

Abstract
It is known that Ras mutations, together with loss of PKC, are apoptotic in various types of mammalian cells. The mechanism of how aberrant Ras transmits this apoptotic signaling remains unclear. Using three V12-Ha-ras loop mutants that preferentially bind to and activate one of Ras effectors, we tested the role of Ras downstream pathways in the induction of apoptosis in rat lung epithelia, human lung or prostate cancer cells. After PKC inhibition, the activation of PI3K/Akt renders the susceptibility of cells to apoptosis. We also demonstrate that the amount of ROS is moderately increased in the cells ectopically expressing V12C40 and dramatically elevated by suppression of PKC, which leads to apoptosis through the activation of UPR. Thus, our study suggests that after PKC abrogation, PI3K functions downstream of Ras to perturb the state of cellular redox and signals to ER stress-regulated apoptotic machinery.
Keywords: apoptosis, Ras, PI3K/Akt, JNK, ROS, ER, UPR
INTRODUCTION
Ras proteins are small guanine nucleotide binding proteins and play an important role in regulating cell proliferation, differentiation, and apoptosis (Barbacid, 1987; Bos, 1989; Wang et al., 2002; Hingorani and Tuveson, 2003). Upon mitogenic stimulation, Ras is transiently activated through a guanine nucleotid exchange mechanism, in which Ras functions as an intrinsic GTPase (Toguski and McCormick, 1993). In the GTP-associated state, Ras interacts with various downstream effectors to promote various biological and physiological activities in cells. JNK (c- Jun kinase) and p38 kinase are stress-related kinases and also regulated by Ras (Woodgett et al., 1996). Studies demonstrated that oncogenic Ras is a key element in human cancer development (Barbacid, 1987; Bos, 1989; Wang et al., 2002; Hingorani and Tuveson, 2003). Despite involved in the regulation of cell growth, it has been shown that oncogenic Ras can initiate apoptosis, depending upon circumstances (Derijard et al., 1994; Fukadawa et al., 1995; Chen and Faller, 1995; Koo et al., 1999; Schwieger et al., 2001; Chen et al., 2001; Xia et al., 2007). For example, abnormal increase of Ras activity rendered Swiss3T3 cells susceptibility to apoptosis (Schwieger et al., 2001). Various types of cells expressing v-ras are able to undergo apoptosis via activation of JNK or PI3K pathway following suppression of protein kinase C (PKC) (Chen and Faller, 1995; Koo et al., 1999; Ma et al., 2002; Xia et al., 2007). Although the effort has been made, the molecular signaling pathways contributing to the induction of apoptosis mediated by Ras have not been fully elucidated. Because Ras effector loop mutant genes, the protein products of which can specifically activate one of effectors, are useful tools for dissecting Ras-mediated, growth-related signaling (Kang and Krauss, 1996; Joneson et al., 1996; Marshall, 1996; Rodriguez-Viciana et al., 1997), we in this study employed these mutant constructs to investigate the apoptotic signaling mediated by Ras.
The ER (endoplasmic reticulum) serves as the site for protein synthesis or processing, and plays a critical role in the maintenance of homeostasis (Aridor and Balch, 1994; Kaufman, 2002). The unfolded protein response (UPR) coordinates with the folding capacity of ER proteins for adapting changes through accelerating the degradation process (McCullough et al., 2001; Li, et al., 2006). Persistent ER stress often activates the apoptotic machinery via the UPR (Kaufman, 2002; Zhang and Kaufman 2003; Schroder and Kaufman, 2005). Activation of oncogenes, including Ha-ras and c-myc, has been shown to be able to trigger the UPR, leading to transformation, premature senescence or apoptosis depending upon experimental settings (Barsyte-Lovejoy, 2004; Benanti and Galloway, 2004). The oxidizing folding of proteins often accompanies with generation of ROS, a persistently high level of which elicits UPR-induced apoptosis (Schroder and Kaufman, 2005). Therefore, it appears that apoptosis mediated by the UPR is a mechanism to preserve cell integrity by eliminating unwanted cells, but also plays a crucial role in some diseases under pathological conditions, such as diabetes (Zhang and Kaufman 2003).
ROS functions as intracellular signal transducers and regulates a numerous biological processes in various types of cells (Adler et al., 1999). In Ras-mediated transformation, moderately increase of ROS play an important role in growth promotion (Irani and Goldschmidt-Clemont, 1998). It has also been reported that ROS and ROS-mediated signaling are able to induce apoptosis in cells expressing v-ras (Liou et al., 2000). In lymphocytes or endothelial cells, activation of PI3K has been reported to cause programmed cell death via upregulating ROS production (Mostefai et al., 2008). GADD153/CHOP is a member of C/EBP (CCAAT/enhancer binding protein) family that can be induced and activated by ER-stress. In ER stress-induced apoptosis, GADD153 is activated by ROS and dimerizes with other C/EBP family members to exercise its function (Ron and Habener, 1992). Although the study has linked the oncogenic Ras-mediated apoptosis to persistent increase of ROS, the mechanism remains unclear.
In this study, to explore the molecular mechanisms of the apoptotic process induced by oncogenic ras under PKC suppression condition, ras effector mutant genes were transiently introduced into rat lung epithelial or human cancer cells to determine which Ras loop mutants confer the susceptibility of the cells to apoptosis. Our data demonstrated that following abrogation of PKC, PI3K/Akt signaling pathway augmented the level of intracellular ROS that in turn signaled to the ER stress-mediated apoptotic machinery via the UPR. Furthermore, concurrent activation of PI3K/Akt and JNK caused a full execution of apoptosis.
MATERIALS AND METHODS
CELL CULTURES AND REAGENTS
Rat lung epithelial RLE and human prostate cancer PC3 cells were purchased from ATCC (American Type Culture Collection, Rockville, MD) and cultured in Dulbecco modified Eagle medium (DMEM) containing 10% newborn calf serum (Gibco, Gaithersburg, MD). Human lung cancer H5810 cells were obtained from Dr. Q. Yu (Boston University School of Medicine) and cultured in the same growth medium indicated above. The retroviral vector MSCV with V12ras, C40ras, G37ras, and S35ras were transiently infected into cells. For the suppression of GADD153, 30 :M of sense or antisense oligos (20 bp of oligos from the start codon) were added into cell cultures for 48 h or longer.
Anti-Ras, Akt, and p-Akt antibodies were purchased from Cell Signaling Technology. Anti-PERK, p-PERK, ATF6, GADD153 and IRE1 antibodies are bought from BD Transduction Laboratories. Other antibodies used in the study were from Santa Cruz Biotechnology. All the inhibitors were purchased from Calbiochem, except KP372-1 (Echelon).
DNA FRAGMENTATION ANALYSIS
A flow cytometric analysis was performed using a FACScan (Becton Dickenson). The data analysis was performed using the Cell-Fit software program (Becton Dickenson). Cell-Fit receives data from the flow cytometer and provides real-time statistical analysis, computed at one second intervals, and also discriminates doublets or adjacent particles. Following treatments, cells were fixed in 70% cold ethanol and stained with 0.1 mg/ml propidium iodide containing 1.5 mg/ml RNase. DNA contents of cells were then tested by a flow cytomer. Cells with sub-G0-G1 DNA contents were counted as apoptotic cells.
ANNEXIN V ASSAY
Following treatments, cells (1 × 106) were washed twice with cold PBS and stained with Annexin V-FITC using the Annexin kit (BD Biosciences) to detect apoptotic cells using a flow cytometer.
PKC ENZYMATIC ASSAY
Following treatments, PKC activity in cell lysates was analyzed using PKC assay kit (Upstate Inc.) that contains a specific substrate peptide for PKC and an inhibitor mixture to block other serine/threonine kinases (such as the inhibitors of PKA and calmodulin-dependent kinases). Subsequently, 32P-incorporating substrates were separated from the residual [32P] ATP using p81 phosphocellulose papers and the radioactivity incorporated into the substrates was measured by a scintillation counter.
IMMUNOBLOTTING ANALYSIS
After treatments, cells were lysed in the lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton-X114, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, containing 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A) on ice for 30 min. The total protein concentrations in the cell lysates were normalized, separated on a 10% SDS-PAGE gel, and visualized by corresponding antibodies.
MEASUREMENT OF RAL ACTIVATION
After the treatments, cells were lysed in a buffer containing 25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Iegpal CA-630, 0.25% sodium deoxycholate, 10% glycerol and 25 mM NaF (Chen et al., 2001). Proteins were normalized to 1 mg/ml, and precipitated by mixing with 20 mg of Ral-RBD overnight at 4° C. The complexes were washed and separated on a 10% SDS-PAGE gel and immunoblotted with an anti-pan-Ras antibody (Oncogene Research Products).
MEASUREMENT OF ROS
Treated or untreated cells were washed with ice-cold PBS and resuspended in 5 μg/ml of 2′, 7′-dichlorodihydrofluorescein diacetate (DCF) (Molecular Probes). The samples were incubated for 10 min at room temperature and analyzed immediately by a flow cytometer.
RESULTS
EFFECT OF RAS DOWNSTREAM PATHWAYS ON APOPTOSIS
JNK functions downstream of Ras to induce apoptosis via activation of caspase 8, the inhibition of JNK only partially blocked apoptosis, which suggests the involvement of other effectors (Chen et al., 2001). To further explore the mechanisms, rat lung epithelial RLE cells were transiently infected with V12ras (V12-Ha-ras) or the effector loop mutant genes. Among these mutants, V12S35Ras is able to bind to Raf; V12C40Ras preferentially activates PI3K; and V12G37Ras interacts with RalGDS only (Rodriguez-Viciana et al., 1997; Kang and Krauss, 1996). The expression level of Ras and the loop mutants in the cells with or without treated with GO6976 (a PKC inhibitor) was determined by Western blot (Fig. 1a). The transfectants expressed high levels of the proteins, which was not altered by GO6976 treatment. The ability of GO6976 to inhibit PKC activity was analyzed using a PKC enzymatic kit (Fig. 1b). A slightly increase in PKC activity was detected in RLE cells after ectopically expressed V12-ras, which might be due to the accommodation of the cells to aberrantly high Ras activity. The addition of GO6976 blocked the ability of PMA (a PKC activator) to upregulate PKC. The activation status of Ras effectors in the cells expressing the loop mutants was also tested (Fig. 1c). The phosphorylated Akt was detected in RLE/V12C40ras cells by the anti-p-Akt antibody. In RLE/V12S35ras cells, ERK1/2 was activated. Consistently, the association of Ral with the RBD-GST fusion protein was only present in the cells expressing V12G37ras.
Figure 1.
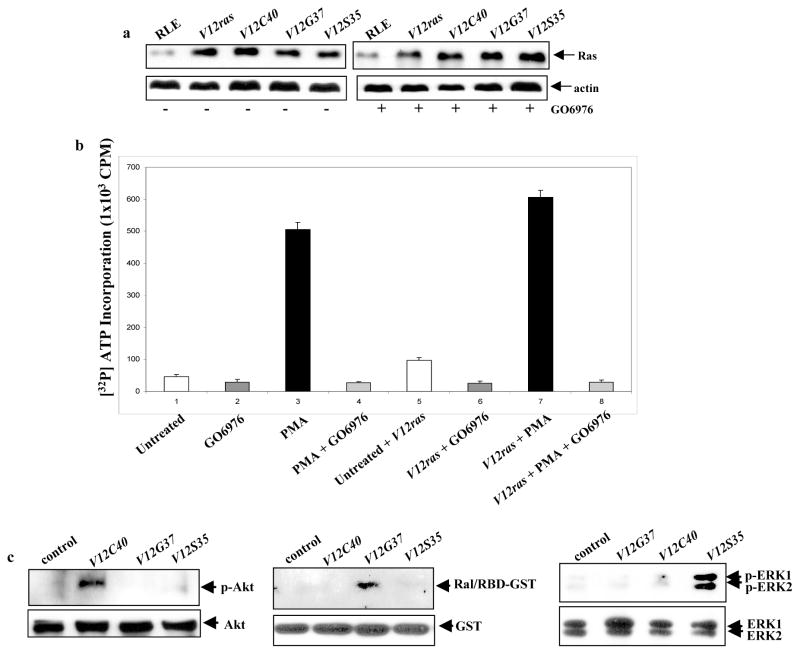
Induction of apoptosis in cells ectopically expressing v-ras or the loop mutant genes. a. After infected with V12ras, V12C40ras, V12G37ras or V12S35ras, RLE cells were treated with 1.0 μM of GO6976 for 24 h. Immunoblotting was performed for the expression of Ras. Equal loading of total proteins was verified by β-actin expression. b. RLE cells, with or without expressing V12ras, were treated with GO6976 (1.0 μM) for 15 min and then PMA (0.1 μM) for 30 min. The PKC activity was then tested by a PKC enzymatic assay. c. The activation of the Ras loop mutants in RLE cells was tested by immunoblotting using the corresponding antibodies or Ral/RBD-GST fusion protein. Equal loading of total proteins was verified by Akt, GST or ERK1/ERK2 expression. c. After treated with GO6976 (1.0 μM for 48h), the percentages of DNA fragmentation in RLE cells was analyzed by a flow cytometer. Error bars represent the standard deviation over five independent experiments. d. The percentage of the cells stained with Annexin V was measured following the same treatment indicated above. Error bars represent the standard deviation over five independent experiments.
To determine which Ras downstream effectors, in response to PKC inhibition, transmits the apoptotic signaling, DNA fragmentation and Annexin V assays were conducted, respectively (Fig. 1d and e). Both assays showed that about 40% of RLE/V12ras cells had fragmented DNA following GO6976 treatment, which was partially blocked by wortmannin (a PI3K inhibitor), KP372-1 (an Akt inhibitor) and JNK inhibitor, indicating the involvement of these effectors in this process. The addition of GO6976 caused about 20% of RLE/JNK1 cells to undergo apoptosis, which is consistent with our previous observation (Chen et al., 2001). A similar percentage of RLE/V12C40 cells were apoptotic following GO6976 treatment, which was completely suppressed by the addition of wortmannin or KP327-1, but not by JNK inhibitor. It appears that PI3K/Akt and JNK function separately for the induction of apoptosis. The suppression of PKC did not induce apoptosis in RLE/V12G37ras or RLE/V12S35ras cells.
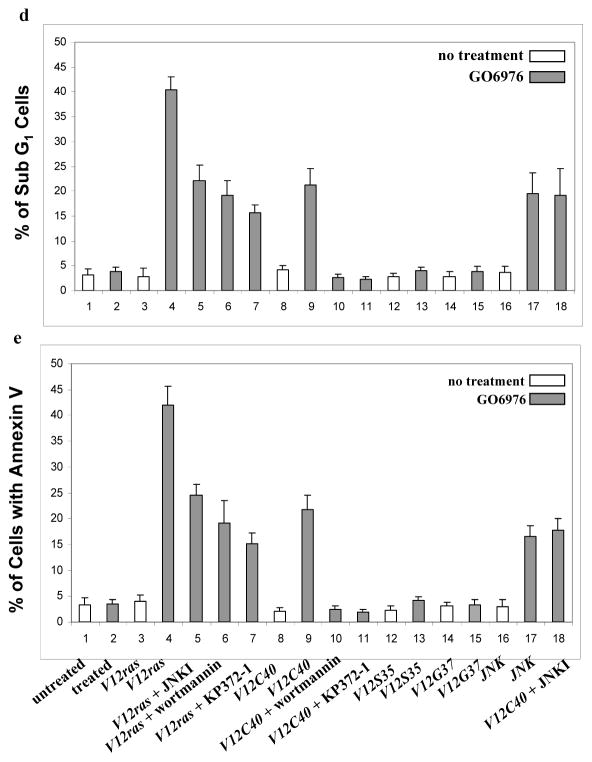
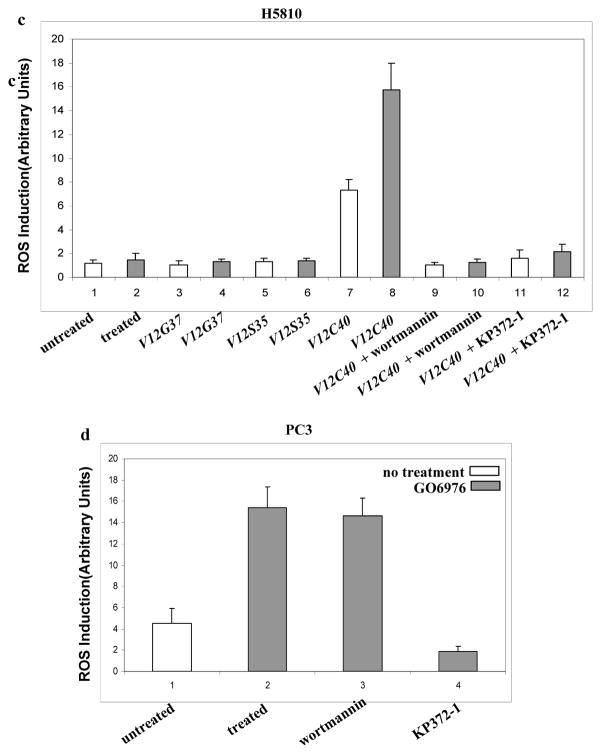
To further determine the role of Ras effectors in the induction of apoptosis, human lung (H5810) or prostate (PC3) cancer cells were used. After introduced v-ras and the mutant genes, a high amount of Ras and its mutant proteins was detected in H5810 cells (Figs. 2a and b). DNA fragmentation assay showed that H5810/v-ras were highly susceptible to apoptosis following PKC suppression, while the same treatment had no role on parental cells (Fig. 2c). Consistently, about 20% of H5810/V12C40 cells had fragmented DNA in response to GO6976 treatment, which was blocked by wortmannin or KP372-1. Apoptosis did not occur in the lung cancer cells expressing other loop mutant genes.
Figure 2.
Induction of apoptosis in human lung H5810 or prostate PC3 cancer cells. a. After infected H5810 cells with V12ras, immunoblotting was performed to detect Ras expression. Equal loading of total proteins was verified by β-actin expression. b. ras loop mutant genes were infected into H5810 cells and the expressions were tested by immunoblotting. Equal loading of total proteins was verified by β-actin expression. c. H5810 cells expressing V12ras, and the loop mutant genes were treated with wortmannin (1.0 μM) or KP372-1 (1.0 μM) for 15 min, prior adding GO6976 (1.0 μM for 48h). Subsequently, DNA fragmentation assay was conducted. Error bars represent the standard deviation over five independent experiments. d. PC3 or prostate epithelial PrEC cells were treated with wortmannin (1.0 μM) or KP372-1 (1.0 μM) for 15 min, prior adding GO6976 (1.0 μM, 48h). Subsequently, DNA fragmentation assay was conducted. Error bars represent the standard deviation over five independent experiments.
It is known that human prostate cancer PC3 cells harbor an activated Akt (Majumder and Sellers, 2005). To further explore the role of Akt signaling in the induction of apoptosis, DNA fragmentation was examined in PC3 cells (Fig. 2d). GO6976 treatment had no effect on normal human prostate epithelial PrEC cells, but a similar percentage of PC3 cells, like H5810/V12C40 cells, underwent apoptosis, which was blocked by KP372-1, but not by wortmannin. The data suggest that PI3K and Akt are key effectors in the induction of apoptosis following PKC suppression, and Akt functions downstream of PI3K.
ACTIVATION OF PI3K/Akt, AFTER PKC INHIBITION, PERTURBS THE CELLULAR REDOX STATE
ROS has been shown to be required for cell growth and however, a persistently high amount of ROS is able to induce apoptosis (Adler et al., 1999). Furthermore, oncogenic Ras causes a slight elevation of ROS in cells for growth promotion (Irani and Goldschmidt-Clemont, 1998). ROS production was tested in our experimental setting (Figs. 3a and b). The amount of ROS was moderately increased in RLE/V12ras or RLE/V12C40 cells, but dramatically elevated after the treatment with GO6976, which is similar to that induced by H2O2. GO6976-mediated ROS production in RLE/V12ras or RLE/V12C40 cells was suppressed by the addition of wortmannin, KP372-1 or NAC (a ROS inhibitor), respectively.
Figure 3.
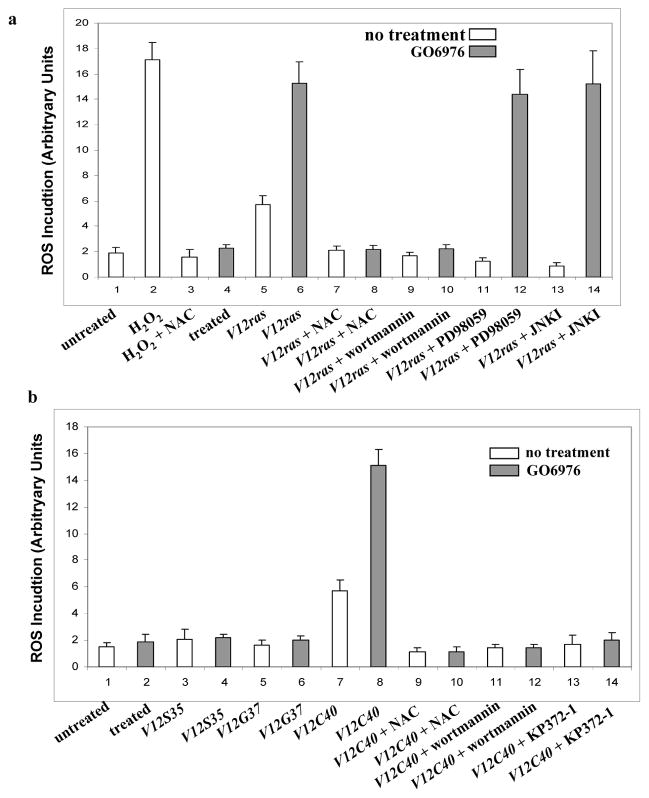
Perturbation of the redox state in the cells expressing V12ras or V12C40ras. a–d. The cells expressing ras or its loop mutant genes were treated with NAC (5.0 mM), wortmannin (1.0 μM), PD98059 (10 μM), JNK inhibitor (5.0 μM) or KP372-1 (1.0 μM) for 15 min, prior adding GO6976. Subsequently, the cells were stained with DCF, and analyzed for ROS levels using a flow cytometer. Error bars represent the standard deviation over five independent experiments.
ROS induction was also tested in human lung H5810 or prostate PC3 cancer cells (Fig. 3c and d). Consistently, the production was upregulated in H5810/ V12C40 or PC3 cells after treated with GO6976, but not altered in the cells expressing other loop mutant genes. Wortmannin or KP372-1 blocked ROS induction. Overall, the data suggest that downregulation of PKC has additive effect on Ras-mediated ROS accumulation and PI3K/Akt signaling is key event in this process, which underscores the importance of Ras in the regulation of oxidation. It also indicates the importance of PKC in maintaining the equilibrium of the intracellular redox in cells with hyperactive Ras.
PI3K TRIGGERS UPR IN RESPONSE TO PKC DOWNREGULATION
The activation of Ras downstream effectors (such as PI3K) has been reported to cause ER stress via the activation of ROS (Ron and Habener, 1992; Denoyells et al., 2006). Persistent ER stress activates the UPR, which often switches on the apoptotic machinery (Mostefai et al., 2008). PERK is an ER transmembrane kinase that becomes phosphorylated in response to ER stress. BiP functions as an adaptor to sequester the inactive forms of UPR sensors (such as PERK, ATF6 or IRE1) that dissociate from BiP under the condition of ER stress (Ron, 2006). To test whether the UPR was activated in our experimental setting, the status of PERK in the cells was tested by immunoblotting using the ant-p-PERK antibody (Fig. 4a). The protein was not phosphorylated in untreated cells. However, phosphorylated PERK, after PKC suppression, was seen in RLE/V12ras or RLE/V12C40 cells, but not in the cells expressing other loop mutant genes. The involvement of PI3K and Akt in PERK activation was also tested in RLE/V12ras cells. The phosphorylation of the protein, after the treatment with GO6976, was blocked by wortamannin or KP372-1, but not by PD98059 or JNK inhibitor (Fig. 4b). Since BiP is dissociated from UPR sensors in response to ER stress, the expression level of unbound BiP, after treated with GO6976, was examined (Fig. 4c). After immunoprecipitated with anti-PERK, ATF6 and IRE1 antibodies, the immunocomplexes were taken away and the supernatants were blotted with an anti-BiP antibody. A very low amount of unbounded BiP was detected in the supernatants collected from untreated cells. In comparison, a high amount of the protein was present in the supernatants collected from RLE/V12ras or RLE/V12C40 cells. These results indicate that ER stress occurs in RLE/V12ras, RLE/V12C40 and PC3 cells after PKC inhibition, in which Ras, via PI3K/Akt pathway, activates the UPR.
Figure 4.
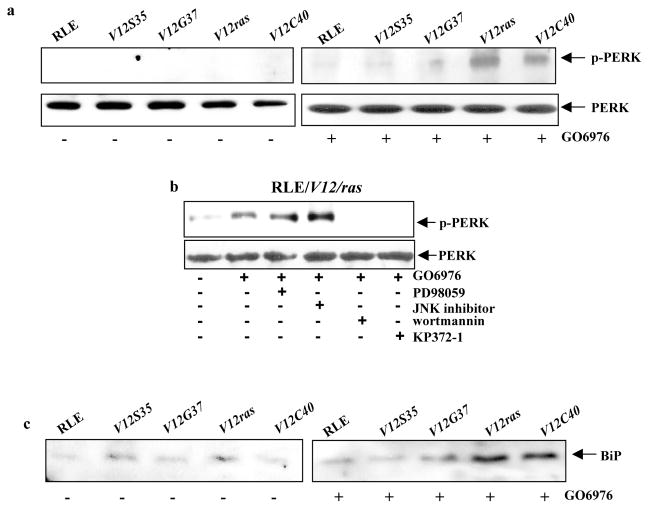
Activation of ER stress regulators following downregulation of PKC. a. After treated with GO6976, the expression of p-PERK in the cells ectopically expressing V12ras or the loop mutant genes was examined by immunoblotting. Equal loading of total proteins was verified by PERK expression. b. Cells were treated with various inhibitors for 15 min prior to adding GO6976 and then immunoblotted for p-PERK. Equal loading of total proteins was verified by PERK expression. c. Following PKC suppression, RLE cells expressing v-ras or the loop mutant genes were immunoprecipitated with anti- PERK, ATF6 or IRE1 antibodies. After took out the immuno-complexes, equal amount of the supernatants were immunoblotted for the expression of BiP.
PI3K-MEDIATED UPR LEADS TO APOPTOSIS VIA UPREGULATING GADD153
GADD153 is an ER stress-inducible protein and has been shown to participate in the induction of UPR-mediated apoptosis (Ron and Habener, 1992; Ron, 2006). This led us to test if GADD153 was involved in the apoptotic process observed here. Immunoblotting analysis was performed following GO6976 treatment in the cells expressing V12ras or the loop mutant genes (Fig. 5a). A baseline expression of GADD153 was observed in untreated cells. After treated with GO6976, the level of the protein was increased in RLE/V12ras or RLE/V12C40 cells, but not in other transfectants. The effect of PI3K or Akt on GADD153 induction during the apoptotic process was also examined in RLE/V12ras cells. The addition of wortamannin or KP372-1, but not by of PD98059 or JNK inhibitor, suppressed the expression of GADD153 following PKC downregulation (Fig. 5b). Subsequently, the role of GADD153 in the induction of apoptosis was tested. The antisense oligos was able to block GADD153 expression (Fig. 5c). The effect of the oligos on the induction of apoptosis was then tested (Figs. 5d). The addition of GADD153 sense oligo had no effect on apoptosis triggered by PKC suppression in the cells. On the contrary, the antisense oligo partially blocked the apoptotic process in RLE/V12ras cells and completely suppressed apoptosis in RLE/V12C40 cells. A similar blocking effect was observed after treated H5810/V12ras, H5810/V12C40 or PC3 cells with the antisense oligos (data not shown).
Figure 5.
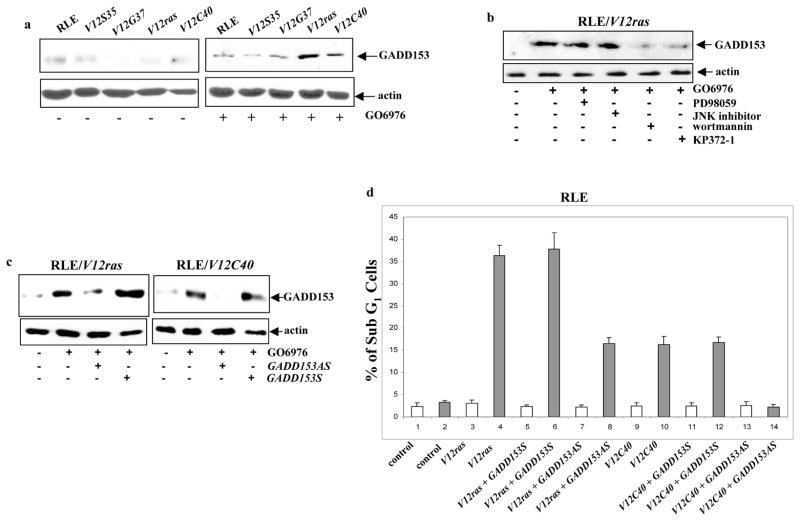
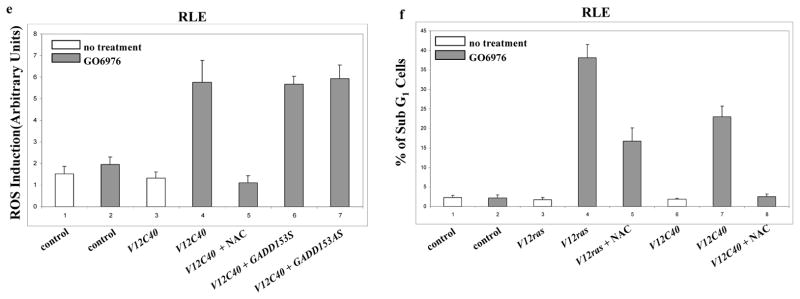
Role of GADD153 in the induction of apoptosis. a. After suppression of PKC, Immunoblotting was performed to test the expression of GADD153 in the cells. Equal loading of total proteins was verified by β-actin expression. b. Cells were treated with various inhibitors for 15 min prior to adding GO6976 and then immunoblotted for the expression of GADD153. Equal loading of total proteins was verified by β-actin expression. c. RLE cells expressing either V12ras or V12C40 were treated with GO6976 in the presence of GADD153 sense or antisense oligo. Subsequently, the expression of GADD153 was tested by immunoblotting. Equal loading of total proteins was verified by β-actin expression. d. After added GADD153 sense or antisense oligo, RLE cells expressing ras or the loop mutant genes were treated with GO6976. Subsequently, DNA fragmentation assay was performed. Error bars represent the standard deviation over 5 independent experiments. e. In the presence or absence of GADD153 sense or antisense oligos, the production of ROS in RLE/V12C40 cells was measured. Error bars represent the standard deviation over five independent experiments. f. The effect of NAC on the apoptotic process in RLE/V12C40 cells was measured by DNA fragmentation assay. Error bars represent the standard deviation over five independent experiments.
To determine whether GADD153 is downstream of ROS in this apoptotic process, RLE cells ectopically expressing V12ras or the loop mutant genes were cultured in the presence of GADD153 sense or antisense oligos for 48 h, and the levels of ROS in the cells were measured after GO6976 treatment (Fig. 5e). The increase level of ROS in GO6976 treated RLE/V12C40 cells was inhibited by NAC, but was not affected by the addition of GADD153 sense or antisense oligo, indicating that GADD153 is probably downstream of ROS. The effect of the suppression of ROS on the induction of apoptosis was also examined (Fig. 5f). The addition of NAC partially blocked the cell death process in RLE/V12ras cells and completely suppressed apoptosis in RLE/V12C40 cells. The similar results were obtained from the lung cancer cells expressing V12ras or V12C40 and PC3 cells (data not shown). Together, these data suggest that after suppression of PKC, PI3K/Akt functions downstream of Ras to accelerate ROS production and further trigger UPR activation, leading to apoptosis via upregulating GADD153 (Fig. 6).
Figure 6.
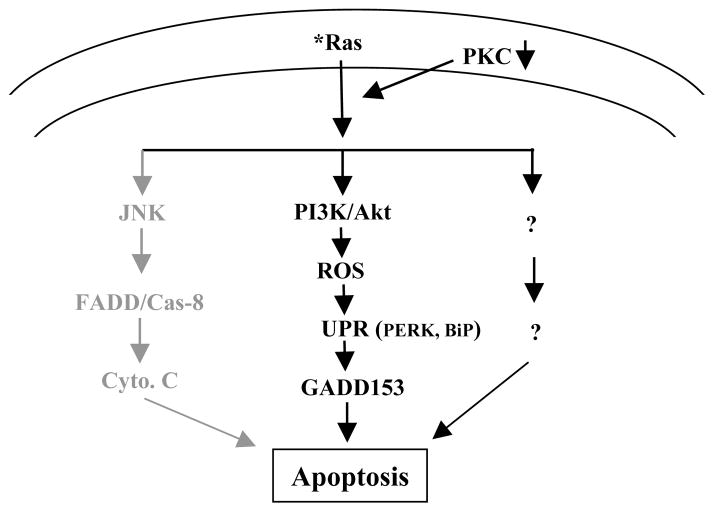
Stochastic signaling pathways regulated by oncogenic Ras in the induction of apoptosis. The pathway governed by JNK in grey color was reported previously. The signal pathway regulated by PI3K/Akt in dark color is presented in this study.
DISCUSSION
It has been demonstrated that oncogenic Ras is able to induce apoptosis once endogenous PKC activity is suppressed (Chen and Faller, 1995; Koo et al., 1999; Schwieger et al., 2001; Xia et al., 2007). Although studies showed that JNK participates in apoptosis induced by mutated Ras under the condition of loss of PKC, the genetic or chemical inhibitors of JNK only partially protect cells from undergoing apoptosis, indicating the involvement of other effectors (Chen et al., 2001). Our current study, using ras loop mutant genes, demonstrated the role of PI3K in the induction of apoptosis. In this apoptotic process, PI3K/Akt perturbs the intracellular redox equilibration following PKC suppression and further induces ER stress, leading to apoptosis via activating the UPR (Fig. 6). Our study also suggests that PKC is crucial for cells to cope with hyperactive Ras for the maintenance of homeostasis and further survival promotion.
It is known that ectopic expression of oncogenic ras in mammalian cell lines triggers a wide spectrum of cellular responses, leading to transformation. These transformation-related responses are responsible for the oncogenic role of Ras in the development of human malignancy. However, the ability of Ras to induce apoptosis under certain circumstances has drawn a lot attention for its therapeutic potential. Ras has been shown to be able to bind to multiple effector proteins and subsequently activate distinct downstream signaling pathways (Kang and Krauss, 1996; Rodriguez-Viciana et al., 1997). The effector loop mutants of Ras that are defective for interacting with specific effectors while remaining competent for binding to and activating other downstream signaling transducers have been widely used to study Ras-mediated cell cycle progression, proliferation and transformation (Barbacid, 1987; Bos, 1989; Wang et al., 2002; Hingorani and Tuveson, 2003). Recently, although JNK or PI3K has been shown to function downstream of Ras for the induction of apoptosis under the condition of PKC suppression in different types of cells (Xia et al., 2007; Chen et al., 2001), the mechanism of which remains unknown. Therefore, we exploited the effector loop mutants and assessed the contribution of each Ras pathway for the induction of apoptosis after PKC inhibition. Our study identified that PI3K/Akt function as apoptotic factors in this process by elevating ROS and further activating ER stress-mediated UPR. It is well known that one of the functions of the ER is to serve as the surveillance machinery that ensures proteins to be timely and properly modified, folded or degraded. Once overwhelming mis-folded proteins are accumulated, the UPR is activated to either adapt changes through adjusting protein degradation-related machineries, or switch on cell death program to eliminate unwanted cells. Because it has been suggested that protein folding and ROS production are closely interconnected, it is possible that ROS, as a byproduct of a hyperactive Ras, is further accumulated by loss of PKC in our experimental setting, which triggers the UPR to induce apoptosis.
PI3K converts phosphatidylinositol 4, 5-bisphosphate to phosphatidylinositol 3, 4, 5-trisphosphate, which in turn recruits and activates a variety of regulatory proteins through associations with their pleckstrin and phox homology domains (Ron, 2006). Studies demonstrated that NADPH oxidase regulates ROS production via converting phosphatidylinositol 4, 5-bisphosphate to phosphatidylinositol 3, 4, 5-trisphosphate (Frey et al., 2006). It has been observed that the inhibition of PI3K/Akt dramatically desensitized different murine fibroblasts overexpressing v-ras to apoptosis after downregulation of PKC (Xia et al., 2007). In the present study, using various Ras loop mutant genes, we further elucidated the molecular signaling pathway governed by PI3K/Akt in the induction of apoptosis following PKC inhibition. Since PI3K and Akt are often activated in a variety of human tumors (such as advanced prostate cancer) (Barbacid, 1987; Bos, 1989; Wang et al., 2002; Hingorani and Tuveson, 2003), the results from current study will facilitate better designs to target these molecules for development of new therapeutics.
ROS have been demonstrated to act as intracellular signal transducers of growth factors. In response to abnormally persistent ligations of growth factor receptors, a moderate increase of intracellular ROS was induced, which is necessary for cellular transformation by altering the structure of the cytoskeleton and further inducing a transformed phenotype in a Rac-depependent fashion (Irani and Goldschmidt-Clemont, 1998). In PC12 cells, Ras has been shown to upregulate ROS production after epidermal growth factor stimulation (Mills et al., 1998). Furthermore, studies demonstrated that oncogenes (such as myc or ras), through perturbing the state of intracellular redox, are able to cause DNA single strand breaks and subsequently disrupt genetic integrity to promote cell transformation (Khwaja and Tatton, 1999). Despite regulating cell proliferation and transformation, a high level of ROS has also been suggested to play an obligatory role in apoptosis induced by various apoptotic stimuli. In TNFα-induced programmed cell death, NF-κB and JNK cooperate to stimulate ROS production that leads to caspase cascade and mitochondrial depolarization (Khwaja and Tatton, 1999). Our data demonstrated that although ROS production is elevated in cells expressing v-ras under normal growth conditions, inhibition of PKC severely disrupts the equilibrium of the redox state. It indicates that PKC is an important factor for cells harboring an aberrant Ras to survive.
ER stress is induced by alternations in proteins and lipid metabolism, which causes the accumulation of unfolded or misfolded proteins (McCullough et al., 2001; Li et al., 2006). The activation of ER stress programmes, such as the UPR, can elicit cytoprotective or cytotoxic reactions, depending upon stimuli, cell types and cellular contexts. Our study demonstrated that PI3K and Akt function as intermediators to connect Ras signaling to the ER. By perturbing the status of redox, it appears that activated PI3K or Akt, upon PKC suppression, utilizes genetic or epigenetic mechanisms (such as the UPR) that surpass the buffering capacity of the ER to trigger a suicide program in the cells.
Taken together, the results of our study provide one of the molecular pathways indicating how mutated Ras utilizes its downstream effectors to activate the cell death program under the condition of loss of PKC. This apoptotic action requires the collaboration of multiple effectors. Since many human cancers contain aberrant Ras or PI3K/Akt, the concept to target oncogenic Ras or its effectors for the development of new cancer therapeutics is very tempting. Thus, a better understanding of the mechanism of each downstream signaling pathway for the induction of apoptosis becomes undoubtedly important.
Acknowledgments
We thank Drs. Z. Luo and J. Xiao (Boston University School of Medicine, Boston, MA) for various constructs and reagents. This study is supported by NIH (RO1CA100498) and DOD (W81XWH-04-1-0246).
References
- Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6084–6086. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- Aridor M, Balch WE. Integration of endoplasmic reticulum signaling in health and disease. Nat Med. 1999;5:745–751. doi: 10.1038/10466. [DOI] [PubMed] [Google Scholar]
- Barbacid M. Ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- Barsyte-Lovejoy D, Mao DY, Penn LZ. c-Myc represses the proximal promoters of GADD45a and GADD153 by a post-RNA polymerase II recruitment mechanism. Oncogene. 2004;23:3481–3486. doi: 10.1038/sj.onc.1207487. [DOI] [PubMed] [Google Scholar]
- Benanti JA, Galloway DA. The normal response to Ras: senescentce or transformation? Mole Cell Biol. 2004;24:2842–2852. doi: 10.1128/MCB.24.7.2842-2852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–645. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- Chen CY, Faller DV. Direction of Ras-generated signals towards cell growth or apoptosis is determined by protein kinase C and Bcl-2. Oncogene. 1995;20:1487–1498. [PubMed] [Google Scholar]
- Chen CY, Juo P, Liou J, Yu Q, Blenis J, Faller DV. The recruitment of Fas-associated death domain/caspase-8 in Ras-induced apoptosis. Cell Growth Differ. 2001;12:297–306. [PubMed] [Google Scholar]
- Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR. Anti-oncotenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053–1063. doi: 10.1038/ncb1471. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T. JNK1: a protein kinase stimulated by UV light and Ha-ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Frey RS, Gao X, Javaid K, Siddiqui SS, Rahman A, Malik AB. Phosphatidylinositol 3-kinase gama signaling through protein kinase C theta induces NADPH oxidase-mediated oxidant generation and NF-kB activation in endothelial cells. J Biol Chem. 2006;281:16128–16138. doi: 10.1074/jbc.M508810200. [DOI] [PubMed] [Google Scholar]
- Fukadawa K, Rulong S, Resau J, Pinto S, Woude GF. Overexpression of mos oncogene product in Swiss 3T3 cells induces apoptosis preferentially during S-phase. Oncogene. 1995;10:1–8. [PubMed] [Google Scholar]
- Hingorani SR, Tuveson DA. Ras redux: rethinking how and where Ras acts. Curr Opin Genet Dev. 2003;13:6–13. doi: 10.1016/s0959-437x(02)00017-5. [DOI] [PubMed] [Google Scholar]
- Irani K, Goldschmidt-Clemont PJ. Ras, superoxide and signal transduction. Biochem Pharmacol. 1998;55:1339–1346. doi: 10.1016/s0006-2952(97)00616-3. [DOI] [PubMed] [Google Scholar]
- Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- Kang J-S, Krauss RS. Ras induces anchorage-independent growth by subverting multiple adhesion-regulated cell cycle events. Mole Cell Biol. 1996;16:3370–3380. doi: 10.1128/mcb.16.7.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A, Tatton L. Resistance to the cytotoxic effects of tumor necrosis factor alpha can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J Biol Chem. 1999;274:36817–36823. doi: 10.1074/jbc.274.51.36817. [DOI] [PubMed] [Google Scholar]
- Koo HM, Gray-Goodrich M, Kohlhagen G, McWilliams MJ, Jeffers M, Vaigro-Wolff A. The ras oncogene-mediated sensitization of human cells to topoisomerase II inhibitor-induced apoptosis. J Natl Cancer. 1999;91:236–244. doi: 10.1093/jnci/91.3.236. [DOI] [PubMed] [Google Scholar]
- Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and MOXA by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- Liou JS, Chen CY, Chen JS, Faller DV. Oncogenic Ras mediates apoptosis in response to protein kinase C inhibition through the generation of reactive oxygen species. J Biol Chem. 2000;275:39001–39011. doi: 10.1074/jbc.M007154200. [DOI] [PubMed] [Google Scholar]
- Ma P, Magut M, Chen X, Chen CY. p53 is necessary for the apoptotic response mediated by a transient increase of Ras activity. Mole Cell Biol. 2002;22:2928–2938. doi: 10.1128/MCB.22.9.2928-2938.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder PJ, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465–7474. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz L-O, Aw T-Y, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl-2 and perturbing the cellular redox state. Mole Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EM, Takeda K, Yu ZX, Ferrans V, Katagiri Y, Jiang H. Nerve growth factor treatment prevents the increase in superoxide produce by epidermal growth factor in PC12 cells. J Biol Chem. 1998;273:22165–22168. doi: 10.1074/jbc.273.35.22165. [DOI] [PubMed] [Google Scholar]
- Mostefai HA, Agouni A, Carusio N, Mastronardi ML, Heymes C, Henrion D. Phosphatidylinositol-3-kinase and xanthine oxidase regulate nitric oxide and reactive oxygen species productions by apoptotic lymphocyte microparticles in endothelial cells. J Immunol. 2008;180:5028–5035. doi: 10.4049/jimmunol.180.7.5028. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Ron D. Cell biology. Stressed cells cope with protein overload. Science. 2006;313:52–53. doi: 10.1126/science.1130469. [DOI] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Gene Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Schwieger A, Bauer L, Hanusch J, Sers C, Schafer R, Bauer G. Ras oncogene expression determines sensitivity for intercellular induction of apoptosis. Carcinogenesis. 2001;22:1385–1392. doi: 10.1093/carcin/22.9.1385. [DOI] [PubMed] [Google Scholar]
- Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, Sun P. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mole Cell Biol. 2002;22:3389–3403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR, Avruch J, Kyriakis J. The stress activated protein kinase pathway. Cancer Surv. 1996;27:127–138. [PubMed] [Google Scholar]
- Xia S, Forman LW, Faller DV. Protein kinase Cδ is required for survival of cells expressing activated p21Ras. J. Biol. Chem. 2007;282:13199–13210. doi: 10.1074/jbc.M610225200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Unfolding the toxicity of cholesterol. Nat Cell Biol. 2003;5:769–770. doi: 10.1038/ncb0903-769. [DOI] [PubMed] [Google Scholar]