

Abstract
The sympathetic nervous system regulates many essential physiological systems, and its dysfunction is implicated in cardiovascular diseases. Mechanisms that control the strength of sympathetic output are therefore potential targets for the management of these disorders. Here we show that neurotrophins rapidly potentiate cholinergic transmission between cultured rat sympathetic neurons. We found that brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), acting at the p75 receptor, increased the amplitude of excitatory postsynaptic currents (EPSCs). We observed increased amplitude but not frequency of miniature synaptic currents after p75 activation, suggesting that p75 acts postsynaptically to modulate transmission at these synapses. This neurotrophic modulation enhances cholinergic EPSCs via sphingolipid signaling. Application of sphingolactone-24, an inhibitor of neutral sphingomyelinase, blocked the effect of BDNF, implicating a sphingolipid pathway. Furthermore, application of the p75-associated sphingolipid second messengers C2-ceramide and d-erythro-sphingosine restricted to the postsynaptic cell mimicked BDNF application. Postsynaptic blockade of ceramide production with fumonisin, a ceramide synthase inhibitor, blocked the effects of BDNF and d-erythro-sphingosine, implicating ceramide or ceramide phosphate as the active signal. Together these data suggest that neurotrophin signaling, which occurs in vivo via release from sympathetic neurons and target tissues such as the heart, acutely regulates the strength of the sympathetic postganglionic response to central cholinergic inputs. This pathway provides a potential mechanism for modulating the strength of sympathetic drive to target organs such as the heart and could play a role in the development of cardiovascular diseases.
Keywords: nerve growth factor, TrkA, cardiac myocyte, cardiovascular disease
neurons convey information via synaptic transmission, with functional consequences that are highly dependent on neurotransmitter identity and synaptic strength. Neurotrophic factors regulate these synaptic properties in many neural systems, contributing to the plasticity that underlies critical functions such as learning and memory (for reviews see Black 1999; Carvalho et al. 2008; Lu 2004; Luther and Birren 2009a; Poo 2001). This form of adaptive plasticity is not limited to the neurons of the central nervous system; neurotrophin signaling regulates maturation and the synaptic properties of peripheral neurons, and it is implicated in cardiovascular diseases and other disorders.
Sympathetic neurons modulate cardiac function via release of norepinephrine onto their cardiac targets in vivo. When cultured together in vitro, sympathetic neurons also form synaptic connections onto cardiac myocytes and release norepinephrine in response to evoked activity, increasing myocyte beat rate (Conforti et al. 1991). Sympathetic neurotransmitter release at this synapse is regulated by neurotrophins through two classes of receptors: the tropomyosin-related receptor tyrosine kinases (Trk) and the p75 receptor. The p75 receptor interacts with all neurotrophins with similar affinity, while the different members of the Trk family have specific affinities to different neurotrophin family members (Reichardt 2006). Target-derived nerve growth factor (NGF), acting through the TrkA receptor, promotes the noradrenergic properties of sympathetic neurons (Chun and Patterson 1977; Lockhart et al. 1997) and regulates the number of postsynaptic contacts (Lockhart et al. 2000). Sympathetic neurons show neurotransmitter plasticity in culture and in vivo (Potter et al. 1986; Schotzinger and Landis 1988), and several factors, including leukemia inhibitory factor and ciliary neurotrophic factor, induce a switch to a cholinergic phenotype, which would be expected to greatly influence sympathetic regulation of target function (Habecker et al. 1997; Patterson and Chun 1977; Rohrer 1992; Slonimsky et al. 2003).
In addition to this long-term developmental regulation of peripheral sympathetic connections, neurotrophins acutely modulate the excitatory and inhibitory properties of sympathetic synapses onto cardiac myocytes through the opposing actions of TrkA and the p75 neurotrophin receptor (Lockhart et al. 1997; Yang et al. 2002). Activation of TrkA by NGF causes potentiation of excitatory noradrenergic transmission at neuron-myocyte synapses in cocultures of cardiac myocytes and sympathetic neurons (Lockhart et al. 1997; Yang et al. 2002), while activation of p75 promotes inhibitory acetylcholine release (Yang et al. 2002). Thus neurotrophic regulation of sympathetic synapses onto heart cells takes place both at the level of long-term changes in synaptic properties and at the level of acute presynaptic regulation of transmitter release.
Ultimately, sympathetic output in vivo is determined by the activity of excitatory cholinergic input from preganglionic spinal neurons and synaptic connections between sympathetic neurons themselves within peripheral ganglia (Kawai 1996; Kawai and Senba 1995; Ostberg et al. 1976). Neurotrophic signaling acts at a developmental timescale to regulate this output by modulating the number of preganglionic synapses onto sympathetic neurons (Causing et al. 1997; Sharma et al. 2010). Mice that overexpress brain-derived neurotrophic factor (BDNF) in sympathetic neurons show an increase in the density of preganglionic synapses onto neurons in superior cervical ganglia (SCGs), and this density is decreased in SCGs of BDNF-knockout mice (Causing et al. 1997). In addition, target-derived NGF, acting through TrkA, positively regulates neuron-to-neuron synapse formation in cultures of sympathetic neurons as well as in vivo and inhibits synapse formation via p75 activation (Sharma et al. 2010). Thus target-dependent neurotrophin signaling acts during development to regulate synapse formation both at the level of innervated somatic tissues such as the heart and blood vessels and at the level of central input driving sympathetic activity. However, while neurotrophin signaling plays a role in acutely regulating functional plasticity at peripheral targets, the question of whether rapid neurotrophin-dependent changes in sympathetic drive also take place at peripheral ganglia has not been addressed.
Here we use a model cell culture system that recapitulates many of the synaptic properties seen in the sympathetic nervous system in vivo to test the hypothesis that neurotrophic signaling regulates the strength of synaptic connections between sympathetic neurons. We show that neurotrophins rapidly potentiate transmission at neuron-to-neuron synapses that form between sympathetic neurons that are cocultured with cardiac myocytes. This suggests a role for target-derived neurotrophin signaling in the regulation of intraganglionic and possibly preganglionic transmission. Interestingly, this modulation takes place via p75 neurotrophin receptor activation, with no effect from TrkA signaling. We demonstrate that neurotrophic modulation of these cholinergic synapses involves a postsynaptic mechanism by showing that inhibition of downstream p75 signaling in the postsynaptic cell blocks BDNF potentiation and activation of p75 second messenger cascades in the postsynaptic cell mimics the effects of BDNF application. We cannot rule out a potential presynaptic contribution to this signaling that may work in concert with the observed postsynaptic mechanism. Synaptic modulation at these neuronal synapses is distinct from neurotrophic modulation of sympathetic synapses onto cardiac targets, where effects on synaptic transmission are presynaptic and modulated via both TrkA and p75 signaling (Locke and Nerbonne 1997; Yang et al. 2002). Pathological changes in target neurotrophin signaling and sympathetic activity are implicated in cardiovascular diseases (Govoni et al. 2011); thus this work suggests a potential link between these neurotrophic pathways, regulation of sympathetic activation, and heart disease.
METHODS
Cell culture.
All protocols involving animals were approved by the Brandeis University Institutional Animal Care and Use Committee. Cell culture was performed as described previously (Lockhart et al. 1997; Luther and Birren 2006). Superior cervical sympathetic ganglia were dissected from postnatal rats (P0 to P2) and incubated at 37°C for 1 h in minimum essential medium (GIBCO BRL, Invitrogen, Carlsbad, CA) containing 325 U/ml collagenase (Worthington, Lakewood, NJ) and 5.5 U/ml dispase (GIBCO BRL). Cardiac myocytes were isolated by removing the bottom third of heart ventricles and chopping them finely with small scissors. Heart tissue was incubated in a buffered saline solution containing 365 U/ml collagenase (Worthington) for 1 h at 37°C. The buffered saline solution for collection and incubation of heart tissue contained (in mM) 116.5 NaCl, 19.8 HEPES, 1 NaH2PO4, 5.6 glucose, 5.4 KCl, and 1 MgSO4 and was adjusted to pH 7.4 with NaOH. The myocytes and neurons were dispersed by passing repeatedly through fire-polished glass pipettes and, after removal of enzyme, were transferred to separate uncoated plastic culture dishes and incubated for 2 h at 37°C. The less adherent sympathetic neurons and myocytes were then rinsed from the dishes with growth medium and counted. Neurons were plated at 10,000 cells per dish and myocytes were plated at 50,000 cells per dish on glass-bottomed plates (MatTek, Ashland, MA) coated with collagen (50 μg/ml; BD Biosciences, Bedford, MA) and mouse laminin (5 μg/ml; BD Biosciences). Cultures were maintained in modified L15CO2 medium (Hawrot and Patterson 1979; Lockhart et al. 1997) supplemented with 10% fetal bovine serum (Omega Scientific, Tarzana, CA), 6 μg/ml dextrose, 2 mM glutamine (Invitrogen), 100 U/ml penicillin-100 μg/ml streptomycin (Invitrogen), 1 μg/ml 6,7-dimethyl-5,6,7,8-tetrahydropterine (DMPH4, Calbiochem, San Diego, CA), 5 μg/ml glutathione (Sigma, St. Louis, MO), and 100 μg/ml l-ascorbic acid. NGF (5 ng/ml, Upstate, Lake Placid, NY) was added to support neuronal survival, and 1 μM cytosine arabinofuranoside (Ara-C, Sigma) was added to inhibit cell division. Half of the medium was exchanged with fresh NGF-containing growth medium three times weekly.
Electrophysiology.
Synaptic recordings were made with an Axopatch 200B amplifier (Axon Instruments, Union City, CA). Patch electrodes (2–4 MΩ) were pulled from borosilicate glass (Warner Instruments, Hamden, CT) on a model P-97 Flaming/Brown horizontal micropipette puller (Sutter Instrument, Novato, CA). Data were acquired with the pCLAMP8 software suite and a Digidata 1320A digitizer board (Axon Instruments) and were digitized at 10 kHz and low-pass filtered at 2 kHz. Recordings were made at a holding potential of −60 mV, which is near the normal resting potential for these cells. Recordings of miniature synaptic events were made at 35–36°C with a QE-1 heated culture dish platform (Warner Instruments); all other recordings were made at 25°C. Recordings were made in external saline solution consisting of (in mM) 150 NaCl, 3 KCl, 2 MgCl2, 5 Na-HEPES, 2 CaCl2, and 11 dextrose, pH 7.4 and adjusted to 330 mosmol/kgH2O with sucrose. The pipette solution contained (in mM) 100 K gluconate, 30 KCl, 1 MgSO4, 1 CaCl2, 10 EGTA, 10 HEPES, 3 Na, and 2 K2ATP, pH 7.4 and adjusted to 290 mosmol/kgH2O with sucrose.
Series resistance (Rs) was monitored throughout recordings but not compensated. Since changes in Rs could systematically alter recorded synaptic current [excitatory postsynaptic current (EPSC)] amplitude by introducing voltage-clamp errors, we examined Rs in a group of six control cells that showed variation of Rs throughout recording. In control recordings we found no systematic relationship between EPSC amplitude and Rs at normally observed values. The mean EPSC amplitude was −54 ± 6 pA at low 12 ± 2 MΩ Rs versus −58 ± 7 pA at high 32 ± 3 MΩ Rs values (amplitude not significant, Rs P < 0.001, paired t-test). These data do not preclude that Rs could affect measured EPSC kinetic values, but they allow us to draw conclusions about EPSC amplitude over the Rs values included for analysis. Data from cells with Rs >40 MΩ were discarded, although Rs was generally much lower, with a mean value of 21 ± 1 MΩ at the end of recording (n = 100 cells). Neurons with membrane potentials depolarized above −47 mV were not included for analysis (Luther and Birren 2009b). Unitary synaptic events were detected with the Clampfit template search function and were scanned by eye to eliminate overlapping compound synaptic events and action currents. For histogram plots, equal numbers of events from each cell for each experimental condition (specified in Figs. 2–4, 6, 7) were chosen randomly from the collected data and compared with control. Data were randomized by aligning to a list of random numbers generated in a Microsoft Excel spreadsheet. We found variability in the baseline amplitude of spontaneous cholinergic EPSCs between batches of cultures (ranging approximately from −40 pA to −100 pA). However, we used each cell as its own internal control, allowing analysis between experimental conditions. Furthermore, even in culture sets with high baseline amplitude we still observed significant enhancement of EPSC amplitude after intracellular sphingolipid application (data not shown), implying that all cultures were examined within a dynamic range of activity.
Fig. 2.

Brain-derived neurotrophic factor (BDNF) increased cholinergic EPSC amplitude. A: voltage-clamp traces recorded at −60 mV showing unitary synaptic currents recorded from a cultured sympathetic neuron. Synaptic currents were larger after 15-min application of 100 ng/ml BDNF (right). B: cumulative percent histogram plot showing a shift toward larger amplitudes of EPSCs after BDNF application (n = 11 cells; 550 and 616 EPSCs for control and BDNF, respectively; P < 0.001). The EPSC potentiation is also illustrated in a bar plot of mean amplitude (inset; −67.4 ± 3.2 pA vs. −90 ± 13 pA for control and BDNF, respectively, n = 11, ***P < 0.001). C: cumulative histogram plot showing that the p75 function-blocking antibody REX blocked the BDNF-induced EPSC enhancement (n = 5 cells; 300 and 255 EPSCs for control and BDNF/REX, respectively). EPSC amplitudes were not altered by BDNF application after incubation with the REX antibody as shown by a bar plot (inset; −53 ± 5 pA vs. −58.7 ± 3.5 pA for control and BDNF/REX conditions, respectively, n = 5). D: cumulative percent histogram plot showing no increase in EPSC amplitude after 10- to 15-min application of saline without BDNF, suggesting that the EPSC amplitude increase was not due to a time-dependent recording artifact (n = 11 cells; 517 and 550 EPSCs for control and saline, respectively, P = 0.17). No difference was seen in mean EPSC amplitudes before and after saline application as shown by a bar plot (inset; −67 ± 8 pA vs. −69 ± 8 pA for control and saline conditions, respectively, n = 11).
Fig. 3.

Nerve growth factor (NGF) potentiated EPSC amplitude via p75 activation. A: the neurotrophin NGF potentiated EPSC amplitude as seen in the shift of the cumulative percent histogram plot for NGF compared with control (n = 6 cells; 294 and 330 EPSCs for control and NGF, respectively, P < 0.001). Mean amplitudes were also significantly different as seen in a bar plot (inset; −61.5 ± 6.3 pA vs. −72.4 ± 8.2 pA for control and after application of 50 ng/ml NGF, respectively; n = 6; **P = 0.005). B: NGF/αNGF30, which blocks NGF activation of p75, did not alter the EPSC amplitude as seen in the cumulative percent histogram plot (n = 8 cells, 440 and 408 EPSCs for control and after NGF/αNGF, respectively) and the bar plot of mean EPSC amplitude (inset; −52 ± 4 pA vs. −49.6 ± 3.9 pA for control and NGF/αNGF, respectively, n = 8). C: coapplication of NGF with the anti-NGF antibody αNGF30 blocks NGF activation of p75, resulting in only TrkA activation. We have previously shown that TrkA increases sympathetic tonic firing and spike output. A plot of spike output vs. stimulus amplitude (normalized to threshold) shows that NGF/αNGF30 (molar ratio 1:2 NGF/αNGF30, 50 ng/ml NGF) caused an increase in spike output over the amplitude range tested (n = 24 and n = 10 for saline control and NGF/αNGF30, respectively; P < 0.001, 2-way ANOVA). NGF/αNGF30 promoted tonic firing as seen by a plot of % of spikes that occurred in the second half of the stimulus, a measurement that we previously have shown represents firing pattern (inset; n = 24 and n = 10 for saline control and NGF/αNGF30, respectively; ***P < 0.001, t-test).
Fig. 4.

Application of BDNF caused an increase in amplitude but did not influence the frequency of miniature (m)EPSCs. A: miniature cholinergic synaptic currents were recorded in the presence of 500 nM tetrodotoxin to block action potentials and at 35–36°C to increase mEPSC frequency. Voltage-clamp traces recorded from a sympathetic neuron at −60 mV show that application of 100 ng/ml BDNF increased the amplitude of mEPSCs. B: cumulative percent histogram plot showing that mEPSC amplitude distribution was shifted toward larger values in BDNF (n = 6 cells; 120 and 102 EPSCs for control and BDNF, respectively, P < 0.01). The mEPSC amplitude increase is also evident in a bar plot of mean amplitude (inset; −56 ± 14 pA vs. −67 ± 14 pA for control and BDNF, respectively, n = 6, *P = 0.01). C: BDNF did not influence mEPSC frequency as seen in a cumulative percent histogram plot and a bar plot of mean values (inset) (n = 6 cells; 114 and 96 data points for control and BDNF and 3.7 ± 0.9 Hz vs. 3.3 ± 1.1 Hz for control and BDNF, respectively).
Fig. 6.

Pharmacological blockade of sphingolipid signaling occluded the effect of BDNF on EPSC amplitude. A: cumulative percent histogram plot showing that the neutral sphingomyelinase inhibitor 25 μM sphingolactone-24 blocked the BDNF enhancement of EPSC amplitude (n = 6 cells; 606 and 306 EPSCs for sphingolactone-24 and sphingolactone-24/BDNF, respectively). This is also seen in a mean amplitude bar plot (inset; −128.6 ± 21.3 pA vs. −120.3 ± 23.6 pA for sphingolactone-24 and sphingolactone-24/BDNF, respectively; n = 6). B: cumulative percent histogram plot showing that postsynaptically applied fumonisin B1 (10 μM), a blocker of endogenous ceramide production, blocked the ability of BDNF to increase EPSC amplitude (n = 8 cells; 384 and 336 EPSCs for fumonisin and fumonisin/BDNF, respectively). This is also reflected in a bar plot of mean amplitudes before and after a 10- to 15-min 100 ng/ml BDNF application in the presence of postsynaptic fumonisin B1 (inset; −43 ± 4 pA vs. −45.8 ± 6.5 pA for control and BDNF, respectively, n = 8). C: plot of spike output vs. stimulus amplitude (normalized to threshold) showing that cytosolic fumonisin B1 application (10 μM) caused an increase in spike discharge in response to square-wave current injections and blocked the previously reported BDNF-dependent decrease in spike output (n = 24 for saline control, n = 5 for internal fumonisin B1 alone, and n = 11 for internal fumonisin B1 and bath-applied 100 ng/ml BDNF; P < 0.001, 2-way ANOVA, saline was different from both other groups). Inset: current-clamp trace showing large overshooting action potentials generated in response to a 400-pA current injection (500% threshold for this cell) in a cell 40 min after establishing the whole cell recording with fumonisin B1 in the recording electrode, suggesting that cells remained healthy during cytosolic fumonisin perfusion.
Fig. 7.

Selective application of C2-ceramide or d-erythro-sphingosine in the postsynaptic cell mimicked the effects of bath-applied BDNF. A: inclusion of 25 μM C2-ceramide or 1 μM d-erythro-sphingosine in the recording pipette delivered the compound to the postsynaptic cell via cytosolic dialysis. Left: cytosolic application of vehicle alone did not effect EPSC amplitude as seen in a bar plot of mean amplitudes (−53.3 ± 8.4 pA vs. −54 ± 5 pA for <10 min and 15–30 min after going whole cell, respectively, n = 8). Center: bar plot of mean EPSC amplitude shows that postsynaptic C2-ceramide caused a time-dependent amplitude increase when amplitude was measured within the first 10 min of the recording compared with 15–30 min after the establishment of the recording (−71.5 ± 18.8 pA vs. −92 ± 24 pA for <10 min and 15–30 min after going whole cell, respectively; n = 8, *P = 0.026). Right: sphingosine significantly increased EPSC amplitude when applied intracellularly (−64 ± 6 pA vs. −94 ± 17 pA for <10 min and 15–30 min after going whole cell, respectively; n = 7; *P = 0.041). B and C: cumulative percent histogram plots showing after 15–30 min of cytosolic application of C2-ceramide (B; n = 8 cells; 408 and 440 EPSCs for <10 and >15 min C2-ceramide, respectively) or sphingosine (C; n = 7; 574 and 357 EPSCs for <10 and >15 min sphingosine, respectively) increased EPSC amplitude compared with events within the first 10 min of recording, evident as a shift in the plot (P < 0.001 for both groups).
Baseline control activity was recorded for 10–15 min. Pharmacological agents were applied for 10–15 min, after which data were collected for the experimental condition for another 10–15 min. We did not see evidence for a washout of the BDNF effect in cells recorded for up to 15 min after removal of the neurotrophin; however, this does not rule out the possibility that a longer wash period would show reversal of the drug effect. We therefore also performed control experiments using saline solution applied over the same time course and compared synaptic events from the first 10–15 min of recording to events recorded after 30 min to rule out a possible effect of time of recording due to internal cytosolic dialysis.
Pharmacological agents.
The nicotinic cholinergic antagonist hexamethonium bromide was obtained from Sigma and diluted directly in recording saline solution to 100 μM. NGF (Upstate) and BDNF (provided by Regeneron Pharmaceuticals, Tarrytown, NY) were divided into aliquots and frozen in growth medium and added directly to the external saline solution at a final concentration of 50 or 100 ng/ml, respectively. The irreversible inhibitor of neutral sphingomyelinase (nSMASE), sphingolactone-24, was dissolved to 25 mM in DMSO and then diluted to 25 μM directly into medium or physiological saline solution (Wascholowski and Giannis 2006). We preincubated cultures in growth medium containing 25 μM sphingolactone-24 for 1–2 h prior to recording. C2-ceramide (Enzo Life Sciences, Farmingdale, NY) was dissolved to 200 mM in DMSO, complexed 1:1 with fatty acid-free bovine serum albumin (Sigma) to prevent precipitation, and then diluted to a final concentration of 25 μM (of C2-ceramide, 0.01% DMSO) in recording or internal pipette solution. We found no significant effect of the solvent/bovine serum albumin vehicle on synaptic activity (see results). To selectively activate the TrkA signaling pathway, we used a monoclonal antibody directed against NGF that blocks activation of p75 without affecting NGF-dependent TrkA activation. A complex of molar ratio 2:1 (αNGF30-NGF, 50 ng/ml NGF) was applied dissolved in external saline solution. d-Erythro-sphingosine was purchased from Avanti Polar Lipids (Alabaster, AL), dissolved in DMSO, stored frozen, and then diluted into internal pipette solution (1 μM and 0.025% DMSO final dilution). The NGF30 monoclonal antibody was kindly provided to us by Dr. Uri Saragovi (Saragovi et al. 1998). To block the activity of p75 we used the function-blocking antibody REX diluted 1:500 in recording saline, kindly provided by Dr. Louis Reichardt (Luther and Birren 2009b; Weskamp and Reichardt 1991). Fumonisin B1, which blocks endogenous ceramide production, was obtained from Enzo Life Sciences and was applied at 10 μM diluted in recording or internal pipette solution.
Statistical analysis.
Data records were analyzed with Clampfit 8 (pCLAMP 8, Axon Instruments). Data are expressed as means ± SE, and differences were considered statistically significant at P < 0.05. Statistical comparisons were made with SigmaStat 2.0 (Systat Software, Chicago, IL) and Clampfit 8. Unless otherwise specified in the text, the paired t-test was used to compare control and drug conditions when data were generated in the same cells and were normally distributed. Paired data for nonnormally distributed data sets were compared with the nonparametric Wilcoxon signed-rank test. EPSC amplitude data shown in cumulative percent histograms were compared with the Kolmogorov-Smirnov test.
RESULTS
Cholinergic synaptic activity in sympathetic neurons in long-term coculture with cardiac myocytes.
We observed spontaneous network activity in rat SCG neurons cocultured with cardiac myocytes for prolonged culture periods (14–98 days, average 26.5 ± 1.1 days; n = 107). The observed network activity consisted of bursts of action potentials and excitatory synaptic events that were dependent on neuron-to-neuron cholinergic synaptic transmission. Bath application of the nicotinic cholinergic antagonist hexamethonium (100 μM) caused a nearly complete and reversible decrease in both synaptic events and cell spiking in the neuron-myocyte cocultures (Fig. 1, A and B; n = 6). This network activity was not observed in the first 3 days of culture (Vega et al. 2010), although we have previously observed evoked noradrenergic transmission at that time (Lockhart et al. 1997). This suggests a time-dependent shift toward cholinergic signaling between neurons in culture, consistent with a late acquisition of cholinergic properties (Furshpan et al. 1986; Potter et al. 1986).
Fig. 1.
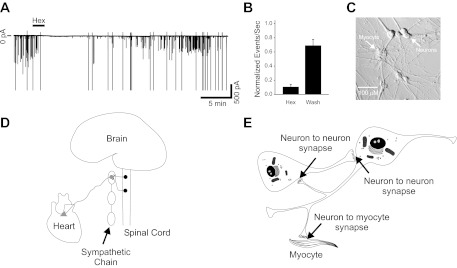
Sympathetic neurons displayed ongoing activity driven by cholinergic synaptic transmission when cultured with cardiac myocytes. A: voltage-clamp trace showing action currents (larger amplitude) and synaptic currents (smaller amplitudes) recorded at a holding potential of −60 mV. Activity was blocked by application of the nicotinic cholinergic antagonist hexamethonium (Hex; 100 μM, 2 min). B: number of synaptic events normalized to control showing that a 2-min application of 100 μM hexamethonium blocked 89.4 ± 3.7% of activity (n = 6). Action currents were excluded from excitatory postsynaptic current (EPSC) analysis. This effect was reversible, showing a 69 ± 9% recovery after 10–20 min in control saline (wash). C: phase image of a sympathetic neuron-cardiac myocyte coculture after 5 wk in culture. Neurons project widely throughout the culture dish, contacting other neurons and cardiac myocytes. D: schematic depiction of the sympathetic nervous system in vivo. Ganglionic sympathetic neurons are located in peripheral ganglia connected through the sympathetic chain along the axis of the spinal cord. Cholinergic preganglionic neurons of the spinal cord project to and innervate the sympathetic neurons, driving their activity. Sympathetic neurons form synapses within the ganglia themselves and project out to innervate tissues (such as the heart) throughout the body. E: schematic representation of our model coculture system. In culture sympathetic neurons form synapses both onto one another and onto cocultured cardiac myocytes.
Sympathetic drive to target tissues such as the heart in vivo is ultimately driven by excitatory cholinergic input from preganglionic neurons to postganglionic neurons at peripheral ganglia (shown schematically in Fig. 1D). When cultured together in vitro, sympathetic neurons form synapses onto one another in addition to forming synapses on to cocultured target cells such as cardiac myocytes (Fig. 1, C and E).
Regulation of synaptic transmission by brain-derived neurotrophic factor.
Neurotrophins modulate many sympathetic neuronal properties including neuronal growth (Bibel and Barde 2000), membrane electrical properties, and transmitter expression (Luther and Birren 2006, 2009a; Slonimsky et al. 2003; Yang et al. 2002). We previously showed that neurotrophins rapidly modulate neurotransmission at sympathetic synapses onto cocultured cardiac myocytes, and that BDNF causes a shift from excitatory noradrenergic to inhibitory cholinergic transmission (Slonimsky et al. 2003; Yang et al. 2002). Since cholinergic transmission plays a critical functional role in driving sympathetic activity in vivo, this suggests that BDNF signaling could provide a mechanism for regulation of sympathetic function within the ganglia, as well as at the level of innervated tissue.
We therefore investigated whether neurotrophin signaling acutely modulates neuron-to-neuron cholinergic transmission in spontaneously active cultures containing neurons and cardiac myocytes. We recorded spontaneous EPSCs from neurons in long-term coculture in voltage clamp at a holding potential of −60 mV (near the normal resting potential for these cells) and selected unitary events for analysis (see methods). Acute bath application of BDNF (100 ng/ml for 10–15 min) caused an increase in the amplitude of cholinergic synaptic EPSCs (Fig. 2, A and B; n = 11). We found that application of BDNF after a 1-h preincubation of cultures with the p75 function-blocking antibody REX (1:500) blocked the potentiating effect of BDNF, suggesting that BDNF acts through the p75 receptor to increase EPSC amplitude (Fig. 2C; n = 5).
We did not see evidence for a washout of the BDNF effect after removal of the neurotrophin within the time frame of our experiments (see methods). Therefore we wanted to rule out the possibility that the increase in amplitude was due to a time-dependent recording artifact (i.e., washout of intracellular molecules due to whole cell recording configuration). We observed no change in EPSC amplitude when saline vehicle was applied for 10–15 min in the absence of BDNF (Fig. 2D; n = 11).
We found no change in the waveform of cholinergic EPSCs after application of BDNF. We measured the 10–90% rise and decay times as well as the half-amplitude width, which did not differ after BDNF treatment (data not shown, n = 11). While this suggests that postsynaptic channel kinetics were unaltered by BDNF, these measurements may not represent true channel kinetics as synaptic events are likely to be filtered because of the large size and well-developed neuritic arborizations of the neurons.
Neurotrophins act through p75 but not TrkA signaling to influence cholinergic EPSC amplitude.
Sympathetic neurons express p75 and the NGF-specific TrkA receptor but do not express TrkB, a specific BDNF receptor (Bamji et al. 1998; Dixon and McKinnon 1994; Wyatt and Davies 1995). Therefore, BDNF only activates the p75 pathway in sympathetic neurons (Singh et al. 2008; Yang et al. 2002). In contrast, NGF activates both the p75 and TrkA signaling systems (Singh et al. 2008; Yang et al. 2002). We therefore hypothesized that NGF would potentiate EPSC amplitude via activation of the p75 signaling pathway and furthermore wanted to test the possibility that TrkA activation could also influence synaptic function.
We found that bath application of NGF (50 ng/ml) also potentiated EPSC amplitude (Fig. 3A; n = 6) and that this potentiation was blocked by inclusion of a monoclonal antibody directed against NGF (αNGF30) that selectively inhibits NGF's interaction with the p75 receptor but allows activation of TrkA (Saragovi et al. 1998) (Fig. 3B; n = 8). Thus signaling via TrkA was not sufficient to enhance EPSC amplitude, suggesting that the effects of NGF at these synapses are mediated via p75 activation.
We previously showed that the neurotrophins BDNF and NGF differentially activate p75 and TrkA, leading to dramatically different effects on sympathetic intrinsic electrical properties (Luther and Birren 2009b) and on neurotransmission at neuron-cardiac myocyte synapses in vitro (Slonimsky et al. 2003; Yang et al. 2002). NGF, when bath applied with the anti-NGF antibody αNGF30, acts as an NGF-dependent TrkA-specific ligand (Saragovi et al. 1998). We confirmed the actions of the NGF/αNGF30 antibody in promoting TrkA-specific NGF signaling by measuring spike output of sympathetic neurons in the presence of NGF and αNGF30. We previously showed that TrkA activation in these neurons increased spike firing and shifted the firing toward a tonic pattern while p75 signaling resulted in decreased firing and a shift toward a phasic pattern (Luther and Birren 2009b). We found that application of NGF/αNGF30 significantly increased spike output in response to a square-wave current pulse over a range of amplitudes consistent with results described previously for TrkA activation (Luther and Birren 2009b). This indicated that we were indeed pharmacologically activating TrkA with the NGF/αNGF30 treatment (Fig. 3C; n = 24 and n = 10 for saline control and NGF/αNGF30, respectively). Furthermore, NGF/αNGF30 caused an increase in the percentage of spikes occurring in the second half of the square-wave stimulus, a measure that we previously showed is a reliable indicator of tonic firing (Fig. 3C, inset; n = 24 and n = 10 for saline control and NGF/αNGF30, respectively).
The lack of an effect of NGF/αNGF30 on cholinergic neuron-to-neuron transmission as measured by EPSC amplitude and frequency, combined with the significant effects of both BDNF and NGF alone, suggests that the effects of neurotrophins on cholinergic transmission at sympathetic neuronal synapses are mediated via p75 signaling and that TrkA activation does not modulate cholinergic EPSC amplitude.
BDNF increases amplitude but not frequency of miniature synaptic currents.
Changes in EPSC amplitude are ascribed to increased quantal content or increased postsynaptic sensitivity (Reimer et al. 1998). We tested the locus of BDNF action by examining the effect of BDNF application on the generation of action potential-independent transmitter release. Miniature synaptic currents were recorded in the presence of the sodium channel blocker tetrodotoxin (TTX, 500 nM) to block activity. Under these conditions of action potential blockade we observed infrequent small unitary events at room temperature, which became much more frequent at 35–36°C; we therefore performed these experiments at the elevated temperature (Fig. 4A). The miniature EPSCs (mEPSCs) reflected cholinergic transmission since, similar to the results seen for spontaneous activity (see Fig. 1), 100 μM hexamethonium reversibly blocked their occurrence (86 ± 8% block, n = 5).
Bath application of BDNF (100 ng/ml for 10–15 min) caused an increase in the amplitude of the miniature postsynaptic events (mEPSCs) recorded in voltage clamp at a holding potential of −60 mV (Fig. 4, A and B). We selected unitary events for further analysis (see methods) and found that the mean mEPSC amplitude increased in BDNF (Fig. 4B; n = 6). In contrast to the significant increase in mEPSC amplitude, BDNF had no effect on frequency, suggesting that presynaptic release probability was not affected (Fig. 4C; n = 6).
Blockade of endogenous ceramide production occludes BDNF-mediated amplitude increase.
We previously showed that acute regulation of sympathetic firing properties and synaptic transmission between neurons and myocytes was mediated via the ceramide signaling pathway (Luther and Birren 2009b; Yang et al. 2002). Sphingolipids, such as ceramide, are important regulators of multiple cellular processes (Hannun et al. 2001; Hannun and Obeid 2008). Ceramide is liberated from sphingomyelin contained in the cell membrane and is freely interconverted between other active metabolites including ceramide 1-phosphate (C1P), sphingosine, and phosphorylated sphingosine (Fig. 5) (Bornancin 2011; Gomez-Munoz et al. 2010; Hannun and Obeid 2008; Kihara et al. 2007). Activation of p75 via neurotrophins has been shown previously to liberate ceramide via activation of nSMASE (Fig. 5) (Brann et al. 2002; Zhang et al. 2002).
Fig. 5.

Schematic of sphingolipid signaling and pharmacological manipulations used in this study. Sphingomyelin is a major lipid component of the cell membrane. Ceramide species are generated from sphingomyelin via the action of sphingomyelinase (SMASE), which is increased by p75 activation. Ceramide is freely interconverted between various signaling lipids in a manner dependent upon the activity of each enzyme. For example, on activation of SMASE intracellular ceramide levels increase, leading to increased sphingosine levels via the action of ceramidase, which converts ceramide to sphingosine. Conversely, increased sphingosine levels will lead to increased ceramide via sphingosine conversion by ceramide synthase. Application of the sphingomyelinase inhibitor sphingolactone-24 blocks the endogenous generation of ceramide and therefore also reduces the other sphingolipid signaling species. Fumonisin B1 blocks the activity of ceramide synthase, inhibiting the conversion of sphingosine to ceramide. However, ceramidase will convert intracellular ceramide to sphingosine; therefore fumonisin B1 application leads to a depletion of ceramide and an increase in sphingosine species.
We hypothesized that EPSC amplitude was potentiated by BDNF via activation of nSMASE leading to increased intracellular ceramide. We tested this by preincubating cultures in the presence of the irreversible inhibitor sphingolactone-24 (25 μM) (Wascholowski and Giannis 2006). We found that inhibition of nSMASE activity blocked the BDNF effect on EPSC amplitude (Fig. 6A; n = 6). Sphingolactone-24 application had no progressive effect on EPSC amplitude in the absence of BDNF, suggesting that a BDNF effect was not masked by an inhibitor-dependent progressive decrease in amplitude (−106 ± 25 pA vs. −104 ± 25 pA during the first 10 min and after 20 min of recording, respectively; n = 7).
Intracellular ceramide is generated from sphingosine via ceramide synthase and is converted back via ceramidase (Fig. 5) (Hannun and Obeid 2008). These metabolites are freely interconvertible, with their intracellular levels being dependent on the activities of the respective metabolic enzymes (Hannun and Obeid 2008). Therefore, blockade of ceramide synthase leads to increased sphingosine species at the expense of ceramides. We predicted that if BDNF acts postsynaptically to increase EPSC amplitude by activating p75 and causing nSMASE-dependent ceramide accumulation (specifically in the postsynaptic cell), then postsynaptic cytosolic perfusion with the ceramide synthase inhibitor fumonisin B1 (Desai et al. 2002) should occlude the BDNF effect, since accumulating ceramide will be converted to sphingosine. If BDNF acted presynaptically, or through postsynaptic mechanisms other than or in addition to the generation of ceramide, then postsynaptic fumonisin B1 perfusion should not alter, or only attenuate, the BDNF effect. We addressed this hypothesis by including 10 μM fumonisin B1 in the pipette recording solution and measuring spontaneous EPSCs after bath application of BDNF. We recorded action potential-dependent synaptic currents in voltage clamp at a holding potential of −60 mV with no TTX and at room temperature. We compared mean EPSC amplitude at the time of the establishment of the recording (<10 min after obtaining a whole cell recording) to the amplitude after extensive cytosolic dialysis (15–30 min of recording).
We first wanted to know whether internal fumonisin B1 perfusion itself would alter synaptic properties or influence cell health. We found that intracellular fumonisin B1 perfusion did not influence baseline EPSC amplitude. The mean amplitude was −84.7 ± 14.5 pA versus −82.5 ± 10.3 pA in cells internally perfused with fumonisin B1 for up to 10 min and after 15 min, respectively (n = 13). However, internal fumonisin B1 perfusion occluded the BDNF-dependent EPSC amplitude increase, consistent with a role for postsynaptic ceramide accumulation in mediating this effect (Fig. 6B; n = 8). Interestingly, neither sphingolactone-24 nor fumonisin B1 influenced EPSC amplitude on its own, suggesting that basal sphingolipid levels are insufficient to modulate synaptic properties.
The lack of a BDNF effect could be due either to blockade of p75-dependent ceramide accumulation or possibly to fumonisin B1-induced toxicity. However, current-clamp recordings showed that the fumonisin B1-perfused cells had resting potentials and input resistances similar to those found for control cells examined in current clamp [resting potential −57 ± 2 mV vs. −60 ± 1 mV and input resistance 436 ± 37 MΩ vs. 491 ± 54 MΩ for fumonisin B1 > 10 min (n = 11) and control (n = 24) cells, respectively], suggesting that the cells were healthy. Furthermore, action potential generation in response to suprathreshold square-wave current injections was normal in fumonisin-perfused cells, showing large overshooting spikes (see Fig. 6C, inset). Fumonisin B1-treated cells, in the presence or absence of BDNF, generated significantly higher spike output than control cells, consistent with our previous report of a C2-ceramide-dependent decrease in spike output (Fig. 6C; P < 0.001, 2-way ANOVA, n = 24 and n = 11, respectively, for control and fumonisin B1) (Luther and Birren 2009b). Together, these experiments demonstrate an effect of sphingolactone-24 and internal fumonisin B1 in blocking the functional effects of p75-mediated ceramide production without toxicity.
Postsynaptic cytosolic perfusion of C2-ceramide and d-erythro-sphingosine mimics BDNF effect on amplitude.
Our data suggest that BDNF acts through a p75-mediated sphingolipid signaling pathway to increase synaptic current amplitude. We directly tested the hypothesis that postsynaptic sphingolipid signaling regulates EPSC amplitude by delivering C2-ceramide or another component of the ceramide signaling pathway, d-erythro-sphingosine, directly to the postsynaptic cell by including either substance in the patch pipette recording solution (Dobrowsky et al. 1994; Hannun et al. 2001). We then recorded action potential-dependent EPSCs as described above. If the amplitude increase is due to a sphingolipid-mediated increase in presynaptic vesicle transmitter content in the absence of postsynaptic changes, then isolated postsynaptic application of C2-ceramide or d-erythro-sphingosine should show no effect on amplitude. If the increase in amplitude is due to a sphingolipid-mediated increase in postsynaptic sensitivity, then C2-ceramide or d-erythro-sphingosine perfusion should mimic BDNF application.
We found that postsynaptic cytosolic perfusion with 25 μM C2-ceramide or 1 μM d-erythro-sphingosine caused a significant increase in EPSC amplitude, consistent with a postsynaptic increase in transmitter sensitivity. We recorded synaptic currents in voltage clamp at a holding potential of −60 mV with no TTX and at room temperature. We compared mean EPSC amplitude at the time of the establishment of the recording (<10 min after going whole cell) to the amplitude after significant cytosolic dialysis had presumably occurred (15–30 min of recording). We did this for C2-ceramide, d-erythro-sphingosine, and the vehicle control (see methods). We observed no effect with vehicle alone between the first 10 min of the recording and the period between 15 and 30 min (Fig. 7A, left) but observed a significant increase in EPSC amplitude for recordings with C2-ceramide in the pipette (Fig. 7, A and B; n = 8). We found that intracellular cytosolic application of d-erythro-sphingosine, a signaling sphingolipid in the ceramide metabolic pathway, also caused a significant increase in EPSC amplitude (Fig. 7, A and C).
Sphingosine and ceramide are freely interconvertible and can be converted to other active metabolites; thus these experiments do not reveal which molecule is the essential signal. We therefore performed recordings with both d-erythro-sphingosine and fumonisin B1 in the recording solution and compared data from the first 10 min to data after significant intracellular dialysis. Fumonisin B1 inhibits ceramide synthase, blocking the conversion of d-erythro-sphingosine to ceramide or C1P. We found that fumonisin B1 blocked the potentiating effect of intracellular d-erythro-sphingosine, suggesting that d-erythro-sphingosine or related compounds such as sphingosine 1-phosphate do not mediate the p75 effect on EPSC amplitude (−93 ± 16 pA vs. −99 ± 15 pA for first 10 min and between 15 and 30 min after achieving whole cell configuration, respectively; n = 6). These data suggest instead that ceramide or C1P is important for mediating the p75-dependent increase in EPSC amplitude.
DISCUSSION
Here we show that neurotrophic factors, acting through the p75 receptor, potentiate cholinergic transmission between sympathetic neurons via a postsynaptic mechanism. This work has two important implications for understanding mechanisms for acute modulation of sympathetic drive to the heart. First, our experiments suggest that specific target interactions result in distinct neurotrophic responses at sympathetic synapses onto neuronal targets versus heart cells. Furthermore, our finding of neurotrophin-mediated postsynaptic modulation of cholinergic transmission at synapses onto sympathetic neurons suggests that local ganglionic or target-derived neurotrophins could modulate sympathetic drive by acute regulation of central cholinergic inputs and intraganglionic synapses. Overall, these findings argue that peripheral postganglionic sympathetic neurons have modulatory functions beyond acting as a relay for central cholinergic inputs, suggesting that local synaptic effects within the sympathetic ganglion regulate the output of central sympathetic drive.
Sympathetic neurons are contacted by cholinergic spinal neurons (Purves and Lichtman 1978) and make connections onto one another in vivo (Kawai 1996) and in culture (Fig. 1). In addition, these neurons form synaptic connections within the heart and onto cultured cardiac myocytes (Landis 1976). Several studies show that these two classes of synapses, neuronal and myocyte, display different properties despite both being formed by sympathetic neurons. An example of this is that individual neurons grown in microcultures with cardiac myocytes form autaptic connections that are morphologically distinct from synapses on myocytes despite being formed by the same neuron (Landis 1976).
Here we provide evidence that neuron-neuron and neuron-myocyte synapses also differ in their acute responses to neurotrophin signaling. NGF, acting through TrkA, rapidly increases the strength of noradrenergic transmission at neuromuscular synapses, as evidenced by a potentiation of beat rate induced by neuron stimulation (Lockhart et al. 1997; Slonimsky et al. 2003). In contrast, we found that TrkA activation had no influence on spontaneous EPSC amplitude at neuron-neuron synapses (Fig. 3). Since norepinephrine acutely inhibits cholinergic transmission at these neuronal synapses (Kawai and Senba 1995, 1997; Vega et al. 2010), this suggests that unlike the neuron-myocyte synapse, norepinephrine release onto neuronal targets is not potentiated or induced by TrkA activation.
Neurotrophin signaling has both common and target-specific effects on cholinergic transmission. BDNF rapidly induces activity-dependent acetylcholine release at neuron-myocyte synapses, resulting in a reduction of myocyte beat rate through muscarinic signaling (Slonimsky et al. 2003; Yang et al. 2002). The effects of BDNF are mediated through the p75 receptor, as shown by the inability of the Trk inhibitor K252a to block the actions of BDNF and the ability of C2-ceramide, a second messenger downstream of p75, to mimic those actions (Lockhart et al. 1997; Yang et al. 2002). BDNF also potentiated cholinergic transmission at neuron-neuron synapses through a p75-dependent mechanism that was mimicked by application of C2-ceramide or d-erythro-sphingosine (Fig. 7), similar to its actions at neuron-myocyte synapses.
Our findings also provide evidence for differences in the mechanism of neurotrophic modulation at neuronal and myocyte targets. While p75 signaling promotes the presynaptic release of acetylcholine at neuron-myocyte connections (Yang et al. 2002), we found evidence that regulation of neuron-neuron synapses occurred via a postsynaptic mechanism. BDNF increased the amplitude of miniature postsynaptic currents (mEPSCs) but did not change mEPSC frequency (Fig. 4). Change in mEPSC amplitude is classically attributed to a postsynaptic change in sensitivity to neurotransmitter (Reimer et al. 1998). Thus our mEPSC data are consistent with a postsynaptic change in sensitivity. Although these data suggest that BDNF effects are predominately postsynaptic, we cannot rule out the possibility of a presynaptic component. Indeed, BDNF potentiates mEPSC amplitude by ∼20%, while EPSC amplitude is potentiated by >30%, raising the possibility that additional mechanisms contribute to this regulation.
A postsynaptic site of action for BDNF is further supported by our observation that restriction of second messengers C2-ceramide and d-erythro-sphingosine to the postsynaptic neuron mimicked the effects of neurotrophins and that blocking postsynaptic C2-ceramide production by inclusion of fumonisin B1 in the patch pipette occluded the actions of bath-applied BDNF. Together these data show that neurotrophins act on the postsynaptic neuron through the p75 neurotrophin receptor to potentiate cholinergic transmission between sympathetic neurons in neuron-myocyte cocultures, defining a target-specific difference in synaptic properties.
Neurotrophins activate sphingolipid signaling pathways, resulting in the generation of ceramide (Dobrowsky et al. 1994, 1995). We found that blockade of nSMASE with sphingolactone-24 or intracellular application of fumonisin B1, an inhibitor of ceramide synthase, blocked the effect of bath-applied BDNF, suggesting that sphingolipid signaling mediates the BDNF effect. We found that both C2-ceramide and another component of the ceramide pathway, d-erythro-sphingosine, potentiated EPSC amplitude. However, intracellular sphingosine is freely converted to ceramide via ceramide synthase (Hannun et al. 2001; Hannun and Obeid 2008), and we found that blockade of this conversion with intracellular fumonisin B1 blocked the effect of sphingosine, suggesting that ceramide or C1P mediates the BDNF effect on EPSC amplitude. It is interesting that neither sphingolactone-24 nor fumonisin alone influenced EPSC amplitude. This is consistent with our previous finding that blockade of baseline p75 signaling did not influence neuron excitability, suggesting that basal sphingolipid levels are insufficient to modulate neuronal electrical properties (Luther and Birren 2009b).
Ceramides are implicated in the activation of several signaling pathways that include protein phosphatase 2A (Dobrowsky et al. 1993), ceramide-activated kinase (Liu et al. 1994), Ras-activating guanine nucleotide exchange factor Vav (Gulbins et al. 1994), and atypical protein kinase C isoforms (Zhang et al. 2011), any of which could act as a downstream mediator of neurotrophin-dependent EPSC enhancement. Furthermore, neurotrophin activation of p75 can result in the activation of many distinct signaling cascades in addition to ceramide that could contribute to signaling in this system (Gentry et al. 2004; Reichardt 2006). An examination of other p75-dependent signaling pathways for possible roles in the regulation of synaptic and electrical properties will be interesting in future experiments.
A possible mechanism for the observed increase in EPSC amplitude could involve an increase in the number of postsynaptic receptors. Sympathetic neurons express a variety of nicotinic channel subtypes (Skok 2002), and expression is regulated by target-derived neurotrophin signaling (Yeh et al. 2001; Zhou et al. 2001). This raises the possibility that the effects we observe could result from a neurotrophin-dependent increase in channel insertion into the membrane, and, despite the short time frame of these effects, a role for new transcription cannot be ruled out. Interestingly, ceramide signaling regulates rapid channel insertion into the membrane in a CHO cell line stably expressing adult murine nicotinic acetylcholine receptors independent of protein kinase Cζ or protein phosphatase 2A (Gallegos et al. 2008). This finding raises the possibility that such a mechanism could play a role in this system as well.
We have shown that p75 activation enhances the M-type potassium current (Luther and Birren 2009b), and since this current is active at rest we considered the possibility that neurotrophin modulation of this current led to our observed synaptic results. However, this particular channel is unlikely to explain the observed increase in EPSC amplitude since this would be expected to cause a decrease in input resistance, which would also decrease the apparent amplitude of EPSCs. While our present experimental design suggests a postsynaptic increase in ACh sensitivity, further experiments to examine downstream signaling systems, transcription, and ACh receptor surface expression will be required to fully elucidate the molecular mechanisms involved.
Neurotrophic modulation of sympathetic neurons and other neurons can involve signaling through either Trk or p75 receptors and can have either acute or long-term developmental effects. Long-term effects of neurotrophins include regulation of cholinergic innervation density of sympathetic ganglia, which is positively regulated by BDNF released from sympathetic neurons and mediated via TrkB signaling (Causing et al. 1997). Neurotrophins are also produced by somatic tissues such as the heart (Bierl and Isaacson 2007; Randolph et al. 2007), which regulate sympathetic innervation density of peripheral targets (Hassankhani et al. 1995). Retrograde signaling by target-derived NGF has been implicated in setting the density of preganglionic to ganglionic innervation in sympathetic ganglia (Sharma et al. 2010). Interestingly, while TrkA signaling promotes synapse formation, p75 signaling negatively regulates synapse number over developmental time. This is in contrast to the acute p75-mediated potentiation that we observe at these synapses, suggesting discrete actions for neurotrophins in regulating the properties of cholinergic synapses at different timescales.
Plastic changes in synaptic properties are strongly implicated in allowing adaptive and maladaptive changes in nervous system function through mechanisms that may involve neurotrophin signaling. Changes in neurotrophin signaling in heart tissues occur coincident with changes in sympathetic function in many cardiovascular diseases. In a rat model of myocardial infarction increased cardiac NGF is associated with increased sympathetic nerve sprouting (Hasan et al. 2006), and changes in norepinephrine release and reuptake were observed coincident with alterations in cardiac neurotrophin production in a rat model of congestive heart failure (Kreusser et al. 2008). These findings suggest that maladaptive and beneficial sympathetic responses mediated via target-derived neurotrophin signaling could be important factors in cardiovascular health. This work sheds light on how neurotrophin signaling could lead to changes in sympathetic drive, which has been strongly implicated in cardiovascular disease-related mortality (Malpas 2010).
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grants RO1 NS-066977 and R01 NS-057305 and P30 NS-45713 for Core Facilities for Neurobiology at Brandeis University.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.A.L., J.E., and S.J.B. conception and design of research; J.A.L. and J.E. performed experiments; J.A.L. and J.E. analyzed data; J.A.L. and S.J.B. interpreted results of experiments; J.A.L. prepared figures; J.A.L. drafted manuscript; J.A.L., J.E., and S.J.B. edited and revised manuscript; J.A.L., J.E., and S.J.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Uri Saragovi for kindly providing us with αNGF30 antibody and Dr. Kiran Nataraj for critical reading of this manuscript.
REFERENCES
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911–923, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14: 2919–2937, 2000 [DOI] [PubMed] [Google Scholar]
- Bierl MA, Isaacson LG. Increased NGF proforms in aged sympathetic neurons and their targets. Neurobiol Aging 28: 122–134, 2007 [DOI] [PubMed] [Google Scholar]
- Black IB. Trophic regulation of synaptic plasticity. J Neurobiol 41: 108–118, 1999 [PubMed] [Google Scholar]
- Bornancin F. Ceramide kinase: the first decade. Cell Signal 23: 999–1008, 2011 [DOI] [PubMed] [Google Scholar]
- Brann AB, Tcherpakov M, Williams IM, Futerman AH, Fainzilber M. Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. J Biol Chem 277: 9812–9818, 2002 [DOI] [PubMed] [Google Scholar]
- Carvalho AL, Caldeira MV, Santos SD, Duarte CB. Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br J Pharmacol 153, Suppl 1: S310–S324, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron 18: 257–267, 1997 [DOI] [PubMed] [Google Scholar]
- Chun LL, Patterson PH. Role of nerve growth factor in the development of rat sympathetic neurons in vitro. I. Survival, growth, and differentiation of catecholamine production. J Cell Biol 75: 694–704, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Tohse N, Sperelakis N. Influence of sympathetic innervation on the membrane electrical properties of neonatal rat cardiomyocytes in culture. J Dev Physiol 15: 237–246, 1991 [PubMed] [Google Scholar]
- Desai K, Sullards MC, Allegood J, Wang E, Schmelz EM, Hartl M, Humpf HU, Liotta DC, Peng Q, Merrill AH., Jr Fumonisins and fumonisin analogs as inhibitors of ceramide synthase and inducers of apoptosis. Biochim Biophys Acta 1585: 188–192, 2002 [DOI] [PubMed] [Google Scholar]
- Dixon JE, McKinnon D. Expression of the trk gene family of neurotrophin receptors in prevertebral sympathetic ganglia. Brain Res Dev Brain Res 77: 177–182, 1994 [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis. Modulation by co-expression of p75NTR with Trk receptors. J Biol Chem 270: 22135–22142, 1995 [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem 268: 15523–15530, 1993 [PubMed] [Google Scholar]
- Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science 265: 1596–1599, 1994 [DOI] [PubMed] [Google Scholar]
- Furshpan EJ, Landis SC, Matsumoto SG, Potter DD. Synaptic functions in rat sympathetic neurons in microcultures. I. Secretion of norepinephrine and acetylcholine. J Neurosci 6: 1061–1079, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos CE, Pediconi MF, Barrantes FJ. Ceramides modulate cell-surface acetylcholine receptor levels. Biochim Biophys Acta 1778: 917–930, 2008 [DOI] [PubMed] [Google Scholar]
- Gentry JJ, Barker PA, Carter BD. The p75 neurotrophin receptor: multiple interactors and numerous functions. Prog Brain Res 146: 25–39, 2004 [DOI] [PubMed] [Google Scholar]
- Gomez-Munoz A, Gangoiti P, Granado MH, Arana L, Ouro A. Ceramide-1-phosphate in cell survival and inflammatory signaling. Adv Exp Med Biol 688: 118–130, 2010 [DOI] [PubMed] [Google Scholar]
- Govoni S, Pascale A, Amadio M, Calvillo L, D'Elia E, Cereda C, Fantucci P, Ceroni M, Vanoli E. NGF and heart: is there a role in heart disease? Pharmacol Res 63: 266–277, 2011 [DOI] [PubMed] [Google Scholar]
- Gulbins E, Coggeshall KM, Baier G, Telford D, Langlet C, Baier-Bitterlich G, Bonnefoy-Berard N, Burn P, Wittinghofer A, Altman A. Direct stimulation of Vav guanine nucleotide exchange activity for Ras by phorbol esters and diglycerides. Mol Cell Biol 14: 4749–4758, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habecker BA, Symes AJ, Stahl N, Francis NJ, Economides A, Fink JS, Yancopoulos GD, Landis SC. A sweat gland-derived differentiation activity acts through known cytokine signaling pathways. J Biol Chem 272: 30421–30428, 1997 [DOI] [PubMed] [Google Scholar]
- Hannun YA, Luberto C, Argraves KM. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry 40: 4893–4903, 2001 [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139–150, 2008 [DOI] [PubMed] [Google Scholar]
- Hasan W, Jama A, Donohue T, Wernli G, Onyszchuk G, Al-Hafez B, Bilgen M, Smith PG. Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res 1124: 142–154, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassankhani A, Steinhelper ME, Soonpaa MH, Katz EB, Taylor DA, Andrade-Rozental A, Factor SM, Steinberg JJ, Field LJ, Federoff HJ. Overexpression of NGF within the heart of transgenic mice causes hyperinnervation, cardiac enlargement, and hyperplasia of ectopic cells. Dev Biol 169: 309–321, 1995 [DOI] [PubMed] [Google Scholar]
- Hawrot E, Patterson PH. Long-term culture of dissociated sympathetic neurons. Methods Enzymol 58: 574–583, 1979 [DOI] [PubMed] [Google Scholar]
- Kawai Y. Ultrastructure of neuronal circuitry in sympathetic ganglia. Microsc Res Tech 35: 146–156, 1996 [DOI] [PubMed] [Google Scholar]
- Kawai Y, Senba E. Correlation between dendrodendritic synapses of adrenergic type and synaptically evoked hyperpolarization in the sympathetic ganglion of adult rats. Neuroscience 68: 925–935, 1995 [DOI] [PubMed] [Google Scholar]
- Kawai Y, Senba E. Noradrenaline-mediated lateral inhibition in the sympathetic ganglion. Neurosci Lett 238: 87–89, 1997 [DOI] [PubMed] [Google Scholar]
- Kihara A, Mitsutake S, Mizutani Y, Igarashi Y. Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res 46: 126–144, 2007 [DOI] [PubMed] [Google Scholar]
- Kreusser MM, Buss SJ, Krebs J, Kinscherf R, Metz J, Katus HA, Haass M, Backs J. Differential expression of cardiac neurotrophic factors and sympathetic nerve ending abnormalities within the failing heart. J Mol Cell Cardiol 44: 380–387, 2008 [DOI] [PubMed] [Google Scholar]
- Landis SC. Rat sympathetic neurons and cardiac myocytes developing in microcultures: correlation of the fine structure of endings with neurotransmitter function in single neurons. Proc Natl Acad Sci USA 73: 4220–4224, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Mathias S, Yang Z, Kolesnick RN. Renaturation and tumor necrosis factor-alpha stimulation of a 97-kDa ceramide-activated protein kinase. J Biol Chem 269: 3047–3052, 1994 [PubMed] [Google Scholar]
- Locke RE, Nerbonne JM. Role of voltage-gated K+ currents in mediating the regular-spiking phenotype of callosal-projecting rat visual cortical neurons. J Neurophysiol 78: 2321–2335, 1997 [DOI] [PubMed] [Google Scholar]
- Lockhart ST, Mead JN, Pisano JM, Slonimsky JD, Birren SJ. Nerve growth factor collaborates with myocyte-derived factors to promote development of presynaptic sites in cultured sympathetic neurons. J Neurobiol 42: 460–476, 2000 [PubMed] [Google Scholar]
- Lockhart ST, Turrigiano GG, Birren SJ. Nerve growth factor modulates synaptic transmission between sympathetic neurons and cardiac myocytes. J Neurosci 17: 9573–9582, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. Acute and long-term synaptic modulation by neurotrophins. Prog Brain Res 146: 137–150, 2004 [DOI] [PubMed] [Google Scholar]
- Luther JA, Birren SJ. Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol 96: 946–958, 2006 [DOI] [PubMed] [Google Scholar]
- Luther JA, Birren SJ. Neurotrophins and target interactions in the development and regulation of sympathetic neuron electrical and synaptic properties. Auton Neurosci 151: 46–60, 2009a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Birren SJ. p75 and TrkA signaling regulates sympathetic neuronal firing patterns via differential modulation of voltage-gated currents. J Neurosci 29: 5411–5424, 2009b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 90: 513–557, 2010 [DOI] [PubMed] [Google Scholar]
- Ostberg AJ, Raisman G, Field PM, Iversen LL, Zigmond RE. A quantitative comparison of the formation of synapses in the rat superior cervical sympathetic ganglion by its own and by foreign nerve fibres. Brain Res 107: 445–470, 1976 [DOI] [PubMed] [Google Scholar]
- Patterson PH, Chun LL. The induction of acetylcholine synthesis in primary cultures of dissociated rat sympathetic neurons. I. Effects of conditioned medium. Dev Biol 56: 263–280, 1977 [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci 2: 24–32, 2001 [DOI] [PubMed] [Google Scholar]
- Potter DD, Landis SC, Matsumoto SG, Furshpan EJ. Synaptic functions in rat sympathetic neurons in microcultures. II. Adrenergic/cholinergic dual status and plasticity. J Neurosci 6: 1080–1098, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D, Lichtman JW. Formation and maintenance of synaptic connections in autonomic ganglia. Physiol Rev 58: 821–862, 1978 [DOI] [PubMed] [Google Scholar]
- Randolph CL, Bierl MA, Isaacson LG. Regulation of NGF and NT-3 protein expression in peripheral targets by sympathetic input. Brain Res 1144: 59–69, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361: 1545–1564, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer RJ, Fon EA, Edwards RH. Vesicular neurotransmitter transport and the presynaptic regulation of quantal size. Curr Opin Neurobiol 8: 405–412, 1998 [DOI] [PubMed] [Google Scholar]
- Rohrer H. Cholinergic neuronal differentiation factors: evidence for the presence of both CNTF-like and non-CNTF-like factors in developing rat footpad. Development 114: 689–698, 1992 [DOI] [PubMed] [Google Scholar]
- Saragovi HU, Zheng W, Maliartchouk S, DiGugliemo GM, Mawal YR, Kamen A, Woo SB, Cuello AC, Debeir T, Neet KE. A TrkA-selective, fast internalizing nerve growth factor-antibody complex induces trophic but not neuritogenic signals. J Biol Chem 273: 34933–34940, 1998 [DOI] [PubMed] [Google Scholar]
- Schotzinger RJ, Landis SC. Cholinergic phenotype developed by noradrenergic sympathetic neurons after innervation of a novel cholinergic target in vivo. Nature 335: 637–639, 1988 [DOI] [PubMed] [Google Scholar]
- Sharma N, Deppmann CD, Harrington AW, St Hillaire C, Chen ZY, Lee FS, Ginty DD. Long-distance control of synapse assembly by target-derived NGF. Neuron 67: 422–434, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, Park KJ, Hong EJ, Kramer BM, Greenberg ME, Kaplan DR, Miller FD. Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat Neurosci 11: 649–658, 2008 [DOI] [PubMed] [Google Scholar]
- Skok VI. Nicotinic acetylcholine receptors in autonomic ganglia. Auton Neurosci 97: 1–11, 2002 [DOI] [PubMed] [Google Scholar]
- Slonimsky JD, Yang B, Hinterneder JM, Nokes EB, Birren SJ. BDNF and CNTF regulate cholinergic properties of sympathetic neurons through independent mechanisms. Mol Cell Neurosci 23: 648–660, 2003 [DOI] [PubMed] [Google Scholar]
- Vega A, Luther JA, Birren SJ, Morales MA. Segregation of the classical transmitters norepinephrine and acetylcholine and the neuropeptide Y in sympathetic neurons: modulation by ciliary neurotrophic factor or prolonged growth in culture. Dev Neurobiol 70: 913–928, 2010 [DOI] [PubMed] [Google Scholar]
- Wascholowski V, Giannis A. Sphingolactones: selective and irreversible inhibitors of neutral sphingomyelinase. Angew Chem Int Ed Engl 45: 827–830, 2006 [DOI] [PubMed] [Google Scholar]
- Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron 6: 649–663, 1991 [DOI] [PubMed] [Google Scholar]
- Wyatt S, Davies AM. Regulation of nerve growth factor receptor gene expression in sympathetic neurons during development. J Cell Biol 130: 1435–1446, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Slonimsky JD, Birren SJ. A rapid switch in sympathetic neurotransmitter release properties mediated by the p75 receptor. Nat Neurosci 5: 539–545, 2002 [DOI] [PubMed] [Google Scholar]
- Yeh J, Ferreira M, Ebert S, Yasuda RP, Kellar KJ, Wolfe BB. Axotomy and nerve growth factor regulate levels of neuronal nicotinic acetylcholine receptor alpha3 subunit protein in the rat superior cervical ganglion. J Neurochem 79: 258–265, 2001 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kays J, Hodgdon KE, Sacktor TC, Nicol GD. Nerve growth factor enhances the excitability of rat sensory neurons through activation of the atypical protein kinase C isoform, PKMzeta. J Neurophysiol 22: 222–223, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J Physiol 544: 385–402, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Deneris E, Zigmond RE. Nicotinic acetylcholine receptor subunit proteins alpha7 and beta4 decrease in the superior cervical ganglion after axotomy. J Neurobiol 46: 178–192, 2001 [DOI] [PubMed] [Google Scholar]