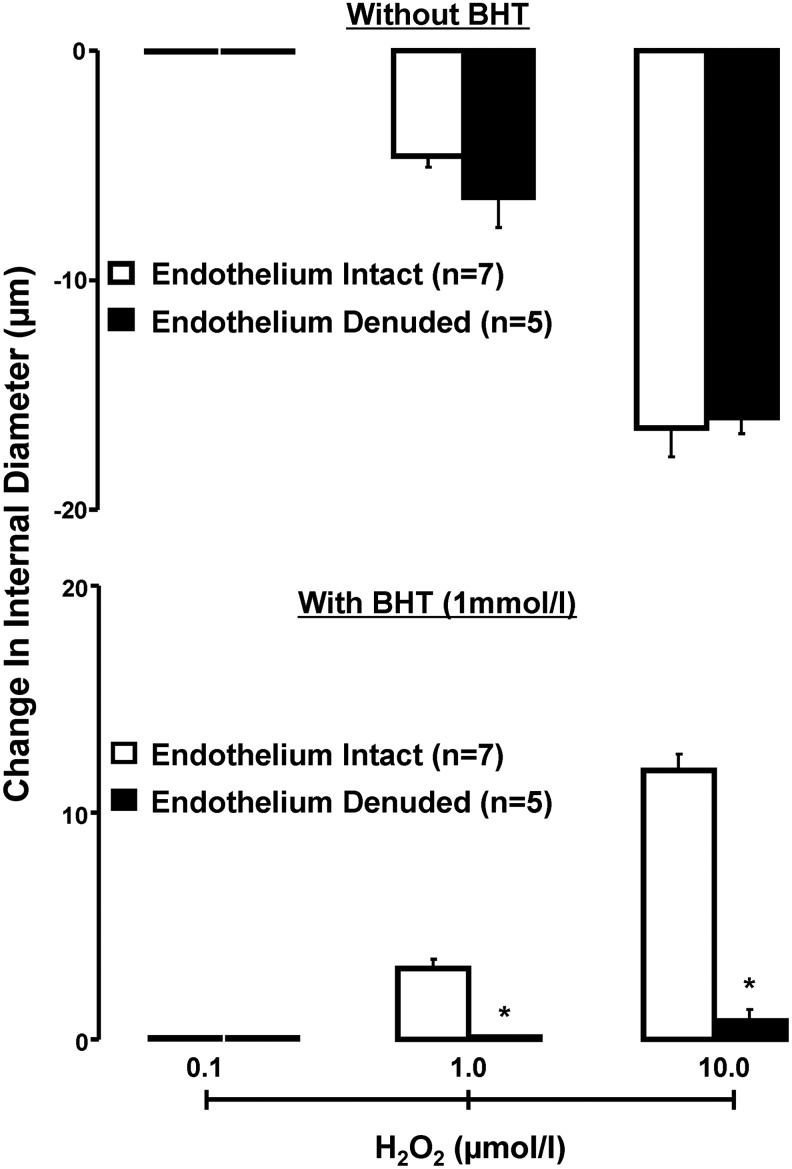
Effect of H2O2 on change in ID, over baseline, of renal interlobular in the presence or in the absence of endothelium, in arteries not exposed and exposed to BHT (1 mM). Results are means±SE. *p<0.05 versus endothelium intact.
Redox-dependent H2O2-induced vascular constriction: contribution of TP receptor activation
Earlier reports have linked oxidative stress to mechanisms of vasoconstriction involving TP receptor stimulation (13, 43). The notion that redox-dependent H2O2-induced vasoconstriction relies on TP receptor activation was addressed by comparing the constrictor action of the oxidant in pressurized RIA pretreated and not pretreated with indomethacin—to inhibit COX-dependent synthesis of constrictor prostanoids, CGS-13080—to selectively inhibit thromboxane synthase, or SQ29548, to effect blockade of TP receptors. As depicted in Figure 4A, the reduction of ID elicited by H2O2 in arteries not pretreated with antioxidants was blunted (p<0.05) in preparations bathed in Krebs' buffer (KB) containing indomethacin, CGS-13080, or SQ29548. These observations suggest dependence of vasoconstrictor action of H2O2 on a TP receptor agonist manufactured via a pathway involving COX and thromboxane synthase activities, most likely TxA2. However, data presented in Figure 4B show that vascular thromboxane B2 (TxB2) synthesis, an estimate of TxA2, is neither stimulated by H2O2 nor suppressed by BHT or tempol. Yet, as illustrated in Figure 4C, the sensitivity of pressurized RIA to the constrictor action of a synthetic agonist for TP receptor, U46619, was significantly enhanced in vessels pretreated with H2O2 (EC50 values: 0.63±0.07 nM in controls vs. 0.17±0.08 nM with H2O2, p<0.05). This sensitizing action of H2O2 was not observed in arteries pretreated with PEG-SOD (EC50 values: 0.49±0.05 nM in PEG-SOD vs. 0.47±0.04 nM with PEG-SOD+H2O2, p<0.05), suggesting that its expression requires a pro-oxidant setting. Accordingly, the constrictor action of H2O2 in renal arteries without antioxidant protection is linked to TP receptor activation, relying on a mechanism involving augmented sensitivity of the vessels to TP receptor stimulation rather than increased vascular production of TxA2. That pretreatment with H2O2 does not sensitize RIA to the constrictor action of α-1 adrenergic receptor stimulation with phenylephrine (EC50 values: 9.3±0.99 and 10.7±2.2 nM, in arteries with and without H2O2 pretreatment, respectively) is keeping in line with the possibility that the sensitizing action of H2O2 on TP receptor agonist-induced vasoconstriction is specific.
FIG. 4.
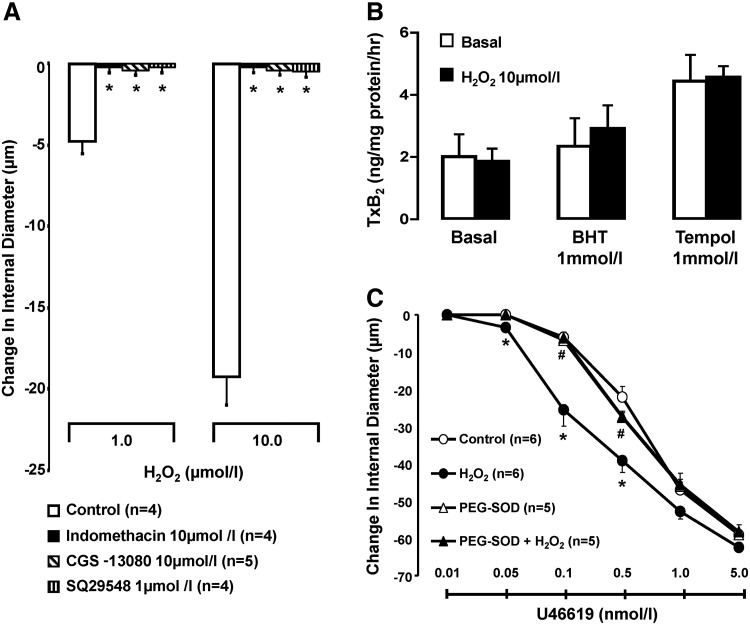
Vascular effects of H2O2 in the absence or presence of modulators of the COX-TxA2-TP receptor pathway. (A) Effect of H2O2 on change in ID, over baseline, of renal interlobular arteries, in arteries not exposed and exposed to treatments as shown. Results are means±SE. *p<0.05 versus control. (B) Effect of H2O2 on vascular TxB2 synthesis in renal interlobar arteries, in the arteries exposed and not exposed to BHT or tempol; Results are means±SE. (C) Effect of H2O2 on concentration–response curve of a TP agonist (U46619), in arteries concurrently exposed or not exposed to pegylated SOD. Results are means±SE; *p<0.05 versus vehicle; #p<0.05 versus H2O2. EC50 is presented in the text. TP, thromboxane; TxB2, thromboxane B2.
Redox-dependent H2O2-induced vascular dilation: contribution of HO-derived CO
The hypothesis that redox status conditions the response of resistance arteries to H2O2 via mechanisms involving vascular production of HO-derived CO was addressed by examining the effect of exogenous H2O2 on CO released from isolated small artery segments incubated in oxygenated KB containing and not containing antioxidant agents. Complementary studies were conducted in pressurized RIA bathed in KB containing antioxidants, to contrast the ability of H2O2 to elicit vasodilation in preparations exposed and not exposed to an inhibitor of HO activity. Dilatory responsiveness to H2O2 also was studied in RIA taken from rats pretreated with antisense oligodeoxynucleotides (AS-ODN) targeting HO-1 and HO-2 or with the corresponding scrambled oligodeoxynucleotides (SAS-ODN).
Figure 5 depicts the effect of H2O2 on release of CO from MA without and with antioxidant pretreatment, in the absence and presence of the HO inhibitor chromium mesoporphyrin (CrMP). In preparations not exposed to CrMP, H2O2 enhanced (p<0.05) the release of CO from arteries pretreated with BHT or BV, although it failed to do so in vessels without antioxidant pretreatment or pretreated with tempol. H2O2 also induced CO release from MA pretreated with DES (500 μM) (from 135.5±16.1 to 227.7±39.0 pmol/mg protein/h, n=7, p<0.05, but not from arteries pretreated with PEG-SOD (163.2±26.1 vs. 156.4±47.6 pmol/mg protein/h, n=6). Release of CO from arteries exposed to CrMP was diminished (p<0.05) by about 50%, and did not increase by further challenge with H2O2, either in the absence or presence of antioxidants (Fig. 5). That H2O2-induced CO release from arteries pretreated with BHT or BV was prevented by CrMP implies that the oxidant promotes release of only HO-derived CO. Relevant to this point, the release of CO-induced by H2O2 from MA pretreated with BHT is not increased in endothelium-denuded preparations (47.4±11.0 vs. 67.7±8.5 pmol/mg protein/h, n=6). It is also noteworthy that estimates of HO activity in arterial homogenates revealed no stimulatory effect of H2O2, either in the absence (729±41 vs. H2O2: 817±78 pmol/mg protein/h, n=7) or in the presence of the antioxidant BV (807±72 vs. 887±83 pmol/mg protein/h, n=4). Hence, estimates of HO activity in cell-free vascular homogenates are not necessarily a reflection of HO-derived CO release from intact vessels afforded antioxidant protection.
FIG. 5.
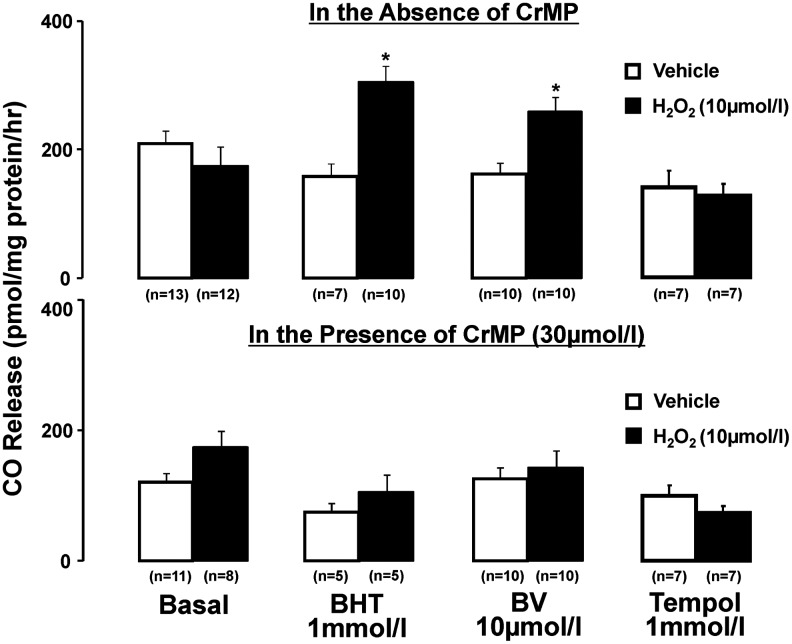
Effect of H2O2 on vascular CO generation in arteries not exposed or concurrently exposed to the HO inhibitor, CrMP. Experiments conducted in the absence or presence of antioxidants, as shown. Results are means±SE. *p<0.05 versus respective vehicle in the presence of antioxidant, either BHT or BV. CO, carbon monoxide; CrMP, chromium mesoporphyrin; HO, heme oxygenase.
Complementary studies also revealed no effect of H2O2 on HO-1 or HO-2 protein expression, in arteries incubated for 90 min in oxygenated KB containing 1 mM BHT (HO-1/β-actin ratio: 0.56±0.14 vs. 0.54±0.03, without and with H2O2 (10 μM), respectively; HO-2/β-actin ratio: 0.62±0.10 vs. 0.60±0.04, without and with H2O2, respectively). Accordingly, the action of H2O2 to promote vascular release of HO-derived CO appears not to depend on enhanced HO isoform protein expression.
Figure 6 illustrates the result of experiments comparing the effect of H2O2 on the ID of pressurized RIA, bathed in KB containing an antioxidant (BHT, BV, or DMTU), with and without concurrent treatment with an inhibitor of HO, CrMP, or stannous mesoporphyrin (SnMP). The ability of H2O2 to increase the ID of such vessels was blunted by pretreatment with CrMP or SnMP. The aforementioned observations linking H2O2-induced vasodilation to release of HO-derived CO were corroborated in a study examining the effect of H2O2 on the ID of BHT- (1 mM) treated RIA taken from rats pretreated with AS-ODN, or corresponding SAS-ODN, targeting HO-1 and HO-2. Shown in Figure 7A–C, respectively, arteries from rats pretreated with HO-1 and HO-2 AS-ODN in combination featured, relative to control data in arteries from rats pretreated with the corresponding SAS-ODN, diminished expression of HO-1 (HO-1/β-actin ratio: 0.25±0.03 vs. 0.61±0.13) and HO-2 protein (HO-2/β-actin ratio: 0.35±0.10 vs. 0.63±0.06, n=4, p<0.05), along with attenuation of H2O2-induced CO release and dilation.
FIG. 6.
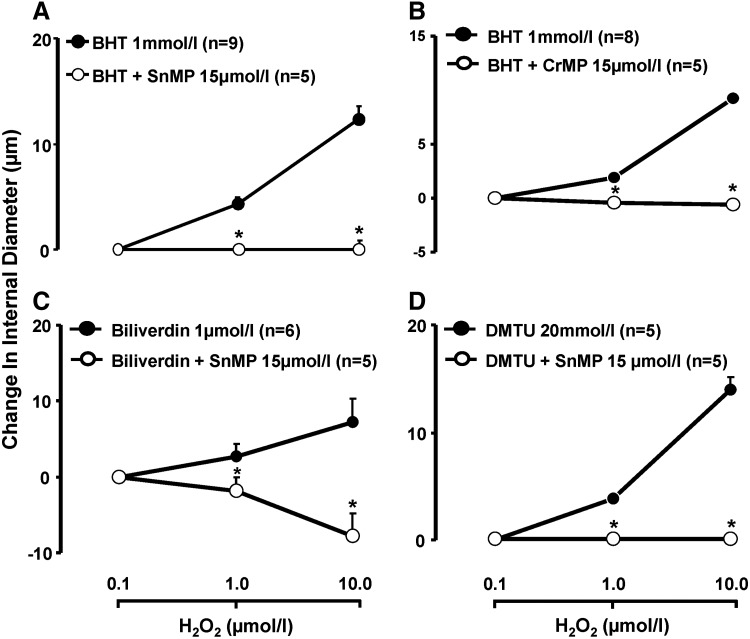
Antioxidant-conditioned vasodilation to H2O2 in the absence or in the presence of HO inhibitor. Effect of H2O2 on change in ID, over baseline, of renal interlobular (A, C, D) or mesenteric resistance arteries (B), in arteries exposed to various antioxidants, and not exposed or concurrently exposed to HO inhibitors. Results are means±SE. *p<0.05 versus without HO inhibitor.
FIG. 7.
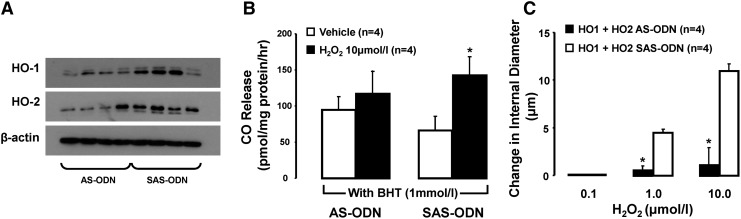
Effect of H2O2 on CO release and change in ID in arteries from rats treated with either SAS-ODN or AS-ODN for HO1 and HO2. (A) Western blot analysis of HO1 and HO2 expression in renal interlobar arteries from rats treated with either AS or SAS-ODN for HO1 and HO2. (B) Effect of H2O2 on vascular CO release in mesenteric resistance arteries exposed to BHT. Results are means±SE; *p<0.05 versus vehicle treated arteries from HO1+HO2 SAS-ODN. (C) Effect of H2O2 on change in ID in renal interlobular arteries exposed to BHT (1 mM). Results are means±SE; *p<0.05 versus arteries from HO1+HO2 SAS-ODN. AS-ODN, antisense oligodeoxynucleotides; SAS-ODN, scrambled oligodeoxynucleotides.
Discussion
H2O2 is a mediator of oxidative stress via complex mechanisms involving promotion of lipid peroxidation, stimulation of cellular oxidases that increase 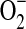 levels, and generation of OH· via the Fenton reaction (1, 45). The first key finding of our study is that exogenous H2O2 constricts pressurized isolated rat resistance arteries, RIA and MA, a response that is prevented or in some cases converted to dilation by pretreatment with antioxidant agents. Importantly, not all antioxidants affect the same change in the vascular response to H2O2: pretreatment with PEG-SOD and tempol prevent H2O2-induced vasoconstriction without uncovering a dilatory response, whereas pretreatment with BHT, DES, BV, and DMTU prevents H2O2-induced vasoconstriction, but also uncovers a vasodilator response. The differential effect of these antioxidants on vascular responsiveness to H2O2 may be a consequence of differences in the profile of ROS targeted by the antioxidants in question. Hence, while the primary target of PEG-SOD and tempol is
levels, and generation of OH· via the Fenton reaction (1, 45). The first key finding of our study is that exogenous H2O2 constricts pressurized isolated rat resistance arteries, RIA and MA, a response that is prevented or in some cases converted to dilation by pretreatment with antioxidant agents. Importantly, not all antioxidants affect the same change in the vascular response to H2O2: pretreatment with PEG-SOD and tempol prevent H2O2-induced vasoconstriction without uncovering a dilatory response, whereas pretreatment with BHT, DES, BV, and DMTU prevents H2O2-induced vasoconstriction, but also uncovers a vasodilator response. The differential effect of these antioxidants on vascular responsiveness to H2O2 may be a consequence of differences in the profile of ROS targeted by the antioxidants in question. Hence, while the primary target of PEG-SOD and tempol is 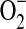 (34), the primary targets of BHT (24), DES (2), BV (38, 39), and DMTU (22) are free radicals downstream of H2O2, including OH·.
(34), the primary targets of BHT (24), DES (2), BV (38, 39), and DMTU (22) are free radicals downstream of H2O2, including OH·.
That H2O2-induced vasoconstriction is impeded by a diverse group of antioxidants suggests a critical connection between the constrictor action and the generation of a pro-oxidant milieu. This conclusion is in alignment with the finding that arterial vessels challenged with H2O2 display increased levels of 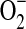 and lipid peroxides. Also, all antioxidants that prevent H2O2-induced vasoconstriction also prevent the associated rise in lipid peroxides. That H2O2-induced vasodilation is uncovered by pretreatment with BHT, DES, BV, or DMTU, but not with PEG-SOD or tempol, suggests that the dilatory action is linked to elimination of a free radical other than
and lipid peroxides. Also, all antioxidants that prevent H2O2-induced vasoconstriction also prevent the associated rise in lipid peroxides. That H2O2-induced vasodilation is uncovered by pretreatment with BHT, DES, BV, or DMTU, but not with PEG-SOD or tempol, suggests that the dilatory action is linked to elimination of a free radical other than 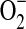 , which interferes with the expression of such an action. All in all, the emerging conclusion is that the response of resistance arteries to exogenous H2O2 is conditioned by the redox status: constriction versus blunted constriction or dilation in vascular preparations not afforded and afforded protection from oxidative stress.
, which interferes with the expression of such an action. All in all, the emerging conclusion is that the response of resistance arteries to exogenous H2O2 is conditioned by the redox status: constriction versus blunted constriction or dilation in vascular preparations not afforded and afforded protection from oxidative stress.
The second key finding of our study is that the redox-dependent vasoconstrictor action of H2O2 is linked to TP receptor activation, relying on a mechanism involving enhanced sensitivity of the vessels to TP receptor stimulation. Other investigators have reported a connection between the constrictive action of H2O2 and constrictor prostanoids (13). We found that pretreatment of vascular preparations with agents that inhibit COX, thromboxane synthase, or TP receptors eliminates constrictor responsiveness to H2O2. This implies a primary role of TxA2 as a mediator of this constrictor response. However, in our studies, H2O2 did not increase vascular TxB2 production; rather, it enhanced the sensitivity of the arteries to a synthetic agonist of TP receptors, U46619. Relevant to this conclusion, previous investigators have shown that H2O2 can result in an immediate and sustained increase in the cell surface expression of TP receptors (3). This effect has been linked to oxidative stress-mediated increased TP recycling to the plasma membrane, thus increasing functional TP expression (42, 44). Increasing the plasma membrane TP receptor density should enhance the number of spare receptors available for activation by endogenous agonists. Consequently, TP receptor occupancy needed for the half-maximal response (EC50) should be lowered without affecting the maximal response. These conclusions are indeed corroborated by our results showing the lowering of EC50 for the TP agonist in the presence of exogenous H2O2. Even though the precise molecular events leading to TP-sensitization remain unknown, consideration should be given to the possibility that this is linked to oxidant-induced activation of PKC signaling, as previously suggested (14).
Upon pharmacological blockade of vasoconstriction, we expected H2O2-induced vasodilation to prevail. However, neither antioxidants targeting O2− (PEG-SOD & tempol) nor agents interfering with the synthesis or actions of constrictor prostanoids allowed expression of H2O2-induced vasodilation, even in the face of blunted vasoconstriction. These observations, in addition to the findings that H2O2-induced vasodilation necessitates pretreatment with BHT, BV, DES, or DMTU, suggest that exclusion of radicals downstream of H2O2, rather than interference with the expression of vasoconstriction, enables H2O2 to elicit vasodilation.
The third key finding of our study is that the redox-dependent vasodilatory action of H2O2 is linked to a mechanism involving enhanced vascular production of HO-derived CO. Two reciprocal lines of evidence substantiate this relationship: first, the experimental conditions found to favor expression of H2O2-induced vasodilation; for example, pretreatment of the arterial vessels with antioxidants such as BHT, BV, and DES also enables exogenous H2O2 to increase the release of vascular CO. Second, experimental interventions that interfere with H2O2-induced release of CO; for example, pretreatment with inhibitors of HO activity or HO protein expression effectively prevents the oxidant from eliciting a vasodilatory response. Accordingly, the redox-depended vasodilatory action of H2O2 may be regarded as a manifestation of the associated increase in vascular production of HO-derived CO. That only arterial vessels pretreated with BHT, BV, or DES are stimulated by H2O2 to release HO-derived CO implies that this response is also redox dependent, necessitating an antioxidant setting for its expression. These results suggest that free radicals, other than O2−, somehow prevent H2O2 from increasing HO-dependent CO; however, our study falls short from addressing the mechanism underlying this redox-dependent stimulatory action of H2O2 on vascular CO release. According to previous reports, higher concentrations of H2O2 have been shown increase HO activity in purified enzyme systems and in renal homogenates, via acting as an electron donor (23, 29). However, in the present study, a stimulatory action of H2O2 on HO activity, measured in arterial homogenates in vitro, could not be documented, either in the absence or presence of a vasodilation promoting antioxidant. However, one cannot exclude the possibility that the ability of exogenous H2O2 to increase vascular release of HO-derived CO relies on stimulation of HO activity via engagement of second-messenger systems that are only operational when cellular integrity is preserved. In this regard, interaction between H2O2-activated kinases (6–8, 30) and kinase-dependent modulation of HO (5, 47) may link H2O2 to stimulation of HO-dependent CO release in arterial vessels.
Thus, as summarized in Figure 8, pleiotropism observed to exogenous H2O2 with regard to the ID of resistance arteries is redox dependent. In the absence of an exogenous antioxidant, H2O2-induced redox imbalance leads to vasoconstriction that in turn is dependent upon vascular TP sensitization. All antioxidants, in spite of their diverse specificity, attenuate oxidative stress and prevent this redox-dependent vasoconstriction. Importantly, vasodilation to H2O2 is only uncovered when oxidants downstream of H2O2 are quenched and involves increased HO-dependent CO generation. These observations lead us to conclude that a free radical, other than O2−, interferes with the mechanisms linking H2O2 to stimulation of HO-dependent CO generation.
FIG. 8.
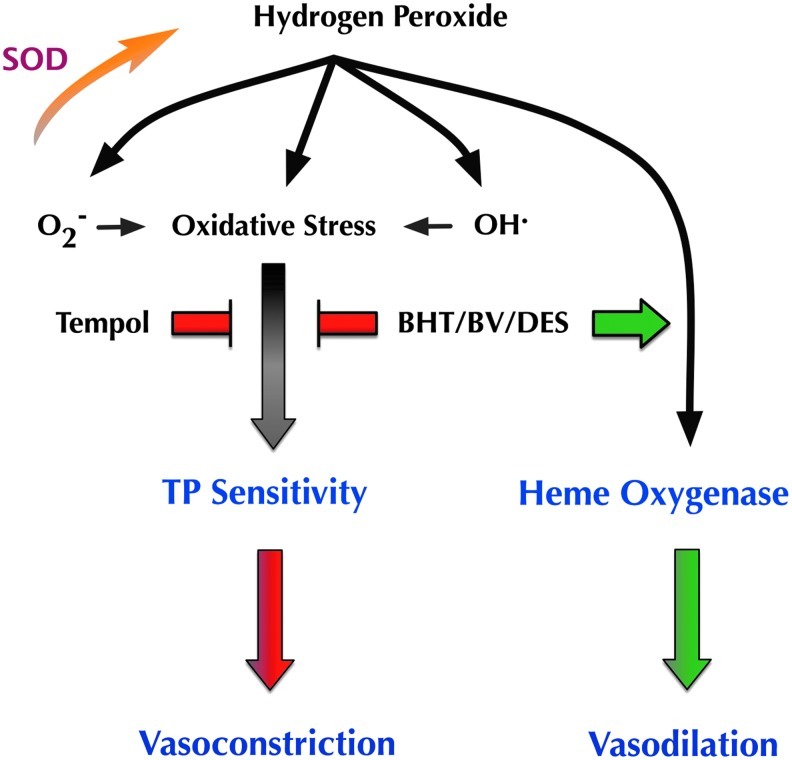
A schematic representation of pleiotropic vascular effects of H2O2, as derived from our hypothesis and results. In the absence of exogenous antioxidants, H2O2 promotes a pro-oxidant setting and induces TP-dependent vasoconstriction. This effect is abrogated by a wide spectrum of antioxidants, targeting radicals both up and downstream of H2O2. Conversely, vasodilation to H2O2 is reliant upon HO-derived products and is only uncovered in the presence of antioxidants targeting radicals downstream of H2O2. These results imply that a radical other than O2− somehow interferes with the ability of H2O2 to stimulate HO product generation and to bring about vasodilation.
Materials and Methods
Animals
The Institutional Animal Care and Use Committee of the New York Medical College approved all animal protocols. Male Sprague-Dawley rats (250–300 g; Charles River) were anesthetized (pentobarbital sodium, 60 mg/kg, intraperitoneally), and the kidneys and the intestines were removed and placed on a dish filled with oxygenated ice-cold KB (composition in mM: 118.5 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25.0 NaHCO3, and 11.1 mM dextrose). RIA and mesenteric arteries (3rd order) were isolated for various experiments as described in the Results section. To investigate the effects of HO inhibition on vascular effects of H2O2, antisense oligonucleotides, HO2-AS-ODN and HO1-AS-ODN (complementary to rat HO2 and HO1 mRNA, respectively), and their scrambled controls were synthesized by Sigma-Genosys; each oligodeoxynucleotide was phosphorothioated on the first three bases of the 3′ end and was purified by high-pressure liquid chromatography. The sequence of HO-1 AS-ODN is 5′-GGCGCTCCATCGCGGGACTG-3′ and targets bases +10 to −9 of HO-1 mRNA, and the sequence of HO-2 AS-ODN is 5′-TCTGAAGACATTGTTGCTGA-3′ and targets bases +11 to −9 of HO-2 mRNA. The sequence of HO-1 S-ODN is 5′-TCCAGCGGCGTCAGCGGTGC-3′, and the sequence of HO-2 S-ODN is 5′-GATCTGACTTCAAGTGATTG-3′. The effectiveness of HO-1 AS-ODN and HO-2 AS-ODN to reduce tissue expression of HO-1 and HO-2, respectively, was documented previously (19). Oligonucleotides encapsulated in liposomes (1:1 molar ratio) prepared using DOTAP liposomal transfection reagent (Roche Diagnostics) were followed by injection into the tail vein of rats, as described before (19). Animals were sacrificed 36 h postinjection, and renal and mesenteric arteries were collected for further studies.
Vascular function studies
RIAs or MAs, were dissected into segments 1 to 2 mm in length and mounted on a myograph (model CH/200/Q; Living System Instrumentation). All residual blood was gently flushed from vessel lumen before experiments. One end of the vessel was mounted on a glass micropipette and connected to a pressure servo-controller (model CH/200/Q; Living System Instrumentation). The opposite end of the vessel was tied to a micropipette connected to a stopcock. A video camera (Javelin), leading to a video caliper (Texas A & M), monitor (Javelin) and recorder, was used to record the vessel chamber. Vessels were superfused throughout the experiment with gassed (95% O2–5% CO2) KB at 1 ml/min, at 37°C. Intraluminal pressure was gradually increased to 80 mmHg and allowed to equilibrate for 1 h. Drugs were added, at indicated concentrations, to the superfusion buffer and allowed to equilibrate for 15 min; changes in ID were then recorded in response to increasing concentrations of H2O2, over a period of 20–25 min. Data are reported as ID (μm) or change in ID (μm) over baseline.
For experiments involving endothelium free arteries, the vascular endothelium of RIA was removed via passage of an air bubble through the lumen of the artery. Endothelium removal was confirmed by failure of these arteries to dilate in response to increasing concentrations of acetylcholine (Ach). Arteries demonstrating Ach-dependent dilation were excluded from the study.
Assessment of vascular TxB2
Freshly dissected renal interlobar arteries were washed and pretreated with and without indicated antioxidants. Following a 15-min preincubation, arteries were removed and placed in fresh vials containing or not containing various antioxidants in the absence or presence of H2O2 (10 μM). Incubations were done in closed vials at 37°C in oxygenated KB for 60 min, so as to allow detection of low concentrations of eicosanoids. After incubation, the reaction was stopped by acidification with acetic acid (∼pH 4.0), and internal standard mix (I.S for TxB2- d4PGE2) was added to the buffer, followed by extraction with ethyl acetate. TxB2 was identified and quantified with a Q-trap 3200 linear ion trap quadrupole liquid chromatography/tandem mass spectrometry equipped with a Turbo V ion source operated in a negative electrospray mode (Applied Biosystems), as previously described (36). Data are normalized to total protein and are presented as ng/mg protein/h.
Assessment of vascular CO release
Freshly dissected small mesenteric arteries (2nd and 3rd order) were pretreated with indicated antioxidants, with untreated arteries kept as controls. Following a 15-min preincubation, 10 μM of H2O2 was added to the vials, and the vials were then sealed and incubated at 37°C for 90 min in oxygenated KB. Experiments were performed in arteries concurrently exposed and not exposed to CrMP (30 μM), to differentiate between HO-dependent and independent sources of CO. After incubation, gas-sealed vials were placed on ice to stop the reaction, and headspace gas was analyzed for CO with C13O16 added as an internal standard. CO measurements were performed using an Agilent 5890 GC-MS, as previously described (19, 31). Data are normalized to total protein and are presented as pmoles/mg protein/h.
Assessment of HO activity in vascular homogenates
Freshly dissected small mesenteric and renal interlobar arteries were homogenized in sucrose–Tris buffer (sucrose, 255 mM, Tris–HCl, 20 mM) with NP-40 (1%) and mammalian protease inhibitor cocktail (Sigma Aldrich). After protein estimation, 0.15 mg protein of the arterial homogenate was incubated with various antioxidants for 15 min. After this, homogenates were incubated with 10 μM of H2O2, 30 μM heme, and 2 mM NADPH, in the absence or presence of an HO inhibitor, CrMP (50 μM). Vials were then sealed and incubated at 37°C for 90 min. After incubation, gas-sealed vials were placed on ice to stop the reaction, and headspace gas was analyzed for CO with C13O16 added as an internal standard. CO measurements were performed using an Agilent 5890 GC-MS, as previously described (19, 31). HO activity is calculated by subtracting CO levels obtained in the presence of CrMP from those obtained without. Data are normalized to total protein and are presented as pmoles/mg protein/h.
Assessment of HO protein expression
Freshly dissected renal interlobar and small mesenteric arteries were paced in oxygenated KB containing 1 mM of BHT. After a 15-min preincubation, arteries were incubated with and without H2O2 (10 μM) at 37°C for 90 min. HO-1 and HO-2 protein expression was assessed in these arteries by immunoblotting, as previously described (9). Assessment of HO protein expression in SD rats treated with either AS-ODN or S-ODN for HO-1 & 2 was performed in freshly dissected renal interlobar arteries from these animals.
Assessment of the redox state
Renal interlobar and small mesenteric arteries were preincubated in the absence or presence of described antioxidants for a period of 15 min, followed by an incubation with and without H2O2 (10 μM) at 37°C for 20 min in oxygenated KB. Subsequently, arteries were snap-frozen in liquid nitrogen before being analyzed for TBARS (using a Cayman Chemicals, Inc. kit # 10009055), as per manufactures' protocol. Data are normalized to total protein and are presented as nmoles/mg protein.
For assessment of effects of H2O2 on arterial SOD activity, mesenteric and renal arteries were incubated with and without H2O2 (10 μM) at 37°C for 20 min in oxygenated KB. Arteries were snap-frozen in liquid nitrogen and analyzed for SOD activity (Cayman Chemicals, Inc. kit # 706002), as per the manufacturers' protocol. Data are normalized to total protein and are presented as U/mg protein.
Lucigenin chemiluminescence was employed for superoxide detection, as previously described (25). Briefly, renal interlobar arteries were incubated in oxygenated KB at 37°C for 20 min with and without H2O2 (10 μM). Arteries were then transferred to preblanked lucigenin vials (5 μM) and analyzed in a scintillation counter. Data are normalized to total protein and are presented as CPM/μg protein.
Data analysis
Data are expressed as means±SEM for the given number (n) of experiments. Results were analyzed by ANOVA with Tukey–Kramer post hoc analysis, or by Student t-test. The null hypothesis was rejected at p<0.05.
Abbreviations Used
- BHT
butylated hydroxytoluene
- BV
biliverdin
- CO
carbon monoxide
- COX
cyclooxygenase
- CrMP
chromium mesoporphyrin
- DES
desferroxamine
- DMTU
dimethyl thiourea
- HO
heme oxygenase
- H2O2
hydrogen peroxide
- ID
internal diameter
- KB
Krebs' buffer
- PEG-SOD
PEGylated superoxide dismutase
- RIA
renal interlobular
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- SnMP
stannous mesoporphyrin
- TP
thromboxane
- TxA2
thromboxane A2
- TxB2
thromboxane B2
Acknowledgments
This work was supported by the National Institutes of Health grants 2PO1HL034300 (AN) and DK068134, HL55601, and HL34300 (NGA). All authors had full access to the data and take responsibility for its integrity. All authors have read and agree with the manuscript as written. We thank Jennifer Brown for her outstanding editorial assistance in the preparation of the manuscript.
Author Disclosure Statement
The authors declare no competing financial interests.
References
- 1.Ardanaz N. Pagano PJ. Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp Biol Med (Maywood) 2006;231:237–251. doi: 10.1177/153537020623100302. [DOI] [PubMed] [Google Scholar]
- 2.Avshalumov MV. Chen BT. Rice ME. Mechanisms underlying H(2)O(2)-mediated inhibition of synaptic transmission in rat hippocampal slices. Brain Res. 2000;882:86–94. doi: 10.1016/s0006-8993(00)02835-3. [DOI] [PubMed] [Google Scholar]
- 3.Ball SK. Field MC. Tippins JR. Regulation of thromboxane receptor signaling at multiple levels by oxidative stress-induced stabilization, relocation and enhanced responsiveness. PLoS One. 2010;5:e12798. doi: 10.1371/journal.pone.0012798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barlow RS. White RE. Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. Am J Physiol. 1998;275:H1283–H1289. doi: 10.1152/ajpheart.1998.275.4.H1283. [DOI] [PubMed] [Google Scholar]
- 5.Boehning D. Moon C. Sharma S. Hurt KJ. Hester LD. Ronnett GV. Shugar D. Snyder SH. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–137. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- 6.Brennan JP. Bardswell SC. Burgoyne JR. Fuller W. Schroder E. Wait R. Begum S. Kentish JC. Eaton P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. 2006;281:21827–21836. doi: 10.1074/jbc.M603952200. [DOI] [PubMed] [Google Scholar]
- 7.Burgoyne JR. Madhani M. Cuello F. Charles RL. Brennan JP. Schroder E. Browning DD. Eaton P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 8.Cai H. Davis ME. Drummond GR. Harrison DG. Induction of endothelial NO synthase by hydrogen peroxide via a Ca(2+)/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arterioscler Thromb Vasc Biol. 2001;21:1571–1576. doi: 10.1161/hq1001.097028. [DOI] [PubMed] [Google Scholar]
- 9.Cao J. Sodhi K. Inoue K. Quilley J. Rezzani R. Rodella L. Vanella L. Germinario L. Stec DE. Abraham NG. Kappas A. Lentiviral-human heme oxygenase targeting endothelium improved vascular function in angiotensin II animal model of hypertension. Hum Gene Ther. 2011;22:271–282. doi: 10.1089/hum.2010.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cseko C. Bagi Z. Koller A. Biphasic effect of hydrogen peroxide on skeletal muscle arteriolar tone via activation of endothelial and smooth muscle signaling pathways. J Appl Physiol. 2004;97:1130–1137. doi: 10.1152/japplphysiol.00106.2004. [DOI] [PubMed] [Google Scholar]
- 11.Fujimoto S. Asano T. Sakai M. Sakurai K. Takagi D. Yoshimoto N. Itoh T. Mechanisms of hydrogen peroxide-induced relaxation in rabbit mesenteric small artery. Eur J Pharmacol. 2001;412:291–300. doi: 10.1016/s0014-2999(00)00940-7. [DOI] [PubMed] [Google Scholar]
- 12.Gao YJ. Hirota S. Zhang DW. Janssen LJ. Lee RM. Mechanisms of hydrogen-peroxide-induced biphasic response in rat mesenteric artery. Br J Pharmacol. 2003;138:1085–1092. doi: 10.1038/sj.bjp.0705147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao YJ. Lee RM. Hydrogen peroxide induces a greater contraction in mesenteric arteries of spontaneously hypertensive rats through thromboxane A(2) production. Br J Pharmacol. 2001;134:1639–1646. doi: 10.1038/sj.bjp.0704420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupte SA. Okada T. Tateyama M. Ochi R. Activation of TxA2/PGH2 receptors and protein kinase C contribute to coronary dysfunction in superoxide treated rat hearts. J Mol Cell Cardiol. 2000;32:937–946. doi: 10.1006/jmcc.2000.1134. [DOI] [PubMed] [Google Scholar]
- 15.Hayabuchi Y. Nakaya Y. Matsuoka S. Kuroda Y. Hydrogen peroxide-induced vascular relaxation in porcine coronary arteries is mediated by Ca2+-activated K+ channels. Heart Vessels. 1998;13:9–17. doi: 10.1007/BF02750638. [DOI] [PubMed] [Google Scholar]
- 16.Iida Y. Katusic ZS. Mechanisms of cerebral arterial relaxations to hydrogen peroxide. Stroke. 2000;31:2224–2230. doi: 10.1161/01.str.31.9.2224. [DOI] [PubMed] [Google Scholar]
- 17.Jansen T. Daiber A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front Pharmacol. 2012;3:30. doi: 10.3389/fphar.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin N. Rhoades RA. Activation of tyrosine kinases in H2O2-induced contraction in pulmonary artery. Am J Physiol. 1997;272:H2686–H2692. doi: 10.1152/ajpheart.1997.272.6.H2686. [DOI] [PubMed] [Google Scholar]
- 19.Kaide J-I. Zhang F. Wei Y. Jiang H. Yu C. Wang WH. Balazy M. Abraham NG. Nasjletti A. Carbon monoxide of vascular origin attenuates the sensitivity of renal arterial vessels to vasoconstrictors. J Clin Invest. 2001;107:1163–1171. doi: 10.1172/JCI11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katusic ZS. Vanhoutte PM. Superoxide anion is an endothelium-derived contracting factor. Am J Physiol. 1989;257:H33–H37. doi: 10.1152/ajpheart.1989.257.1.H33. [DOI] [PubMed] [Google Scholar]
- 21.Kinobe R. Ji Y. Nakatsu K. Peroxynitrite-mediated inactivation of heme oxygenases. BMC Pharmacol. 2004;4:26. doi: 10.1186/1471-2210-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinugawa S. Tsutsui H. Hayashidani S. Ide T. Suematsu N. Satoh S. Utsumi H. Takeshita A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res. 2000;87:392–398. doi: 10.1161/01.res.87.5.392. [DOI] [PubMed] [Google Scholar]
- 23.Kruger AL. Peterson SJ. Schwartzman ML. Fusco H. McClung JA. Weiss M. Shenouda S. Goodman AI. Goligorsky MS. Kappas A. Abraham NG. Up-regulation of heme oxygenase provides vascular protection in an animal model of diabetes through its antioxidant and antiapoptotic effects. J Pharmacol Exp Ther. 2006;319:1144–1152. doi: 10.1124/jpet.106.107482. [DOI] [PubMed] [Google Scholar]
- 24.Lambert CR. Black HS. Truscott TG. Reactivity of butylated hydroxytoluene. Free Radic Biol Med. 1996;21:395–400. doi: 10.1016/0891-5849(96)00050-0. [DOI] [PubMed] [Google Scholar]
- 25.Lamon BD. Zhang FF. Puri N. Brodsky SV. Goligorsky MS. Nasjletti A. Dual pathways of carbon monoxide-mediated vasoregulation: modulation by redox mechanisms. Circ Res. 2009;105:775–783. doi: 10.1161/CIRCRESAHA.109.197434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee MY. Griendling KK. Redox signaling, vascular function, and hypertension. Antioxid Redox Signal. 2008;10:1045–1059. doi: 10.1089/ars.2007.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leffler CW. Busija DW. Armstead WM. Mirro R. H2O2 effects on cerebral prostanoids and pial arteriolar diameter in piglets. Am J Physiol. 1990;258:H1382–H1387. doi: 10.1152/ajpheart.1990.258.5.H1382. [DOI] [PubMed] [Google Scholar]
- 28.Lucchesi PA. Belmadani S. Matrougui K. Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J Hypertens. 2005;23:571–579. doi: 10.1097/01.hjh.0000160214.40855.79. [DOI] [PubMed] [Google Scholar]
- 29.Matsui T. Nakajima A. Fujii H. Matera KM. Migita CT. Yoshida T. Ikeda-Saito M. O(2)- and H(2)O(2)-dependent verdoheme degradation by heme oxygenase: reaction mechanisms and potential physiological roles of the dual pathway degradation. J Biol Chem. 2005;280:36833–36840. doi: 10.1074/jbc.M503529200. [DOI] [PubMed] [Google Scholar]
- 30.Rathore R. Zheng YM. Li XQ. Wang QS. Liu QH. Ginnan R. Singer HA. Ho YS. Wang YX. Mitochondrial ROS-PKCepsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+ ]i in pulmonary artery smooth muscle cells. Biochem Biophys Res Commun. 2006;351:784–790. doi: 10.1016/j.bbrc.2006.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez F. Lamon BD. Gong W. Kemp R. Nasjletti A. Nitric oxide synthesis inhibition promotes renal production of carbon monoxide. Hypertension. 2004;43:347–351. doi: 10.1161/01.HYP.0000111721.97169.97. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Martinez MA. Garcia-Cohen EC. Baena AB. Gonzalez R. Salaices M. Marin J. Contractile responses elicited by hydrogen peroxide in aorta from normotensive and hypertensive rats. Endothelial modulation and mechanism involved. Br J Pharmacol. 1998;125:1329–1335. doi: 10.1038/sj.bjp.0702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato A. Sakuma I. Gutterman DD. Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol. 2003;285:H2345–H2354. doi: 10.1152/ajpheart.00458.2003. [DOI] [PubMed] [Google Scholar]
- 34.Schnackenberg CG. Welch WJ. Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32:59–64. doi: 10.1161/01.hyp.32.1.59. [DOI] [PubMed] [Google Scholar]
- 35.Schultz D. Harrison DG. Quest for fire: seeking the source of pathogenic oxygen radicals in atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:1412–1413. doi: 10.1161/01.atv.20.6.1412. [DOI] [PubMed] [Google Scholar]
- 36.Sodhi K. Inoue K. Gotlinger K. Canestraro M. Vanella L. Kim DH. Manthati VL. Koduru SR. Falck JR. Schwartzman ML. Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther. 2009;331:906–916. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Somers MJ. Harrison DG. Reactive oxygen species and the control of vasomotor tone. Curr Hypertens Rep. 1999;1:102–108. doi: 10.1007/s11906-999-0080-z. [DOI] [PubMed] [Google Scholar]
- 38.Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. 2004;6:841–849. doi: 10.1089/ars.2004.6.841. [DOI] [PubMed] [Google Scholar]
- 39.Stocker R. McDonagh AF. Glazer AN. Ames BN. Antioxidant activities of bile pigments: biliverdin and bilirubin. Methods Enzymol. 1990;186:301–309. doi: 10.1016/0076-6879(90)86123-d. [DOI] [PubMed] [Google Scholar]
- 40.Tang XD. Garcia ML. Heinemann SH. Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol. 2004;11:171–178. doi: 10.1038/nsmb725. [DOI] [PubMed] [Google Scholar]
- 41.Thakali K. Davenport L. Fink GD. Watts SW. Cyclooxygenase, p38 mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase MAPK, Rho kinase, and Src mediate hydrogen peroxide-induced contraction of rat thoracic aorta and vena cava. J Pharmacol Exp Ther. 2007;320:236–243. doi: 10.1124/jpet.106.110650. [DOI] [PubMed] [Google Scholar]
- 42.Valentin F. Field MC. Tippins JR. The mechanism of oxidative stress stabilization of the thromboxane receptor in COS-7 cells. J Biol Chem. 2004;279:8316–8324. doi: 10.1074/jbc.M306761200. [DOI] [PubMed] [Google Scholar]
- 43.Wang D. Chabrashvili T. Wilcox CS. Enhanced contractility of renal afferent arterioles from angiotensin-infused rabbits: roles of oxidative stress, thromboxane prostanoid receptors, and endothelium. Circ Res. 2004;94:1436–1442. doi: 10.1161/01.RES.0000129578.76799.75. [DOI] [PubMed] [Google Scholar]
- 44.Wilson SJ. Cavanagh CC. Lesher AM. Frey AJ. Russell SE. Smyth EM. Activation-dependent stabilization of the human thromboxane receptor: role of reactive oxygen species. J Lipid Res. 2009;50:1047–1056. doi: 10.1194/jlr.M800447-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995:82–83. doi: 10.1016/0378-4274(95)03532-x. 969–974. [DOI] [PubMed] [Google Scholar]
- 46.Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol. 2009;296:H539–H549. doi: 10.1152/ajpheart.01167.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xi Q. Tcheranova D. Basuroy S. Parfenova H. Jaggar JH. Leffler CW. Glutamate-induced calcium signals stimulate CO production in piglet astrocytes. Am J Physiol Heart Circ Physiol. 2011;301:H428–H433. doi: 10.1152/ajpheart.01277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y. Shi W. Cui N. Wu Z. Jiang C. Oxidative stress inhibits vascular K(ATP) channels by S-glutathionylation. J Biol Chem. 2010;285:38641–38648. doi: 10.1074/jbc.M110.162578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang ZW. Zheng T. Wang J. Zhang A. Altura BT. Altura BM. Hydrogen peroxide induces contraction and raises [Ca2+ ]i in canine cerebral arterial smooth muscle: participation of cellular signaling pathways. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:646–653. doi: 10.1007/s002109900128. [DOI] [PubMed] [Google Scholar]
- 50.Zembowicz A. Hatchett RJ. Jakubowski AM. Gryglewski RJ. Involvement of nitric oxide in the endothelium-dependent relaxation induced by hydrogen peroxide in the rabbit aorta. Br J Pharmacol. 1993;110:151–158. doi: 10.1111/j.1476-5381.1993.tb13785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]