

Abstract
Familial hypertrophic cardiomyopathy (FHC) is one of the most common genetic causes of heart disease. Approximately 15% of FHC-related mutations are found in cTnT [cardiac troponin (cTn) T]. Most of the cTnT FHC-related mutations are in or flanking the N-tail TNT1 domain that directly interacts with overlapping tropomyosin (Tm). We investigate two sets of cTnT mutations at opposite ends of TNT1, mutations in residue 92 in the Tm–Tm overlap region of TNT1 and mutations in residues 160 and 163 in the C-terminal portion of TNT1 adjacent to the cTnT H1–H2 linker. Though all the mutations are located within TNT1, they have widely different phenotypes clinically and biophysically. Using a complete atomistic model of the cTn–Tm complex, we identify mechanisms by which the effects of TNT1 mutations propagate to the cTn core and site II of cTnC, where calcium binding and dissociation occurs. We find that mutations in TNT1 alter the flexibility of TNT1, which is inversely proportional to the cooperativity of calcium activation of the thin filament. Further, we identify a pathway of propagation of structural and dynamic changes from TNT1 to site II of cTnC, including TNT1, cTnT linker, I-T arm, regulatory domain of cTnI, the D-E linker of cTnC, and site II cTnC. Mutationally induced changes at site II of cTnC alter calcium coordination that corresponds to biophysical measurements of calcium sensitivity. Finally, we compare this pathway of mutational propagation with that of the calcium activation of the thin filament and find that they are identical but opposite in direction.
Keywords: molecular dynamics, thin filament, troponin, cardiac, calcium signaling
Introduction
Familial hypertrophic cardiomyopathy (FHC) is a dominant “disease of the sarcomere” and is one of the most frequently occurring cardiac genetic disorders.1–4 In 1990, the original studies establishing the genetic basis for FHC were published.5,6 Since then, over 700 individual mutations in components of the cardiac sarcomere have been associated with FHC pathogenesis including thick and thin filaments and associated proteins.7–10 The clinical phenotypes and prognoses associated with these mutations differ widely.11,12 Thus, one over-riding goal of FHC research is to link genotype to phenotype.13,14 Many of the basic molecular mechanisms that underlie these clinically relevant differences remain unknown. Understanding these mechanisms of molecular pathogenesis will provide valuable information regarding the heterogeneity of FHC-related mutation phenotypes.
The cardiac thin filament is a multimeric structure with each fundamental unit composed of seven actin monomers, one tropomyosin (Tm) dimer, and a single cardiac troponin (cTn) complex composed of three independent subunits: cTnC, the calcium binding subunit; cTnI, the inhibitory subunit; and cTnT, the Tm binding and cTnI–cTnC complex binding subunit, as diagrammed in Fig. 1. The cTnT–Tm interaction occurs along an extended region spanning the head-to-tail region of the contiguous array of Tm within the thin filament. cTnT is also thought to promote the ordered assembly of the cTn–Tm complex onto the actin filament.15 The physiologic role of the cardiac thin filament involves both calcium-independent and calcium-dependent regulatory roles in myofilament activation.15 As a result, cTn acts as a calciumactivated regulator of actomyosin interactions by means of allosteric regulation of Tm dynamics on actin.15,16
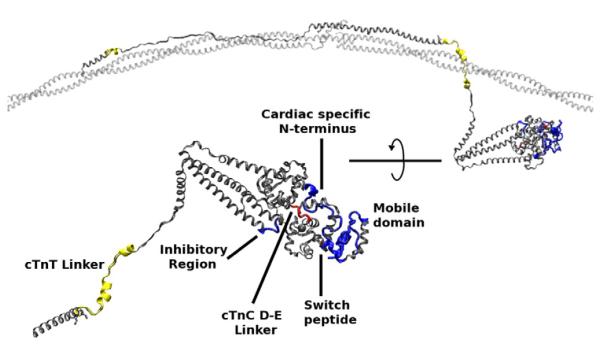
Fig. 1.
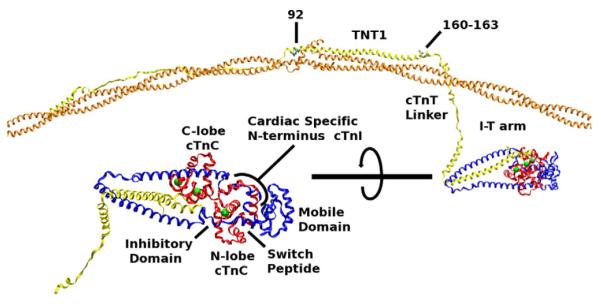
Orientation to the cTn–Tm–Tm complex used in our simulations. Orange, overlapping Tm; yellow, cTnT; blue, cTnI; red, cTnC; green, calcium ion. The lower left image is the cTn core rotated and enlarged. Tm is positioned upon an actin scaffold. The edges of Tm are held in position by constraining their alpha carbons allowing only the side chains and overlapping regions of Tm to move freely. All of cTn moves freely with the exception of two atoms (the alpha carbons of cTnT 205 and 277).
cTnT consists of two domains (largely historically) and a linker that connects them. The N-terminus includes a highly helical domain, TNT1, where approximately 75% of all cTnT FHC-linked mutations occur or are adjacent to, including those that we discuss below.17,18 It is well accepted that the N-tail of cTnT including TNT1 is required for cooperative activation of the thin filament;19,20 the C-terminal domain TNT2 is necessary for cTnT–cTnI–cTnC calcium-sensitive interactions.19,21 In addition, studies suggest that flexibility of TNT1 is important in the interactions between cTn and Tm.18,22–24 There is a flexible linker between TNT1 and TNT2. Despite the cTnT linker’s obvious role in calcium signal propagation, little is known about its structure due to its hypervariability.25
cTnI consists of three domains: the cardiac specific N-terminus, the helices preceding and comprising the I-T arm, and the calcium-dependent regulatory regions. The cardiac specific N-terminus consists of approximately 30 residues that are unique to cTn and contains phosphorylation sites (serines 23 and 24) that are responsible for beta-adrenergic stimulation of the heart.26–28 The calcium-dependent regulatory region regulates actomyosin interaction and consists of the switch peptide domain, the inhibitory domain, and the mobile domain.29–32
The I-T arm is a coiled coil consisting of two helices, one from cTnI and the other from cTnT. It plays a key role in calcium signaling and represents the most stable region of the cTn complex in terms of subunit–subunit interactions.25,33–35 The stability of the I-T arm allows both helices to rotate and translate as a single entity, which is critical to signal transduction through the cTn complex.25,33
cTnC consists of two globular domains connected by a flexible linker. The N-lobe has one low-affinity calcium binding site (site II), which is ultimately responsible for regulating the contraction and relaxation of the heart. The C-lobe consists of two high-affinity calcium binding sites (sites III and IV). The link between the two lobes is located between helix D of the N-lobe and helix E of the C-lobe; hence, it is referred to as the D-E linker. In cTn, the D-E linker is mobile and flexible, which is necessary for the calcium-dependent regulation of cTn.25,34
Site II cTnC is a canonical E-F hand that consists of 12 residues in specific sequence whereby calcium is coordinated by oxygens of cTnC side chains and backbone and a water bridge36 (see Supporting Information, “Site II E-F hand sequence alignment”). In a canonical E-F hand, six residues in positions 1, 3, 5, 7, 9, and 12 are responsible for calcium coordination. These correspond to residues 65, 67, 69, 71, 73, and 76 of site II in cTnC. Computational work on a fragment of cTn in explicit solvation demonstrated that site II cTnC is sensitive to perturbations. These perturbations changed the manner in which calcium is coordinated by surrounding oxygens including gains and losses of coordination with nearby oxygens in the loop.37
Calcium activation of the thin filament, therefore, requires molecular information to be transferred from the site of calcium binding (site II of cTnC) to Tm. This causes a shift in equilibrium between the three states of cTn–actin–myosin interactions: open (calcium bound to cTnC, strong actomyosin binding, and cross-bridge interactions result in force production), closed (calcium bound to cTnC, weak actomyosin binding, and cross-bridge interactions do not result in force production), and blocked (calcium not bound to cTnC, no actomyosin interactions, and no cross-bridge interactions to produce force).38,39
In our previous work, we used a complete atomistic model of the cTn–Tm complex to identify statistically significant changes that were transmitted through the complex.33 Our aim in this paper is to link molecular mechanisms of FHC-linked mutations in TNT1 of cTnT to their respective physiological phenomena in analogous experiments in order to identify (1) mechanisms by which TNT1 mutations communicate with the cTn core and (2) how different mutations in nearby locations display different biophysical effects on cooperativity of calcium binding and calcium activation of the thin filament. Elucidating the biophysical alterations and mechanistic links between specific cTnT mutations and their biomolecular phenotypes will not only further our understanding of the fundamental biology of thin filament function but also provide new insights into the pathogenesis of thin filament-related cardiomyopathies. This work is a part of our integrative approach toward investigating FHC-related mutations.14
In the current work, we focus our investigation on the fundamental biophysical effects of TNT1 mutations R92L, R92W, ΔE160, E163K, and E163R. These mutations in humans present with a large degree of clinical heterogeneity, making genotype–phenotype correlations difficult.13 ΔE160 is associated with a high incidence of sudden cardiac death.7,11 R92L is associated with varying degrees of cardiac hypertrophy and a low incidence of sudden cardiac death. Patients with the R92W mutation exhibit a complex progressive cardiomyopathy and a high incidence of sudden cardiac death.40–42 E163K and E163R are difficult to accurately characterize due to small samples.7 Furthermore, biophysical studies of these mutations have demonstrated that they display different biophysical effects despite being in the same domain of cTnT.18,43–45 We have found that our atomistic model of the cTn–Tm complex successfully captures mutant-specific fundamental molecular mechanisms of these FHC-related changes.
Results and Discussion
Our simulations yielded thermodynamically and structurally stable complexes over 1-ns simulations (see Supporting Information, “rmsd data”). We are capable of linking structural and dynamic mutational effects of FHC-related TNT1 mutations in our simulations with observed physiological changes. This sheds unique insights into the molecular mechanisms by which these mutations cause disease. We will first discuss cTnT R92 mutations, then we will compare and contrast these findings with cTnT E160 and E163 mutations.
Mutations R92L and R92W
Mutations involving R92 are located in the over-lapping Tm region of TNT1, an area recognized as critical to cooperative actin binding and the regulatory function of cTn.18,19,22 A primary function of cTnT is to bind Tm, and our simulations show that R92L and R92W cause the interaction between cTnT and Tm to decrease compared to wild type (WT) as shown in Table 1. Average structures of WT, R92L, and R92W complexes are superimposed in Fig. 2. TNT1 is enlarged and rotated to highlight a noticeable shift in the average position of TNT1 for mutants R92L and R92W resulting from this decreased interaction. Palm et al. found that cTnT fragments of residues 70–170 cTnT containing these same mutations had a decreased affinity for Tm. They hypothesized that this was the result of altered interactions with overlapping Tm.18 We demonstrate here in atomic detail that the decreased interaction between cTnT and Tm resulting from mutations R92L and R92W is the likely mechanism of the decreased affinity of cTnT to Tm observed by Palm et al.
Table 1.
Average number of cTnT atoms within 4 Å of Tm using cTnT residues 70–190
| cTnT variant | Number of atoms |
|---|---|
| WT | 463 |
| R92L | 425 |
| R92W | 437 |
| ΔE160 | 380 |
| E163K | 442 |
| E163R | 452 |
Fig. 2.
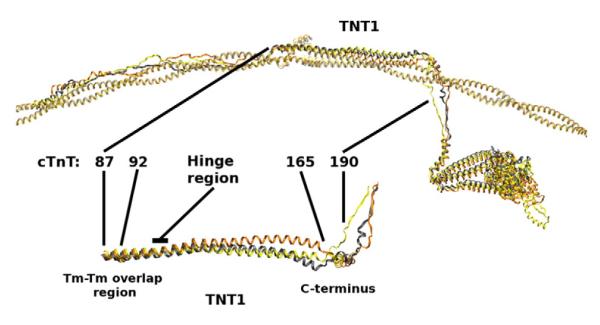
cTnT R92L and R92W mutations compared with WT. Top: Average structures of cTn with overlapping Tm–Tm for WT (gray), R92L (yellow), and R92W (orange) aligned with respect to Tm. Bottom: TNT1 is enlarged and rotated to highlight a noticeable shift in the average position of TNT1 for mutants R92L and R92W that results from decreased interactions between TNT1 and Tm.
As one follows TNT1 from the Tm–Tm overlap region to the C-terminal region (from left to right inFig. 2), WT, R92L, and R92W average structures are similar until the hinge region (residues 105–110 of cTnT) at which point R92L and R92W deviate from WT. We interpret this as a result of mutationally induced increased flexibility in the TNT1 region, a phenomenon we observed in our earlier work with fragments of cTnT26,46,47 but now shown in the context of the entire cTn complex in conjunction with Tm. This increased flexibility is also reflected in the rgyr (radius of gyration) of residues 70–170 of R92L and R92W cTnT shown in Fig. 3. Compared to our earlier results,47 our current results show a much smaller increase in flexibility in TNT1. We attribute this difference to the inclusion of Tm in our current simulations, which underlines the role of Tm as the “stabilizer” of the cTn complex. The presence of Tm decreases the movements of TNT1; however, TNT1 still appears to fluctuate despite its interactions with Tm. R92L and R92W mutations increase TNT1’s flexibility even in the presence of Tm. In earlier work, we correlated the change in flexibility of these mutations with calcium cooperativity using fragments.26,46 We demonstrate here that the inverse relationship between flexibility and cooperativity of calcium activation of the thin filament exists in our current model in the context of the entire cTn–Tm complex, as shown in Fig. 3. The increase in flexibility of TNT1 caused by R92L and R92W mutations correlates with a decrease in the cooperativity of calcium activation of thin filaments containing the same mutations measured with regulated in vitro motility (R-IVM).
Fig. 3.
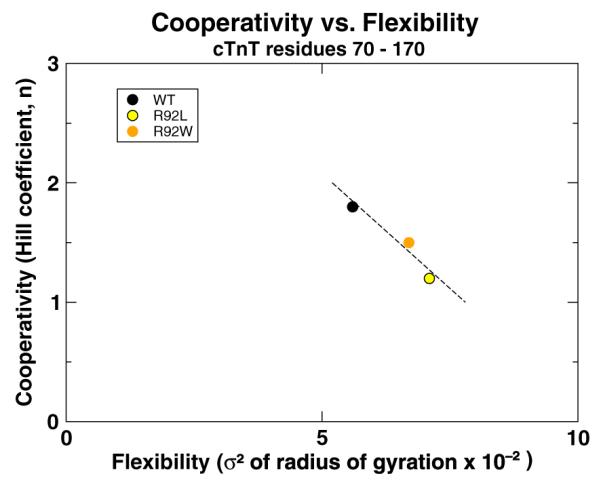
The comparison of cooperativity of calcium activation of the thin filament measured with R-IVM experiments26 and flexibility of TNT1 measured from our simulations shows a correlation between the two properties (r=−0.94). The flexibility of TNT1 was calculated by measuring the variance (σ2) of the rgyr of residues 70–170 of cTnT. The broken line is a best-fit line to highlight the inverse nature of the relationship between flexibility and cooperativity. Data are provided in Supporting Information, “rgyr and R-IVM data”.
The changes we observe due to R92L and R92W mutations are not localized to the TNT1 region. With our model, we are able to observe how mutationally induced changes in TNT1 propagate to the cTn core via the cTnT linker region. Figure 4 shows that the increase in flexibility of R92L and R92W mutations causes a decrease in secondary structure in the vicinity of cTnT residue 190 that alters I-T arm dynamics. The dynamic changes in the I-T arm also result in changes of secondary structure in the calcium-dependent regions of cTnI and cTnC (see Supporting Information, “Secondary structural changes”) and in the average location of site II calcium ions as seen in Fig. 4. Sites III and IV remain relatively stable. Of particular interest is a decrease in secondary structure of the D-E linker of cTnC, which apparently alters the dynamics of helices C and D of the calcium binding site II in cTnC as shown in Fig. 4. This affects normal interactions between calcium and the coordinating oxygens of site II in cTnC as shown in Fig. 5. These changes demonstrate mutationally induced alterations in calcium handling at the molecular level and provide molecular details as to how TNT1 mutations alter calcium binding and cooperativity at a significant distance.26 There are other models of calcium coordination,37,48 but the power of our model is its ability to monitor calcium–oxygen distances in the context of the complete cTn–Tm complex. The small changes we find in calcium binding for R92L and R92W concur with relatively small changes in calcium sensitivity we found earlier in experiments using R-IVM.26
Fig. 4.
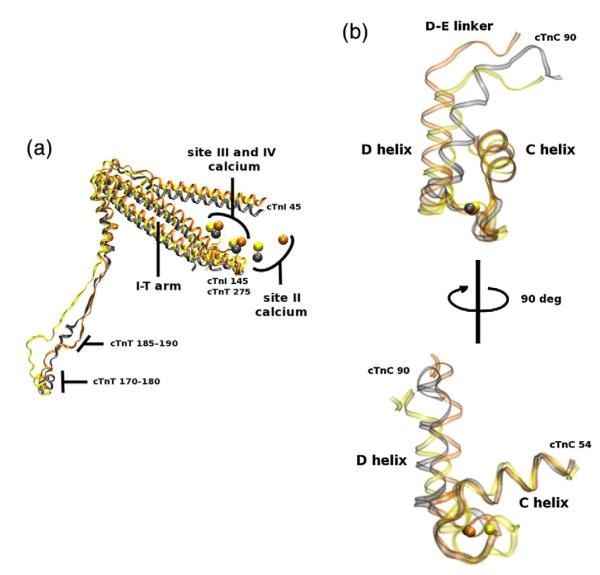
(a) cTn core of WT (gray), R92L (yellow), and R92W (orange) aligned with respect to Tm (data not shown). This image is a portion of Fig. 2 rotated and enlarged. The portions shown are cTnT residues 170–288, cTnI 45–135, and the calcium ions. There is a change in secondary structure noticeable in cTnT residues 185–190 and a noticeable shift in the I-T arm and site II calcium ions. (b) Site II cTnC of WT (gray), R92L (yellow), and R92W (orange) aligned with respect to the C helix of cTnC. This image highlights the mutationally induced effects experienced at site II. There is a minor though noticeable perturbation in the dynamics of site II and the average position of calcium. The backbone is left transparent to better observe the calcium positions, and the top image is rotated 90° to yield the bottom image for better perspective of the entire E-F hand. The overall change at site II of the R92 mutations is small compared to those observed in E160 and E163 mutations below.
Fig. 5.
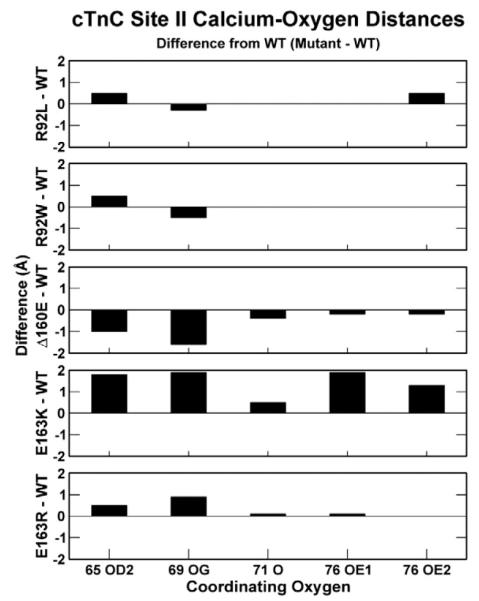
Average distances between coordinating oxygens of site II and calcium ion. The values are graphed as the difference from WT distances (mutation minus WT); therefore, a positive value means that oxygen and calcium are farther apart from one another than WT and vice versa. The coordinating oxygens of a canonical E-F hand correspond to oxygens of cTnT residues 65 (Asp), 67 (Asp), 69 (Ser), 71 (Thr), 73 (Asp), and 76 (Glu). Two residues (cTnT 67 and 73) require a water bridge to interact with calcium. Since our model uses implicit solvation, our simulations are only capable of capturing interactions between calcium and oxygens from four of these residues (cTnT 65, 69, 71, and 76). Atom labeling legend: 65 OD2 is the delta oxygen of residue 65 cTnC, aspartic acid; 69 OG is the gamma oxygen of residue 69 cTnC, serine; 71 O is the carbonyl oxygen of residue 71 cTnC, threonine; 76 OE1/OE2 are the epsilon oxygens of residue 76 cTnC, glutamic acid. Data are provided in Supporting Information, “Calcium coordination data”.
In summary, R92L and R92W mutations reduce cTnT–Tm interactions resulting in changes in cooperativity of calcium activation of the thin filament and small changes in calcium binding affinity. The flexibility of TNT1 is increased due to cTnT R92L and R92W mutations and is inversely proportional to the cooperativity. These changes propagate to site II of cTnC by means of conformational and dynamic changes through the cTn complex beginning with the cTnT linker. These changes alter the dynamics of site II of cTnC resulting in relatively small calcium binding changes at the molecular level. This concurs with experimental data showing large changes in calcium cooperativity and relatively small changes in calcium sensitivity of R92L and R92W using R-IVM.26 This also builds upon previous evidence by Jin et al. demonstrating that seemingly remote portions of the N-tail of cTnT can significantly affect the conformation and function of the cTn core.19,49,50
Mutations ΔE160, E163K, and E163R
Mutations involving E160 or E163, as shown in Fig. 6, are in the C-terminal region of TNT1 immediately adjacent to the cTnT linker and are responsible for linking the site of calcium binding to Tm. We believe that the proximity of mutations ΔE160, E163K, and E163R to the cTnT linker amplifies their mutationally induced effects.
Fig. 6.
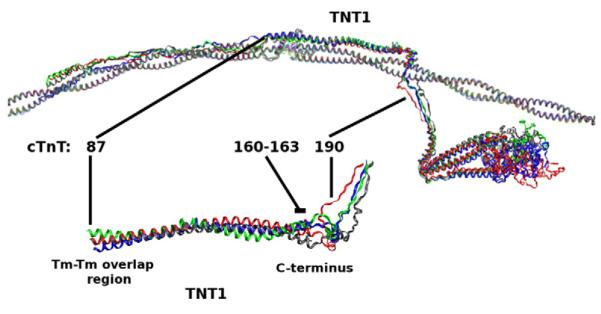
cTnT ΔE160, E163K, E163R mutations compared with WT. Top: Average structures of cTn with overlapping Tm–Tm for WT (gray), ΔE160 (red), E163K (blue), and E163R (green) aligned with respect to Tm. Bottom: TNT1 is enlarged and rotated to highlight a noticeable shift in the average position of TNT1 for mutants ΔE160, E163K, and E163R that results from decreased interactions between TNT1 and Tm.
Mutations ΔE160, E163K, and E163R also decrease the interaction between cTnT and Tm compared to WT, as shown in Table 1. This directly affects how the C-terminus of TNT1 interacts with Tm and the dynamics of the cTnT linker, as shown inFig. 6. It appears that a normal function of the distal linker region is to maintain appropriate interactions between cTnT and Tm in order to properly transmit a signal from cTnC to Tm, a function that is altered by 160E, E163K, and E163R mutations.
There is an increase in the flexibility of TNT1 as a result of mutations E163K and E163R similar to the increase in flexibility we observed in mutations R92L and R92W. Based on the correlation between flexibility and cooperativity we described above, we expected that filaments containing these mutations would show a large decrease in cooperativity, a finding that appears to be true based on preliminary R-IVM data.51 ΔE160 simulations resulted in a slight decrease in TNT1 flexibility compared to WT; therefore, little to no change in cooperativity should be expected, also consistent with preliminary findings.51 We correlated our data for ΔE160, E163K, and E163R simulations with respective R-IVM data and found that the same relationship exists between flexibility and cooperativity for these mutations and the R92 mutations described above; flexibility of TNT1 is indirectly proportional to the cooperativity of activation of the thin filament, as shown in Fig. 7.
Fig. 7.
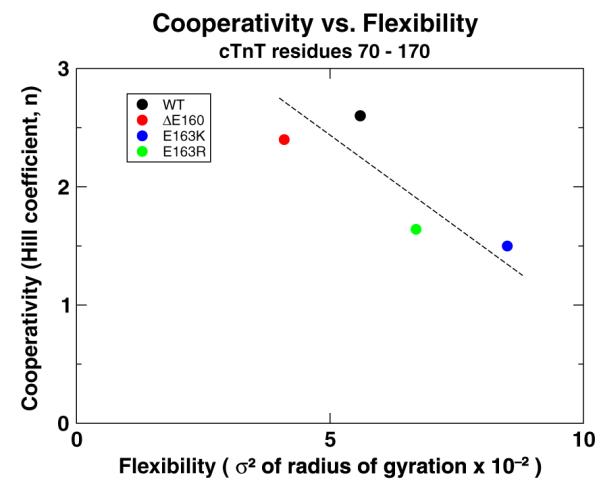
The comparison of cooperativity of calcium activation of the thin filament measured with R-IVM experiments51 and flexibility of TNT1 measured from our simulations. This shows an indirect correlation between the two properties (r=−0.74). The flexibility of TNT1 was calculated by measuring the variance (σ2) of the rgyr of residues 70–170 of cTnT. The broken line is a best-fit line to highlight the inverse nature of the relationship between flexibility and cooperativity. Data are provided in Supporting Information, “rgyr and R-IVM data”.
With the dramatic decrease in the number of interacting atoms between cTnT and Tm for ΔE160,one would expect the TNT1 flexibility of ΔE160 mutants to increase; however, the opposite is true as shown in Fig. 7. The nature of the deletion mutation results in a tightening of the helix at the C-terminus of TNT1 that both stiffens TNT1 and pulls on the cTnT linker. This results in dramatically different physical behavior between the deletion mutation ΔE160 and adjacent substitution mutations E163K and E163R. Our earlier simulations of cTnT fragments containing only residues 70–170 cTnT with neither the cTnT linker nor the remainder of cTn and Tm failed to capture these initial mutational effects. Because of this, we believe that the mass of the cTn core and the torque the cTnT linker imposes on the long helix of TNT1 play critical roles in the function and assembly of the thin filament. Harada et al. studied myofibrils containing the cTnT ΔE160 mutation and observed dramatic effects on thin filament assembly and function including a disruption in TNT1–Tm binding,52 whereas Palm et al.’s work with filaments containing only a fragment of residues 70–170 cTnT failed to notice any difference between ΔE160 and E163K mutants and WT characteristics, concluding that these mutations “must cause disease by other mechanisms.”18 Simulations of the mutations ΔE160, E163K, and E163R that we performed on fragments of cTn only containing cTnT residues 70–170, the same fragment of cTnT used by Palm et al., also showed little to no changes in behavior compared to WT; however, our current, complete model is capable of demonstrating mechanisms by which these mutations affect cTn function.
Though ΔE160, E163K, and E163R mutations initiate changes throughout the cTn complex in a manner similar to that of the R92 mutations, the structural changes we observe as a result of E160 and E163 mutations are greater (see Supporting Information, “Secondary structural changes”). In particular, a greater effect is seen in the D-E linker of cTnC and site II of cTnC for ΔE160, E163K, and E163R mutations than for the R92 mutations, as shown in Fig. 8. As a result, the coordinating oxygens in site II of cTnC of mutants E163K and E163R tend to move farther away from calcium, as shown in Fig. 5. The movement of these oxygens in this low-affinity calcium binding site results in further diminished affinity for bound calcium. Therefore, in order to activate the same number of cTn–Tm–Tm complexes within thin filaments containing mutations E163K and E163R compared to WT thin filaments, we require a higher concentration of calcium surrounding the thin filaments. This agrees with preliminary R-IVM experimental data showing a decrease in calcium sensitivity for thin filaments with these same mutations.51 ΔE160, however, causes structural changes of a different nature in the linker region and subsequently through the cTn core, as seen in Fig. 8. These changes allow the oxygens of site II to more readily interact with calcium, thus decreasing its ability to dissociate from calcium. ΔE160, therefore, has a greater affinity for bound calcium in site II cTnC than E163K, E163R, and WT. These measurements correspond to calcium sensitivity of thin filaments in unloaded systems such as those of R-IVM.
Fig. 8.
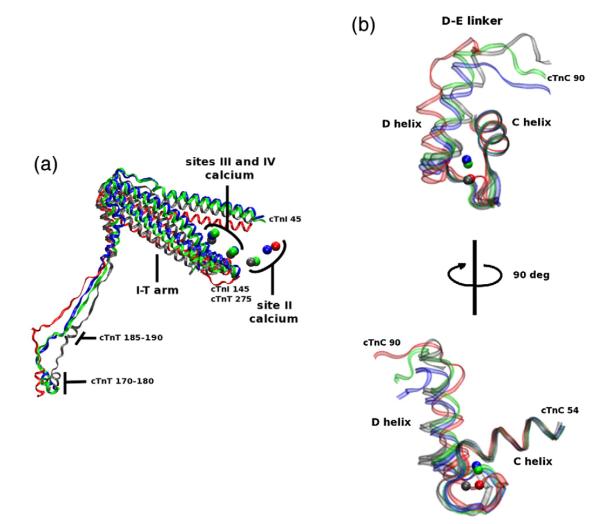
a) cTn core of WT (gray), ΔE160 (red), E163K (blue), and E163R (green) aligned with respect to Tm (data not shown). This image is a portion of Fig. 6 rotated and enlarged. The portions shown are cTnT residues 170–288, cTnI 45–135, and the calcium ions. The decrease in secondary structure noticeable in cTnT residues 185–190. The shift in the I-T arm and site II calcium ions are similar for E163K and E163R but different from ΔE160. (b) Site II cTnC of WT (gray), ΔE160 (red), E163K (blue), and E163R (green) aligned with respect to the C helix of cTnC. The backbone is left transparent to better observe the calcium positions, and the top image is rotated 90° to yield the bottom image for better perspective of the entire E-F hand. This image highlights the mutationally induced effects experienced at site II. There is a noticeable perturbation in the dynamics of site II and the average positions of calcium. These changes result in an increase in the distances between coordinating oxygens in site II and calcium for E163K and E163R mutations and a decrease in distance for ΔE160.
While one might imagine that WT would have the maximum site II affinity for calcium, this does not appear to be the case. In the case of the WT complex, it appears that oxygens in site II interact with calcium in a manner that allows it to escape at an optimum rate. Physiologically, the escape of calcium from site II is required for relaxation of the heart. E163K and E163R mutations increase the probability of this escape; ΔE160 decreases the probability of such an escape. We found this to be of particular interest because impaired relaxation is prominent in cases of FHC.53 While it is not possible to scale our findings up to whole heart physiology, our model appears to provide insight into a molecular mechanism of calcium-dissociation impairment.
Kobayashi and Solaro have shown that changes in calcium sensitivity due to mutations in cTnI result from changes in the dissociation of calcium from cTnC site II (kd).54 Norman et al. demonstrated that changes in calcium binding affinity of cTnC have a direct effect on cTn’s role as the modulator of forcegenerating actomyosin interactions.55 Beginning from similar states of bound calcium, effects from FHC-related mutations in our models propagate through the cTn complex ultimately causing oxygen–calcium distances to fluctuate in site II cTnC. While the average changes in R92L and R92W oxygen–calcium distances are rather small, the changes we observe in mutants ΔE160, E163K, and E163R are rather large. Both of these findings concur with R-IVM measurements of calcium sensitivity.26,51 Therefore, we believe that we are observing the molecular mechanism by which kd is altered in FHC-related mutations.
In summary, ΔE160, E163K, and E163R mutations cause changes in the interactions between cTnT and Tm. In the cases of E163K and E163R, this results in an increase in the flexibility of TNT1. In the case of ΔE160, this results in a decrease in flexibility of TNT1. These substitution and deletion mutations have very different effects on the cTn core. The substitution mutations, E163K and E163R, cause changes in site II of cTnC that dramatically decrease its affinity for bound calcium while the opposite changes occur as a result of the deletion mutation, ΔE160, hence its increased affinity for bound calcium.
A common pathway of propagation
Despite their differences, TNT1 mutations appear to share a common pathway of propagation through the cTnT linker and I-T arm to the cTn core. Certain regions of the cTn core and complex appear structurally sensitive to TNT1 mutations, including the cardiac specific N-terminus, inhibitory domain, switch peptide, and mobile domain of cTnI and the D-E linker of cTnC, which connects the N- and Clobes of cTnC (see Supporting Information,“Secondary structural changes”). These regions are superimposed on the average WT structure in Fig. 9 and are similar to the locations of calcium-dependent changes throughout the cTn complex as a function of calcium binding to site II of cTnC.33 There is no observable change in the structure of the I-T arm or the helices of cTnI and cTnT that directly lead to it. In calcium signaling, we and others observed that the I-T arm is a stable coiled coil that rotates and translates as a single entity.25,33,35 In the case of the mutations we have discussed, this same property appears to transmit mutational effects from the cTnT linker to cTnC and cTnI. Therefore, the regions of structural and dynamic changes that we identified as responsible for calcium signal transduction are the same as those responsible for propagating mutational effects. It should not be surprising that the signaling mechanism by which TNT1 mutations communicate with cTn core is the same as the mechanism by which calcium signaling in cTnC communicates with overlapping Tm but opposite in direction.
Fig. 9.
Common pathway of mutationally induced changes in secondary structure. WT average structure (gray) with regions that are structurally sensitive to TNT1 mutations highlighted. The highlighted regions are colored as a function of their subunit: yellow, cTnT; blue, cTnI; and red, cTnC.
Summary
The majority of cTnT FHC-related mutations are in or flanking TNT1. We demonstrated that mutations within TNT1 can have distinctly different molecular mechanisms of disease despite being located in the same domain. Though their pathways of propagation may appear similar, their fundamental mechanisms of alteration in sarcomeric function differ. This sheds light on the difficulty of reasonably classifying similar FHC-related mutations and the difficulty of genotype–phenotype matching for this disease, a central issue in the field.13 Our model has shown two fundamental mechanisms of FHC-related TNT1 mutations: (1) mutationally induced changes in flexibility of TNT1, which are inversely proportional to the cooperativity of calcium activation of the thin filament, and (2) mutational effects on cTnC that alter site II’s affinity for bound calcium.
The cTn complex appears to be a calcium-triggered dynamic filter consisting of a connected network of dynamic and structurally sensitive regions that are also sensitive to FHC hotspot mutations. Our future work will aim to see how TNT1 mutations affect these calcium-dependent changes in cTn in the calcium-depleted state as well. A computational model such as the one we have presented earlier33 and employed here may be useful to categorize FHC mutations based on their molecular mechanisms. This model may also prove to be a useful tool for characterizing newly discovered mutations within cTn. Long-term application of our high-resolution model may allow for more rigorous molecular genotype–phenotype correlation.
Methods
Molecular dynamics simulations were performed with CHARMM version 33b156,57 on an IBM iDataplex x3650 Linux cluster. All simulations were performed using GBSW implicit solvation.58,59 The simulations performed in this work were based on our existing model.33 We constructed a thin filament consisting of cTn and over-lapping Tm in the closed state oriented to an actin backbone by synthesizing numerous existing atomistic models including the following: an atomistic model of the cTn core complex from Protein Data Bank (PDB) ID 1J1E;25 an atomistic model of Tm based on the Lorenz–Holmes model†‡; an overlapping model of Tm based on PDB ID 2Z5I;22 cTnT–Tm interactions based on PDB ID 2Z5H;22 an atomistic model of the thin filament including actin, nonoverlapping Tm, and the cTn core;60 an atomistic model of chicken fast skeletal troponin from PDB ID 1YTZ;61 and an atomistic model of the cardiac specific N-terminus of cTnI from PDB ID 2JPW.27 Improvements in the current model are included in the supporting information (see Supporting Information, “Components of complete cTn Model” and “Substitutions to contributing structures”). Missing regions of cTn were constructed using secondary structure prediction (PSIPRED).62,63 Though various overlapping Tm structures exist,22,64,65 only the incorporation of PDB IDs 2Z5H and 2Z5I into our model produced a structurally stable model.
Mutations were introduced into cTnT as necessary, and simulations were then performed in an identical manner as previously described.33 Briefly, the thin filament was constructed in the closed state by constraining carbons of the nonoverlapping Tm in the closed positions on actin. Actin is then removed. Actin is not included in the simulations due to computational expense. cTn and overlapping Tm were allowed to move freely while nonoverlapping regions of Tm remained stationary due to constraints. Two cTn atoms (the alpha carbons of cTnT 205 and 277) were held in place with harmonic constraints of 2 kcal/mol/Å to simulate interactions between cTn and actin. With the cTn core oriented to actin, the N-tail of cTnT was docked to overlapping Tm in a flexible manner. This was accomplished through the use of distance constraints over 30 ps while gradually reducing the distance constraints to 0. With the system in thermodynamic equilibrium, we allowed the system to evolve for 50 ps.
The WT and mutant models were then subjected to identical minimization, heating, and equilibration conditions. They were minimized by alternating the steepest descent and adopted basis Newton–Raphson methods until energy optimized (final gradient less than 0.0001 kcal/mol/Å) then heated gradually from 0 K to 300 K over 30 ps. The system was again allowed to equilibrate for 30 ps, then the system was evolved for 1 ns. Root-mean-square deviation (rmsd) analyses for the cTnT–Tm region and the cTn core were performed over the production run to ensure structural stability. A schematic of the method by which we constructed and performed our simulations is diagrammed in Fig. 10.
Fig. 10.
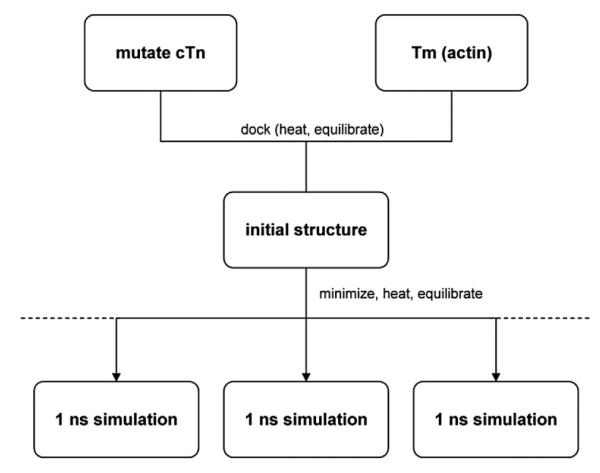
Schematic of simulation method. After the initial cTn structure was mutated, the mutant cTn and Tm were gradually docked. The resulting structure was then minimized, heated, and equilibrated. Multiple simulations were run with varying initial conditions (the broken lines representing the potential for additional simulations with varied initial conditions in the future). Each 1-ns production run took approximately 2 weeks.
While 1-ns simulations may seem relatively short, simulations of a system of this size are at the limit of current computational possibilities and preclude its explicit solvation. It has been shown that multiple short simulations are more likely indicative of molecular behavior than a single long simulation;66 therefore, we ran three separate simulations for each variant with various initial conditions chosen by varying random seeds. Our quantitative results for WT and mutants are the averages of their three respective 1-ns simulations. Graphically, we present average structures from single simulations.
We thus produced equilibrium simulations of thin filaments for WT and cTnT mutants R92L, R92W, ΔE160, E163K, and E163R. Each set of simulations consisted of three separate simulations whose only differences were their seeds for the random number generator. The simulations were visualized with VMD.67 rmsd and rgyr analyses were performed using CHARMM version 33b1.56,57 Only the backbone atoms (amine N, alpha C, and carboxyl C) were used in calculating rmsd and rgyr. Secondary structure analyses were performed with STRIDE.68 A Pearson correlation between molecular dynamics and R-IVM results was performed with the statistical software package R.2.11.1 for Windows.
In order to investigate mutant effects on calcium binding, we used our WT simulations as a baseline. We identified the oxygens with which calcium coordinated in site II by searching for any atoms that were within 4 Å of site II calcium during the course of our WT simulations. Due to our use of implicit solvation, we cannot observe water bridge interactions required by residue 73 cTnC. As a result, coordination with residue 67 is quickly lost in our WT simulations. No new oxygen–calcium interactions occurred during the course of WT or mutant simulations. We therefore measured the distances between site II calcium and the oxygens with which we found calcium to be coordinated: OD2 of residue 65, OG of residue 69, O of residue 71, and OE1 and OE2 of residue 76 during the course of WT and mutant simulations, as shown in Fig. 11.
Fig. 11.
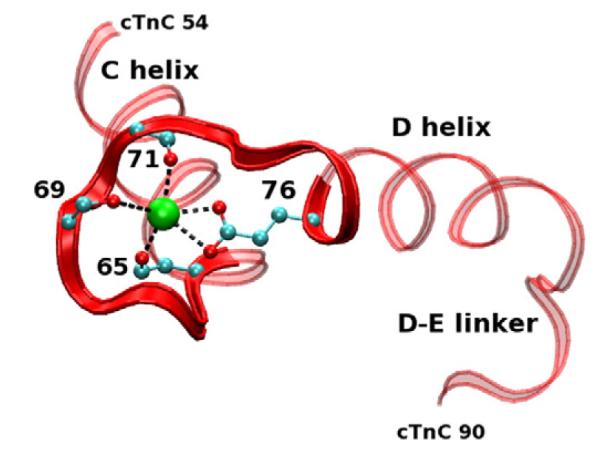
Site II cTnC average structure from a WT simulation. Red ribbon, cTnC; green, calcium ion. Portions of the side chains with coordinating oxygens are shown with carbons (cyan spheres) and oxygens (red spheres). Our model is capable of measuring five of the interactions between oxygen and calcium due to our use of implicit solvation.
We then compared the results of our analyses with experimental results. Our results are the most comparable with R-IVM studies, which measure functional characteristics of the thin filament, such as cooperativity of calcium activation and calcium sensitivity, under no load conditions. Even with this experimental system, we recognize the limitations of our model. For example, without actin or myosin being simulated, it would be imprudent to compare our computational results with filament sliding speeds measured with R-IVM because they are measurements of actomyosin kinetics. Similarly, we obviously cannot scale our findings above the thin filament level such as to fibers and whole heart experiments. Our present methodology is a bottom-up approach in investigating fundamental biophysical effects of FHC-related mutations.
Supplementary Material
Acknowledgements
This work has been supported in part by National Institutes of Health grants HL075619 (to J.C.T.), GM068036 (to S.D.S.), and HL107046-01 (to J.C.T. and S.D.S.).
Abbreviations
- FHC
familial hypertrophic cardiomyopathy
- cTn
cardiac troponin
- Tm
tropomyosin
- WT
wild type
- R-IVM
regulated in vitro motility
- R-IVM
regulated in vitro motility
- PDB
Protein Data Bank
Footnotes
http://www.mpimf-heidelberg.mpg.de/holmes; accessed on 24 Mar 2009.
ftp://149.217.48.3/pub/holmes; accessed on 24 Mar 2009.
Supplementary Data Supplementary data to this article can be found online at doi:10.1016/j.jmb.2012.05.008
References
- 1.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, et al. Alphatropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 2.Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 1997;16:379–382. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 4.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat. Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 5.Tanigawa G, Jarcho JA, Kass S, Solomon SD, Vosberg HP, Seidman JG, Seidman CE. A molecular basis for familiar hypertrophic cardiomyopathy: an alpha/beta cardiac myosin heavy chain hybrid gene. Cell. 1990;62:991–998. doi: 10.1016/0092-8674(90)90273-h. [DOI] [PubMed] [Google Scholar]
- 6.Geisterfer-Lowrance AAT, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 7.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to clinics. J. Cardiovasc. Electrophysiol. 2008;19:104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 8.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat. Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima-Taniguchi C, Matsui H, Nagata S, Kishimito T, Yamauchi-Takihara K. Novel missense mutation in alpha-tropomyosin gene found in Japanese patients with hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 1995;27:2053–2058. doi: 10.1016/0022-2828(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 10.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat. Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 11.Watkins H, Seidman JG, Seidman CE. Familial hypertrophic cardiomyopathy: a genetic model of cardiac hypertrophy. Hum. Mol. Genet. 1995;4:1721–1727. doi: 10.1093/hmg/4.suppl_1.1721. [DOI] [PubMed] [Google Scholar]
- 12.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Phys. Rev. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 13.Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, et al. Research priorities in hypertrophic cardiomyopathy: report of a working group of the National Heart, Lung, and Blood Institute. Circulation. 2010;122:1130–1133. doi: 10.1161/CIRCULATIONAHA.110.950089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tardiff JC. Thin filament mutations: developing an integrative approach. Circ. Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 17.Gomes AV, Barnes JA, Harada K, Potter JD. Role of troponin T in disease. Mol. Cell. Biochem. 2004;263:115–129. doi: 10.1023/B:MCBI.0000041853.20588.a0. [DOI] [PubMed] [Google Scholar]
- 18.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys. J. 2001;81:2827–2837. doi: 10.1016/S0006-3495(01)75924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biesiadecki BJ, Chong SM, Nosek TM, Jin JP. Troponin T core structure and the regulatory NH2-terminal variable region. Biochemistry. 2007;46:1368–1379. doi: 10.1021/bi061949m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaertl S, Lehrer SS, Geeves MA. Separation and characterization of the two functional regions of troponin involved in muscle thin filament regulation. Biochemistry. 1995;34:15890–15894. doi: 10.1021/bi00049a003. [DOI] [PubMed] [Google Scholar]
- 21.Pearlstone JR, Smillie LB. Effects of troponin-I plus C on the binding of troponin-T and its fragments to alpha-tropomyosin. J. Biol. Chem. 1983;258:2534–2542. [PubMed] [Google Scholar]
- 22.Murakami K, Stewart M, Nozawa K, Tomii K, Kudou N, Igarashi N, et al. Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc. Natl Acad. Sci. USA. 2008;105:7200–7205. doi: 10.1073/pnas.0801950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinkle A, Tobacman LS. Folding and function of the troponin tail domain. Effects of cardiomyopathic troponin T mutations. J. Biol. Chem. 2003;278:506–513. doi: 10.1074/jbc.M209194200. [DOI] [PubMed] [Google Scholar]
- 24.Palm T, Greenfield NJ, Hitchcock-DeGregori SE. Tropomyosin ends determine the stability and functionality of overlap and troponin T complexes. Biophys. J. 2003;84:3181–3189. doi: 10.1016/S0006-3495(03)70042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 26.Manning EP, Guinto PJ, Tardiff JC. Correlation of molecular and functional effects of mutations in cardiac troponin T linked to familial hypertrophic cardiomyopathy: an integrative in silico/in vitro approach. J. Biol. Chem. 2012;287:14515–14523. doi: 10.1074/jbc.M111.257436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J. Mol. Biol. 2007;373:706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 28.Solaro RJ, Moir AJG, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 29.Galinska A, Hatch V, Craig R, Murphy AM, Eyk JEV, Wang CLA, et al. The C terminus of cardiac troponin I stabilizes the Ca2+-activated state of tropomyosin on actin filaments. Circ. Res. 2010;106:705–711. doi: 10.1161/CIRCRESAHA.109.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mudalige WAKA, Tao TC, Lehrer SS. Ca2+-dependent photocrosslinking of tropomyosin residue 146 to residues 157–163 in the C-terminal domain of troponin I in reconstituted skeletal muscle thin filaments. J. Mol. Biol. 2009;389:575–583. doi: 10.1016/j.jmb.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman RMB, Sykes BD. Isoform-specific variation in the intrinsic disorder of troponin I. Proteins. 2008;73:338–350. doi: 10.1002/prot.22063. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman RMB, Blumenschein TMA, Sykes BD. An interplay between protein disorder and structure confers the Ca2+ regulation of striated muscle. J. Mol. Biol. 2006;361:625–633. doi: 10.1016/j.jmb.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 33.Manning EP, Tardiff JC, Schwartz SD. A model of calcium activation of the thin filament. Biochemistry. 2011;50:7405–7413. doi: 10.1021/bi200506k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowlessur D, Tobacman LS. Low temperature dynamic mapping reveals unexpected order and disorder in troponin. J. Biol. Chem. 2010;285:38978–38986. doi: 10.1074/jbc.M110.181305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowlessur D, Tobacman LS. Troponin regulatory function and dynamics revealed by H/D exchange-mass spectrometry. J. Biol. Chem. 2010;285:2686–2694. doi: 10.1074/jbc.M109.062349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson MR, Chazin WJ. Structures of EF-hand calcium binding proteins: diversity in the organization, packing and response to calcium binding. BioMetals. 1998;11:297–318. doi: 10.1023/a:1009253808876. [DOI] [PubMed] [Google Scholar]
- 37.Varughese JF, Baxley T, Chalovich JM, Li Y. A computational and experimental approach to investigate bepridil binding with cardiac troponin. J. Phys. Chem. B. 2011;115:2392–2400. doi: 10.1021/jp1094504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poole KJV, Lorenz M, Evans G, Rosenbaum G, Pirani A, Craig R, et al. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fiber diagrams from non-overlap muscle. J. Struct. Biol. 2006;155:273–284. doi: 10.1016/j.jsb.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 39.McKillop DFA, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N. Engl. J. Med. 1995;332:1058–1064. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 41.Forissier JF, Carrier L, Farza H, Bonne G, Bercovici J, Richard P, et al. Codon 102 of the cardiac troponin T gene is a putative hot spot for mutations in familial hypertrophic cardiomyopathy. Circulation. 1996;94:3069–3073. doi: 10.1161/01.cir.94.12.3069. [DOI] [PubMed] [Google Scholar]
- 42.Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H. Sudden death due to troponin T mutations. J. Am. Coll. Cardiol. 1997;29:549–555. doi: 10.1016/s0735-1097(96)00530-x. [DOI] [PubMed] [Google Scholar]
- 43.Lombardi R, Bell A, Senthil V, Sidhu J, Noseda M, Roberts R, Marian AJ. Differential interactions of thin filament proteins in two cardiac troponin T mouse models of hypertrophic and dilated cardiomyopathies. Cardiovas. Res. 2008;79:109–117. doi: 10.1093/cvr/cvn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haim TE, Dowell C, Diamanti T, Scheuer J, Tardiff JC. Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J. Mol. Cell. Cardiol. 2007;42:1098–1110. doi: 10.1016/j.yjmcc.2007.03.906. [DOI] [PubMed] [Google Scholar]
- 45.Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J. Biol. Chem. 2004;279:14488–14495. doi: 10.1074/jbc.M309355200. [DOI] [PubMed] [Google Scholar]
- 46.Guinto PJ, Manning EP, Schwartz SD, Tardiff JC. Computational characterization of mutations in cardiac troponin T known to cause familial hypertrophic cardiomyopathy. J. Theor. Comput. Chem. 2007;6:413–419. doi: 10.1142/S0219633607003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ertz-Berger B, He H, Dowell C, Factor S, Haim T, Nunez S, et al. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc. Natl Acad. Sci. USA. 2005;102:18219–18224. doi: 10.1073/pnas.0509181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spyracopoulos L, Li MX, Sia SK, Gagne SM, Chandra M, Solaro RJ, Sykes BD. Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry. 1997;36:12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 49.Wei B, Jin JP. Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch. Biochem. Biophys. 2011;505:144–154. doi: 10.1016/j.abb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM, Jin JP. Restricted N-terminal truncation of cardiac troponin T: a novel mechanism for functional adaptation to energetic crisis. J. Physiol. 2008;586:3537–3550. doi: 10.1113/jphysiol.2008.153577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore RK, Guinto PJ, Dowell-Martino C, Tardiff JC. Functional and structural changes induced by cTnT-related FHC mutations in TNT1 alter actomyosin binding interactions. 2009 Biophysical Society Meeting Abstracts. Biophys. J. Supplement. 2010;6a Abstract, 34-Plat. [Google Scholar]
- 52.Harada K, Takahashi-Yanaga F, Minakami R, Morimoto S, Ohtsuki I. Functional consequences of the deletion mutation deletion Glu160 in human cardiac troponin. T. J. Biochem. 2000;127:263–268. doi: 10.1093/oxfordjournals.jbchem.a022603. [DOI] [PubMed] [Google Scholar]
- 53.Maron BJ, Spirito P, Green KJ, Wesley YE, Bonow RO, Arce J. Noninvasive assessment of left ventricular diastolic function by pulsed Doppler echocardiography in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1987;10:733–742. doi: 10.1016/s0735-1097(87)80264-4. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi T, Solaro RJ. Increased Ca2+ affinity of cardiac thin filaments reconstituted withcardiomyopathy-related mutant cardiac troponin. I. J. Biol. Chem. 2006;281:13471–13477. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 55.Norman C, Tal JA, Tikunova SB, Davis JP. Modulation of the rate of cardiac muscle contraction by troponin C constructs with various calcium binding affinities. Am. J. Physiol.: Heart Circ. Physiol. 2007;293:H2580–H2587. doi: 10.1152/ajpheart.00039.2007. [DOI] [PubMed] [Google Scholar]
- 56.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 58.Chen J, Im W, Brooks CL. Balancing solvation and intramolecular interactions: toward a consistent generalized Born force field. JACS. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Im W, Lee MS, Brooks CL. Generalized Born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 60.Pirani A, Vinogradova MV, Curmi PMG, King WA, Fletterick RJ, Craig R, et al. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J. Mol. Biol. 2006;357:707–717. doi: 10.1016/j.jmb.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 61.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R. Ca2+-regulated structural changes in troponin. Proc. Natl Acad. Sci. USA. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Ward JS, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005:W36–W38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 64.Frye J, Klenchin VA, Rayment I. Structure of the tropomyosin overlap complex from chicken smooth muscle: insight into the diversity of N-terminal recognition. Biochemistry. 2010;49:4908–4920. doi: 10.1021/bi100349a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greenfield NJ, Huang YJ, Swapna GV, Bhattacharya A, Rapp B, Singh A, et al. Solution NMR structure of the junction between tropomyosin molecules: implications for actin binding and regulation. J. Mol. Biol. 2006;364:80–96. doi: 10.1016/j.jmb.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 66.Caves LS, Evanseck JD, Karplus M. Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci. 1998;7:649–666. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Humphrey W, Dalke A, Schulten K. VMD—Visual Molecular Dynamics. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 68.Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.