

Abstract
Complex diseases are caused by perturbations of biological networks. Genetic analysis approaches focused on individual genetic determinants are unlikely to characterize the network architecture of complex diseases comprehensively. Network medicine, which applies systems biology and network science to complex molecular networks underlying human disease, focuses on identifying the interacting genes and proteins which lead to disease pathogenesis. The long biological path between a genetic risk variant and development of a complex disease involves a range of biochemical intermediates, including coding and non-coding RNA, proteins, and metabolites. Transcriptomics, proteomics, metabolomics, and other –omics technologies have the potential to provide insights into complex disease pathogenesis, especially if they are applied within a network biology framework. Most previous efforts to relate genetics to –omics data have focused on a single –omics platform; the next generation of complex disease genetics studies will require integration of multiple types of –omics data sets in a network context. Network medicine may also provide insight into complex disease heterogeneity, serve as the basis for new disease classifications that reflect underlying disease pathogenesis, and guide rational therapeutic and preventive strategies.
I. Overview of Network Medicine
Most major public health problems, such as coronary artery disease, diabetes mellitus, stroke, and chronic obstructive pulmonary disease, are complex diseases, which are likely influenced by multiple genetic and environmental factors operating within a developmental context. Genome-wide association and DNA resequencing studies have identified some susceptibility loci for complex diseases, but our understanding of the functional role of these loci in the etiology and pathogenesis of these conditions remains woefully incomplete. In addition to defining the etiological mechanisms for disease, another key challenge in complex disease genetics is to understand disease heterogeneity. Transcriptomics, metabolomics, proteomics, and other –omics technologies have the potential to provide insights into complex disease pathogenesis and heterogeneity, especially if they are applied within a network biology framework.
Network medicine is the rapidly developing field which applies systems biology and network science methods to human disease (Barabasi et al., 2011). Networks can be used to visualize and analyze a broad range of biological processes, with nodes in the network representing a biological entity (e.g., gene, protein, disease) and edges representing the relationships between entities (e.g., physical interactions, transcriptional activation, correlations in gene expression levels). A holistic approach is used to relate the interactome network—the complete set of macromolecular interactions between genes and their products--to disease (Vidal et al., 2012). Single genetic variants are unlikely to explain complex disease phenotypes, because perturbations of biological networks, not isolated genes or proteins, confer disease risk. Robustness to perturbation, a key feature of biological networks, is manifested as persistence of ‘normal’ network properties, such as average path length, connectedness, or function, upon removal of vertices or edges (Newman, 2010), the equivalent of the ‘knock-out’ experiment for a biological system. Cells have evolved to be robust to perturbations in order to withstand the inherent stochastic effects in gene expression and somatic mutation; cells are buffered to withstand these insults (Koonin et al., 2006).
Multiple approaches have been used to define cellular interactome networks, including capturing reported biological relationships from the scientific literature, computational predictions of biochemical or physical interactions, and high throughput experimental strategies (e.g., yeast two-hybrid systems or affinity purification/mass spectrometry) of protein-protein interactions in model organisms (Vidal et al., 2011). Network motifs are characteristic network patterns or subgraphs associated with specific biological functions. Vidal and colleagues cite multiple lines of evidence that cellular networks underlie genotype-phenotype relationships in human disease (Vidal et al., 2011), including the following: a) global disease networks demonstrate aggregation of disease classifications and co-morbidities, suggesting that diseases are not independent from one another; b) prediction of new disease genes from cellular network models (e.g., genes for ataxia syndromes) (Lim et al., 2006) provides evidence that this approach will be more generally successful; and c) network perturbations by pathogens may act as surrogates for human genetic variants by influencing local and global properties of cellular networks. They point out that network perturbations resulting from genetic variation may range from complete removal of a key gene (e.g., nonsense mutation) to alteration of specific protein interactions (i.e., ranging from eliminating a key node or edge of the network, or altering the strength of interaction between two or more nodes).
In this review, we will use selected recent examples to demonstrate the potential utility of network medicine approaches in human disease. We will do so by emphasizing the role of multiple –omics approaches to provide insights into complex diseases.
II. Challenges of Complex Disease Genetics
The recent identification of many common genetic variants associated with complex diseases using genome-wide association studies (GWAS) followed an era of largely irreproducible results from candidate gene case-control studies in almost every complex disease. GWAS is still a prominent genetic epidemiological study design, although exome sequencing has been quite successful at identifying causal variants in Mendelian diseases in very small sample sizes (Hoischen et al., 2010; Ng et al., 2010), and whole genome sequencing is rapidly becoming affordable as a means by which to identify both rare and common genetic determinants of complex diseases. Complex disease genetic research was transformed by the development of low-cost, high-throughput SNP genotyping arrays, which provided reasonably comprehensive coverage of common variants in the human genome and which made GWAS a feasible study design. Hundreds of genetic variants have now been associated with complex diseases at stringent levels of statistical significance (Lander, 2011), using what we have termed “First Generation Genetic Studies” (Figure 1). Despite these successes, the odds ratios for the identified genetic determinants have been surprisingly low, and the percentage of genetic variation explained by GWAS signals has generally been modest—most heritability for these traits has remained unexplained (Manolio et al., 2009). There are several potential explanations for this missing heritability, including the role of rare genetic variants and interactions (gene-gene and gene-environment), which are summarized in Table 1. Yang and colleagues (Yang et al., 2010) argued that the most likely contributors to the missing heritability from GWAS are: 1) most genetic determinants of modest effect size have not yet been identified at the stringent statistical significance levels used in GWAS; 2) causative variants are not in complete linkage disequilibrium (LD) with the GWAS signals; and 3) causative variants are more likely to have lower allele frequencies than ’normal’ biological variants. Adjusting for these three factors using quantitative genetic analysis, they estimated that relatively common variants could account for nearly all of the heritability for height. Very large sample sizes will be required to detect large numbers of such common variants of modest effect (Park et al., 2010). For example, GWAS of plasma lipids in more than 100,000 Caucasian individuals identified 95 loci of genome-wide significance which account for 25–30% of the genetic variance in these traits (Teslovich et al., 2010). Nevertheless, much of the genetic variation for complex diseases remains unexplained, despite using very large sample sizes in GWAS.
Figure 1.
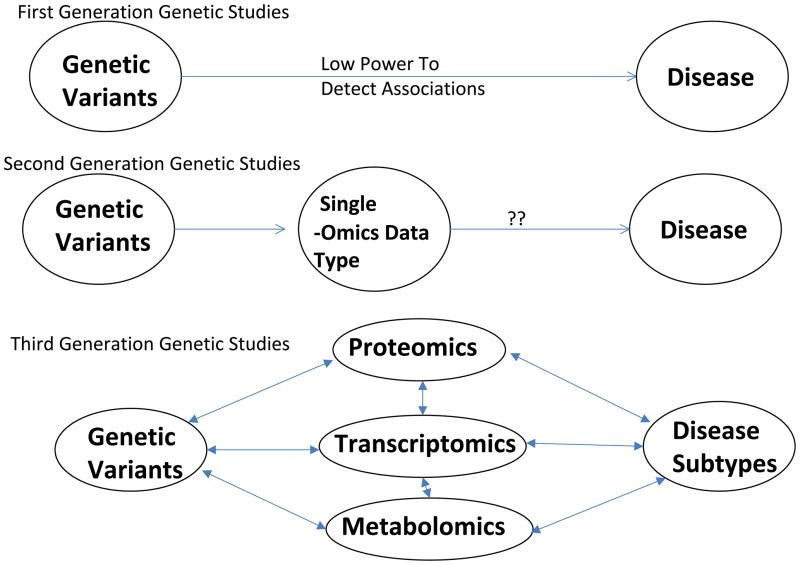
Development of Complex Disease Genetic Studies. First Generation Genetic Studies attempt to relate genetic variants, such as SNPs, directly to a complex disease; these approaches have low power to detect associations and even lower power to detect gene-gene and gene-environment interactions. Second Generation Genetic Studies assess for associations between genetic variants and a single type of –omics data, such as transcriptomics, metabolomics, or proteomics. These studies have provided useful insights into the genetic determinants of these biological intermediate phenotypes, but relating these findings to disease susceptibility has been more difficult. Third Generation Genetic Studies, which will require new methodological development, will explore the relationships between genetic variants and multiple types of -omics data in a network context, potentially with models that address the phenotypic and genetic heterogeneity of complex diseases.
Table 1.
Potential Explanations for Incomplete Genetic Architecture of Complex Diseases
| Explanation | Rationale | Comments |
|---|---|---|
| Common Genetic Variants | More common variants are likely to be found in GWAS with larger sample sizes. | Effect sizes of known GWAS loci may be underestimated since functional variants have often not yet been found. |
| Rare Genetic Variants | Resequencing studies (e.g. whole exome, whole genome) could identify rare genetic determinants of large effect size. | Limited evidence for rare variants of major effect in complex diseases accounting for large amount of genetic variation |
| Interactions | Gene-gene and gene-environment interactions are likely important for complex diseases. | Limited evidence for statistical interactions in complex diseases; network-based approaches may be helpful to identify these interactions. |
| Inaccurate Heritability Estimates | Heritability estimates are typically performed assuming that gene-gene and gene-environment interactions are not present. | Limiting pathway model suggests that epistasis could account for missing heritability in complex diseases (Zuk et al., 2012) |
| Phenotypic and Genetic Heterogeneity | Most complex diseases are likely syndromes with multiple potentially overlapping disease subtypes. | Improvements in phenotyping of complex diseases will be required to understand genetic architecture. |
Part of the unexplained genetic variation in complex diseases likely does relate to the failure to identify the functional genetic variant or variants within the vast majority of GWAS loci. Most of these GWAS loci are found within non-coding regions (Manolio, 2010), which may contain regulatory elements for nearby or distant genes. For example, identification of functional variants in non-coding regions has been reported for colorectal cancer (Pomerantz et al., 2009), lipoprotein levels (Musunuru et al., 2011), COPD (Zhou et al., 2012), and coronary artery disease (Harismendy et al., 2011). Further work will be required to identify such functional variants. Studying the functional variants in isolation, however, will likely not provide a complete picture of the genetic architecture of complex diseases; evaluating these genetic variants, genes, and gene products within a network context will be required.
Since determinants of disease phenotypes are so abundant, it is not surprising that genotype-phenotype associations have modest effect sizes; transcription, translation, and post-translational modifications are regulated processes that affect genotype-phenotype relationships, along with gene-gene and gene-environment interactions (Loscalzo, 2007). The application of network approaches in genetics has also been termed “systems genetics” (Nadeau & Dudley, 2011), with a major focus on the interactions between genes, known as epistasis. Epistasis is defined by non-additive contributions of two genetic loci to a phenotype. Despite their likely great importance, epistatic interactions have been difficult to identify in genetic studies of complex disease. Zuk and Lander developed a genetic model that suggested that epistatic effects could account for missing heritability in complex diseases (Zuk et al., 2012). They argued that there is not necessarily a large amount of missing heritability in complex diseases; rather, the denominator of total narrow sense heritability, which is based on assuming additive genetic contributions without interactions, is likely wrong. They term this concept, “phantom heritability,” and point out that biological processes often depend on the rate-limiting value of multiple inputs, which is consistent with a network medicine perspective. Using a limiting pathway model in which a trait depends on the rate-limiting value of k inputs, when k > 1, there can be substantial missing heritability. Epistasis is likely common, but very difficult to detect since effects are small. They point out that the limiting pathway model may not be correct, but it shows that phantom heritability can exist. Network approaches incorporating multiple –omics platforms have the potential to identify these epistatic effects.
In one of the few examples successfully demonstrating epistasis in human complex disease, Emily and colleagues performed gene-gene interaction tests for seven complex diseases in the Wellcome Trust Case-Control Consortium (Emily et al., 2009). Studying 125 billion SNP pairs with a 500,000 SNP chip is problematic statistically, since p-values < 10−13 are required for statistical significance; moreover, performing such a large number of pairwise comparisons is computationally extremely intensive. They prioritized SNPs based on the protein-protein interaction network in the STRING database, and only assessed markers in genes expected to interact biologically. They found 71,000 potential protein-protein interactions in STRING, and identified all SNPs located +/− 100 kb from genes related to those interactions. They used a likelihood ratio test only for those 71,000 potential interactions and adjusted for multiple testing after accounting for nonindependence of tests—a less conservative approach than Bonferroni correction. They found four significant pairwise interactions--one each for Crohn’s disease, hypertension, rheumatoid arthritis, and bipolar disorder.
Although human complex disease studies using traditional genetic approaches have found limited evidence for epistasis, studies of microorganisms have been more successful. Hinkley and colleagues studied the key drug targets for HIV treatment--the protease and reverse transcriptase enzymes (Hinkley et al., 2011). HIV rapidly evolves drug resistance mutations (due to the high mutation rate and large population size within an infected individual), but genetic events leading to resistance have been difficult to identify—potentially due to epistasis. For this study, they defined epistasis as the impact of one genetic variant depending on the presence/absence of variants elsewhere in the HIV genome. They assessed replicative capacity of 70,081 clinical HIV isolates exposed to 15 antiviral drugs for resistance testing using a test vector in which the HIV-1 envelope gene was replaced by a luciferase expression cassette, and they sequenced amplification products of protease and reverse transcriptase genes from these assays to identify nonsynonymous SNPs. They applied a new statistical approach to assess for epistasis: generalized kernel ridge regression to accommodate non-normality and large sample size, which penalizes against parameters with low explanatory power. They compared effects of models for replicative capacity with only main effects of nonsynonymous variants with models also including pairwise epistatic interactions, and they validated results using the Stanford HIV Drug Resistance Database. A large number of variants were found, with 1859 nonsynonymous SNPs in the protease and reverse transcriptase genes. The model including epistatic effects had an average of 18.3% better predictive power than the model without epistatic effects. Moreover, amino acid variants predicted to have large effects on viral fitness in the epistasis model correlated strongly with high frequency variants in the Stanford database of HIV patient samples. They found that intragenic interaction effects were generally greater than intergenic effects, and the strongest interactions were between nearby variants within the same protein structural domain. Limitations of the study were that only pairwise interactions were considered; studying higher order interactions dramatically increases the number of parameters that need to be estimated. This study demonstrated that including environmental perturbations (e.g., different drug treatments) can be very helpful in revealing gene-gene interactions.
III. –Omics Approaches in Network Medicine and Genetics
The long biological path between a genetic risk variant and development of a complex disease involves a range of biochemical intermediates, such as mRNA, proteins, and metabolites. These intermediate phenotypes, which can be captured using high throughput assays, have been used to gain insight into the biological networks relevant for complex diseases. We will review some key examples of the application of network approaches to three major types of –omics data: transcriptomics, proteomics, and metabolomics. We will also review genetic studies of single –omics approaches, which we describe as Second Generation Genetic Studies (Figure 1). These studies have identified genetic influences on the levels of biological intermediates; however, relating those genetic determinants of an –omics type to complex disease susceptibility has been less successful.
Transcriptomics
Multiple methods have been used to determine network structure from gene expression microarray data, including Boolean networks, differential equations, and Bayesian networks (Djebbari & Quackenbush, 2008). Inferring network structure from gene expression analyses of human tissues has been challenging for several reasons. First, the sample size of assayed tissues is typically much smaller than the number of analyzed genes, which limits the identification of an optimal network. Second, some of the most promising methods, including Bayesian networks (which use directed acyclic graphs), are computationally extremely intensive. Third, static comparisons between groups are typically used, although changes in gene expression are dynamic. Therefore, approaches to limit the network search space have been developed. For example, Djebbari and Quackenbush proposed an approach to create transcriptomic networks with the use of seed genes from the literature or from protein-protein interaction networks (Djebbari & Quackenbush, 2008). By analyzing leukemia gene expression data sets, they found that use of prior network information from the literature or protein-protein interaction networks improved their ability to identify gene expression network relationships. To help to overcome the limitations imposed by small sample sizes and variable effect sizes, they used a non-parametric bootstrapping approach to assess confidence in their network models.
Gene expression networks of human cells or tissues are phenomenological, reflecting incomplete modeling and dynamic observations. They are correlative but not necessarily causal networks (Koonin et al., 2006). Stochastic effects may also have important effects on gene expression. Burga and colleagues modeled incomplete penetrance in C. elegans and showed that stochastic effects in gene expression of an ancestral gene duplication and in induction of molecular chaperones led to differential effects of inherited mutations (Burga et al., 2011). They used a genetic network interaction model to demonstrate that if one gene of a partly redundant gene pair is inactivated, the phenotype depends on the stochastic expression variation of the remaining gene.
MicroRNA and epigenetic marks such as DNA methylation and histone acetylation are often key regulators of gene expression. Parikh and colleagues defined a disease module for pulmonary arterial hypertension within the macromolecular network based on previously studied genes and then used this disease module to identify microRNA species likely to be involved in disease pathogenesis (Parikh et al., 2012). miR-21 was identified as a candidate microRNA regulating key pulmonary arterial hypertension pathways that was validated in multiple in vitro and in vivo models.
Integrative genomic studies have been widely performed to relate SNPs to gene expression levels. For example, Small and colleagues studied metabolic syndrome, which includes obesity, insulin resistance, hypertension, type 2 diabetes mellitus, and hyperlipidemia (Small et al., 2011). Environmental factors are clearly important for metabolic syndrome, but component phenotypes of this syndrome are highly heritable. Many GWAS signals have been found for individual components of the metabolic syndrome, but no genetic loci were previously found to be key regulators of the entire syndrome. KLF14 encodes a transcription factor gene; SNPs approximately 14 kb upstream from KLF14 had been previously associated with type 2 diabetes mellitus and HDL levels, and a cis-eQTL influencing KLF14 gene expression in adipose tissue was reported in that same region. They performed microarray gene expression analysis in adipose tissue from 776 female twins, and then performed trans-eQTL analysis between the KLF14 cis-eQTL SNP and adipose gene expression levels. The most significant SNPs from this analysis were then related to metabolic syndrome phenotypes in a very large consortium GWAS. Although a single key SNP was not identified and functional studies were not performed, conditioning trans- associations on rs4731702 eliminated other significant associations. They found 10 genes with genome-wide significant trans-gene expression associations to this KLF14 SNP; 7/10 of these associations were replicated in gene expression data from 589 other adipose tissue samples, and 5/10 genome-wide significant transgene expression association genes were associated with a broad range of metabolic syndrome traits, including all of the key features of metabolic syndrome. Integration of gene expression and genetic variation data is clearly important to build pathways; but this is a cautionary tale in that such large sample sizes for gene expression studies were needed in a relevant tissue type. Every gene in which expression correlated with disease did not have SNPs associated with disease phenotypes. It is clearly essential to have functional variants within the study population to find a genetic association.
Several studies have performed integrative genomic analyses within a network context. Barrenas and colleagues hypothesized that after defining disease modules using both the protein-protein interaction network and differential gene expression data from relevant tissues, modules with the most interconnected disease genes would have more GWAS disease SNPs (Barrenas et al., 2012). They used publicly available gene expression and GWAS data for 13 complex diseases, then studied their specific disease of interest, seasonal allergic rhinitis. They defined core susceptibility modules as genes with high interconnectivity; they then created a disease-specific core susceptibility module for each disease and a global core susceptibility module for all 13 diseases. They found enrichment of GWAS genes in the disease-specific core susceptibility modules, while the group of all differentially expressed genes was not enriched for GWAS genes. They also found evidence for susceptibility to multiple complex diseases (a total of 145 diseases) in the global core susceptibility module genes derived from their 13 main diseases. Although they only had gene expression data for 12 seasonal allergic rhinitis subjects, they found that disease-specific core susceptibility module genes from this gene expression network for seasonal allergic rhinitis were more likely to contain significant GWAS SNPs than other genes.
Chu and Raby developed a stepwise approach that starts by building a Gaussian graphical network model based on gene expression levels, then examines regulation of specific target genes (Chu et al., 2009). Subsequently, genetic association testing is performed conditional on the developed graphical network. They applied this approach in asthma, focusing on the subset of genes showing variable gene expression across samples of CD4 positive lymphocytes from asthmatics. Subsequently, they extended their approach to compare differences in gene-gene connectivity patterns across disease states (Chu et al., 2011). Posterior odds ratios between disease groups give a quantitative measure of differences in network connectivity. Simulations suggest that specificity is high but sensitivity is not sufficient to detect these differences. A key feature of their Gaussian graphical model approach is use of partial correlation coefficients, which distinguish direct and indirect interactions.
Proteomics
Studies of proteomics have obvious biological relevance and can provide complementary information to transcriptomics. However, developing robust, high throughput proteomic assays has been technically challenging. New proteomic technology and analytical approaches, such as selected reaction monitoring (SRM, a mass spectrometric technique), have been developed for quantitative proteomic assessment (Brusniak et al., 2012). Traditional tandem mass spectrometric approaches require extensive bioinformatic searches to match peptide fragments against protein databases. At a recent NIH workshop on proteomics (September, 2011), it was noted that dynamic aspects of proteins can be assessed with quantitative measurements of protein levels and determination of post-translational modifications and splicing effects which alter protein levels (Vidal et al., 2012). Recently optimized proteomic approaches have allowed identification of ~10,000 proteins from human cell lines. Multiple peptides for SRM analysis have been prepared for most human proteins. Current protein interaction networks are incomplete, but developing such networks could be a major step forward in understanding disease etiology (Charloteaux et al., 2011). Static protein interaction maps will need to be extended to understand dynamic responses to perturbation.
Proteomics has been linked to genetics in several ways. In some studies, protein levels have been used as phenotypes for genetic analysis. Naitza and colleagues used several protein biomarkers of inflammation, including IL-6, MCP-1, and CRP, as phenotypes for genome-wide association analysis in Sardinia. They performed GWAS in 4,694 individuals, with replication in 1,392 subjects (Naitza et al., 2012). They also genotyped the ImmunoChip and MetaboChip in the full cohort. Not surprisingly, some of the significant associations to protein levels were within or near the coding gene for that protein; for example, a SNP in CRP was a significant determinant of CRP levels. Other associations recapitulated known biological relationships, such as the associations of a SNP near CCR2, the receptor for MCP-1, with MCP-1 levels, and a SNP in the IL-6 receptor gene (IL6R) with IL-6 levels. Other significant associations that were not in expected regions may reveal novel biological relationships; for instance, a SNP near the Inducible T-cell co-stimulator ligand (ICOSLG) and autoimmune regulator (AIRE) genes was significantly associated with CRP levels.
Another approach for integrating proteomics with genetics is to use the global network of protein-protein interactions to limit the genetic search space for association studies—thus reducing the number of statistical tests performed—as mentioned above by Emily and colleagues (Emily et al., 2009). Alternatively, GWAS (or other types of genetic information) can be used to assist in defining disease-related network modules within the protein-protein interaction network. For example, Jia and colleagues (Jia et al., 2011) developed dmGWAS, a dense module searching approach for GWAS in protein-protein interaction networks. They picked the single most significant SNP to represent the gene in the interaction analysis and used a seed gene-based approach to build the disease-related network. A potential advantage of this approach is that it uses all of the GWAS data, not just the top SNPs from the GWAS.
Metabolomics
Metabolomic studies can provide complementary information to other –omics approaches, with high throughput assay systems now having been developed. In a pivotal early investigation, Jeong and colleagues demonstrated impressive conservation of metabolic network structure across phylogenetic groups (Jeong et al., 2000). More recently, Krumsiek and colleagues used a Gaussian graphical modeling approach to build networks from metabolomic data for 151 metabolites in 1020 KORA Study subjects (Krumsiek et al., 2011). Network modules related to the seven major metabolite classes that they measured were identified. Moreover, they found that known metabolic relationships from fatty acid biosynthesis were captured in their network model, suggesting that the metabolomic measurements accurately reflect the underlying biochemical pathways which generate the metabolites.
Suhre and colleagues recently reported a GWAS of metabolomic measurements. They performed metabolomic profiling of fasting serum for > 250 metabolites from 60 biochemical pathways in 2,820 individuals from two European studies (including the KORA Study) (Suhre et al., 2011). They used existing genome-wide SNP genotyping data and 37,000 individual metabolites and metabolite ratios in a screening stage with log-transformed metabolite values and adjustment for age, gender, and family structure. They found 37 loci that were genome-wide significant in meta-analysis; in 25 loci, effect size per allele was > 10%. At 30 loci, the associated SNP mapped to a protein biochemically related to the metabolite (e.g., synthesis, degradation). At 15 loci, a SNP in an associated locus was associated with a complex disease or drug response. For example, fibrinopeptide A-alpha peptides were associated with three loci (ABO, ALPL, and FUT2)— genes that are functionally linked through blood groups. Importantly, ABO SNPs are associated with many complex diseases including venous thromboembolism and acute myocardial infarction (Reilly et al., 2011).
IV. Building Disease Networks from Multiple –Omics Data
Most efforts to relate genetics to –omics data have focused on a single –omics platform. Third Generation Genetics Studies (Figure 1) will require integration of multiple types of –omics data in a network context. A positive step in this direction was performed by Guan and colleagues (Guan et al., 2010); they applied machine learning techniques to genomic assessments of gene expression and protein levels in order to identify genes potentially involved in a disease phenotype. This approach can create lists of candidate genes and help to narrow focus to a single gene when there are multiple genes within a locus of interest. They used support vector machines to analyze murine functional data and a Bayesian network approach to create a network of functional relationships among all genes in the mouse, using protein-protein interactions, phylogenetic profiles, and gene expression data. From this functional network, they used two supervised learning approaches to relate genotype to phenotype—support vector machine classification and a summed weight approach—which was tested on 1,157 diverse phenotypes. They selected two genes for bone mineral density (TIMP2 and ABCG8) that were not found using previous quantitative genetic approaches (but were supported by gene expression, physical interaction, and phylogenetic information), and did functional studies to validate their potential biological roles.
Ultimately, the comprehensive integration of genetics with multiple –omics approaches will be essential. Chen and colleagues performed multiple -omics analysis longitudinally in a single individual, including whole genome DNA sequencing as well as genomic, transcriptomic, proteomic, metabolomic, and autoantibody profiling in what they label an integrative personal -omics profile (iPOP) (Chen et al., 2012). This individual had two viral infections and developed type 2 diabetes mellitus during more than one year of close observation. Whole genome sequencing revealed 26 Mb of sequence not annotated in the hg19 reference sequence and many other regions that were not included at all in hg19; these results emphasized that the reference genomic sequence is not yet complete. A Luminex panel was used to assay protein levels of 51 cytokines and showed increases in pro-inflammatory cytokines during viral infections. Overall, they found marked dynamic changes over time in multiple -omics assessments, an important observation since most studies only sample a single time point. They performed integrated analyses of transcriptomics, proteomics, and metabolomics. Some pathways were only detected using one of the approaches; for example, the insulin secretion pathway was only seen using proteomics.
Complex diseases are often heterogeneous syndromes rather than discrete disease entities. For example, stroke includes several key clinical subtypes, including large vessel, cardioembolic, and lacunar. A recent GWAS of ischemic stroke in 3,548 cases and 5,972 controls did not reveal novel genome-wide significant signals when all ischemic strokes were considered together (Bellenguez et al., 2012); however, a SNP in HDAC9 was strongly associated with large vessel stroke. This study showed significant heterogeneity in association between stroke subtypes. Inclusion of comprehensive phenotyping using clinical and imaging approaches will be essential to complement multiple –omics profiling in the dissection of complex diseases.
Further efforts to integrate genetics with multiple –omics data types will likely lead to new disease classifications that reflect underlying disease pathogenesis (Loscalzo et al., 2007); such approaches may divide complex diseases into biologically meaningful subtypes, but they could also lead to merging of diseases which have been classified separately based on specific organ involvement. As shown in Figure 2, improved phenotyping will be essential to this effort. Pathobiology-based disease classification will also provide the opportunity for targeted treatment. Just as multiple genetic factors likely interact in a network context to cause disease, treatment strategies which target multiple members of the disease network will likely be required to treat complex diseases optimally.
Figure 2.
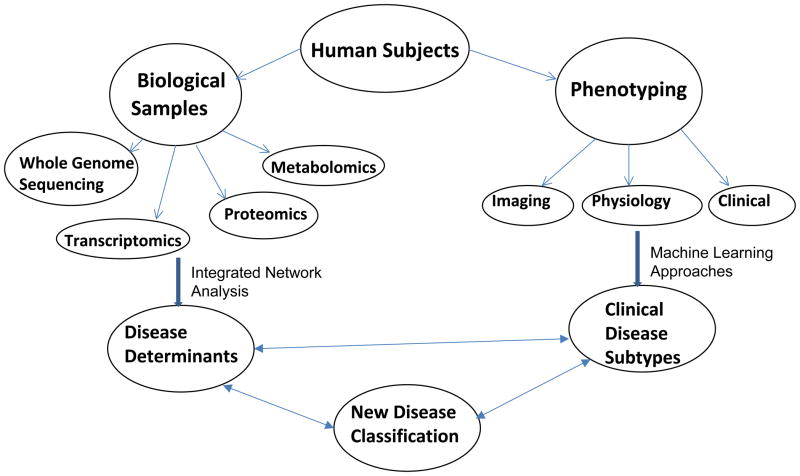
Potential Approaches to Reclassify Complex Diseases in Network Medicine. Although the optimal approach to integrate multiple types of biological and clinical information into a new classification for disease remains speculative, we have outlined a potentially useful framework. After an appropriate study population is obtained, collection of biological samples and comprehensive phenotyping will be required. Multiple –omics assessments will be performed, including whole genome sequencing, genome-wide gene expression and proteomic analysis, and metabolomic assessment. These data types will be integrated using network-based approaches to identify determinants of the complex disease. In parallel, comprehensive phenotyping will be performed; machine learning (and other) approaches may be used to define disease subtypes. In an iterative process, the disease determinants and disease subtypes will be refined to create a pathophysiology-based disease classification.
Acknowledgments
This work was supported by NIH grants HL113264, HL075478, HL105339, and HL083069 (EKS) and by NIH grants HL061795, HL048743, HL107192, HL070819, and HL108630 (JL).
Footnotes
Disclosures:
Edwin K. Silverman received grant support and consulting fees from GlaxoSmithKline for studies of COPD genetics. EKS received honoraria for consulting fees from AstraZeneca. EKS received consulting fees from Merck.
References
- Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nature reviews Genetics. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrenas F, Chavali S, Couto Alves A, Coin L, Jarvelin MR, Jornsten R, Langston MA, Ramasamy A, Rogers G, Wang H, Benson M. Highly interconnected genes in disease-specific networks are enriched for disease-associated polymorphisms. Genome Biol. 2012;13(6):R46. doi: 10.1186/gb-2012-13-6-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellenguez C, Bevan S, Gschwendtner A, Spencer CC, Burgess AI, Pirinen M, Jackson CA, Traylor M, Strange A, Su Z, Band G, Syme PD, Malik R, Pera J, Norrving B, Lemmens R, Freeman C, Schanz R, James T, Poole D, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. 2012;44(3):328–333. doi: 10.1038/ng.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusniak MY, Chu CS, Kusebauch U, Sartain MJ, Watts JD, Moritz RL. An assessment of current bioinformatic solutions for analyzing LC-MS data acquired by selected reaction monitoring technology. Proteomics. 2012;12(8):1176–1184. doi: 10.1002/pmic.201100571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burga A, Casanueva MO, Lehner B. Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature. 2011;480(7376):250–253. doi: 10.1038/nature10665. [DOI] [PubMed] [Google Scholar]
- Charloteaux B, Zhong Q, Dreze M, Cusick ME, Hill DE, Vidal M. Protein-protein interactions and networks: forward and reverse edgetics. Methods Mol Biol. 2011;759:197–213. doi: 10.1007/978-1-61779-173-4_12. [DOI] [PubMed] [Google Scholar]
- Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, Cheng Y, Clark Mj, Im H, Habegger L, Balasubramanian S, O’huallachain M, Dudley Jt, Hillenmeyer S, Haraksingh R, Sharon D, Euskirchen G, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148(6):1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JH, Lazarus R, Carey VJ, Raby BA. Quantifying differential gene connectivity between disease states for objective identification of disease-relevant genes. BMC Syst Biol. 2011;5:89. doi: 10.1186/1752-0509-5-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JH, Weiss ST, Carey VJ, Raby BA. A graphical model approach for inferring large-scale networks integrating gene expression and genetic polymorphism. BMC Syst Biol. 2009;3:55. doi: 10.1186/1752-0509-3-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebbari A, Quackenbush J. Seeded Bayesian Networks: constructing genetic networks from microarray data. BMC Syst Biol. 2008;2:57. doi: 10.1186/1752-0509-2-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emily M, Mailund T, Hein J, Schauser L, Schierup MH. Using biological networks to search for interacting loci in genome-wide association studies. Eur J Human Genet. 2009;17(10):1231–1240. doi: 10.1038/ejhg.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Ackert-Bicknell CL, Kell B, Troyanskaya OG, Hibbs MA. Functional genomics complements quantitative genetics in identifying disease-gene associations. PLoS Computat Biol. 2010;6(11):e1000991. doi: 10.1371/journal.pcbi.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG, Frazer KA. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470(7333):264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkley T, Martins J, Chappey C, Haddad M, Stawiski E, Whitcomb JM, Petropoulos CJ, Bonhoeffer S. A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nat Genet. 2011;43(5):487–489. doi: 10.1038/ng.795. [DOI] [PubMed] [Google Scholar]
- Hoischen A, Van Bon BW, Gilissen C, Arts P, Van Lier B, Steehouwer M, De Vries P, De Reuver R, Wieskamp N, Mortier G, Devriendt K, Amorim MZ, Revencu N, Kidd A, Barbosa M, Turner A, Smith J, Oley C, Henderson A, Hayes IM, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42(6):483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407(6804):651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Jia P, Zheng S, Long J, Zheng W, Zhao Z. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics. 2011;27(1):95–102. doi: 10.1093/bioinformatics/btq615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Wolf Y, Karev G. Power Laws, Scale-Free Networks, and Genome Biology. Landes Bioscience; Georgetown, Texas, USA: 2006. [Google Scholar]
- Krumsiek J, Suhre K, Illig T, Adamski J, Theis FJ. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst Biol. 2011;5:21. doi: 10.1186/1752-0509-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470(7333):187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, Barabasi AL, Vidal M, Zoghbi HY. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125(4):801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Loscalzo J. Association studies in an era of too much information: clinical analysis of new biomarker and genetic data. Circulation. 2007;116(17):1866–1870. doi: 10.1161/CIRCULATIONAHA.107.741611. [DOI] [PubMed] [Google Scholar]
- Loscalzo J, Kohane I, Barabasi AL. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363(2):166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, Mccarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AA, Boehnke M, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2011;466(7307):714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH, Dudley AM. Genetics. Systems genetics. Science. 2011;331(6020):1015–1016. doi: 10.1126/science.1203869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naitza S, Porcu E, Steri M, Taub DD, Mulas A, Xiao X, Strait J, Dei M, Lai S, Busonero F, Maschio A, Usala G, Zoledziewska M, Sidore C, Zara I, Pitzalis M, Loi A, Virdis F, Piras R, Deidda F, et al. A genome-wide association scan on the levels of markers of inflammation in Sardinians reveals associations that underpin its complex regulation. PLoS Genet. 2012;8(1):e1002480. doi: 10.1371/journal.pgen.1002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MEJ. Networks: An Introduction. Oxford University Press; New York, New York, USA: 2010. [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, Shaik RS, Waxman AB, Zhang YY, Maron BA, Hartner JC, Fujiwara Y, Orkin SH, Haley KJ, Barabasi AL, Loscalzo J, Chan SY. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation. 2012;125(12):1520–1532. doi: 10.1161/CIRCULATIONAHA.111.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42(7):570–575. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, Yao K, Kehoe SM, Lenz HJ, Haiman CA, Yan C, Henderson BE, Frenkel B, Barretina J, Bass A, Tabernero J, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41(8):882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, Burnett MS, Devaney JM, Knouff CW, Thompson JR, Horne BD, Stewart AF, Assimes TL, Wild PS, Allayee H, Nitschke PL, Patel RS, Martinelli N, Girelli D, Quyyumi AA, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377(9763):383–392. doi: 10.1016/S0140-6736(10)61996-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, Thorsteindottir U, Shin SY, Richards HB, Soranzo N, Ahmadi KR, Lindgren CM, Stefansson K, Dermitzakis ET, Deloukas P, Spector TD, Mccarthy MI. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. 2011;43(6):561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wagele B, Altmaier E, Deloukas P, Erdmann J, Grundberg E, Hammond CJ, De Angelis MH, Kastenmuller G, Kottgen A, Kronenberg F, Mangino M, Meisinger C, Meitinger T, Mewes HW, Milburn MV, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477(7362):54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Chan DW, Gerstein M, Mann M, Omenn GS, Tagle D, Sechi S, Apweiler R, Bader J, Barnes S, Bettauer R, Chan MM, Coon J, Dolinski K, Dowell BL, Edmonds CG, Gaudet S, Gerszten R, Kagan J, Mazurchuk R, et al. The human proteome - a scientific opportunity for transforming diagnostics, therapeutics, and healthcare. Clin Proteomics. 2012;9(1):6. doi: 10.1186/1559-0275-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell. 2011;144(6):986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, Mcevoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, Goddard ME, Visscher PM. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Baron RM, Hardin M, Cho MH, Zielinski J, Hawrylkiewicz I, Sliwinski P, Hersh CP, Mancini JD, Lu K, Thibault D, Donahue AL, Klanderman BJ, Rosner B, Raby BA, Lu Q, Geldart AM, Layne MD, Perrella MA, Weiss ST, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum Mol Genet. 2012;21(6):1325–1335. doi: 10.1093/hmg/ddr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109(4):1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]