

Abstract
Pancreatic ductal adenocarcinoma (a.k.a. pancreatic cancer) remains one of the most feared and clinically challenging diseases to treat despite continuous improvements in therapies. The genetic landscape of pancreatic cancer demonstrates near ubiquitous activating mutations of KRAS, and recurrent inactivating mutations of CDKN2A, SMAD4 and TP53. To date, attempts to develop agents to target KRAS to specifically kill cancer cells has been disappointing. In this regard, an understanding of cellular metabolic derangements in pancreatic cancer could lead to novel therapeutic approaches. Like other cancers, pancreatic cancer cells rely on fuel sources for homeostasis and proliferation; as such, interrupting the use of two major nutrients, glucose and glutamine, may provide new therapeutic avenues. In addition, KRAS-mutant pancreatic cancers have been documented to depend on autophagy, and the inhibition of autophagy in the preclinical setting has shown promise. Herein, the conceptual framework for blocking the pancreatic fuel supply is reviewed.
Introduction
While inherited and acquired mutations are believed to cause pancreatic cancer (see review by Iacobuzio-Donahue et al in this issue 1), common mutations in canonical oncogenes, such as AKT, MYC, PI3K, and RAS, and tumor suppressors, including TP53 and PTEN also alter cancer metabolism to enable cancer cells to survive and proliferate in the hypoxic and nutrient deprived tumor microenvironment (see review by Feig et al in this issue 2)3-10. Oncogenes and tumor suppressors alter metabolism through transcriptional and post-transcriptional mechanisms. Mutations in genes encoding metaboIic enzymes, such as succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase (IDH) have also been linked to tumorigenesis, underscoring the intimate connections between metabolism and cancer11, 12. oss of function SDH or FH mutations result in accumulation of precursors such as succinate or fumarate. Mutated IDH1 or IDH2, on the other hand, are neomorphic enzymes that reverse the chemical reaction to produce 2-hydroxyglutarate from α-ketoglutarate 13. These accumulated metabolic intermediates all have the ability to inhibit α-ketoglutarate-dependent dioxgenases, which are involved in histone or DNA demethylation and in prolyl hydroxylases that modulate the hypoxia inducible factors (HIF)14,15, 16. In this regard, mutations in enzymes cause epigenetic deregulation that contributes to tumorigenesis.
In order to survive and proliferate under adverse tumor microenvironment with limited nutrients and oxygen, cancer cells must rely on their ability to reprogram canonical biochemical pathways to provide the necessary bioenergetics and precursors of proteins, nucleic acids and membrane lipids 17-19 (Figure 1). In addition to rewiring metabolism, tumor cells can also activate autophagy, a process that permits recycling of cellular constituents as internal fuel sources when external nutrient supplies are limited20-22. While nutrient rich conditions inhibit autophagy through activation of mTOR, nutrient-deprived conditions decreases ATP production, resulting in activated AMPK that stimulates autophagy 23, 24 (Figure 1). As such, cancer cells have multiple mechanisms for metabolic adaptation to the tumor microenvironment.
Figure 1.
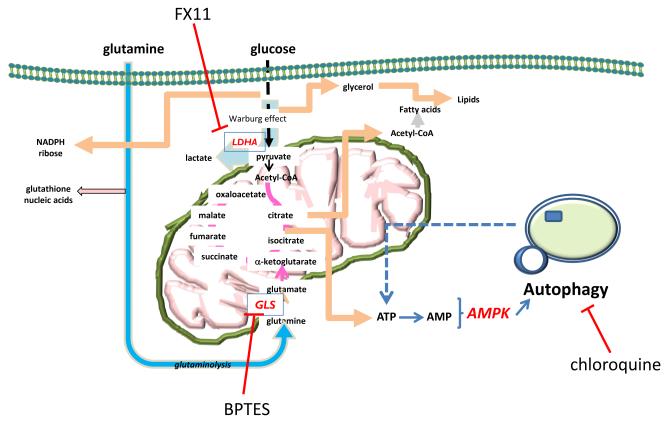
Diagram depicting the metabolism of glucose and glutamine via glycolysis and the tricarboxylic acid cycle. Both substrates contribute to the production of ATP, which when depleted would activate AMP kinase (AMPK) that in turn triggers autophagy. Autophagy increases lysosomal recycling of cellular constituents to recover ATP. LDHA; lactate dehydrogenase. GLS; glutaminase.
Proliferating cancer cells transport glucose and glutamine into the cell as major nutrient sources for the production of ATP and building blocks for macromolecular synthesis 3, 4, 6 (Figure 1). The mitochondrion serves not only as the cellular powerhouse, producing ATP efficiently from glucose and glutamine, but it is also a hub for the production of key intermediates involved in nucleic acid, fatty acid, and heme synthesis. The proliferating cancer cells, hence, also produce toxic by-products that must be eliminated or extruded for cell survival. Reactive oxygen species from the mitochondrion are neutralized; superoxide is converted by SOD to hydrogen peroxide, which is neutralized by catalase via its conversion to water and oxygen25-27. Lactate and carbon dioxide, produced from glucose and glutamine catabolism, are exported through monocarboxylate transporters or neutralized and extruded by carbonic anhydrase 28. Although this review focuses on blocking cancer fuel supply as a promising new approach to pancreatic cancer therapy, therapeutic opportunities may also exist in blocking the exhaust pipes which eliminate metabolic toxic by-products from cancer cells.
Characteristic features of metabolism in cancers
Aberrant metabolism is now considered one of the hallmarks of cancer 29. As a result of genetic alterations and tumor hypoxia, cancer cells reprogram metabolism to meet increased energy demand for enhanced anabolism, cell proliferation, and protection from oxidative damage and cell death signals. The molecular underpinnings for the reprogrammed metabolism of cancer cells have been linked to activation of oncogenes or loss of tumor suppressors, which can function independently of, or through constitutive stabilization of the hypoxia inducible factor, HIF-1α. Activated Ras and AKT oncogenes can increase glycolysis, enhancing the conversion of glucose to lactate 3. The MYC oncogene, which encodes the transcription factor Myc, increases the expression of glycolytic genes, thereby enhancing glycolysis and production of lactate.
Glycolysis is a multi-enzymatic pathway that catabolizes glucose to pyruvate via over a dozen different enzymes with several apparent rate-limiting steps. Phosphofructose kinase provides the first rate-limiting step where fructose-1-phosphate, derived from glucose, is converted to fructose-1,6-bisphosphate. The other rate-limiting step is the conversion of phosphoenolpyruvate to pyruvate by pyruvate kinase (muscle form) PKM. Recent studies have raised an intense interest in the embryonic M2 isoform of pyruvate kinase (PKM2), which is highly expressed in human tumors. A study by Gao et al.30 showed that dimeric, not tetrameric PKM2 which localizes the cellular nucleus, activates transcription of MEK5 by phosphorylating stat3 at Y705 using PEP as a phosphate donor. This protein kinase activity of PKM2 plays a role in promoting cell proliferation, revealing an important link between metabolism alteration and gene expression during tumor transformation and progression. The xenografted adenocarcinoma cells, which carry a mutant form of PKM2 that preferentially forms dimers, grow more rapidly and appear more aggressive than cells carrying wild-type PKM2. Although the dynamics of PKM2 oligomeric states and their roles in cancer remain confusing, these studies suggest thatperturbation of glycolysis via manipulation of PKM2 could have a significant therapeutic effect.
Alteration of glucose metabolism in cancer is known as aerobic glycolysis or the Warburg effect3,5,31, which describes the ability of cancer cells to avidly metabolize glucose to lactate, even in the presence of oxygen. A manifestation of the Warburg effect is the increased 18F-fluorodeoxyglucose (FDG) import by pancreatic cancers as determined by positron emission tomography (PET) scans. However, only about 50% of pancreatic cancers have positive clinical PET, suggesting additional complexities and the possibility that pancreatic cancers could use other fuels or resort to other metabolic survival modes 32, 33.
In addition to glucose, proliferating cancer cells also rely on glutamine as a major source of energy and building blocks. In fact, MYC, which is frequently amplified or over-expressed in pancreatic cancer 34, has been mechanistically linked to the regulation of glutamine metabolism 35. In this regard, it is reasonable to surmise that PET negative pancreatic cancers may use glutamine as a major nutrient source.
Applying metabolomics technologies, with the use of NMR and mass spectrometer-based Stable Isotope Resolved Metabolomics (SIRM) with 13C labeled glucose and glutamine, a study by Le et al. 36 documents that cancer cells use either glucose or glutamine, depending on the availability of the nutrients. This flexibility of cancer metabolism enables cancer cells to proliferate and survive even under the hypoxic and nutrient deprived conditions which are often encountered in the tumor microenvironment. Moreover, in hypoxia, we observed the enhanced conversion of glutamine to glutathione, an important reducing agent for control of the accumulation of mitochondrial reduced oxygen species (ROS).Most importantly, this study uncovered a previously unsuspected glucose-independent glutamine-driven TCA cycle. Cancer cells subjected to glucose deficiency and/or hypoxia would benefit from such glucose-independent TCA cycle activity in the tumor microenvironment. Cell growth and survival can be sustained by glutamine metabolism alone. In addition to the links between oncogenes and tumor suppressors to altered cancer cell metabolism, mutations in specific tricarboxylic acid cycle enzymes contribute to tumorigenesis of familial or spontaneously acquired cancers. The study by Mullen et al. 37 uncovered other metabolic reprogrammed pathways in cancer cells which have mutations in complex I or complex III of the electron transport chain (ETC). These are often found in patient-derived renal carcinoma cells with mutations in FH, and also in pharmacologically ETC-inhibited cells with normal mitochondria.These cancer cells use glutamine-dependent reductive carboxylation generating acetyl-CoA for lipid synthesis. These processes use mitochondrial and cytosolic isoforms of NADP+/NADPH-dependent isocitrate dehydrogenase. The ETC-deficient cancer cells, like fumarate hydratase (FH) mutations cannot grow without glutamine. Hence, depending on the genetic makeup, a cancer cell could be distinctively addicted to glucose or glutamine.
Given that pancreatic cancers uniquely display increased levels of fibrotic stroma, the pancreatic cancer cell environment could be highly nutrient deficient 38, 39. In this nutrient-deprived state, increased AMPK activity could trigger autophagy 21. The process of self-eating or autophagy provides starved cells a means to survive by recycling cellular component as bioenergetic substrates for energy production and building blocks. As such, certain metabolic hubs should be exploitable for therapy, particularly if a specific cancer type is ‘addicted’ to that pathway. Metabolic enzymes are readily pharmacologically targetable; in fact, many historical clinically effective chemotherapeutic drugs were termed ‘anti-metabolites.’ Seeking drugs that directly inhibit these new metabolic targets involved in cancer cell energy metabolism while sparing normal cells is among the most desirable goal to improve cancer therapy, especially for the treatment of pancreatic cancer.
Blocking pancreatic cancer fuel supply
Addiction to glucose could be exploited through targeted inhibition of enzymes involved in glycolysis. One such target is the glycolytic pathway, in which the most consistent abnormality is its ultimate step of conversion of pyruvate to lactic acid by lactate dehydrogenase A (LDHA) to regenerate nicotinamide adenine dinucleotide (NAD+) that is required for the further glycolytic conversion of glucose to pyruvate to generate ATP. Recent studies 40,41 have targeted this phenotype of altered metabolism for therapy. The first drug-like small molecule (called FX11) which inhibits LDHA was used as proof of concept for targeting aerobic glycolysis in cancer. This compound, 7-Benzyl-2,3-dihydroxy-6-methyl-4-n-propyl-1-napthoic acid, has shown an anti-tumorigenic effect in mice models of human lymphoma and pancreatic cancer through the increased production of reactive oxygen species and cell death 40. By blocking LDHA, FX11 diminishes the ability of malignant cells to metabolize pyruvate to lactate, and halts the regeneration of nicotinamide adenine dinucleotide (NAD+) for glycolysis processing. This study also showed a strong synergy effect in vitro and in vivo with the use of FX11 in combination with FK866, an inhibitor of NAD+ biosynthesis, which accentuates NAD+ depletion.
Besides the Warburg effect, cancer cells also maintain mitochondrial oxidation of glutamine by glutaminase that converts glutamine to glutamate, which enters the TCA cycle as 2-oxoglutarate. Two recent studies targeted glutaminolysis in cancer by a specific glutaminase inhibitor, BPTES (bis-2-[5-(phenylacetamido)-1,3,4-thiadiazol-2-yl]ethyl sulfide) 42. The first study by Seltzer et al. 43 reported a preferable inhibition of mutant IDH1 cell growth by BPTES as compared to the wild type enzyme. Mutation at the R132 residue of isocitrate dehydrogenase 1 (IDH1), creates a novel enzyme function that produces 2-hydroxyglutarate (2-HG) from α-ketoglutarate (α-KG) which is from glutamate, a product of glutamine via glutaminase. Cancer cells with mutant IDH1 become addicted to glutamine and heavily depend on glutaminase. The addition of exogenous α-KG rescued growth suppression of mutant IDH1 cells by BPTES, which lowered glutamate and α-KG levels, inhibited glutaminase activity, and increased glycolytic intermediates. However 2-HG levels were unaffected by BPTES. This presents a potential therapeutic opportunity.The study by Wang et al. 44 provided another aspect of targeting mitochondrial glutaminase activity inhibiting oncogenic transformation. They demonstrated that glutaminase activity, which is dependent on Rho GTPases and NF-κB activity, increased in transformed fibroblasts and breast cancer cells. Targeting glutaminase activity by BPTES to inhibit oncogenic transformation had thus been demonstrated, through a connection between Rho GTPase activation and cellular metabolism, to be a promising way forward. The importance of hypoxic glutamine metabolism in the study by Le et al. was also underscored by the anti-proliferative therapeutic effect of BPTES on neoplastic cells in vitro and in a tumor xenograft model in vivo 36.
In addition to the proof-of-concept studies suggesting the feasibility of targeting glucose or glutamine metabolism in pancreatic cancer, it has been noted that pancreatic cancers display significant autophagic activities for survival 45-49. In fact, Ras-transformed cells depend on autophagy for survival 50. Hence, inhibition of autophagy with the anti-malarial agent chloroquine has resulted in significant preclinical responses of pancreatic cancer xenografts and allografts in treated mice as compared to control 49, 51. Chloroquine also diminishes pancreatic tumorigenesis in a transgenic model 49. These studies suggest that two related and widely used agents with extremely favorable safety profiles, chloroquine or hydroxychloroquine, could have profound clinical effects. Indeed, there are now clinical trials testing this concept in pancreatic cancer. As for targeting glycolysis or glutaminolysis, the field is awaiting pharmaceutical companies to develop clinically safe highly potent drugs to be tested in the clinic. Notwithstanding the inherent challenges for drug development, the concept of blocking the pancreatic cancer fuel supply provides a reasonable framework for the development of what is hoped to be a new class of anti-cancer agents.
Future direction
Although cancer cells can exhibit unique metabolic pathways, they also use the classic metabolic pathways of normal cells. This presents a great challenge to directly target metabolic pathways, especially metabolic enzymes as drug targets. The success of small molecular agents depends on how much cancer cells are “addicted” to the fuel nutrition versus normal cells, as shown in studies mentioned above. The combination of multi-omics technologies will give a functional perspective of cancer progression, beyond genes and protein expression profiles. The extensive data obtained from metabolic flux will be mined to identify specific pathways active in the tumor compared with untransformed cells. These data are essential for understanding the metabolic activity in tumor tissue, especially how cancer cells can respond to different environmental conditions and how they respond to therapy in order to detect likely responders and non-responders to a particular chemotherapeutic agent. This is highly desirable because it is important to avoid the use of cytotoxic drugs that have no benefit. The ultimate goal is to characterize and enable targeted selection of patients based upon predicted metabolic responses. The metabolic signatures that correlate with sensitivity of pancreatic cancers to metabolic inhibitors will also be used in the future to help us combine existing drugs to target multiple metabolic pathways and attack specific attributes of each patient’s cancer. In fact, metformin, which inhibits NADH dehydrogenase and mitochondrial respiration, has preclinical activity against pancreatic cancer xenografts (Kumar et al., unpublished) and is used in clinical trials. However, parameters that predict resistance or response remain poorly understood. Hence, defining the metabolic pathways of pancreatic cancer through metabolomics will pave the way for prediction of response and the identification of new enzyme targets for pancreatic cancer therapies. A major technical challenge in this arena is the heterogeneity of the pancreatic tumor tissue which is laced with an extensive fibrotic stroma comprising of host immune cells.
The potential for cancer cells to reprogram their metabolism to bypass the targeted enzymatic step may occur and hence pose a challenge to metabolic therapy. This issue is especially significant given the interconnectivity of the cellular metabolic network. In combination with appropriate computational tools (e.g., flux balance analysis), metabolomics offers a powerful way to identify possible “metabolic escape routes”. With the insight of this guide, we will eliminate these escape routes through rational combination therapies, e.g. in combination with current anti-metabolites. This will allow for more cost-effective and personalized cancer treatment.
Further complicating attempts at developing pancreatic cancer therapies, there is emerging evidence of pancreatic cancer stem cells in pancreatic cancers and these cells are responsible for drug resistance to standard chemotherapy, resulting in relapse and metastasis of pancreatic adenocarcinoma (see review by Penchev et al in this issue 52). Therefore, recognizing the heterogeneity of multi-subpopulation of pancreatic tumors and understanding the distinguished metabolic modes of each are critical important for targeting of these subpopulations.
Acknowledgements
Our work is supported by an AACR 107467 Stand-Up-to-Cancer Translational Grant (to A.L., N.V.R, A.M. and C.V.D.); The Sol Goldman Pancreatic Cancer Research fund 80028595 (to A.L.); grants 5R01CA051497-21 and 5R01CA057341-20 (to C.V.D.).
References
- 1.Iacobuzio-Donahue CA, Velculescu VE, Wolfgang CL, Hruban RH. The genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. Clin Cancer Res. 2012;18 doi: 10.1158/1078-0432.CCR-12-0315. xx-xx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feig C, Gopinathan A, Neesse A, Chan D, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18 doi: 10.1158/1078-0432.CCR-11-3114. xx-xx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature reviews Cancer. 2011;11:325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature Reviews Drug Discovery. 2011;10:671–84. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer cell. 2008;13:472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. Systemic Elevation of PTEN Induces a Tumor-Suppressive Metabolic State. Cell. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaglio D, Soldati C, Vanoni M, Alberghina L, Chiaradonna F. Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS One. 2009;4:e4715. doi: 10.1371/journal.pone.0004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 107:8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–82. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- 12.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 13.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012 Feb 15;483(7390):474–8. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–24. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Current opinion in genetics & development. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Current opinion in genetics & development. 2011;21:113–9. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 23.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–4. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology. 2011;13:1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finkel T. Signal transduction by reactive oxygen species. The Journal of cell biology. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finkel T. From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Science signaling. 2012;5:pe10. doi: 10.1126/scisignal.2002943. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Cao L, Finkel T. Oxidants, metabolism, and stem cell biology. Free radical biology & medicine. 2011;51:2158–62. doi: 10.1016/j.freeradbiomed.2011.10.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brahimi-Horn MC, Bellot G, Pouyssegur J. Hypoxia and energetic tumour metabolism. Current opinion in genetics & development. 2011;21:67–72. doi: 10.1016/j.gde.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase m2 regulates gene transcription by acting as a protein kinase. Molecular cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 32.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:4548–54. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma WW, Jacene H, Song D, Vilardell F, Messersmith WA, Laheru D, et al. [18F]fluorodeoxyglucose positron emission tomography correlates with Akt pathway activity but is not predictive of clinical outcome during mTOR inhibitor therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:2697–704. doi: 10.1200/JCO.2008.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birnbaum DJ, Adelaide J, Mamessier E, Finetti P, Lagarde A, Monges G, et al. Genome profiling of pancreatic adenocarcinoma. Genes, chromosomes & cancer. 2011;50:456–65. doi: 10.1002/gcc.20870. [DOI] [PubMed] [Google Scholar]
- 35.Dang CV. MYC on the path to cancer. Cell. 2012 Mar 30;149(1):22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15:110–21. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong SM, Park JY, Hruban RH, Goggins M. Molecular signatures of pancreatic cancer. Archives of pathology & laboratory medicine. 2011;135:716–27. doi: 10.5858/2010-0566-ra.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–20. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 107:2037–42. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J Med Chem. 54:1599–612. doi: 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]
- 42.Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) The Biochemical journal. 2007;406:407–14. doi: 10.1042/BJ20070039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 70:8981–7. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 18:207–19. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grasso D, Garcia MN, Iovanna JL. Autophagy in pancreatic cancer. International journal of cell biology. 2012;2012:760498. doi: 10.1155/2012/760498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Udelnow A, Kreyes A, Ellinger S, Landfester K, Walther P, Klapperstueck T, et al. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PloS one. 2011;6:e20143. doi: 10.1371/journal.pone.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang S, Kimmelman AC. A critical role for autophagy in pancreatic cancer. Autophagy. 2011;7:912–3. doi: 10.4161/auto.7.8.15762. [DOI] [PubMed] [Google Scholar]
- 48.Mukubou H, Tsujimura T, Sasaki R, Ku Y. The role of autophagy in the treatment of pancreatic cancer with gemcitabine and ionizing radiation. International journal of oncology. 2010;37:821–8. doi: 10.3892/ijo_00000732. [DOI] [PubMed] [Google Scholar]
- 49.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes & development. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes & development. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Penchev VR, Rasheed ZA, Maitra A, Matsui W. Heterogeneity and targeting of pancreatic cancer stem cells. Clin Cancer Res. 2012;18 doi: 10.1158/1078-0432.CCR-11-3112. xx-xx. [DOI] [PMC free article] [PubMed] [Google Scholar]