

Abstract
Objective
Atherosclerosis is characterized as a chronic inflammatory condition that involves cholesterol deposition in arteries. Together with scavenger receptor B1 (SR-B1), the ATP-binding cassette transporters ABCA1 and ABCG1 are the major components of macrophage cholesterol efflux. Recent studies have shown that low grade inflammation plays a distinct regulatory role in the expression of SR-B1 and ABCA1/ABCG1. However, the mechanisms linking low grade inflammation and cholesterol accumulation are poorly understood.
Methods and Results
Using primary bone marrow-derived macrophages, we demonstrate that subclinical low dose lipopolysaccharide (LPS) potently reduces the expression of SR-B1 and ABCA1/ABCG1, as well as cholesterol efflux from macrophages through interleukin-1 receptor associated kinase 1 (IRAK-1) and toll-interacting-protein (Tollip). Low dose LPS down-regulates the nuclear levels of retinoic acid receptor RARα leading to their reduced binding to the promoters of SR-B1 and ABCA1/ABCG1. We observe that glycogen synthase kinase 3β (GSK3β) activation by low dose LPS through IRAK1 and Tollip is responsible for reduced levels of RARα and reduced expression of SR-B1 and ABCA1/ABCG1. IRAK-M, on the other hand, counteracts the function of IRAK-1.
Conclusion
Collectively, our data reveal a novel intracellular network regulated by low dose endotoxemia that disrupts cholesterol efflux from macrophages and leads to the pathogenesis of atherosclerosis.
Keywords: low grade inflammation, molecular signal transductions, macrophages, atherosclerosis, membrane transporters
Introduction
Atherosclerosis underlies the pathogenesis of a wide range of cardiovascular diseases. Consequently, atherosclerosis and related cardiovascular complications are among the leading causes of morbidity and mortality in the United States 1-3. Despite significant improvements in clinical diagnosis and patient care, the incidence of atherosclerosis has been increasing in the last decade, potentially due to changing life styles 3. Atherosclerosis-related diseases were estimated to cost more than $500 billion annually in the United States 4, 5. Recent studies indicate that low grade endotoxemia may be one of the culprits for the pathogenesis of atherosclerosis. Both external and internal risk factors including infections and high fat diets can dramatically alter the landscape of gut microbiota, increase mucosal permeability, and as a consequence, raise the levels of circulating bacterial endotoxin lipopolysaccharide (LPS) 6-9. Indeed, slightly elevated levels of blood endotoxin (<100 pg/mL) were observed in humans with chronic cardiovascular diseases, higher body mass indexes (BMI), and in aged adults with inflammatory complications10-12.
In contrast to the effect of high dose LPS (>10ng/mL), which can induce major cytokine storms and septic shock, the trace amount of LPS seen in chronic subclinical endotoxemia (<100 pg/mL) does not induce acute cytokine storm. Instead, subclinical endotoxemia induces mild yet persistent elevation of inflammatory mediators, potentially leading to low grade inflammation and pathogenesis of atherosclerosis. Despite significant pathological implication of low dose endotoxemia, the molecular mechanisms responsible for the effects of low dose endotoxin are not well understood. We reported that low dose LPS fails to activate the classical NFκB pathway 13. Instead, low dose LPS preferentially induces the mild induction of pro-inflammatory mediators through ATF2, C/EBPδ, and mitochondria reactive oxygen species 13, 14. In terms of the intra-cellular signaling components, we observed that the interleukin-1 receptor-associated kinase 1 (IRAK-1) and Toll-interacting-protein (Tollip) are critically required for the effects of low dose LPS, without compromising the effects of high dose LPS 13, 14.
Bacterial endotoxin not only induces pro-inflammatory mediators, but also suppresses the expression of anti-inflammatory mediators 15, 16. Of particular relevance, SR-B1 and ABCA1 are among the anti-inflammatory mediators expressed by macrophages 17, 18. These membrane transporters are essential for cholesterol export from macrophages and maintaining macrophage homeostasis 17, 19. Together with SR-B1, ABCA1 and ABCG1 contribute to the efflux of cholesterol from macrophages and prevent the formation of foam cells. SR-B1 promotes bidirectional cholesterol exchange between cells and lipoproteins including HDL. ABCA1 facilitates cholesterol export to lipid-free apolipoprotinA-I (ApoA-I) and generation of HDL, while ABCG1 acts to export cholesterol to mature HDL particles 19. In the meantime, ABCA1 attenuates TLR4 signaling and alleviates inflammatory response to LPS 17. The expressions of ABCA1 and ABCG1 are controlled by nuclear receptors including RARα 20, 21. We and others have shown that LPS can suppress nuclear receptors and reduce retinoic acid-induced expression of ABCA1 22-25. We also reported that several key intracellular signaling molecules including IRAK-1 are involved in the suppression of RARα by low dose LPS 13.
The molecular mechanisms responsible for the suppression of macrophage cholesterol export by low dose LPS are not well studied. We reported that low dose LPS preferentially reduces levels of nuclear receptor RARα, which is responsible for the expression of ABCA1 and ABCG1, as well as the suppression of pro-inflammatory mediators 13, 23. This current study further examines the mechanisms responsible for reduced cholesterol export in macrophages treated with low dose LPS. Using primary macrophages harvested from wild type (WT), IRAK-1 deficient, Tollip deficient, and IRAK-M deficient mice, we examined the molecular pathways responsible for the reduced expression of cholesterol transporters in macrophages by low dose LPS.
METHODS
Detailed protocols are given in SI Materials and Methods.
Reagents
LPS (Escherichia coli O111:B4) was obtained from Sigma. The antibodies against RARα, GAPDH, SRC3, GSK3β, pGSK3β-Tyr216 and β-actin were purchased from Santa Cruz Biotechnology. ABCA1, ABCG1 and SR-B1 specific antibodies were obtained from Novus Biologicals and the pGSK3β-Ser9, LaminB antibodies were purchased from Cell Signaling. [3H]cholesterol (40 Ci/mmol) was obtained from Perkin-Elmer.
Mice
C57BL/6 Wild type (WT) mice were purchased from the Jackson laboratory. IRAK-1 deficient mice were obtained from James Thomas (University of Texas Southwestern Medical School). Tollip deficient mice were provided by Jürg Tschopp (University of Lausanne at Switzerland). IRAK-M deficient mice were provided by Richard Flavell (Yale University School of Medicine). The WT and GSK3β deficient murine embryonic fibroblast cells were obtained from James Woodgett (Ontario Cancer Institute). All mice were housed and bred at Derring Hall animal facility in compliance with approved Animal Care and Use Committee protocols at Virginia Polytechnic Institute and State University. BMDMs were isolated from the tibias and femurs of mice by flushing the bone marrow with Dulbecco’s modified Eagle’s medium (DMEM). The cells were cultured in untreated tissue culture dishes with 50 ml DMEM containing 30% L929 cell supernatant. On the third day of culture, the cells were fed with an additional 20 ml fresh medium and cultured for another additional 3 days. Cells were harvested with phosphate-buffered saline (PBS), resuspended in DMEM supplemented with 1% fetal bovine serum, and allowed to rest overnight before further treatments.
Statistical analysis
Statistical significance was determined using the unpaired two-tailed Student’s t test. p values less than 0.05 were considered statistically significant. Data are expressed as mean +/- SD.
RESULTS
Low dose LPS decreases the expression of SR-B1, ABCA1, and ABCG1, as well as cholesterol efflux to HDL and ApoA1
We first examined the expression levels of SR-B1, ABCA1, and ABCG1 in murine bone marrow derived macrophages (BMDM) treated with a low dose LPS (50 pg/mL). As shown in Figure 1A, the RNA levels of Sr-b1, Abca1, and Abcg1 decreased significantly in response to low dose LPS treatment. We further examined their protein levels by Western blot analyses. As shown in Figure 1B, the protein levels of SR-B1, ABCA1, and ABCG1 were significantly reduced after low dose LPS treatment (Figure 1B). To determine whether decreased expression of these molecules may translate into compromised cholesterol export in macrophages, we performed cholesterol export assay as we previously described 23. The primary function of the membrane transporters, ABCA1 and ABCG1, involve mediating cholesterol efflux to apoA1 and HDL, respectively, to reduce the buildup of cholesterol within the cells. SR-B1 can facilitate the cholesterol export mediated by ABCA1 and ABCG1. As shown in Figure 1C, the cholesterol exports to both Apo-A1 and HDL were significantly reduced in cells treated with low dose LPS for 18 h. These results indicate that low dose LPS suppresses cholesterol export from macrophages through suppressing the expression of SR-B1, ABCA1, and ABCG1.
Figure 1. Low dose LPS reduces the expression and function of cholesterol transporters.
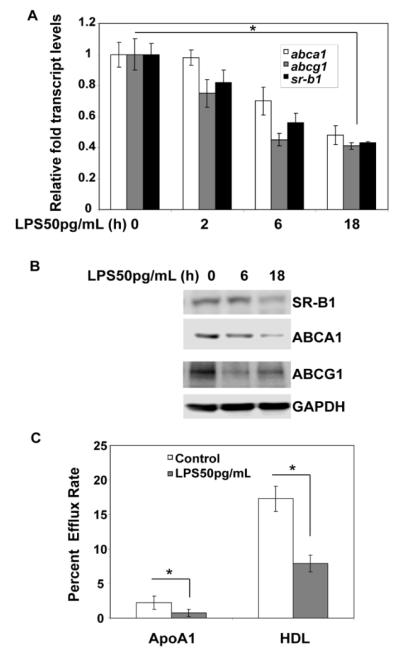
(A) WT BMDM cells were either treated with low dose LPS (50 pg/mL) for the indicated time periods or left untreated for 18 h, and the expression levels of Srb1, Abca1, and Abcg1 transcripts were measured by real-time RT-PCR assays and standardized against Gapdh levels. Each experiment was performed in triplicate. *, p < 0.05 compared with the untreated control samples. Error bars represent SD. (B) WT BMDM cells were either untreated or treated with low dose LPS (50 pg/mL) for 6 h and 18 h followed by Western blot analysis of total cell extracts (70 μg) using SRB1, ABCA1, and ABCG1-specific antibodies. Antibodies against GAPDH were used as the internal loading control. The data are representative of at least three independent experiments.(C) The BMDMs derived from WT mice were radiolabeled with [3H]cholesterol in cell culture medium for 18 h and then treated with or without low dose LPS (50 pg/mL) in the presence of apoA1 (10 μg/ml) or HDL (50 μg protein/ml). Cellular cholesterol efflux was analyzed in liquid scintillation counting assays, and the efflux rates were calculated as described in the supplemental methods. Efflux rates were expressed as means ± standard deviations from three independent experiments, each performed in triplicate. *, p < 0.05 compared with the untreated control samples.
IRAK-1 and Tollip are critically involved in the suppression of macrophage cholesterol export by low dose LPS
Previous studies indicate that IRAK-1 and Tollip are preferentially involved in mediating the low-grade inflammatory effects elicited by low dose LPS 13, 26. Tollip is a novel adaptor molecule selectively associated with IRAK-1 27. Therefore, we tested whether the suppressive effect of low dose LPS on cholesterol export depends upon IRAK-1 and Tollip. As shown in Figure 2, low dose LPS failed to suppress cholesterol efflux activities to either ApoA1 or HDL in IRAK-1 or Tollip deficient macrophages.
Figure 2. Low dose LPS mediated suppression of macrophage cholesterol efflux is IRAK-1 and Tollip-dependent.
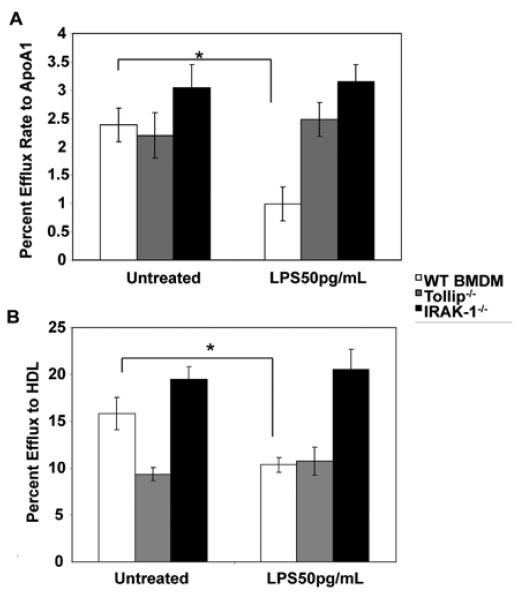
The BMDMs derived from WT, IRAK-1 and Tollip deficient mice were radiolabeled with [3H]cholesterol in cell culture medium for 16 h and then treated with or without low dose LPS (50pg/mL) in the presence of (A) apoA1 (10 μg/ml) or (B) HDL (50 μg protein/ml). Cellular cholesterol efflux was analyzed in liquid scintillation counting assays. Efflux rates were calculated as described in the supplemental methods, and the results are expressed as means ± standard deviations from three independent experiments, each performed in triplicate. *, p < 0.05 compared with the untreated control samples. Error bars represent SD.
To further determine the underlying molecular mechanisms, we tested the involvement of IRAK-1 and Tollip in the expression of key cholesterol exporters including SR-B1, ABCA1, and ABCG1. In consistent with previous reports, we observed that low dose LPS significantly suppressed the transcript and protein expressions of these transporters in WT macrophages (Figure 3A and B). In sharp contrast, low dose LPS failed to suppress the expression of SR-B1, ABCA1, and ABCG1 in IRAK-1 or Tollip deficient macrophages (Figure 3). These data indicate that IRAK-1 and Tollip are involved in the downstream effect of low dose LPS in modulating the expression and function of macrophages cholesterol exporters.
Figure 3. Low dose LPS reduces the expression levels of cholesterol transporters through IRAK-1 and Tollip.
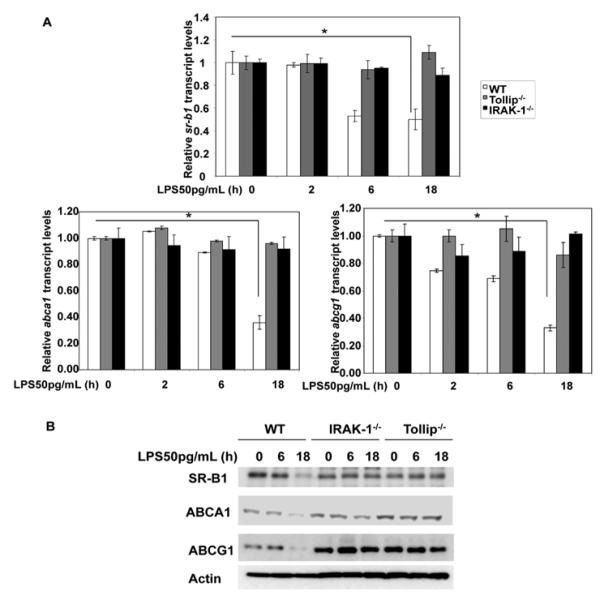
(A) WT, IRAK-1 and Tollip deficient BMDM cells were treated with low dose LPS (50 pg/mL) for the indicated times and the expression levels of Sr-b1, Abca1 and Abcg1 transcripts were measured by real-time RT-PCR assays and standardized against Gapdh levels. Each experiment was performed in triplicate. *, p < 0.05 compared with the untreated control samples. (B) WT, IRAK-1 and Tollip deficient BMDM cells were treated with low dose LPS (50 pg/mL) for the indicated times. Equal protein amounts (70 μg) were subjected to immunoblot analysis with SRB1, ABCA1, and ABCG1-specific antibodies. The same blots were re-probed with anti-ACTIN to control for protein loading. The data are representative of at least three independent experiments.
Low dose LPS suppresses RARα through IRAK-1 and Tollip
We then examined the levels of key transcription factors responsible for the gene transcription of these cholesterol transporters. Earlier studies have shown that ABCA1 expression can be up regulated by members of the nuclear receptor family, such as RARα, LXRα and PPARs 21, 23, 28. Therefore, we examined the nuclear levels of RARα and LXRα in macrophages harvested from WT, IRAK-1 and Tollip deficient mice. As shown in Figure 4A, the nuclear levels of RARα and LXRα were significantly reduced in WT macrophages but not in IRAK-1 and Tollip deficient macrophages. Therefore, low dose LPS mediated suppression of nuclear RARα and LXRα levels was mediated through IRAK1 and Tollip.
Figure 4. IRAK-1 and Tollip are involved in the low dose LPS mediated suppression of RARα.
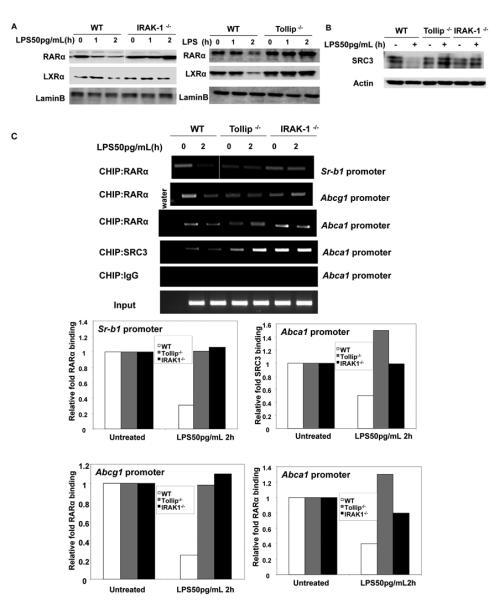
The BMDMs derived from WT, IRAK-1 and Tollip deficient mice were treated with low dose LPS (50pg/mL) for the indicated times followed by nuclear protein extraction. Equal protein aliquots (50 μg) were processed for immunoblotting with (A) anti-RARα and LXRα and (B) anti-SRC3 antibodies. The same blots were re-probed with anti-Lamin B to control for protein loading. (C) WT, IRAK-1 and Tollip deficient BMDMs were either untreated or treated with low dose LPS (50 pg/mL) for 2 h and subjected to a ChIP assay using the specified antibodies. The primers spanning the PPRE and DR4 region on the Sr-b1, Abca1 and Abcg1 promoters were used in the ChIP assays. Data are representative of three independent experiments. The band intensities were quantified using the Fujifilm Multi Gauge software, and the fold suppression is depicted after normalization against input levels.
The transcriptional activity of RARα relies on its ability to recruit co-activators such as steroid receptor coactivator (SRC). SRC-3 facilitates RAR-directed chromatin remodeling and favors the generation of a transcriptionally permissive environment at the responsive promoter elements 29. Thus, we tested whether low dose LPS may also affect the levels of SRC3. As shown in Figure 4B, the levels of SRC3 were significantly reduced in WT BMDMs treated with low dose LPS. In contrast, low dose LPS failed to alter the levels of nuclear SRC-3 in either IRAK-1 or Tollip deficient macrophages.
We further carried out chromatin immunoprecipitation assays to evaluate the binding of nuclear receptors to the endogenous promoters of Sr-b1, Abca1/Abcg1 in WT, IRAK-1 and Tollip deficient macrophages treated with low dose LPS. As shown in Figure 4C, low dose LPS treatment led to reduced binding of RARα to the promoters of Sr-b1, Abca1/Abcg1 in WT but not in IRAK-1 or Tollip-deficient macrophages. Low dose LPS also reduced the recruitment of SRC-3 to the Abca1 promoter. There was no detectable binding of SRC3 to the Abcg1 promoter, indicating that there may be distinct and gene specific regulation of Abca1 and Abcg1.
Low dose LPS suppresses the expression of cholesterol exporters through IRAK-1/Tollip-mediated activation of GSK3β
GSK3β is known to cause degradation and inactivation of both RARα and SRC3 30, 31. Thus, we tested whether low dose LPS may suppress RARα through activation of GSK3β. A previous study indicates that low dose LPS causes GSK3β activation in vivo32. GSK3β is modulated by two distinct phosphorylation events at Serine 9 and Tyrosine 216. Ser9 phosphorylation suppresses, while Tyr216 phosphorylation activates GSK3β activity. We observed that low dose LPS preferentially induced Tyr216 phosphorylation, and suppressed Ser9 phosphorylation of GSK3β (Figure 5A), an indication of GSK3β activation. In contrast, low dose LPS failed to induce Tyr216 in either IRAK-1 or Tollip deficient macrophages (Figure 5A).
Figure 5. Low dose LPS activates GSK3β in IRAK-1 and Tollip dependent manner.
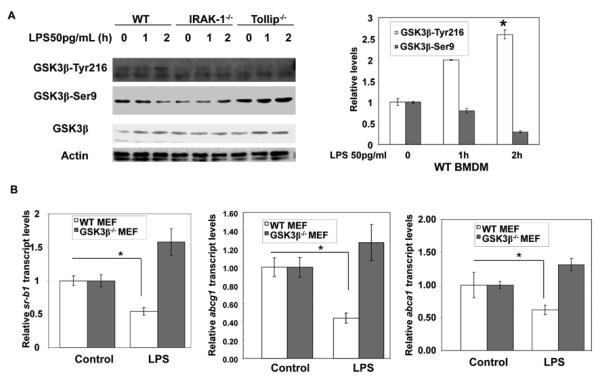
(A) WT, IRAK-1 and Tollip deficient BMDM cells were treated with low dose LPS (50pg/mL) for the indicated times. Whole cell lysates (75 μg) were subjected to immunoblot analyses with total GSK3β, pGSK3β-Ser9 and pGSK3β-Tyr216-specific antibodies. The same blots were re-probed with anti-ACTIN to confirm equal total protein levels. The data are representative of at least three independent experiments. The band intensities were quantified using the Fujifilm Multi Gauge software, and the relative levels were depicted after normalization against β-Actin levels. *, p < 0.05 compared with the untreated control samples. (B) GSK3β+/+ and GSK3β−/− MEFs were left untreated or treated with low dose LPS (50pg/mL) for 18 h, and total RNA was isolated. Expression levels of Srb-1, and Abca1, Abcg1 messages were evaluated by quantitative real-time PCR, and Gapdh was used for normalization. Data are expressed as means ± SD of three independent experiments.
To confirm the role of GSK3β in low dose LPS-mediated suppression of cholesterol exporters, we measured the expression levels of Sr-b1, Abca1/Abg1 in wild type and GSK3β deficient murine embryonic fibroblasts (MEF). As shown in Figure 5B, similar to the effects seen in macrophages, low dose LPS suppressed the expression of these exporters in WT MEF cells. In contrast, LPS failed to suppress these exporters in GSK3β deficient MEF cells.
Severe suppression of cholesterol efflux in IRAK-M deficient cells
Given the fact that IRAK-M counter-acts the function of IRAK-1 33, we tested the expression of cholesterol exporters and cholesterol efflux in IRAK-M deficient macrophages. As shown in Figure 6A, the basal efflux efficiency to either ApoA1 or HDL was significantly lower in IRAK-M deficient macrophages as compared to that in WT macrophages. Low dose LPS further decreased cholesterol efflux in IRAK-M deficient macrophages.
Figure 6. Effect of low dose LPS on the expression and function of cholesterol transporters in IRAKM deficient BMDMs.
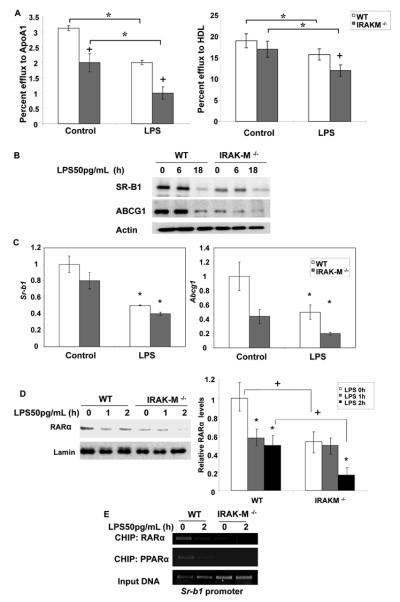
(A) WT and IRAK-M−/− BMDMs were radiolabeled with [3H]cholesterol in cell culture medium for 18 h and then treated with low dose LPS in the presence of apoA1 (10 μg/ml) or HDL (50 μg protein/ml). Cellular cholesterol efflux was analyzed by liquid scintillation counting assays. Efflux rates were calculated as described in the supplemental methods and the results are expressed as means ± standard deviations from three independent experiments, each performed in triplicate. *, p < 0.05 compared with the untreated control samples. +p < 0.05 compared between WT and IRAK-M deficient cells. (B) WT and IRAKM −/− BMDMs were treated with 50 pg/mL LPS for the indicated time points followed by whole-cell protein extraction. The samples (70 μg) were resolved by SDS-PAGE followed by immunoblotting with anti-ABCG1 and SRB1 antibodies. Beta-ACTIN was probed as loading controls. (C) The BMDMs derived from WT and IRAK-M−/− mice were treated with low dose LPS for 18 h followed by total RNA extraction. The resulting cDNAs were used to detect Srb-1 and Abcg1 transcript levels by real-time RT-PCR and standardized against Gapdh levels. Each experiment was performed in triplicate. *, p < 0.05 compared with the untreated control samples. +p < 0.05 compared between WT and IRAK-M deficient cells. (D) The BMDMs derived from WT and IRAK-M deficient mice were treated with low dose LPS (50pg/mL) for the indicated times followed by nuclear protein extraction. Equal protein aliquots (50 μg) were processed for immunoblotting with anti-RARα antibodies. The same blots were re-probed with anti-Lamin B antibodies to control for protein loading. The band intensities were quantified using the Fujifilm Multi Gauge software, and the relative levels were depicted after normalization against Lamin-B levels. *, p < 0.05 compared with the untreated control samples. +p < 0.05 compared between WT and IRAK-M deficient cells. (E) WT and IRAK-M deficient BMDMs were either untreated or treated with low dose LPS (50 pg/mL) for 2 h and subjected to a ChIP assay. The primers spanning the PPAR-response element region on the Sr-b1 promoter and the specified antibodies were used in the ChIP assays. Data are representative of three independent experiments.
We then examined the levels of SR-B1, ABCG1, and ABCA1 in WT and IRAK-M deficient macrophages. We observed that, as compared to the WT cells, the resting protein levels of SR-B1 and ABCG1 were significantly lower in IRAK-M deficient cells (Figure 6B). Low dose LPS further decreased the levels of SR-B1 and ABCG1. Similar effects were seen at the message level through real-time RT-PCR analyses (Figure 6C). The levels of ABCA1, on the other hand, were not significantly altered among WT and IRAK-M deficient macrophages, indicating that these exporter genes may be differentially regulated by IRAK-1 and IRAK-M.
To further determine the molecular mechanism, we measured the levels of RARα in WT and IRAK-M deficient macrophages treated with low dose LPS. As shown in Figure 6D, basal levels of RARα were significantly lower in IRAK-M deficient macrophages and were further decreased upon low dose LPS challenge. We also performed ChIP analyses to determine the recruitment of RARα to the SR-B1 promoter. As shown in Figure 6E, the recruitments of RARα to SR-B1 promoter were significantly reduced in IRAK-M deficient macrophages. Since RARα often form dimers with other nuclear receptors such as PPARα, and that PPARα is also implicated in the transcription of SR-B1 34-36, we further examined the recruitment of PPARα to the promoter of Sr-b1 in WT and IRAK-M deficient cells. Consistently, we observed that low dose LPS caused a dramatic decrease in the association of PPARα to the Sr-b1 promoter in WT cells. On the other hand, the basal levels of PPARα were significantly diminished in IRAK-M deficient cells.
Opposing effects of IRAK-1/Tollip and IRAK-M in the foam cell formation mediated by low dose LPS
To determine the functional consequence of our mechanistic observation, we performed foam cell formation assay using primary macrophages from WT, IRAK-1 deficient, Tollip deficient, and IRAK-M deficient mice. As shown in Figure 7, low dose LPS significantly increased foam cell formation in WT macrophages. In contrast, the effect of low dose LPS was ablated in either IRAK-1 or Tollip deficient macrophages. On the other hand, IRAK-M deficient macrophages exhibited elevated foam cell formation even without the treatment of LPS as compared to WT cells. Low dose LPS further elevated foam cell formation in IRAK-M deficient macrophages.
Figure 7. Low dose LPS induces foam cell formation dependent upon IRAK-1 and Tollip.
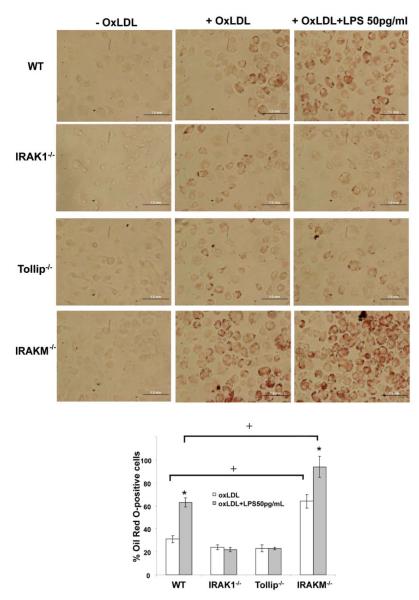
(A) WT, IRAK1 −/−, Tollip −/− and IRAKM −/− BMDMs were plated in 24-well plates and incubated with or without 50 μg/mL OxLDL to induce foam cell formation in the presence or absence of LPS 50 pg/mL. The cells were fixed with 10% phosphate buffered formalin and stained with filtered Oil Red O. Positively stained cells were observed through light microscope and images show representative Oil Red O-stained BMDMs. Magnification, 40X. Black bar represents 1.0 mm. (B) The Oil Red O positively stained foam cells were counted under different viewing fields, and their percentages out of total cells were represented in the figure. *p < 0.05 compared with the untreated sample. +p < 0.05 compared between WT and IRAK-M deficient cells.
Taken together, our data reveal a novel mechanism responsible for the suppression of cholesterol export in macrophages by subclinical dose of endotoxin (Figure 8).
Figure 8. An illustration of potential mechanisms responsible for the suppression of cholesterol exporters in macrophages by low dose LPS.
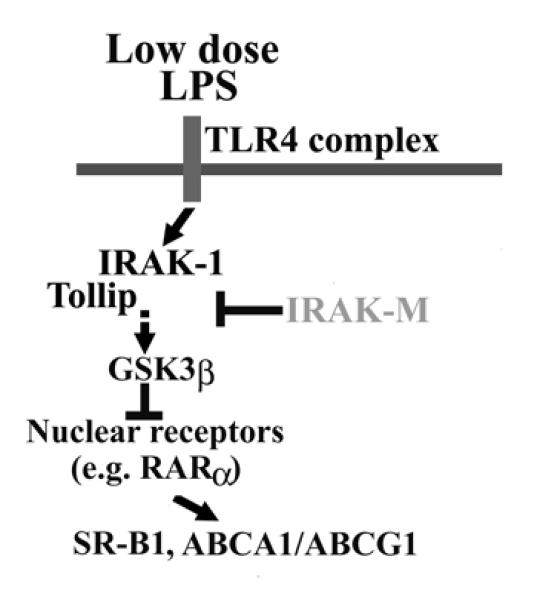
Low dose LPS reduces the expression of SR-B1, ABCA1/ABCG1 through reduction of RARα mediated by IRAK-1/Tollip-induced activation of GSK3β. IRAK-M, on the other hand, counter-acts such effect.
DISCUSSION
Our current report reveals not only the suppressive effects of low dose LPS on cholesterol efflux in macrophages, but also the underlying molecular mechanisms. First, we demonstrated that low dose LPS reduces the expression of key cholesterol exporters including SR-B1, ABCA1 and ABCG1 in macrophages, through an IRAK-1 and Tollip dependent mechanism. Second, IRAK-1 and Tollip suppresses their expression through GSK3β-mediated inhibition of nuclear receptors such as RARα. Third, IRAK-M serves as an antagonist for IRAK-1 and exerts opposing effects on the modulation of SR-B1 and cholesterol export in macrophages (Figure 7).
Our data provide a mechanism explaining the link between subclinical low dose endotoxemia and atherosclerosis. Recent human and animal studies indicate that slightly elevated levels of circulating endotoxin are closely associated with risks of atherosclerosis 7, 9, 37-41. To this regard, it is worth to note that macrophages can be programmed to opposing phenotypes by low versus high dose LPS 42, 43. High dose endotoxin can induce both pro-inflammatory mediators and compensatory anti-inflammatory mediators 44. As a consequence, a transient and resolving inflammatory response may ensue following an acute high dose LPS challenge. In contrast, low dose endotoxin preferentially skews toward mild expression of pro-inflammatory mediators, which may lead to low-grade yet non-resolving inflammatory complications 45. In addition to the chronic skewing of pro-inflammatory environment, we herein demonstrate that subclinical low-dose endotoxin also potently suppresses the expression of key cholesterol exporters including SR-B1, ABCA1/ABCG1, and consequently reduces cholesterol efflux from macrophages. Our functional study further demonstrates increased foam cell formation in macrophages treated with low dose LPS. In consistent with our observation, a previous study demonstrates that double deletion of SR-B1 and ABCA1 further compromises cholesterol efflux in macrophages, significantly induces foam cell formation, and dramatically exacerbates the pathogenesis of atherosclerotic plaques 46.
This is in contrast to the effects of high dose LPS (>100ng/mL), which may either suppress or activate ABCA1 expression depending upon different experimental settings 24, 47. Likewise, high levels of lipids and lipoproteins in humans can also induce the expression of both pro-inflammatory mediators as well as anti-inflammatory and lipid-lowering molecules such as ABCA1 48. This conflicting paradigm developed by the host may be a controlled compensatory response to fine-tune the pro- and anti-inflammatory balance 42, 49. Our study reveals that subclinical low-dose LPS may pose a much severe danger by potently suppressing the anti-inflammatory cholesterol exporters from macrophages, thus depriving the compensatory mechanism to maintain proper homeostasis and resolve inflammation.
Our report underscores the significant role that IRAK-1 and Tollip plays in mediating the effect of low dose LPS. Although both molecules were previously shown to physically associate with LPS receptor TLR4, their functional roles are largely not well defined. IRAK-1 seems to be redundant with either IRAK-2 or IRAK-4 when cells are challenged with a high dose LPS or IL-1β 50, 51. The effect of Tollip is also minimal in cells treated with high dose LPS 26. Instead, our data clearly indicate that IRAK-1 and Tollip are essential for suppressing cholesterol efflux from macrophages treated with subclinical low dose LPS. It is likely that additional regulatory mechanisms other than IRAK-1 and Tollip may be triggered in cells treated with high dose LPS. These additional mechanisms may be involved in the compensatory modulation of macrophage activation and homeostasis.
Our finding reveals that IRAK-1 and Tollip modulate the effect of low dose LPS through GSK3β. This is consistent with previous report that GSK3β is closely associated with the pro-inflammatory skewing of macrophages in vitro and in vivo32, 52. GSK3β is also shown to contribute to the phosphorylation and proteasome-mediated degradation of PPARα and RARα 31, 53. This is in agreement with our finding that low dose LPS suppresses RARα through IRAK-1/Tollip-mediated activation of GSK3β. Our data indicate that IRAK-1 and Tollip sit upstream of GSK3β and are responsible for the tyrosine phosphorylation of GSK3β induced by low dose LPS. However, it is not clear whether IRAK-1 may either directly or indirectly contribute to GSK3β phosphorylation. Given the fact that IRAK-1 is a putative Serine/Threonine kinase, it is likely that IRAK-1 may indirectly lead to GSK3β Tyr216 phosphorylation through another un-known intermediate kinase. Future works are clearly warranted to clarify this important connection.
IRAK-M is known as a general inhibitor of IRAK-1 function 33, 54. Extending upon these observations, our data reveal that IRAK-M counter-acts IRAK-1 and selectively sustains the levels and activities of RARα, as well as the expression of cholesterol exporters SR-B1, ABCA1/ABCG1. Based on our mechanistic findings, it is likely to posit that IRAK-M is beneficial in attenuating the pathogenesis of atherosclerosis and foam cell formation. Indeed, our preliminary data indicate ApoE−/−/IRAK-M−/− mice develop much severe atherosclerosis as compared to ApoE−/− mice when fed with a high fat diet. On the other hand, we previously published that ApoE−/−/IRAK-1−/− mice have attenuated development of atherosclerosis as compared to ApoE−/− mice 55.
This study is limited to the pre-conditioning of macrophages by low dose LPS, and the modulation of signaling pathways responsible for the reduced expression of cholesterol transporters in naïve macrophages. To our knowledge, this is the first attempt to reveal the intracellular signaling network responsible for the novel activation of GSK3β, and suppression of nuclear receptors involved in the expression of ABCA1, ABCG1, and SR-B1 by low dose LPS. The dynamic competition among IRAK-1 and IRAK-M plays a key role in mediating the effect of low dose LPS. It is worth noting that there may be differences involved in the regulation of these transporters in macrophages by low dose LPS. As shown in our data, although IRAK-1 and Tollip are involved in the suppression of ABCA1, ABCG1, and SR-B1, IRAK-M is selectively involved in the suppression of SR-B1 and ABCG1. Other unknown intracellular molecules may also be involved in the differential regulation of these transporters.
In addition to cholesterol transport, low dose LPS may also affect the expression of additional modulators that can potentially affect cholesterol metabolism and transport. Although not a focus of this work, we performed limited study and examined the modulation of ApoE expression by low and high dose LPS. Although high dose LPS (100 ng/mL) suppresses the expression of ApoE, low dose LPS fails to affect ApoE expression (Supplementary Figure I). Future detailed biochemical analyses are required to further dissect the molecular networks involved in the unique regulation of these genes involved in cholesterol metabolism and transport in macrophages.
It is important to note that macrophages can not only be pre-conditioned by low dose LPS, but may also be pre-programmed by other distinct pro-inflammatory stimulants and mediators. For example, macrophages can be pre-programmed by interferon gamma, leptin, IL-4 and adopt either a pro-inflammatory or anti-inflammatory state 56-58. It remains to be tested whether cholesterol, oxLDL, or other lipid mediators may also capable of pre-conditioning macrophages, and affect their subsequent responses to inflammatory signals such as LPS. Conceivably, macrophages are dynamically modulated depending upon the timing and threshold of simultaneous or sequential challenges of multiple stimulants. The modulation of molecules involved in the TLR4 pathway has not been carefully studied and should be systematically examined in the future both in vitro and in vivo. To this regard, we have attempted a systems biology approach employing computational tools to define the dynamic priming and tolerance of macrophages, as well as skewing of T helper cells to distinct states 59, 60. Future single-cell analyses with detailed live-cell kinetics are required to further define this complex yet highly important dynamics in macrophages.
Our in vitro mechanistic data are consistent with in vivo studies published by our group and others. IRAK-1 or IRAK-4 deletion in ApoE deficient background led to reduced atherosclerosis in mice 55, 61. GSK3β activation was recently shown to be critically involved in the pathogenesis of atherosclerosis 62. Known GSK3β inhibitors and emerging small-compound inhibitors for IRAK-1 may serve as attractive candidates for the development of effective drugs for the treatment of atherosclerosis. Future in vivo studies in animals and humans are warranted to define the involvement of Tollip and IRAK-M during the pathogenesis of atherosclerosis associated with low-dose endotoxemia and low-grade inflammation.
Taken together, our signaling study uncovers a dynamic and competing intra-cellular circuit involved in modulating cholesterol efflux in macrophages. Given its dynamic and competing nature, we postulate that host macrophages may either undergo proper cholesterol export or be programmed into non-resolving foam cells when challenged with varying dosages of LPS or other stimulants. Our signaling study sets the stage for future functional works in defining the dynamic modulation of cholesterol efflux from macrophages, and reveals potential molecular targets for the effective intervention of atherosclerosis.
Supplementary Material
Acknowledgments
Sources of Funding This study was supported by funds from the National Institute of Health NIH R01 HL115835, American Heart Association, and Virginia Tech to L.L.
Footnotes
Disclosures None
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet. 2006;367:1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 2.Stone NJ. The clinical and economic significance of atherosclerosis. Am J Med. 1996;101:4A6S–9S. [PubMed] [Google Scholar]
- 3.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: A report from the american heart association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 5.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: A report from the american heart association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 7.Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, Gonen A, Butler S, Witztum JL, Glass CK, Miller YI. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappab and activator protein-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2010;107:56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, Tabita-Martinez J, Sellers KF, Rickels MR, Reilly MP. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2010;59:172–181. doi: 10.2337/db09-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86:1286–1292. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- 10.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goto T, Eden S, Nordenstam G, Sundh V, Svanborg-Eden C, Mattsby-Baltzer I. Endotoxin levels in sera of elderly individuals. Clin Diagn Lab Immunol. 1994;1:684–688. doi: 10.1128/cdli.1.6.684-688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Attas OS, Al-Daghri NM, Al-Rubeaan K, da Silva NF, Sabico SL, Kumar S, McTernan PG, Harte AL. Changes in endotoxin levels in t2dm subjects on anti-diabetic therapies. Cardiovasc Diabetol. 2009;8:20. doi: 10.1186/1475-2840-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maitra U, Gan L, Chang S, Li L. Low-dose endotoxin induces inflammation by selectively removing nuclear receptors and activating ccaat/enhancer-binding protein δ. J Immunol. 2011;186:4467–4473. doi: 10.4049/jimmunol.1003300. [DOI] [PubMed] [Google Scholar]
- 14.Maitra U, Deng H, Glaros T, Baker B, Capelluto DG, Li Z, Li L. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol. 2012;189:1014–1023. doi: 10.4049/jimmunol.1200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirohashi N, Morrison DC. Low-dose lipopolysaccharide (lps) pretreatment of mouse macrophages modulates lps-dependent interleukin-6 production in vitro. Infect Immun. 1996;64:1011–1015. doi: 10.1128/iai.64.3.1011-1015.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shnyra A, Brewington R, Alipio A, Amura C, Morrison DC. Reprogramming of lipopolysaccharide-primed macrophages is controlled by a counterbalanced production of il-10 and il-12. J Immunol. 1998;160:3729–3736. [PubMed] [Google Scholar]
- 17.Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS. Macrophage abca1 reduces myd88-dependent toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–3206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng H, Guo L, Wang D, Gao H, Hou G, Zheng Z, Ai J, Foreman O, Daugherty A, Li XA. Deficiency of scavenger receptor bi leads to impaired lymphocyte homeostasis and autoimmune disorders in mice. Arterioscler Thromb Vasc Biol. 2011;31:2543–2551. doi: 10.1161/ATVBAHA.111.234716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fitzgerald ML, Mujawar Z, Tamehiro N. Abc transporters, atherosclerosis and inflammation. Atherosclerosis. 2010;211:361–370. doi: 10.1016/j.atherosclerosis.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor lxr alpha. Proc Natl Acad Sci U S A. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costet P, Lalanne F, Gerbod-Giannone MC, Molina JR, Fu X, Lund EG, Gudas LJ, Tall AR. Retinoic acid receptor-mediated induction of abca1 in macrophages. Mol Cell Biol. 2003;23:7756–7766. doi: 10.1128/MCB.23.21.7756-7766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baranova I, Vishnyakova T, Bocharov A, Chen Z, Remaley AT, Stonik J, Eggerman TL, Patterson AP. Lipopolysaccharide down regulates both scavenger receptor b1 and atp binding cassette transporter a1 in raw cells. Infect Immun. 2002;70:2995–3003. doi: 10.1128/IAI.70.6.2995-3003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maitra U, Parks JS, Li L. An innate immunity signaling process suppresses macrophage abca1 expression through irak-1-mediated downregulation of retinoic acid receptor alpha and nfatc2. Mol Cell Biol. 2009;29:5989–5997. doi: 10.1128/MCB.00541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between lxr and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 25.Feingold KR, Wang Y, Moser A, Shigenaga JK, Grunfeld C. Lps decreases fatty acid oxidation and nuclear hormone receptors in the kidney. J Lipid Res. 2008;49:2179–2187. doi: 10.1194/jlr.M800233-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Didierlaurent A, Brissoni B, Velin D, Aebi N, Tardivel A, Kaslin E, Sirard JC, Angelov G, Tschopp J, Burns K. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol Cell Biol. 2006;26:735–742. doi: 10.1128/MCB.26.3.735-742.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. Myd88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 28.Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A ppar gamma-lxr-abca1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 29.Brown K, Chen Y, Underhill TM, Mymryk JS, Torchia J. The coactivator p/cip/src-3 facilitates retinoic acid receptor signaling via recruitment of gcn5. J Biol Chem. 2003;278:39402–39412. doi: 10.1074/jbc.M307832200. [DOI] [PubMed] [Google Scholar]
- 30.Wu RC, Feng Q, Lonard DM, O’Malley BW. Src-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129:1125–1140. doi: 10.1016/j.cell.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 31.Si J, Mueller L, Collins SJ. Gsk3 inhibitors enhance retinoic acid receptor activity and induce the differentiation of retinoic acid-sensitive myeloid leukemia cells. Leukemia. 2011;25:1914–1918. doi: 10.1038/leu.2011.171. [DOI] [PubMed] [Google Scholar]
- 32.Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr., Medzhitov R, Flavell RA. Irak-m is a negative regulator of toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 34.Akita N, Tsujita M, Yokota T, Gonzalez FJ, Ohte N, Kimura G, Yokoyama S. High density lipoprotein turnover is dependent on peroxisome proliferator-activated receptor alpha in mice. J Atheroscler Thromb. 2010;17:1149–1159. doi: 10.5551/jat.4820. [DOI] [PubMed] [Google Scholar]
- 35.Genolet R, Wahli W, Michalik L. Ppars as drug targets to modulate inflammatory responses? Curr Drug Targets Inflamm Allergy. 2004;3:361–375. doi: 10.2174/1568010042634578. [DOI] [PubMed] [Google Scholar]
- 36.Lazennec G, Canaple L, Saugy D, Wahli W. Activation of peroxisome proliferator-activated receptors (ppars) by their ligands and protein kinase a activators. Mol Endocrinol. 2000;14:1962–1975. doi: 10.1210/mend.14.12.0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laugerette F, Vors C, Geloen A, Chauvin MA, Soulage C, Lambert-Porcheron S, Peretti N, Alligier M, Burcelin R, Laville M, Vidal H, Michalski MC. Emulsified lipids increase endotoxemia: Possible role in early postprandial low-grade inflammation. J Nutr Biochem. 2011;22:53–59. doi: 10.1016/j.jnutbio.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 38.Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. doi: 10.2174/092986708784872393. [DOI] [PubMed] [Google Scholar]
- 39.Kallio KA, Buhlin K, Jauhiainen M, Keva R, Tuomainen AM, Klinge B, Gustafsson A, Pussinen PJ. Lipopolysaccharide associates with pro-atherogenic lipoproteins in periodontitis patients. Innate Immun. 2008;14:247–253. doi: 10.1177/1753425908095130. [DOI] [PubMed] [Google Scholar]
- 40.Szeto CC, Kwan BC, Chow KM, Lai KB, Chung KY, Leung CB, Li PK. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin J Am Soc Nephrol. 2008;3:431–436. doi: 10.2215/CJN.03600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, Willeit J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: Prospective results from the bruneck study. J Am Coll Cardiol. 1999;34:1975–1981. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 42.Morris M, Li L. Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp (Warsz) 2012;60:13–18. doi: 10.1007/s00005-011-0155-9. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Morrison DC. Lipopolysaccharide-induced selective priming effects on tumor necrosis factor alpha and nitric oxide production in mouse peritoneal macrophages. J Exp Med. 1993;177:511–516. doi: 10.1084/jem.177.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (m2) in human mononuclear cells. J Immunol. 2011;186:7243–7254. doi: 10.4049/jimmunol.1001952. [DOI] [PubMed] [Google Scholar]
- 45.Lassenius MI, Pietilainen KH, Kaartinen K, Pussinen PJ, Syrjanen J, Forsblom C, Porsti I, Rissanen A, Kaprio J, Mustonen J, Groop PH, Lehto M. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34:1809–1815. doi: 10.2337/dc10-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Y, Pennings M, Hildebrand RB, Ye D, Calpe-Berdiel L, Out R, Kjerrulf M, Hurt-Camejo E, Groen AK, Hoekstra M, Jessup W, Chimini G, Van Berkel TJ, Van Eck M. Enhanced foam cell formation, atherosclerotic lesion development, and inflammation by combined deletion of abca1 and sr-bi in bone marrow-derived cells in ldl receptor knockout mice on western-type diet. Circ Res. 2010;107:e20–31. doi: 10.1161/CIRCRESAHA.110.226282. [DOI] [PubMed] [Google Scholar]
- 47.Thompson PA, Gauthier KC, Varley AW, Kitchens RL. Abca1 promotes the efflux of bacterial lps from macrophages and accelerates recovery from lps-induced tolerance. J Lipid Res. 2010;51:2672–2685. doi: 10.1194/jlr.M007435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smoak KA, Aloor JJ, Madenspacher J, Merrick BA, Collins JB, Zhu X, Cavigiolio G, Oda MN, Parks JS, Fessler MB. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell Metab. 2010;11:493–502. doi: 10.1016/j.cmet.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, Vachharajani VT, Yoza BK, McCall CE. Fueling the flame: Bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012 doi: 10.1189/jlb.0212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li S, Strelow A, Fontana EJ, Wesche H. Irak-4: A novel member of the irak family with the properties of an irak-kinase. Proc Natl Acad Sci U S A. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yao J, Kim TW, Qin J, Jiang Z, Qian Y, Xiao H, Lu Y, Qian W, Gulen MF, Sizemore N, DiDonato J, Sato S, Akira S, Su B, Li X. Interleukin-1 (il-1)-induced tak1-dependent versus mekk3-dependent nfkappab activation pathways bifurcate at il-1 receptor-associated kinase modification. J Biol Chem. 2007;282:6075–6089. doi: 10.1074/jbc.M609039200. [DOI] [PubMed] [Google Scholar]
- 52.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. Ifn-gamma suppresses il-10 production and synergizes with tlr2 by regulating gsk3 and creb/ap-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 53.Gopinathan L, Hannon DB, Peters JM, Vanden Heuvel JP. Regulation of peroxisome proliferator-activated receptor-alpha by mdm2. Toxicol Sci. 2009;108:48–58. doi: 10.1093/toxsci/kfn260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, Standiford TJ. Sepsis-induced suppression of lung innate immunity is mediated by irak-m. J Clin Invest. 2006;116:2532–2542. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang D, Fasciano S, Li L. The interleukin-1 receptor associated kinase 1 contributes to the regulation of nfat. Mol Immunol. 2008;45:3902–3908. doi: 10.1016/j.molimm.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adib-Conquy M, Cavaillon JM. Gamma interferon and granulocyte/monocyte colony-stimulating factor prevent endotoxin tolerance in human monocytes by promoting interleukin-1 receptor-associated kinase expression and its association to myd88 and not by modulating tlr4 expression. J Biol Chem. 2002;277:27927–27934. doi: 10.1074/jbc.M200705200. [DOI] [PubMed] [Google Scholar]
- 57.Vaughan TL. L. Inflammatory complication of leptin. Molecular Immunology. 2010;47:2515–2518. doi: 10.1016/j.molimm.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 58.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 59.Fu Y, Glaros T, Zhu M, Wang P, Wu Z, Tyson JJ, Li L, Xing J. Network topologies and dynamics leading to endotoxin tolerance and priming in innate immune cells. PLoS Comput Biol. 2012;8:e1002526. doi: 10.1371/journal.pcbi.1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong TX,J, Li L, Tyson J. A mathematical model for the reciprocal differentiation of t helper 17 cells and induced regulatory t cells. pLoS Computational Biology. 2011;7:e1002122. doi: 10.1371/journal.pcbi.1002122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim TW, Febbraio M, Robinet P, Dugar B, Greene D, Cerny A, Latz E, Gilmour R, Staschke K, Chisolm G, Fox PL, DiCorleto PE, Smith JD, Li X. The critical role of il-1 receptor-associated kinase 4-mediated nf-kappab activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J Immunol. 2011;186:2871–2880. doi: 10.4049/jimmunol.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McAlpine CS, Bowes AJ, Khan MI, Shi Y, Werstuck GH. Endoplasmic reticulum stress and glycogen synthase kinase-3beta activation in apolipoprotein e-deficient mouse models of accelerated atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:82–91. doi: 10.1161/ATVBAHA.111.237941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.