

Abstract
Class switch DNA recombination (CSR) of the immunoglobulin heavy chain (IgH) locus is central to the maturation of the antibody response and critically requires the AID cytidine deaminase. CSR entails changes of the chromatin state and transcriptional activation of the IgH locus upstream and downstream switch (S) regions that are to undergo S-S DNA recombination, induction of AID, and targeting of CSR factors to S regions by 14-3-3 adaptors and as enabled by the transcription machinery and histone modifications. In this Review, we focus on recent advances in CSR induction and targeting. We also outline an integrated model of the assembly of macromolecular complexes that transduce critical epigenetic information to enzymatic effectors of the CSR machinery.
Antibodies (immunoglobulins) are central mediators of immunity1. They directly neutralize pathogens and pathogen products as well as recruit molecular and cellular immune effectors to eradicate infections and tumor cells. IgM, IgD, IgG, IgA and IgE antibody classes are identified by different constant regions in their heavy chains (IgH) and have distinct tissue distribution and efficacy against different types of pathogens. IgMs are secreted as pentamers or hexamers, possess a high avidity for antigens with repetitive motifs (such as those occurring on most microbial pathogens) and mediate complement activation. They, however, cannot pass into the extravascular space due to their large size. By contrast, monomeric IgG, monomeric IgE and monomeric or dimeric IgA can distribute systemically to tissues where they mediate a variety of biological effector functions2. While naïve B cells express only IgM and IgD, selected immunoglobulin classes and/or subclasses (isotypes) of antibodies are elicited during the course of an immune response, depending on the nature of the eliciting antigen and its entry mode. In humans, IgG1 and IgG3 are effective against viruses, IgG2 against encapsulated bacteria3, IgG4 and IgE against large extracellular parasites4, and IgA1 and IgA2 against pathogenic bacteria at the mucosae5.
The constant regions of different immunoglobulin isotypes are encoded by different CH exon clusters, which are organized in the order of Cμ, Cδ, Cγ, Cε and Cα in the IgH locus (FIG. 1). Class switch DNA recombination (CSR) results in the replacement of the expressed CH exon cluster — for example, Cμ for IgM — with Cγ, Cα or Cε, thereby giving rise to IgG, IgA or IgE, respectively, in which the antigen-binding variable region is unaltered6, 7. Of note, IgD is generated not through CSR, but through alternative splicing of the primary transcripts that encode IgM. CSR also gives rise to class-switched B cells, such as IgG+ B cells, which can respond to antigen faster than naive Igμ+ or Igδ+ B cells, probably because the longer cytoplasmic tail of IgG induces stronger B cell receptor (BCR) signaling8. Together with somatic hypermutation (SHM), which inserts mainly point-mutations in the antibody variable region at a high rate to provide a structural substrate for positive selection of higher affinity mutants by antigen9, 10, CSR is central to the maturation of the antibody response elicited by natural infections and vaccines11–13. Defective or aberrant CSR results in diseases ranging from hyper-IgM (HIGM) syndrome, systemic or organ-specific autoimmunity, allergy and asthma associated with atopic IgE, to neoplastic transformation.
Figure 1. CSR entails DNA deletion.
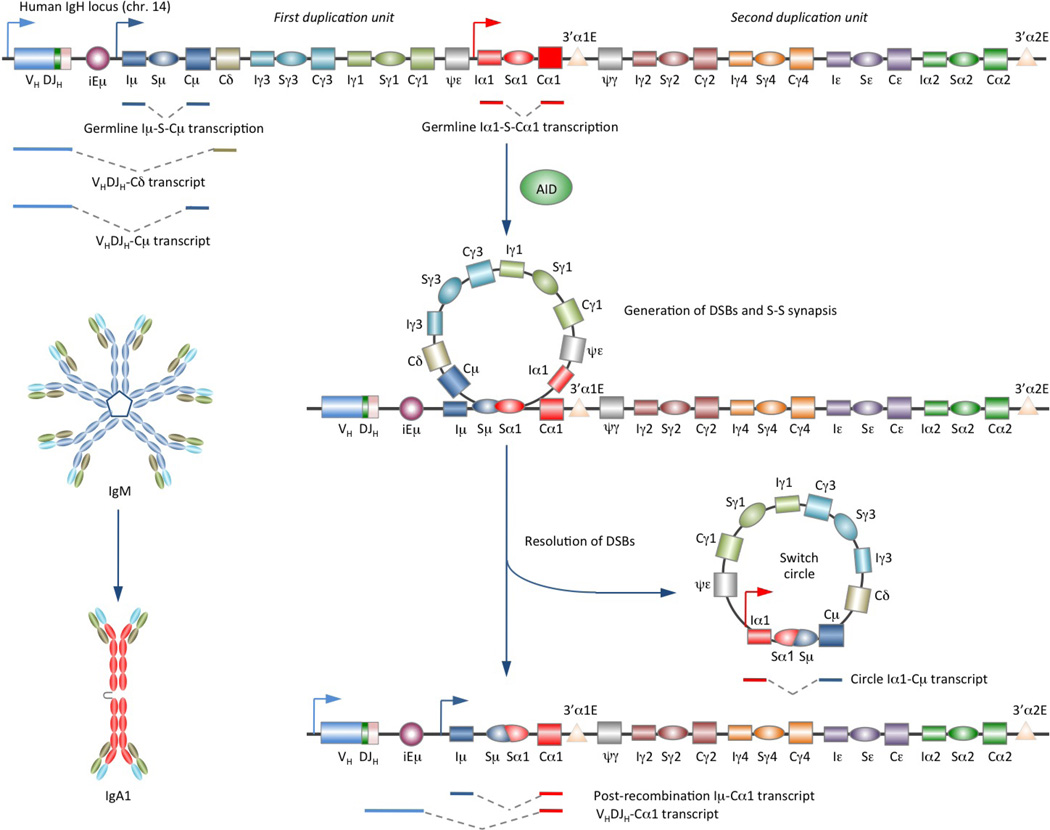
CSR exchanges the gene encoding the heavy chain constant region (CH) with one of a set of downstream CH genes (depicted is CSR from Sμ to Sα1 in the human IgH locus). This deletion-recombination reaction requires AID and involves the generation of double-strand DNA breaks in S regions (lying upstream of the constant-region gene) followed by DSB repair. This leads to juxtaposition of rearranged VDJ DNA with a downstream CH exon cluster and deletion of the intervening sequence between S regions as an extrachromosomal circle.
In this Review, we focus on the molecular mechanisms underlying the induction of CSR and targeting of the CSR machinery to the upstream and downstream S regions that are to undergo S-S DNA recombination by the RNA polymerase II transcriptional machinery and adaptor proteins that specifically bind repeats characteristic of S region DNA. We also discuss the emerging scaffold functions of both enzymatic and non-enzymatic elements in the stabilization of CSR factors on S regions and recent data on the epigenetic induction and regulation in the specification of the S region targets of CSR.
S region DSB generation and resolution
CSR requires transcription that initiates at an intervening (IH) promoter and elongates through the IH exon, switch (S) region and then the CH exon cluster, known as germline IH-S-CH transcription. CSR also requires activation-induced cytidine deaminase (AID)14, which deaminates deoxycytosines in S region DNA, yielding deoxyuracils15 (BOX 1, FIG. 2). Processing of such deoxyuracils results in insertion of double-strand DNA breaks (DSBs) in the upstream (donor) and downstream (acceptor) S regions. CSR then proceeds through DSB resolution, leading to deletion of intervening DNA, which gives rise to an extrachromosomal DNA switch circle, and S-S junctions that bring the downstream CH region DNA closer to the VHDJH region DNA.
Box 1 | Activation-induced cytidine deaminase (AID).
AID is a 198-amino acid protein that initiates CSR and SHM by deaminating deoxycytosines in single-strand DNA and supercoiled double-strand DNA, both of which can arise during transcription. It shares a conserved catalytic domain with other members of the AID/APOBEC family of cytosine/cytidine deaminases. The catalytic domain contains His56, Cys87 and Cys90, which bind Zn2+, and Glu58; the carboxylic acid group of Glu58 serves as a general acid-base catalyst. The APOBEC-like domain of AID binds DNA surrounding deoxycytosines and influences the substrate specificity. The C-terminal domain is essential for AID to mediate CSR and the N-terminal domain is essential for AID to mediate SHM.
Naturally occurring mutations in AID responsible for the autosomal recessive HIGM type 2 (HIGM2) syndrome (the number of cases reported for each mutation is depicted in the box figure) and experimentally generated AID mutations cause defective CSR and/or SHM. AID deamination activity and CSR are virtually abolished by mutations at Arg112 in the APOBEC-like domain and Arg24 in the DNA-binding N-terminal region, which are two positively charged residues frequently mutated in HIGM2 patients. Arg112 is just outside the hotspot recognition loop, which contacts the DNA substrate and determines the substrate specificity of the AID deoxycytosine deamination activity.
The C- and N-terminal domains also function as the nuclear export signal (NES) and nuclear localization signal (NLS), respectively. Active nuclear export of AID results in its dominant cytoplasmic distribution. This together with the instability of nuclear AID limits the levels of AID that can cause genomic damage. AID is phosphorylated at Ser38 and Thr27 to create a binding site for RPA and mediate CSR. Phosphorylation at Thr140 is important for SHM, whereas the role of Ser3 or Tyr184 phosphorylation is unclear. Binding regions in AID to 14-3-3, MDM2 and DNA-PKcs are also depicted.
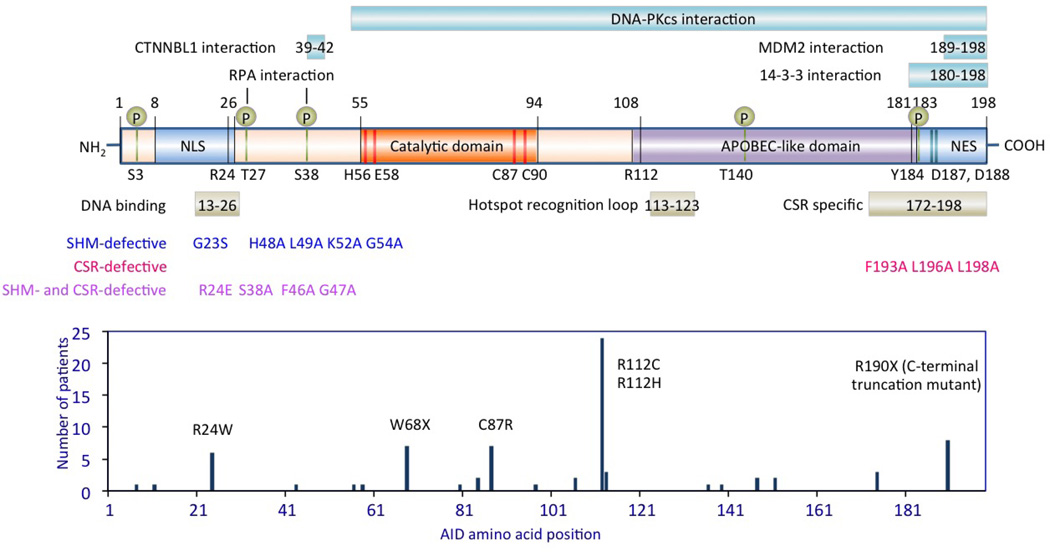
Figure 2. Enzymes and scaffold elements in CSR.
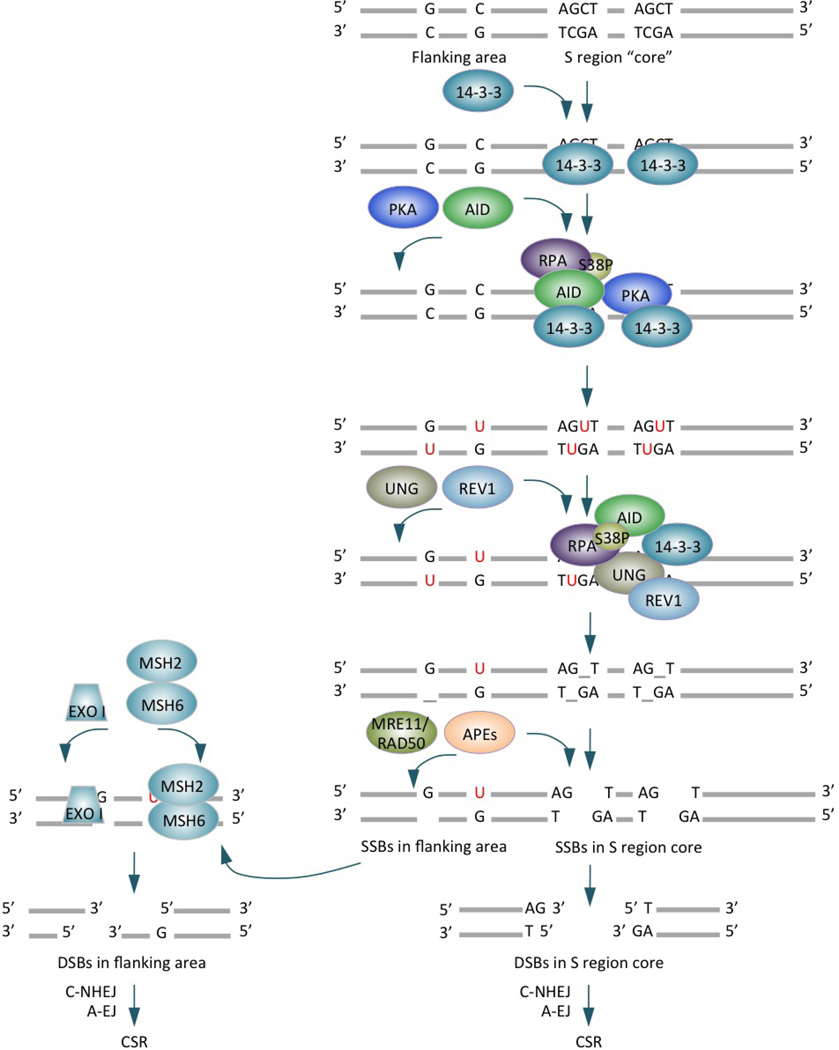
CSR is initiated by AID, which is targeted to S regions by 14-3-3 adaptors that specifically bind 5′-AGCT-3′ repeats in the S region core and recruit AID and PKA to S region DNA. AID is phosphorylated at Ser38 of the N-terminal region by PKA, generating a binding site for RPA. AID also binds 14-3-3 proteins through its C-terminal region (inset). RPA enhances AID deamination of deoxycytosines in transcribed S region DNA. The resulting deoxyuracils are removed by UNG, which would be recruited and stabilized on S regions in a fashion dependent on the scaffold functions of 14-3-3, RPA and REV1 (Zan, H., C.A. White, L. Thomas, J. Zhang, G. Li, E.S. Yu, Z. Xu, T. Mai and P. Casali. 2012. Submitted for publication). RPA and REV1 bind to different regions of UNG. UNG is also stabilized by AID, which indirectly interacts with UNG (likely through 14-3-3, Lam, T. and P. Casali, unpublished observation), within a putative macromolecular complex. Excision of UNG-generated abasic sites by APEs and/or the MRE11–RAD50 lyase results in single stranded breaks, which either directly form double stranded breaks (as occurring in the S region core) or are converted to DSBs in an MSH2- and EXO I-dependent fashion (as occurring in the flanking area). DSB resolution by C-NHEJ or A-EJ leads to formation of S-S junctions and CSR.
To mediate CSR, AID and other factors must be significantly upregulated and then targeted to S regions that will undergo recombination, where they generate DSBs and ultimately promote S-S DNA recombination. The generation of DSBs (which are obligatory CSR intermediates) entails processive AID-mediated deamination of deoxycytosines, particularly those within 5′-AGCT-3′ and 3′-TCGA-5′ tandem repeats in the ‘core’ of the S region, yielding high densities of deoxyuracils in both DNA strands (FIG. 2). Uracils are removed by uracil DNA glycosylase (UNG), a critical element of the base excision repair (BER) pathway, to yield abasic sites. Excision of these abasic sites by apurinic/apyrimidinic endonucleases (APEs) leads to single-strand DNA breaks (SSBs). Proximal SSBs in opposite strands would readily form staggered DSBs6. In addition, staggered DSBs can arise from AID- and UNG-dependent processing of blunt S region DSBs generated by endonuclease G16, 17. Finally, topoisomerase I can introduce nicks in S region cores, likely by cleaving non-B form DNA18.
AID can deaminate DNA flanking the S region core, but less efficiently due to the paucity of 5′-AGCT-3′ repeats, thereby leading to generation of mainly SSBs. Such SSBs can be converted to DSBs in a manner dependent on key mismatch repair (MMR) elements such as MSH2 and MSH6. These element may be recruited to deoxyuracil:deoxyguanine mismatches adjacent to the SSB nicks, where they recruit 5′→3′ exonuclease I (EXO I) to yield DSBs19 (FIG. 2). Thus, MSH2 adds to the main BER pathway (which is dependent on the S region core and UNG) to generate DSBs, as emphasized by the ablation of CSR in B cells lacking both UNG and MSH220 or in B cells lacking both the Sμ core and MSH221.
DSBs in S regions are generated under physiological conditions, but likely trigger the same set of DNA damage response (DDR) factors that repair DSBs generated by extrinsic DNA-damaging agents, such as ionizing irradiation. DSBs are eventually resolved by the classic non-homologous end joining (C-NHEJ) or the alternative end joining (A-EJ) pathway (BOX 2). A prominent role of C-NHEJ, which occurs in the cell cycle G1 phase, in CSR is supported by the predominant presence in the G1 phase of AID-dependent S region DSBs and IgH foci that contain the CSR factor NBS16, 22. Of note, homologous recombination occurs in the G2/M phase and it is unclear when A-EJ occurs. A-EJ, which is mediated by CTIP23 and PARP124 (TABLE 1), functions as a back-up pathway when a core factor of C-NHEJ is absent or occurs when DSBs are generated through the MSH2- and EXO I-dependent pathway in CSR25. Overall, resolution of S region DSBs in CSR is a highly coordinated process that utilizes the DNA repair machinery and maintains genomic integrity.
Box 2 | Double-strand DNA break (DSB) resolution in CSR.
DNA damage sensors, adaptors and effectors in the DNA damage response (DDR) play important roles in the DSB resolution stage in CSR. The MRE11–RAD50–NBS1 complex (MRN) is an early sensor of AID-mediated DSBs and localizes to damage sites, where it recruits and activates ATM. This phosphorylates NBS1 and downstream DDR proteins, thereby strengthening the DDR response. MRN also activates the γH2AX–MDC1–53BP1 pathway; γH2AX and 53BP1 mediate synapsis of upstream and downstream DSBs. MRN can process DSB ends for both C-NHEJ and A-EJ pathways in a manner independent of the MRE11 nuclease activity (this MRE11 nuclease activity is instead required for DSB repair mediated by homologous recombination). In C-NHEJ, DSBs are bound by KU70 and KU86 that form complexes with DNA-PKcs and play scaffold functions to recruit other essential factors, such as the XRCC4–DNA ligase IV complex, to complete end joining process for the formation of S-S junctions. In A-EJ, DSB ends are processed by MRN and CTIP, thereby generating microhomologies between upstream and downstream DSB ends. Ligation of DSBs by DNA ligase(s), which remain(s) to be identified, yields S-S junctions that show higher microhomologies that those formed in C-NHEJ. The choice of C-NHEJ, which is also essential to maintain genome integrity, over A-EJ, which is frequently associated with chromosomal translocations, depends on the expression of 53BP1, which protects DSB ends from resection for A-EJ, and on KU70 and KU86, which inhibit CTIP. Aberrant repair of DSBs can lead to chromosomal translocations and genomic instability.
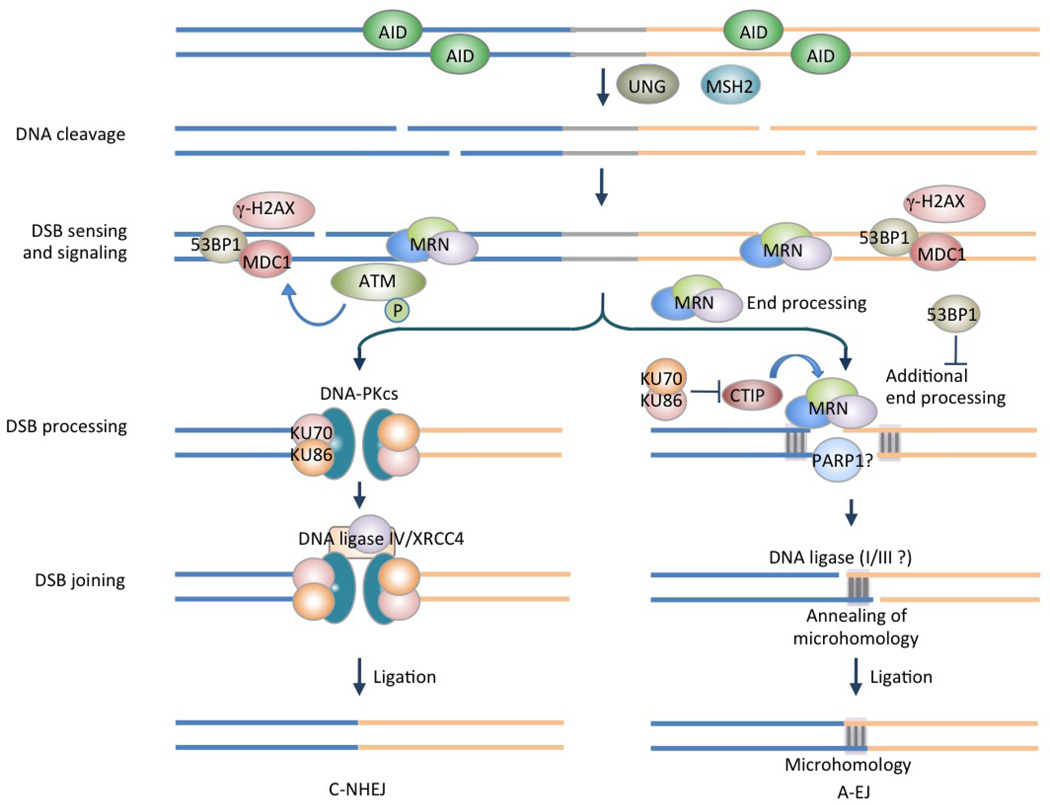
Table 1.
Factors involved in CSR
| Factor | (Putative) function(s) | Refs. |
|---|---|---|
| B cell specific factors | ||
| AID | Initiates CSR by deaminating deoxycytosines; involved in post-cleavage repair | 6, 7 |
| Chromatin modification-/transcription-related factors | ||
| PTIP | Promotes chromatin changes that control accessibility of the IgH locus to CSR | 60 |
| SSRP1 (SPT16) | Regulates histone modification H3K4me3 and DNA cleavage in CSR | 112 |
| AID-binding factors | ||
| 14-3-3 adaptors | Target S regions and recruit/stabilize AID and other CSR factors | 70 |
| CTNNBL1 | Unclear, might promote AID localization or association with RNA spliceosomes | 82 |
| eEF1A | Interacts with AID in the cytoplasm | 43 |
| HSP90 | Stabilizes cytoplasmic AID | 119 |
| KAP1 | Interacts with AID and tethers AID to Sμ | 115 |
| PKA | Phosphorylates AID at Ser38 on S region DNA | 75 |
| PTBP2 | Binds RNA and promotes AID binding to S region | 85 |
| REG-γ | Interacts with AID and promotes AID degradation in the nucleus | 45 |
| RNA exosome | Targets AID to both strands of transcribed duplex DNA | 63 |
| RNA polymerase II | Facilitates AID targeting to transcribed DNA | 80 |
| RPA | Binds to Ser38-phosphorylated AID and likely facilitates conversion of deoxyuracils to DSBs and promotes repair of DSBs | 75 |
| SPT5 | Facilitates the association of AID with RNA polymerase II | 83 |
| SPT6 | Regulates differentially AID functions in CSR and SHM | 84 |
| Base excision repair factors | ||
| APE1/APE2 | Nicks abasic site to generate DSBs in S regions | 6 |
| UNG | Removes AID-generated deoxyuracils and is involved in the DNA repair stage | 20, 104 |
| XRCC1 | Likely participates in A-EJ | 120–122 |
| Mismatch repair factors | ||
| EXO I | Generates DSBs by excising DNA containing deoxyuracil: deoxyguanine mismatches as a 5′→3’ DNA exonuclease. | 123, 124 |
| MLH3 | Contributes to the creation and/or stabilization of microhomologies between DNA ends in upstream and downstream S regions | 125 |
| MSH2, MSH6 | Recognizes deoxyuracil: deoxyguanine mismatches and initiates the process that leads to generation of DSBs; stabilizes AID on S region DNA | 20 |
| PMS2 and MLH1 | Recruit EXO I to promote DNA excision | 6 |
| Endonucleases/nickase | ||
| Endonuclease G | Generates DSBs in S regions in an AID-independent fashion | 17 |
| Topoisomerase 1 | Introduces nicks in S region, resulting in DSBs | 18 |
| DNA damage sensors, adaptors and effectors | ||
| 53BP1 | Stabilizes S region synapsis and protects DSB ends from resection | 126 |
| APLF | Promotes XRCC4/DNA ligase IV complex retention in chromatin in C-NHEJ | 127 |
| ARTEMIS | Mediates resolution of DSBs by C-NHEJ | 128–130 |
| ATM | Mediates DSB recognition and/or repair and S region synapsis | 131, 132 |
| ATR | Is involved in end-joining pathways that require long microhomologies | 133 |
| NHEJ1 | Mediates resolution of DSBs by C-NHEJ | 134 135 |
| CTIP | Promotes microhomology-directed A-EJ | 23 |
| DNA-PKcs | Plays a role in synapsis and DNA end joining | 128 |
| H2AX | Regulates higher order chromatin remodeling that facilitates S region synapsis | 136 |
| KU70, KU86 | Bind DSB ends and recruit downstream factors; are essential for C-NHEJ | 137, 138 |
| MRE11, RAD50, NBS1 | Stabilize DSBs and process DNA termini to facilitate repair by both C-NHEJ and A-EJ; also functions as lyase to nick abasic sites | 139 |
| PARP1 | Facilitates microhomology-directed A-EJ | 24 |
| REV1 | Recruits/stabilizes UNG to S region DNA | 108, 140 |
| RNF8, RNF168 | Propagates DSB signals and facilitates the recruitment of various DDR proteins | 141–143 |
| XRCC4, DNA ligase IV | Catalyzes the DNA ligation step in C-NHEJ | 144, 145 |
53BP1, p53-binding protein 1; A-EJ, alternative end joining; AID, activation-induced cytidine deaminase; APE, apurinic/apyrimidinic endonuclease; APLF, aprataxin and PNKP like factor; ATM, Ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related; CTIP, CTBP-interacting protein; CSR, class switch DNA recombination; CTNNBL1, catenin, beta like 1; DDR, DNA damage response; DNA-PKcs, DNA protein kinase catalytic subunit; DSB: double-strand DNA breaks; eEF1α, eukaryotic translation elongation factor 1α; EXO I, exonuclease I; H2AX, H2A histone family, member X; HSP90, heat shock protein 90; KAP1, KRAB-associated protein 1; KU70, KU autoantigen, 70 kDa; KU86, KU autoantigen, 86 kDa; MLH3, MutL homolog 3; MRE11, meiotic recombination 11; MSH2, MutS homolog 2; MSH6, MutS homolog 6; NBS1, Nijmegen breakage syndrome 1; NHEJ1: nonhomologous end-joining factor 1, also termed XRCC4-like factor (XLF) and CERNUNNOS; PARP1, poly (ADP-ribose) polymerase 1; PKA, protein kinase A; PMS2, postmeiotic segregation increased 2; PTBP2, polypyrimidine tract binding protein 2; PTIP, PAX interaction with transcription-activation domain protein-1; RNF8, ring finger protein 8; RNF168, ring finger protein 168; RPA, replication protein A; SHM, somatic hypermutation; S region, switch region; SSRP1, structure specific recognition protein 1; UNG, uracil DNA glycosylase; XRCC1, X-ray repair complementing defective repair in Chinese hamster cells 1; XRCC4, X-ray repair complementing defective repair in Chinese hamster cells 4.
Induction and regulation of CSR
Induction of CSR requires both “primary” and “secondary” stimuli. Primary CSR-inducing stimuli, whether T-dependent or T-independent, induce expression of AID and other important CSR-related genes through transcription factors, such as nuclear factor-κB (NF-κB; FIG. 3). Secondary CSR-inducing stimuli, that is, interleukin-4 (IL-4), transforming growth factor-β (TGF-β) and interferon-γ (IFN-γ, in mice but not humans), cannot induce AID, but are required for directing switching to IgG, IgA and/or IgE by selecting the acceptor S region(s) through induction of germline IH-S-CH transcription.
Figure 3. CSR-inducing stimuli and interplay of transcription factors.
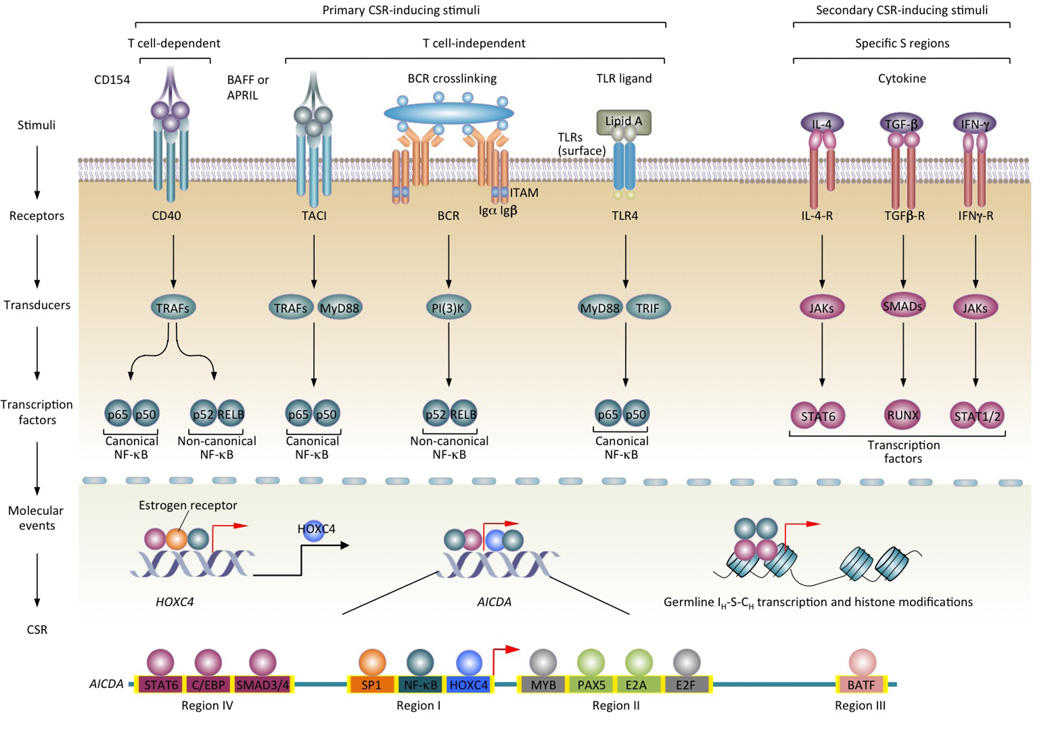
CSR entails induction of AID, germline IH-S-CH transcription and active histone modifications in S regions by primary CSR-inducing stimuli together with secondary stimuli (cytokines). Both T-dependent (CD40) and T-independent (dual TLR–BCR, TACI–BCR or TLR–TACI engagement) CSR-inducing stimuli — through distinct signal transducers — activate NF-κB for induction of HOXC4 (whose expression is enhanced by estrogen) and HOXC4-dependent induction of AID. The ability of primary CSR-inducing stimuli to induce AID and CSR correlates with their activation of both the canonical and non-canonical NF-κB pathways. Cytokines IL-4, TGF-β and IFN-γ do not activate NF-κB, but activate transcription factors specific for IH promoters. These in turn bind to specific IH promoters to initiate germline IH-S-CH transcription, thereby specifying the S regions that are to undergo recombination. A combinatorial interplay of primary and secondary CSR-inducing stimuli-activated transcription factors (teal and plum circles, respectively) results in optimal induction of AID and germline IH-S-CH transcription. Transcription factor-binding sites in Region I through IV of the AID gene locus that have been shown to regulate AID expression are depicted. Binding sites for MYB and E2F (grey circles) negatively regulate AID expression.
During T-dependent antibody responses, CSR occurs mainly in germinal centers and is induced upon engagement of CD40 on B cells by trimeric CD154 expressed by CD4+ follicular T helper (TFH) cells26, 27. CD40 induces high levels of CSR to IgG1 and IgE in the presence of IL-4 and moderate levels of CSR to IgG2a and IgA in the presence of IFN-γ and TGF-β, respectively28. Deficiency in CD154 or CD40 results in HIGM syndrome in humans and greatly reduced IgG, IgA and IgE levels in mice6, indicating a critical role of CD40 in class-switched T-dependent antibody responses. Nevertheless, specific IgG and IgA antibodies can emerge at early stages of an infection before specific T helper cells are activated, suggesting that T-independent and CD40-independent CSR has a role in the generation of these antibodies. In addition, T-independent CSR mediates the generation of IgG antibodies specific for many microbial antigens, such as bacterial polysaccharides. Indeed, T cell- or CD40-deficient mice display normal serum IgG3 levels and can generate IgG antibodies that are protective against a variety of infectious agents, albeit not as efficiently as wild-type mice29.
Deleterious mutations in Toll-like receptors (TLRs), TACI or their downstream signaling molecules result in reduced polysaccharide-specific IgG antibodies and defective immune response to encapsulated bacteria30, suggesting that T-independent CSR is induced by TLRs and/or TACI. However, engagement of TLRs31 or TACI32, 33, with or without IL-4, TGF-β or IFN-γ co-stimulation, induces marginal levels of CSR. The BCR signal, which alone does not induce CSR, synergizes with signals from TLR1–TLR2, TLR4, TLR7 or TLR9 to efficiently induce CSR to IgG3 and, in the presence of IL-4, TGF-β or IFN-γ, to other immunoglobulin isotypes; it also enhances TACI-dependent IgA switching33. The only known microbial component that can directly induce efficient CSR is bacterial lipopolysaccharides (LPS). LPS can engage both TLR4 on mouse B cells through its lipid A moiety and can crosslink the BCR of a large proportion of B cells through its repetitive polysaccharidic moiety (O-antigen)33. The need for dual TLR and BCR engagement for CSR induction emphasizes the role of naturally linked microbe-associated molecular patterns and repetitive microbial antigens in shaping the adaptive B cell response34. Like the BCR signal, the TACI signal synergizes with the signal from TLR7 and TLR9 to induce CSR32, 33.
Overall, CD40 engagement mediates T-dependent CSR, whereas dual TLR–BCR, TACI–BCR or TLR–TACI engagement mediates T-independent CSR, which plays an important role in T-independent antibody responses but also in early stages of T-dependent antibody responses.
Induction of AID
Whether induced in a T-dependent or T-independent manner, CSR requires AID, which is expressed in a B cell lineage-specific and differentiation stage-specific fashion. AID is virtually undetectable in resting mature B cells but is expressed at high levels in switching B cells. In addition, AID is expressed at a low level and can mediate CSR to IgE in immature B cells35. It might also mediate epigenetic programming in primordial germ cells (PGCs)36 and reprogramming in somatic cells37, particularly in light of the recent evidence that AID (and APOBEC family members) can promote demethylation of 5-hydroxymethylcytosines (5hmCs, converted from 5-methylcytosines by TET hydroxylases) when coupled with the BER pathway, which removes 5-hydroxymethyluracils (likely generated by AID-mediated deamination of 5hmCs)38. A tight regulation of AID expression is necessary to avoid chromosomal translocations and maintain genomic integrity in both B cells and non-B cells39–41. This is achieved through fine control of the AID gene (AICDA in the human and Aicda in the mouse) transcription19, 42, post-transcription14 and post-translation (including nuclear/cytoplasmic distribution43 and stability44, 45) regulation and enzymatic function46.
Transcription of the AID gene depends on NF-κB, which is activated in B cells mainly by primary CSR-inducing stimuli, that is, CD40, TLR and BCR33, 34, and TLR and TACI47 (FIG. 3). Of note, CD40, TLRs and TACI, but not BCR, alone can induce AID expression. CD40 engagement and dual TLR–BCR engagement can activate both canonical and non-canonical NF-κB pathways33, 48. Owing to its ability to engage TLR4 and crosslink BCR, LPS activates both pathways to induce AID, as highlighted by the recruitment of the (non-canonical) NF-κB p52 subunit to the AID gene promoter28 and the (canonical) p65 subunit to an upstream enhancer element49. Also, the kinetics of AID induction (peaking at 48 – 60 h after stimulation) mirror the kinetics of activation of the non-canonical NF-κB pathway33, which mediates sustained gene expression to support cell proliferation (required for CSR) and differentiation50. By contrast, the canonical NF-κB pathway is rapidly activated to induce immediate, but typically transient gene expression. Thus, activation of the canonical and non-canonical NF-κB pathways initiates and sustains AID gene transcription.
In addition to NF-κB, which is ubiquitously expressed and broadly regulates many genes51, transcription factors that are expressed in a B cell lineage- and/or differentiation stage-specific fashion42, 49 regulate AID induction. Prominent among these is HOXC4, a homeodomain transcription factor that is highly expressed in B cells and is induced by the same stimuli that induce AID and CSR. HOXC4 directly binds to the AID gene promoter, which is evolutionarily conserved, through a site (5′-ATTT-3′) embedded within a binding site for POU domain-containing transcription factor OCT1 and/or OCT228 (5′-ATTTGAAT-3′). NF-κB and SP1 also bind the same promoter28 (FIG. 3). HOXC4 is upregulated by estrogen, thereby potentiating AID and CSR52, 53, suggesting that estrogen is responsible for the AID dysregulation in systemic lupus erythematosus (SLE) patients, as well as in lupus-prone mice. These mice display increased levels of class-switched (and hypermutated) autoantibodies and chromosomal translocations, which are induced in a HOXC4- and AID-dependent fashion54.
AID induction is influenced by four major evolutionarily conserved regions in the AID gene locus through selected cis-regulating elements, many of which are binding sites of transcription factors that are activated by IL-4 and TGF-β (FIG. 3). For example, signal transducer and activator of transcription 6 (STAT6), which is activated by IL-4, probably binds Region I located upstream of the promoter, and IL-4-activated STAT6 and TGF-β-activated SMAD3 and SMAD4 bind Region IV located 9 Kb upstream of the promoter49. In addition, paired box protein 5 (PAX5) and E2A proteins bind Region II within the first intron49, 55, and the AP1 family transcription factor BATF binds Region III located 17 Kb downstream of the promoter56, 57. These transcription factors likely interplay with NF-κB and HOXC4 on the promoter and enhancer elements, probably through long-range DNA interactions, to mediate the AID induction by primary CSR-inducing stimuli and cytokines.
The AID gene transcription is subjected to negative regulation through four repressive cis-elements in Region II: two putative binding sites for MYB, which is known to activate or repress transcription depending on gene targets, one putative binding site for the E2F transcription factor family, which contains both activators and repressors, and a 350-basepair CT-rich sequence49. Both MYB and inhibitory factors in the E2F family are expressed in naïve B cells and non-B cells and would, therefore, play an important role in silencing AID expression in those cells. In switching B cells, this silencing effect is either reversed or overcome by primary stimuli- and cytokine-activated transcription factors. Finally, AID expression is suppressed by inhibitory E-box protein ID2, which antagonizes PAX5 and E2A42.
Thus, positive and negative transcription factors restrict AID induction to B cells undergoing CSR and/or SHM.
Germline IH-S-CH transcription specifies CSR
Primary stimuli enable CSR but require the cooperation of secondary stimuli to direct switching to predetermined immunoglobulin isotypes. Thus, secondary stimuli (IL-4, TGF-β or IFN-γ) adapt antibody effector functions to different milieus, such as IgA in the mucosae, where TGF-β is produced at high levels58, 59. CSR specification to a given immunoglobulin isotype depends upon induction of germline IH-S-CH transcription that progresses through the related acceptor S region – the donor S region is constitutively transcribed. During transcription elongation, the chromatin accessibility of intronic S regions is greatly increased60, thereby facilitating the targeting of the CSR machinery. This reflects the general principle that an open chromatin state is required for DNA recombination, repair and replication. Germline IH-S-CH transcription would generate single-strand DNA in the non-transcribed strand in S regions for AID-mediated deamination of deoxycytosines61, 62. During transcription, the RNA exosome will allow AID to deaminate deoxycytosines in the transcribed DNA strand, possibly by degrading RNA molecules in RNA:DNA hybrids63.
Germline IH-S-CH transcription is initiated by specific IH promoters in a manner generally dependent on cytokine-activated transcription factors. IL-4 activates STAT6, which binds the Iγ1 and Iε promoters to initiate germline transcription that proceeds through Sγ1 and Sε for CSR to IgG1 and IgE, respectively. TGF-β activates SMAD3, SMAD4 and/or RUNX proteins to promote transcription proceeding through Sγ2b and Sα for CSR to IgG2b and IgA, respectively. IFN-γ activates T-bet, STAT1 and/or STAT2 for the initiation of germline transcription that proceeds through Sγ2a for CSR to IgG2a6, 62. Competition between different IH promoters for the transcription machinery furthers isotype selection. For example, IKAROS mediates suppression of germline Iγ2a-Sγ2a-Cγ2a and Iγ2b-Sγ2b-Cγ2b transcription, thereby downregulating CSR to IgG2a and IgG2b, and promotes class-switching to IgG3 and IgG164.
Primary and secondary stimuli activate distinct sets of transcription factors. The division of roles of these factors, however, is blurred due to their combinatorial interplay on promoters and enhancers in the IgH locus. IL-4, TGF-β or IFN-γ can only induce significant levels of germline IH-S-CH transcription when CD40 or TLR and BCR are also engaged. Such transcription is induced at levels sufficient for CSR only in the presence of a fully functional IgH intronic enhancer (iEμ)65 and the locus control region located 3′ of Cα (3′-LCR)66. This contains several DNase I-hypersensitive enhancer elements that can be activated by NF-κB, HOXC4, OCT1 and/or OCT267, suggesting that these transcription factors likely interact with those activated by IL-4, TGF-β or IFN-γ through long-range DNA “synapsis” involving 3′-LCR and selected IH promoters for full induction of transcription68. The combinatorial interplay of primary and secondary stimuli-induced transcription factors is also important in AID induction. IL-4-activated STAT6 and TGF-β-activated SMAD significantly enhance AID induction by NF-κB, HOXC4, OCT1 and/or OCT2 activated by primary CSR-inducing stimuli.
Thus, while primary CSR-inducing stimuli activate transcription factors that regulate transcription of AID and a variety of genes, secondary stimuli (IL-4, TGF-β or IFN-γ) activate transcription factors that bind to different IH promoter(s) to initiate germline IH-S-CH transcription, thereby specifying the acceptor S regions that are to undergo CSR. Full induction of germline IH-S-CH transcription requires the interplay of both sets of transcription factors.
Targeting of the CSR machinery
The specific targeting of the CSR machinery to the S regions that will undergo recombination relies on: the recurrence at a high frequency of 5′-AGCT-3′ repeats in S regions, but not in the genome at large, that are specifically bound by 14-3-3 adaptors and other CSR elements; the induced open chromatin state of the S regions targets of recombination (as specified by secondary CSR-inducing stimuli), allowing for access of the CSR machinery; and, finally, the stalling of RNA polymerase II during germline IH-S-CH transcription, allowing for high rate deposition on those S regions of the AID hitched by the polymerase.
S regions and 5’-AGCT-3’ repeats
S regions are the specific targets of the CSR machinery, and deletion of Sμ or downstream S regions ablates CSR69. The CSR machinery does not insert DSBs at predetermined sites within S regions. Rather, it can introduce DSBs in virtually any area of an S region (which ranges from 3 Kb to 10 Kb), but with a strong preference for its core. The core of all S regions is highly enriched in 5′-AGCT-3′ repeats. These account for over 45% of Sμ core DNA but less than 1.4% of DNA in the genome, including CH regions70. The S region-specific and frequent recurrence of 5′-AGCT-3′ repeats is conserved across species that use CSR to diversify their antibodies, from frogs to humans70 (SUPPLEMENTARY FIG. 1). High serum levels of specific immunoglobulin isotypes in certain species (for example, IgE in horses and IgG2a in rats) likely reflect adaptation to selection applied by pathogens in host living environments or diets; the high 5′-AGCT-3′ frequencies in S regions of these isotypes are, therefore, suggestive of an evolutionary benefit of 5′-AGCT-3′ tandem repeats to CSR.
CSR is impaired in B cells in which the Sμ core was deleted and is almost abolished in B cells that compound the deletion of the Sμ core with abrogation of MSH221. Furthermore, replacement of the mouse Sγ1 DNA with the 5′-AGCT-3′-rich Sμ DNA from X. laevis promoted CSR to IgG171 and replacement of the full-length mouse Sγ1 region with Sγ1 and Sγ3 cores, which are rich in 5′-AGCT-3′ repeats, was sufficient to mediate CSR69. These observations further support the critical importance of 5′-AGCT-3′ repeats in CSR. In addition, 5′-AGCT-3′ is one iteration of the 5′-RGYW-3′ hotspot of SHM and contains the AID deamination motif 5′-WRC-3′ on both DNA strands72. 5′-AGCT-3′ repeats provide the substrate for AID to generate deoxyuracils at high-densities on both DNA strands and eventually DSBs in the S region core. Given the paucity of 5′-AGCT-3′ repeats in areas flanking the S region core, AID-mediated deamination in those areas, likely dependent on the R-loop formation73, would lead to generation of mainly SSBs, which are converted to DSBs in a MSH2-dependent manner6.
Thus, the 5′-AGCT-3′ core repeats provide the structural substrates for the intrinsic affinity of the CSR machinery for S regions but not for other DNA sequences in the IgH locus or non-immunoglobulin genes in the genome at large.
14-3-3 proteins as targeting adaptors
5′-AGCT-3′ repeats are not merely preferred substrates of AID, but also the specific targets of 14-3-3 adaptor proteins, which do not bind other 5′-RGYW-3′ iterations or G-rich motifs70. 14-3-3 adaptors are recruited to S regions (FIG. 2), and their binding to S regions is blocked by difopein, resulting in diminished CSR. Also, B cells deficient in 14-3-3γ or that express a dominant negative 14-3-3σ mutant are defective in CSR70.
14-3-3 adaptors play an important role in targeting the CSR machinery to S regions by virtue of their ability to bridge proteins with DNA or other proteins74. They directly interact with AID and PKA catalytic subunit (PKA-Cα) and mediate binding of AID and PKA-Cα to the S region(s)70, where PKA phosphorylates AID75 (FIG. 2). Binding of 14-3-3 to AID depends on the C-terminal region of AID70. This region is dispensable for the deamination activity of AID, but is critical for CSR and has been hypothesized to be the binding site for CSR-specific “cofactors” of AID76, 77. The failure of an AID C-terminal truncation mutant (AID1–190), which is found in some patients with HIGM syndrome, to bind 14-3-3 adaptors explains the inability of such an AID mutant to be concentrated on S regions78. Indeed, an enforced “binding” of AID1–190 to 14-3-3γ through physical linking of two peptides could fully rescue CSR in Aicda−/− B cells (Mai, T., E.J. Pone, G. Li, J. Moehlman, Z. Xu and P. Casali. 2012. Submitted for publication). By contrast, linking other peptides, such as those from protein kinase inhibitor α, HIV-1 Rev, HTLV-1 Rex, MAP kinase, or Ran binding protein 1, to AID1–190 failed to do so79, suggesting that 14-3-3 adaptors are the AID (C-terminus-binding) cofactors necessary for CSR.
Thus, the inherent S region targeting by the CSR machinery depends on 14-3-3 adaptors, as highly specific for 5′-AGCT-3′ repeats. 14-3-3 adaptors play an important role in the recruitment/stabilization of AID and PKA to/on S region DNA.
Transcription and CSR targeting
The high density of 5′-AGCT-3′ repeats in all S regions and the inherent high avidity of 14-3-3 adaptors for those repeats ensure that 14-3-3 can bind any S regions. These adaptors (as well as other CSR factors), however, only target the donor and acceptor S regions that will undergo recombination70. This is likely due to an open chromatin state of these S regions that allows for access of CSR factors, as reflected by the (constitutive or induced) germline IH-S-CH transcription of the donor and acceptor S regions.
A direct role for germline IH-S-CH transcription in AID targeting was suggested by the selective association of AID with RNA polymerase II and the transcribed S region DNA80, 81. Accordingly, CSR is impaired in B cells deficient in AID-binding proteins that also function in RNA metabolism63, 82–85, such as SPT5 and SPT6, which regulate transcription, PTBP2, which regulates RNA splicing, and the RNA exosome, which functions in RNA processing (FIG. 4). AID interaction with RNA polymerase II, which is stalled/paused in S regions86, 87, perhaps due to complex secondary DNA conformations (such as cruciform-like structures) made possible by palindromic 5′-AGCT-3′ repeats, is mediated by SPT5, which, like RNA polymerase II, is enriched in S regions83. SPT5, however, is also enriched on the promoter of a significant proportion of actively transcribed non-immunoglobulin genes83, consistent with findings showing genome-wide AID binding to transcribed genes88. Findings of SPT5- and RNA polymerase II stalling-mediated AID recruitment would lend support to the originally proposed model that the (AID-centered) SHM machinery is linked to the transcriptional machinery89 and explain recent data that AID can generate mutations in many transcribed genes90. AID binding to S regions and selected non-immunoglobulin loci, coupled with the ability of the RNA exosome to allow AID to deaminate deoxycytosines in both transcribed and non-transcribed DNA strands63, would also explain AID-mediated generation of DSBs in S regions and, at a much lower frequency, throughout the genome91, 92. DSBs that are not resolved as intra-S or inter S-S region recombinations will provide the substrate for interchromosomal translocations93, 94. These can be deleterious, particularly when the translocation involves oncogenes, such as MYC, as occurs at high frequencies in B cell lymphoma, further emphasizing that AID expression, activity and targeting need to be tightly controlled.
Figure 4. Targeting of the CSR machinery.
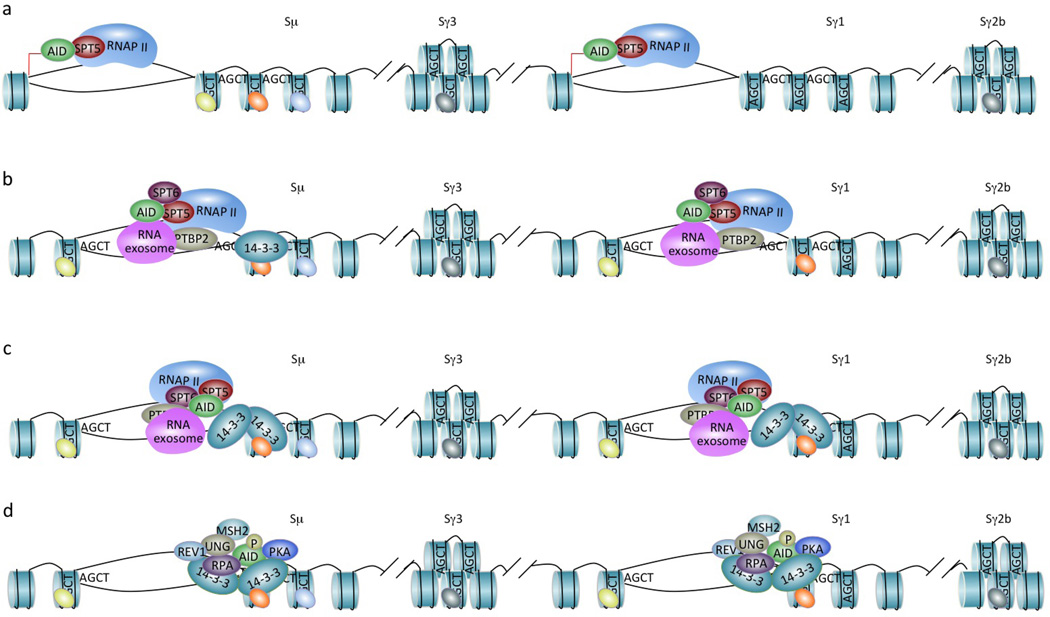
CSR targeting entails the epigenetic regulation of the S regions that are to undergo recombination (depicted is CSR from Sμ to Sγ1 in the mouse IgH locus), high-avidity binding of 14-3-3 adaptors to 5′-AGCT-3′ repeats and scaffold functions of CSR factors. (a) RNA polymerase II and histone modifying enzymes, such as histone methyltransferase and histone acetyltransferase complexes, are recruited to the Iγ1 promoter to initiate germline Iγ1-S-Cγ1 transcription (red arrow) and catalyze histone modifications, respectively (the Iμ promoter undergoes constitutive transcription initiation and histone modifications). Through SPT5, RNA polymerase II associates with AID at the Iμ and Iγ1 promoter, albeit at low levels. (b) During transcription elongation, RNA polymerase II together with SPT5 and, perhaps, SPT6, PTBP2 and the RNA exosome (all within a putative macromolecular complex), stalls in Sμ and Sγ1, and likely plays a role in recruiting/stabilizing histone modifying enzymes, such as histone methyltransferase and histone acetyltransferase complexes that catalyze H3K4me3 (green ovals) and H3K9acS10ph (orange ovals) modifications, respectively. This and loss of repressive histone modifications, such as H3K9me3 (light blue ovals) and H3K27me3 (grey ovals), reflect the open chromatin state of Sμ and Sγ1 that allows the access of AID and other CSR factors. AID, as “hitched” on the transcription machinery, is enriched in S regions due to RNA polymerase II stalling. (c) AID is further concentrated to Sμ and Sγ1 by 14-3-3 adaptors, which access the same Sμ and Sγ1 regions due to their open chromatin state and are recruited/stabilized there through interactions with 5′-AGCT-3′ repeats and likely H3K9acS10ph. The RNA exosome also interacts with AID and allows AID to deaminate both transcribed and non-transcribed S region DNA. (d) 14-3-3, RPA and enzymatic elements AID and REV1 (Zan, H., C.A. White, L. Thomas, J. Zhang, G. Li, E.S. Yu, Z. Xu, T. Mai and P. Casali. 2012. Submitted for publication) all possess scaffold functions that stabilize each other and other CSR factors, including PKA, UNG and MSH2, on S region DNA within a macromolecular complex.
Thus, highly regulated mechanisms must be in place to target AID to intronic S regions, which display the highest density in the genome of bound AID molecules. AID may associate with RNA polymerase II through SPT5, then “rides” on the transcription machinery to “scan” the transcriptome until stalled on S region DNA. There, AID is stabilized by binding to 14-3-3 adaptors (FIG. 4). This is reminiscent of the targeting of p53, which uses a non-core domain to bind “non-specifically” to and quickly slide along DNA until “hitting” a target sequence that is then bound by the p53 core domain with high affinity95. 14-3-3-mediated AID stabilization in S regions may, in turn, contribute to further stalling/pausing and, therefore, enrichment of RNA polymerase II in S regions.
Thus, targeting of the CSR machinery to the S regions that are to undergo recombination is made possible by the open chromatin state of those S regions. This is enabled by germline IH-S-CH transcription and ensures that 14-3-3 adaptors access 5′-AGCT-3′ repeats only in the S regions that are to undergo recombination - all other S regions being inaccessible. Stalling of RNA polymerase II in S regions during transcription allows for deposition of AID, which is further concentrated by 14-3-3.
Epigenetics and protein scaffolding
The recruitment, stabilization and enzymatic function of AID, UNG and other CSR enzymatic elements on S regions require additional interactions, which involve the non-catalytic domains of these enzymes, non-enzymatic adaptors, such as 14-3-3 proteins, and specific histone modifications that are induced in the S region targets of CSR.
Scaffold functions of 14-3-3 and RPA
In CSR, targeting of AID, insertion of DSBs and resolution of DSBs depend on the formation of S region DNA-containing macromolecular complexes. These would be nucleated by the high-avidity binding of 14-3-3 homo- and hetero-dimers or tetramers to 5′-AGCT-3′ tandem repeats in the S region core. Further, as scaffold proteins96, 14-3-3 stabilize the binding of AID and PKA to S region DNA and enhance AID deamination activity70. Within the same macromolecular complex, PKA-mediated phosphorylation of AID at Ser38 in situ recruits RPA88, which enhances AID deamination of deoxycytosines97. RPA also likely function as a scaffold protein to stabilize CSR factors in the post-deamination stage, such as UNG98, MMR factors99 and the MRE11/RAD50/NBS1 complex100 in the generation of DSBs, as well as γ-H2AX101, 53BP1102 and DNA-PKcs103 in the resolution of DSBs. In supporting an important role of RPA scaffold functions in CSR, RPA interacts with the UNG N-terminal region, which is critical for CSR104.
Scaffold functions of AID and REV1
In addition to their deamination activity, AID molecules function as scaffold elements to recruit other CSR factors, such as UNG, MSH2 and MSH6, to S regions and/or stabilize them there. AID molecules are, in turn, stabilized by these factors78. The reciprocal and cooperative stabilization of AID with UNG, MSH2 and MSH6 occurs without a direct interaction of AID with these factors and depends on the AID C-terminal region78. This region also mediates AID binding to 14-3-3, suggesting that 14-3-3 proteins, which can directly interact with UNG (Lam, T. and P. Casali, unpublished observation), bridge AID and UNG to form an AID–14-3-3–UNG complex. This could enhance the processing of AID-inserted deoxyuracils by UNG for the eventual generation of DSBs.
AID and UNG may play a role in stabilizing factors involved in C-NHEJ over those involved in A-EJ in the DSB resolution stage, as suggested by the increased microhomologies, which are hallmarks of A-EJ, in Sμ-Sα junctions in patients expressing the C-terminal truncation mutant AID1–190 or deficient in UNG105. Thus, the AID C-terminal region mediates CSR by targeting AID to S regions through 14-3-3 adaptors, leading to insertion of DSBs, and by mediating scaffold functions that allow for the stabilization of DNA repair factors, which mediate DSB resolution78, 105, 106. These contrasting activities of the AID C-terminal region would provide an explanation for the finding that the levels of S region DSBs in B cells expressing AID1–190 were comparable to those in B cells expressing wildtype AID106.
Like AID, UNG mediates CSR in a fashion dependent on both catalytic and non-catalytic amino acid residues104. In addition to 14-3-3 and RPA, the translesion DNA synthesis polymerase REV1 directly interacts with UNG through the non-catalytic UNG 231WXXF234 motif and recruits UNG to S regions to mediate CSR (Zan, H., C.A. White, L. Thomas, J. Zhang, G. Li, E.S. Yu, Z. Xu, T. Mai and P. Casali. 2012. Submitted for publication). Thus, UNG would be stabilized on S regions by RPA through N-terminal domain, by 14-3-3 through C-terminal domain (in a fashion independent of UNG 231WXXF234 motif) and by REV1 through 231WXXF234. Interestingly, the catalytic activity of REV1 (incorporation of deoxycytosines opposite deoxyuracil and abasic sites) is important in SHM107 but dispensable in CSR108, suggesting that REV1 functions only as a scaffold protein in this process.
Overall, both enzymatic elements and non-enzymatic adaptor proteins exert scaffold functions in CSR through multivalent DNA–protein and protein–protein interactions, thereby cooperatively stabilizing each other as well as other factors on S region DNA (FIG. 4).
Histone modifications in S regions
Adaptor molecules and enzymes are selectively recruited to and stabilized specifically on the S regions that are to undergo recombination because only these regions are in an open chromatin state. This is a reflection of germline IH-S-CH transcription as well as loss of repressive histone modifications, such as H3K27me3, and acquisition of permissive histone modifications, such as H3K4me3, in acceptor S regions (Sμ is “constitutively” marked by such modifications in any IgM-expressing B cell)60, 83, 86, 88, 109–112. These modifications play an important role in CSR in a fashion independent of germline IH-S-CH transcription, as suggested by defective CSR to IgA in mouse CH12 B cells upon siRNA-mediated downregulation of histone methyltransferases and subsequent impairment in H3K4me3 in spite of normal germline Iα-S-Cα transcription112.
Permissive histone modifications are induced by primary CSR-inducing stimuli in acceptor S regions specified by secondary stimuli, such as IL-4 or TGF-β, which alone cannot induce such modifications. The concurrent induction of histone modifications (histone code “writing”) and germline IH-S-CH transcription suggests a coordinated regulation of both processes by transcription factors that specifically bind to the IH promoters (FIG. 4). Such transcription factors may recruit PAX-interacting protein 1 (PTIP), which plays a critical role in initiation of germline IH-S-CH transcription and in generation of multiple histone modifications (H3K4me3, H3K36me3 and H3K27ac, but not H3K4me1) in S regions60. In addition, contrasting with the paucity of permissive or activating histone modifications (particularly H3K4me3) in the body – as compared to promoters – of transcribed genes in the genome at large113, such histone modifications are enriched in S regions at levels higher than those on IH promoters and exons as well as CH exons. This difference could stem from the recruitment/stabilization of histone modifying enzymes by stalling of the RNA polymerase II complex and are, therefore, enriched in S regions during germline IH-S-CH transcription.
According to the “histone code” hypothesis, combinatorial patterns of histone modifications not only encrypt information on the specification of distinct chromatin states but also increase the complexity of chromatin-interacting effectors114, thereby determining specific biological information outputs. High levels of many histone modifications in S regions that will undergo recombination suggest that these modifications play a role in recruiting and stabilizing CSR elements to S regions. Among histone modifications in S regions, H3K9me3 (a nominally repressive histone modification) is bound by HP1, as complexed with KAP1 and AID on Sμ, but not any acceptor S regions; B cells deficient in KAP1 display reduced AID binding to Sμ and defective CSR115. 14-3-3 adaptors bind the combinatorial H3K9acS10ph histone modification, facilitate histone crosstalk116, 117 and likely are readers of specific histone modifications in S regions (FIG. 4). 14-3-3 stabilization on S regions through binding to 5′-AGCT-3′ repeats and H3K9acS10ph would parallel the RAG1–RAG2 complex recruitment to V(D)J recombination centers through binding to recombination signal sequences and H3K4me3118.
Thus, specific histone codes are written in the S regions that are to undergo recombination, resulting from hitching of histone modifying enzymes on RNA polymerase II to reach S regions and enriched there during germline IH-S-CH transcription initiated by specific transcription factors, as activated by the combination of primary and secondary CSR-inducing stimuli. Such histone codes are read by CSR factors, such as 14-3-3 adaptors, thereby further stabilizing 14-3-3 on those S regions. Finally, 14-3-3 as well as other scaffold proteins, such as RPA and REV1, stabilize AID and/or UNG on S region DNA and enhance their enzymatic activity, thereby contributing to the “transduction” of epigenetic information to critical CSR factors.
Conclusions
Primary CSR-inducing stimuli provide the main thrust for CSR by inducing AID and other critical CSR factors as well as histone modifications. Secondary stimuli, i.e., cytokines, specify the downstream (acceptor) S region(s) that will undergo germline IH-S-CH transcription, histone modifications and, eventually, recombination. This division of roles, however, is blurred, as primary and secondary stimuli synergize – through combinatorial interplay of transcription factors – to induce AID, germline IH-S-CH transcription and histone modifications. Better characterization of the recruitment and interplay of these transcription factors would lead to understanding of the dynamics of the IgH locus chromatin opening and accessibility. Induction of open chromatin state and germline IH-S-CH transcription in the donor and acceptor S regions allows for 14-3-3 targeting to those S regions, as underpinned by the inherent high avidity of these adaptors for 5′-AGCT-3′ repeats, which are characteristic of all S regions. 14-3-3 adaptors, through direct interactions with AID and other CSR factors, would nucleate the assembly of macromolecular DNA and protein complexes, within which scaffold enzymatic and non-enzymatic elements further stabilize CSR factors on S regions. The dynamic composition of such macromolecular complexes and nature of histone code readers within the complexes remain to be fully elucidated. The regulation of CSR induction by TLR- and BCR-dependent stimuli and by different cell types needs to be explored, as does the contribution of CSR to the modulation of B cell differentiation. This will likely further our understanding of dysregulation of B cell differentiation and “off-targeting” of AID, which can lead to chromosomal translocations and neoplastic transformation.
Supplementary Material
Acknowledgements
We apologize that owing to space limitations, only a fraction of the relevant literature was cited. We thank all other members of the Casali laboratory for careful evaluation of this manuscript. Work on CSR in the Casali laboratory has been supported by National Institutes of Health grants AI 079705, AI 045011 and AI 060573. T. Mai has also been supported by grant T32 CA 009054.
Glossary terms
- B cell receptor (BCR) signaling
Signals triggered by antigen binding to membrane immunoglobulin of the BCR complex and phosphorylation of CD79a and CD79b of BCR at their ITAM motifs, leading to activation of tyrosine kinases, second messengers and pathways such as NF-κB and MAPK. BCR signaling is important for diverse B cell processes including development, survival, activation, proliferation and differentiation.
- Hyper-IgM syndrome (HIGM)
Syndromes characterized by elevated titers of IgM and decreased or absent titers of IgG, IgA and IgE, resulting from impairment or ablation of CSR due to B cell intrinsic or extrinsic causes.
- Activation-induced cytidine deaminase (AID)
A member of AID/APOBEC cytidine/cytosine deaminase family that catalyzes deoxycytosines deamination to deoxyuracils, thereby initiating immunoglobulin CSR, SHM and gene conversion.
- Uracil DNA glycosylase (UNG)
An enzyme that removes uracil bases from DNA by cleaving N-glycosylic bonds and initiates the basic excision repair pathway that removes deoxyuracils from the genome.
- Base excision repair (BER)
A DNA repair pathway that replaces damaged bases through excision of damaged bases by DNA glycosylases, nicking of abasic sites by apurinic/apyrimidinic endonucleases (APEs), filling-in of single-stranded DNA gap by DNA polymerases (such as polymerase β) and ligation by DNA ligases.
- Endonuclease G (EndoG)
A highly expressed eukaryotic nuclease that cleaves single- and double-strand DNA mainly at deoxyguanines/ deoxycytosines for non-caspase-associated DNA digestion in apoptosis and generation of DSBs in S region DNA in CSR.
- Mismatch repair (MMR)
A DNA repair system that recognizes and repairs mismatched DNA base pairs resulting from DNA damage or erroneous insertions, deletions and misincorporations of bases during DNA replication or recombination.
- Classic non-homologous end joining (C-NHEJ)
A DNA repair process that joins together non-homologous DNA ends using the four core components KU70, KU86, XRCC4 and DNA ligase IV, and additional components such as DNA-PKcs and Artemis.
- Alternative end joining (A-EJ)
A DSB repair process occurring in the absence of certain classical NHEJ components and likely dependent on the MRN complex, CtIP and/or Parp1 to generate a microhomology between DSB ends for end joining.
- Homologous recombination
A DNA recombination mechanism whereby extensive homology between two different DNA regions allows for annealing of complementary strands, each from two different regions, followed by enzymatic resolution of this hybrid intermediate, leading to recombination between the two different DNA regions.
- Primary CSR-inducing stimuli
Stimuli that induce expression of AID, 14-3-3 adaptors and other important CSR-factors through transcription factors, such as NF-κB, thereby enabling CSR. These stimuli are triggered by ligands engaging B cell surface and endosomal receptors: CD40 (T-dependent stimulus), dual TLR-BCR, TACI-BCR or TLR-TACI engagement (T-independent stimuli). Primary stimuli enable secondary CSR-inducing stimuli to induce germline IH-S-CH transcription and histone modifications in selected switch regions.
- Secondary CSR-inducing stimuli
Cytokines IL-4, TGF-β and IFN-γ (in mice but not humans) that cannot induce AID, but are required for directing switching to IgG, IgA and/or IgE by selecting the acceptor S region(s) through induction of germline IH-S-CH transcription and enrichment of specific histone modifications. These stimuli also enhance AID induction, as enabled by primary CSR-inducing stimuli.
- T-dependent antibody response
An antibody response to protein antigens that requires recognition of the antigen by T helper cells and cooperation between antigen-specific B and T cells and leads to the generation of high-affinity antibodies and long-term memory.
- Germinal centers
Organized microanatomical sites in secondary lymphoid organs where B cells, upon activation by specific T helper cells, proliferate, undergo SHM and CSR, and differentiate to memory B cells and plasma cells.
- Toll-like receptors (TLRs)
A family of conserved PRRs that recognize microbe-associated molecular patterns (MAMPs, invariant molecular structures recurring in microbes, but largely absent in metazoans) through leucine-rich repeats (LRRs) in the extracellular or intra-endosomal domain, and activate signaling pathways, leading to activation and differentiation of innate immune cells and B lymphocytes.
- TACI
A TNF receptor family member specific for B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL) that activates signaling pathways (such as the NF-κB pathway) through TRAF adaptors.
- T-independent antibody response
An antibody response to certain polymeric microbial constituents, including those that can directly activate B cell proliferation (type I) and repetitive polysaccharides (type II), in the absence of T cell help.
- Systemic lupus erythematosus (SLE)
A chronic systemic autoimmune disease that is characterized by multi-organ pathology including rashes, arthritis and kidney diseases. It is mediated by antibodies that are specific for double-stranded DNA and other nuclear antigens.
- Chromosomal translocations
Aberrant chromosome structures that result from the breaking and joining of a chromosome (or part of a chromosome) to another chromosome through improper DSB ligation and that frequently occur in cancers.
- RNA exosome
An eukaryotic multi-protein complex comprised of a ring-like nine-subunit core, consisting of six different RNase subunits and three distinct RNA-binding subunits, and three associated subunits that also possess RNase activities. It degrades erroneous RNA molecules, thereby enforcing RNA quality control, and non-erroneous RNA molecules, thereby maintaining their normal half-life and, as recently shown, exposing the DNA strands of transcribed S regions for AID to deaminate.
- 14-3-3 adaptors
A family of seven evolutionarily conserved and highly homologous adaptor proteins that form homo-/heterodimers and/or tetramers and bind a multitude of protein and DNA ligands through either the amphipathic groove or the outer surface. 14-3-3 proteins regulate diverse cell homeostasis events, such as signal transduction, survival, cell cycle progression and DNA replication, and cell differentiation processes, such as CSR.
- 5′-RGYW-3′ hotspot
A DNA motif that is mutated with the highest possibility, as a result of intrinsic preference of the SHM machinery (R: a purine; Y: a pyrimidine; W: A or T).
- R-loop
A nucleic acid structure in which an RNA strand hybridizes with its complementary strand of a double-strand DNA molecule and displaces the other DNA strand that is typically rich in deoxyguanines, thereby forming a loop of up to hundreds of nucleotides.
- Histone modifications
Post-translational chemical modifications, including methylation, acetylation and phosphorylation, of histones that are inserted by histone modifying enzymes and can actively mediate specific biological processes by interacting with selected effector proteins, including scaffold proteins that recruit/stabilize downstream factors. Histone modifications also determine chromatin accessibilities for DNA transactions.
- Scaffold protein
A multi-domain protein or multi-subunit protein oligomer that, by switching the conformation of its ligands, coordinates the spatio-temporal organization of macromolecular complex, leading to the proximity of enzymes with their substrates and, eventually, specific biological information output.
- REV1
A translesion DNA synthesis polymerase that bypasses lesions through its dCMP transferase activity for DNA repair. REV1 also possesses scaffold functions to recruit or stabilize other DNA repair factors.
- Histone code hypothesis
A hypothesis postulating that distinct histone modifications that act alone, sequentially or combinatorially, form a “histone code” that is read by effector proteins to mediate biological processes.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Casadevall A, Pirofski LA. A new synthesis for antibody-mediated immunity. Nat. Immunol. 2011;13:21–28. doi: 10.1038/ni.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wingren C, Hansson U-B, Alkner U. Van Nostrand's Scientific Encyclopedia2007) [Google Scholar]
- 3.Mond JJ, Lees A, Snapper CM. T cell-independent antigens type 2. Annu. Rev. Immunol. 1995;13:655–692. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- 4.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008;8:205–217. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 5.Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interface. Annu. Rev. Immunol. 2011;29:273–293. doi: 10.1146/annurev-immunol-031210-101317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. • A comprehensive review on molecular mechanisms of CSR.
- 7.Honjo T. A memoir of AID which engraves antibody memory on DNA. Nat. Immunol. 2008;9:335–337. doi: 10.1038/ni0408-335. [DOI] [PubMed] [Google Scholar]
- 8.Liu W, Meckel T, Tolar P, Sohn HW, Pierce SK. Intrinsic properties of immunoglobulin IgG1 isotype-switched B cell receptors promote microclustering and the initiation of signaling. Immunity. 2010;32:778–789. doi: 10.1016/j.immuni.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casali P. Somatic recombination and hypermutation in the immune system. In: Krebs JE, Goldstein ES, Kilpatrick ST, editors. Lewin's Genes X. Sudbury, MA: Jones & Bartlett; 2009. pp. 570–623. [Google Scholar]
- 10.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat. Immunol. 2010;11:681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- 11.Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2004;2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- 12.Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 13.Plotkin SA. Vaccines: correlates of vaccine-induced immunity. Clin. Infect Dis. 2008;47:401–409. doi: 10.1086/589862. [DOI] [PubMed] [Google Scholar]
- 14.Delker RK, Fugmann SD, Papavasiliou FN. A coming-of-age story: activation-induced cytidine deaminase turns 10. Nat. Immunol. 2009;10:1147–1153. doi: 10.1038/ni.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, Gramlich HS, Schatz DG, Woodgate R, Wilson DM, 3rd, Gearhart PJ. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat. Immunol. 2011;12:70–76. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zan H, Casali P. AID- and Ung-dependent generation of staggered double-strand DNA breaks in immunoglobulin class switch DNA recombination: a post-cleavage role for AID. Mol. Immunol. 2008;46:45–61. doi: 10.1016/j.molimm.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zan H, Zhang J, Al-Qahtani A, Pone EJ, White CA, Lee D, Yel L, Mai T, Casali P. Endonuclease G plays a role in immunoglobulin class switch DNA recombination by introducing double-strand breaks in switch regions. Mol. Immunol. 2011;48:610–622. doi: 10.1016/j.molimm.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi M, Aida M, Nagaoka H, Begum NA, Kitawaki Y, Nakata M, Stanlie A, Doi T, Kato L, Okazaki IM, Shinkura R, Muramatsu M, Kinoshita K, Honjo T. AID-induced decrease in topoisomerase 1 induces DNA structural alteration and DNA cleavage for class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2009;106:22375–22380. doi: 10.1073/pnas.0911879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32:194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Min IM, Schrader CE, Vardo J, Luby TM, D'Avirro N, Stavnezer J, Selsing E. The Sµ tandem repeat region is critical for Ig isotype switching in the absence of Msh2. Immunity. 2003;19:515–524. doi: 10.1016/s1074-7613(03)00262-0. [DOI] [PubMed] [Google Scholar]
- 22.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat. Struct. Mol. Biol. 2011;18:75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robert I, Dantzer F, Reina-San-Martin B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 2009;206:1047–1056. doi: 10.1084/jem.20082468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eccleston J, Yan C, Yuan K, Alt FW, Selsing E. Mismatch repair proteins MSH2, MLH1, and EXO1 are important for class-switch recombination events occurring in B cells that lack nonhomologous end joining. J. Immunol. 2011;186:2336–2343. doi: 10.4049/jimmunol.1003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008;8:22–33. doi: 10.1038/nri2217. • This and Ref. 26 provide a comprehensive review of B cell differentiation processes.
- 27.McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat. Rev. Immunol. 2012;12:24–34. doi: 10.1038/nri3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, Xu Z, Mai T, Casali P. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat. Immunol. 2009;10:540–550. doi: 10.1038/ni.1725. • This study outlines the AID gene promoter structure and identified important transcription factors that regulate AID induction.
- 29.Pone EJ, Zan H, Zhang J, Al-Qahtani A, Xu Z, Casali P. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: relevance to microbial antibody responses. Crit. Rev. Immunol. 2010;30:1–29. doi: 10.1615/critrevimmunol.v30.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu. Rev. Immunol. 2011;29:447–491. doi: 10.1146/annurev-immunol-030409-101335. [DOI] [PubMed] [Google Scholar]
- 31.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, Shan M, Xiong H, Bussel JB, Chiu A, Puel A, Reichenbach J, Marodi L, Doffinger R, Vasconcelos J, Issekutz A, Krause J, Davies G, Li X, Grimbacher B, Plebani A, Meffre E, Picard C, Cunningham-Rundles C, Casanova JL, Cerutti A. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat. Immunol. 2010;11:836–845. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pone EJ, Zhang J, Mai T, White CA, Li G, Sakakura JK, Patel PJ, Al-Qahtani A, Zan H, Xu Z, Casali P. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nat. Commun. 2012;3:767. doi: 10.1038/ncomms1769. • This and Ref. 34 outline the critical role of BCR signaling in enabling TLRs to efficiently induce T cell-independent CSR and B cell response.
- 34.Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat. Rev. Immunol. 2012;12:282–294. doi: 10.1038/nri3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wesemann DR, Magee JM, Boboila C, Calado DP, Gallagher MP, Portuguese AJ, Manis JP, Zhou X, Recher M, Rajewsky K, Notarangelo LD, Alt FW. Immature B cells preferentially switch to IgE with increased direct Smu to Sepsilon recombination. J. Exp. Med. 2011;208:2733–2746. doi: 10.1084/jem.20111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev. 2010;24:2107–2114. doi: 10.1101/gad.1963010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC, 3rd, Nussenzweig MC, Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nat. Genet. 2008;40:108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- 40.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, Ried T, Nussenzweig A, Nussenzweig MC. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol. Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat. Immunol. 2010;11:820–826. doi: 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Z, Pone EJ, Al-Qahtani A, Park SR, Zan H, Casali P. Regulation of aicda expression and AID activity: relevance to somatic hypermutation and class switch DNA recombination. Crit. Rev. Immunol. 2007;27:367–397. doi: 10.1615/critrevimmunol.v27.i4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasler J, Rada C, Neuberger MS. Cytoplasmic activation-induced cytidine deaminase (AID) exists in stoichiometric complex with translation elongation factor 1alpha (eEF1A) Proc. Natl. Aca. Sci. U.S.A. 2011;108:18366–18371. doi: 10.1073/pnas.1106729108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoufouchi S, Faili A, Zober C, D'Orlando O, Weller S, Weill JC, Reynaud CA. Proteasomal degradation restricts the nuclear lifespan of AID. J. Exp. Med. 2008;205:1357–1368. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uchimura Y, Barton LF, Rada C, Neuberger MS. REG-gamma associates with and modulates the abundance of nuclear activation-induced deaminase. J. Exp. Med. 2011;208:2385–2891. doi: 10.1084/jem.20110856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li G, Pone EJ, Tran DC, Patel PJ, Dao L, Xu Z, Casali P. Iron inhibits activation-induced cytidine deaminase enzymatic activity and modulates immunoglobulin class switch DNA recombination. J. Biol. Chem. 2012 doi: 10.1074/jbc.M112.366732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He B, Qiao X, Cerutti A. CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J. Immunol. 2004;173:4479–4491. doi: 10.4049/jimmunol.173.7.4479. [DOI] [PubMed] [Google Scholar]
- 48.Zarnegar B, He JQ, Oganesyan G, Hoffmann A, Baltimore D, Cheng G. Unique CD40-mediated biological program in B cell activation requires both type 1 and type 2 NF-kappaB activation pathways. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8108–8113. doi: 10.1073/pnas.0402629101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, Honjo T, Nagaoka H. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat. Immunol. 2010;11:148–154. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 50.Smale ST. Hierarchies of NF-kappaB target-gene regulation. Nat. Immunol. 2011;12:689–694. doi: 10.1038/ni.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baltimore D. NF-kappaB is 25. Nat. Immunol. 2011;12:683–685. doi: 10.1038/ni.2072. [DOI] [PubMed] [Google Scholar]
- 52.Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J. Exp. Med. 2009;206:99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mai T, Zan H, Zhang J, Hawkins JS, Xu Z, Casali P. Estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to potentiate HoxC4-mediated activation-induced cytosine deaminase induction, immunoglobulin class switch DNA recombination, and somatic hypermutation. J. Biol. Chem. 2010;285:37797–37810. doi: 10.1074/jbc.M110.169086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White CA, Seth Hawkins J, Pone EJ, Yu ES, Al-Qahtani A, Mai T, Zan H, Casali P. AID dysregulation in lupus-prone MRL/Fas(lpr/lpr) mice increases class switch DNA recombination and promotes interchromosomal c-Myc/IgH loci translocations: Modulation by HoxC4. Autoimmunity. 2011 doi: 10.3109/08916934.2011.577128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat. Immunol. 2003;4:586–593. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- 56.Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, Alt FW, Tang J, Oltz EM, Murphy TL, Murphy KM. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011;12:536–543. doi: 10.1038/ni.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Betz BC, Jordan-Williams KL, Wang C, Kang SG, Liao J, Logan MR, Kim CH, Taparowsky EJ. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J. Exp. Med. 2010;207:933–942. doi: 10.1084/jem.20091548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cerutti A, Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity. 2008;28:740–750. doi: 10.1016/j.immuni.2008.05.001. • This and Ref. 59 are comprehensive reviews T cell-dependent and -independent CSR to IgA in mucosal antibody responses.
- 59.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol. 2010;28:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 60. Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, Filsuf D, Chen HT, Gazumyan A, Yamane A, Cho YW, Sun HW, Ge K, Peng W, Nussenzweig MC, Casellas R, Dressler GR, Zhao K, Nussenzweig A. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329:917–923. doi: 10.1126/science.1187942. • This study has shown that abolishing of chromatin changes results in CSR impairment.
- 61.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 62. Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004;4:541–552. doi: 10.1038/nri1395. • An overview of the role of germline IH-S-CH transcription in regulating AID activity and CSR.
- 63. Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, Alt FW. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. • This study provides important insights into the mechanisms underlying AID deamination of dC on both DNA strands.
- 64.Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. J. Exp. Med. 2009;206:1073–1087. doi: 10.1084/jem.20082311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perlot T, Alt FW. Cis-regulatory elements and epigenetic changes control genomic rearrangements of the IgH locus. Adv. Immunol. 2008;99:1–32. doi: 10.1016/S0065-2776(08)00601-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dunnick WA, Collins JT, Shi J, Westfield G, Fontaine C, Hakimpour P, Papavasiliou FN. Switch recombination and somatic hypermutation are controlled by the heavy chain 3' enhancer region. J. Exp. Med. 2009;206:2613–2623. doi: 10.1084/jem.20091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim EC, Edmonston CR, Wu X, Schaffer A, Casali P. The HoxC4 homeodomain protein mediates activation of the immunoglobulin heavy chain 3' hs1,2 enhancer in human B cells. Relevance to class switch DNA recombination. J. Biol. Chem. 2004;279:42258–42269. doi: 10.1074/jbc.M407496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, Alt FW, Cogne M, Pinaud E, Kenter AL. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hackney JA, Misaghi S, Senger K, Garris C, Sun Y, Lorenzo MN, Zarrin AA. DNA targets of AID evolutionary link between antibody somatic hypermutation and class switch recombination. Adv. Immunol. 2009;101:163–189. doi: 10.1016/S0065-2776(08)01005-5. [DOI] [PubMed] [Google Scholar]
- 70. Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CW, Park SR, Steinacker P, Li Z, Yates JRI, Herron B, Otto M, Zan H, Fu H, Casali P. 14-3-3 adaptor proteins recruit AID to 5'-AGCT-3'-rich switch regions for class switch recombination. Nat. Struct. Mol. Biol. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. • This study has identified 14-3-3 proteins as adaptors that specifically target S region core 5'-AGCT-3' repeats and recruit AID.
- 71.Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, Tian M. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat. Immunol. 2004;5:1275–1281. doi: 10.1038/ni1137. [DOI] [PubMed] [Google Scholar]
- 72.Han L, Masani S, Yu K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2011;108:11584–11589. doi: 10.1073/pnas.1018726108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang FT, Yu K, Balter BB, Selsing E, Oruc Z, Khamlichi AA, Hsieh CL, Lieber MR. Sequence dependence of chromosomal R-loops at the immunoglobulin heavy-chain Smu class switch region. Mol. Cell Biol. 2007;27:5921–5932. doi: 10.1128/MCB.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morrison DK. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell. Biol. 2009;19:16–23. doi: 10.1016/j.tcb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat. Immunol. 2009;10:420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol. Cell. 2003;12:501–508. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 77.Shinkura R, Ito S, Begum NA, Nagaoka H, Muramatsu M, Kinoshita K, Sakakibara Y, Hijikata H, Honjo T. Separate domains of AID are required for somatic hypermutation and class-switch recombination. Nat. Immunol. 2004;5:707–712. doi: 10.1038/ni1086. [DOI] [PubMed] [Google Scholar]
- 78.Ranjit S, Khair L, Linehan EK, Ucher AJ, Chakrabarti M, Schrader CE, Stavnezer J. AID binds cooperatively with UNG and Msh2-Msh6 to Ig switch regions dependent upon the AID C terminus. J. Immunol. 2011;187:2464–2475. doi: 10.4049/jimmunol.1101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geisberger R, Rada C, Neuberger MS. The stability of AID and its function in class-switching are critically sensitive to the identity of its nuclear-export sequence. Proc. Natl. Acad. Sci. USA. 2009;106:6736–6741. doi: 10.1073/pnas.0810808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y, Shimizu A. Transcriptioncoupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 81.Shen HM, Poirier MG, Allen MJ, North J, Lal R, Widom J, Storb U. The activation-induced cytidine deaminase (AID) efficiently targets DNA in nucleosomes but only during transcription. J. Exp. Med. 2009;206:1057–1071. doi: 10.1084/jem.20082678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibodydiversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol. Cell. 2008;31:474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 83. Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, Root DE, Casellas R, Nussenzweig MC. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. • This study has demonstrated that AID is associated with RNAP II through SPT5 on S regions and promoters of certain non-Ig loci.
- 84.Okazaki IM, Okawa K, Kobayashi M, Yoshikawa K, Kawamoto S, Nagaoka H, Shinkura R, Kitawaki Y, Taniguchi H, Natsume T, Iemura SI, Honjo T. Histone chaperone Spt6 is required for class switch recombination but not somatic hypermutation. Proc. Natl. Acad. Sci. USA. 2011;108:7920–7925. doi: 10.1073/pnas.1104423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat. Immunol. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, histone modifications create chromatin accessibility during class switch recombination. J. Exp. Med. 2009;206:1817–1830. doi: 10.1084/jem.20081678. • This study and Ref. 87 have demonstrated that RNA polymerase II and active histone modifications are highly enriched in the S regions that will undergo recombination.
- 87.Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, Gearhart PJ. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J. Exp. Med. 2009;206:1237–1244. doi: 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat. Immunol. 2011;12:62–69. doi: 10.1038/ni.1964. • This study and Refs. 89–94 have demonstrated that AID can bind to non-Ig loci, albeit less efficiencly than to S regions, and cause genome-wide DNA deamination, DSB generation and translocations.
- 89.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 90.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 91.Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Mol. Cell. 2011;41:232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, Mathe E, Liang G, Cobell J, Nakahashi H, Robbiani DF, Nussenzweig A, Hager GL, Nussenzweig MC, Casellas R. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, Casellas R, Nussenzweig MC. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, Neuberg D, Monti S, Giallourakis CC, Gostissa M, Alt FW. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. U.S.A. 2011;108:563–568. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 98.Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE. Post-replicative base excision repair in replication foci. EMBO J. 1999;18:3834–3844. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guo S, Zhang Y, Yuan F, Gao Y, Gu L, Wong I, Li GM. Regulation of replication protein A functions in DNA mismatch repair by phosphorylation. J. Biol. Chem. 2006;281:21607–21616. doi: 10.1074/jbc.M603504200. [DOI] [PubMed] [Google Scholar]
- 100.Oakley GG, Tillison K, Opiyo SA, Glanzer JG, Horn JM, Patrick SM. Physical interaction between replication protein A (RPA) and MRN: involvement of RPA2 phosphorylation and the N-terminus of RPA1. Biochemistry. 2009;48:7473–7481. doi: 10.1021/bi900694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shao R-G, Cao C-X, Zhang H, Kohn KW, Wiold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yoo E, Kim BU, Lee SY, Cho CH, Chung JH, Lee CH. 53BP1 is associated with replication protein A and is required for RPA2 hyperphosphorylation following DNA damage. Oncogene. 2005;24:5423–5430. doi: 10.1038/sj.onc.1208710. [DOI] [PubMed] [Google Scholar]
- 103.Block WD, Yu Y, Lees-Miller SP. Phosphatidyl inositol 3-kinase-like serine/threonine protein kinases (PIKKs) are required for DNA damage-induced phosphorylation of the 32 kDa subunit of replication protein A at threonine-21. Nucleic Acids Res. 2004;32:997–1005. doi: 10.1093/nar/gkh265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Begum NA, Stanlie A, Doi T, Sasaki Y, Jin HW, Kim YS, Nagaoka H, Honjo T. Further evidence for involvement of a noncanonical function of uracil DNA glycosylase in class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2752–2757. doi: 10.1073/pnas.0813252106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kracker S, Imai K, Gardes P, Ochs HD, Fischer A, Durandy AH. Impaired induction of DNA lesions during immunoglobulin class-switch recombination in humans influences end-joining repair. Proc. Natl. Acad. Sci. U.S.A. 2010;107:22225–22230. doi: 10.1073/pnas.1012591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Doi T, Kato L, Ito S, Shinkura R, Wei M, Nagaoka H, Wang J, Honjo T. The C-terminal region of activation-induced cytidine deaminase is responsible for a recombination function other than DNA cleavage in class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2758–2763. doi: 10.1073/pnas.0813253106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med. 2006;203:319–323. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Masuda K, Ouchida R, Li Y, Gao X, Mori H, Wang JY. A critical role for REV1 in regulating the induction of C:G transitions and A:T mutations during Ig gene hypermutation. J. Immunol. 2009;183:1846–1850. doi: 10.4049/jimmunol.0901240. [DOI] [PubMed] [Google Scholar]
- 109.Wang L, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. J. Exp. Med. 2006;203:215–226. doi: 10.1084/jem.20051774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chowdhury M, Forouhi O, Dayal S, McCloskey N, Gould HJ, Felsenfeld G, Fear DJ. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15872–15877. doi: 10.1073/pnas.0808462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kuang FL, Luo Z, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2009;106:5288–5293. doi: 10.1073/pnas.0901368106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2010;107:22190–22195. doi: 10.1073/pnas.1016923108. • This study has shown that inhibition of writing of H3K4me3 results in CSR impairment.
- 113.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 114.Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, Alt FW, Losson R, Reina-San-Martin B. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J. Exp. Med. 2011;208:1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Macdonald N, Welburn JP, Noble ME, Nguyen A, Yaffe MB, Clynes D, Moggs JG, Orphanides G, Thomson S, Edmunds JW, Clayton AL, Endicott JA, Mahadevan LC. Molecular basis for the recognition of phosphorylated and phosphoacetylated histone H3 by 14-3-3. Mol. Cell. 2005;20:199–211. doi: 10.1016/j.molcel.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 117.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 118. Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011;11:251–263. doi: 10.1038/nri2941. • A comprehensive review on epigenetic regulation of V(D)J recombination.
- 119.Orthwein A, Patenaude AM, Affar el B, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J. Exp. Med. 2010;207:2751–2765. doi: 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Saribasak H, Maul RW, Cao Z, McClure RL, Yang W, McNeill DR, Wilson DM, 3rd, Gearhart PJ. XRCC1 suppresses somatic hypermutation and promotes alternative nonhomologous end joining in Igh genes. J. Exp. Med. 2011;208:2209–2216. doi: 10.1084/jem.20111135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, Zhang Y, Chai H, Lee CS, Jankovic M, Saez LM, Nussenzweig MC, McKinnon PJ, Alt FW, Schwer B. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1) Proc. Natl. Acad. Sci. U.S.A. 2012;109:2473–2478. doi: 10.1073/pnas.1121470109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Han L, Mao W, Yu K. X-ray repair cross-complementing protein 1 (XRCC1) deficiency enhances class switch recombination and is permissive for alternative end joining. Proc. Natl. Acad. Sci. U.S.A. 2012;109:4604–4608. doi: 10.1073/pnas.1120743109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bardwell PD, Woo CJ, Wei K, Li Z, Martin A, Sack SZ, Parris T, Edelmann W, Scharff MD. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat. Immunol. 2004;5:224–229. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- 124.Vallur AC, Maizels N. Activities of human exonuclease 1 that promote cleavage of transcribed immunoglobulin switch regions. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16508–16512. doi: 10.1073/pnas.0805327105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu X, Tsai CY, Patam MB, Zan H, Chen JP, Lipkin SM, Casali P. A role for the MutL mismatch repair Mlh3 protein in immunoglobulin class switch DNA recombination and somatic hypermutation. J. Immunol. 2006;176:5426–5437. doi: 10.4049/jimmunol.176.9.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 127.Rulten SL, Fisher AE, Robert I, Zuma MC, Rouleau M, Ju L, Poirier G, Reina-San-Martin B, Caldecott KW. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell. 2011;41:33–45. doi: 10.1016/j.molcel.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 128.Franco S, Murphy MM, Li G, Borjeson T, Boboila C, Alt FW. DNA-PKcs and Artemis function in the endjoining phase of immunoglobulin heavy chain class switch recombination. J. Exp. Med. 2008;205:557–564. doi: 10.1084/jem.20080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Du L, van der Burg M, Popov SW, Kotnis A, van Dongen JJ, Gennery AR, Pan-Hammarstrom Q. Involvement of Artemis in nonhomologous end-joining during immunoglobulin class switch recombination. J. Exp. Med. 2008;205:3031–3040. doi: 10.1084/jem.20081915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rivera-Munoz P, Soulas-Sprauel P, Le Guyader G, Abramowski V, Bruneau S, Fischer A, Paques F, de Villartay JP. Reduced immunoglobulin class switch recombination in the absence of Artemis. Blood. 2009;114:3601–3609. doi: 10.1182/blood-2008-11-188383. [DOI] [PubMed] [Google Scholar]
- 131.Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, Morse HCr, Gearhart PJ, Wynshaw-Boris A, Max EE, Hodes RJ. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J. Exp. Med. 2004;200:1111–1121. doi: 10.1084/jem.20041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. J. Exp. Med. 2004;200:1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pan-Hammarström Q, Lähdesmäki A, Zhao Y, Du L, Zhao Z, Wen S, Ruiz-Perez VL, Dunn-Walters DK, Goodship JA, Hammarström L. Disparate roles of ATR and ATM in immunoglobulin class switch recombination and somatic hypermutation. J. Exp. Med. 2006;203:99–110. doi: 10.1084/jem.20050595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, Dubois RL, Alt FW. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011;469:250–254. doi: 10.1038/nature09604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Du L, Peng R, Bjorkman A, Filipe de Miranda N, Rosner C, Kotnis A, Berglund M, Liu C, Rosenquist R, Enblad G, Sundstrom C, Hojjat-Farsangi M, Rabbani H, Teixeira MR, Revy P, Durandy A, Zeng Y, Gennery AR, de Villartay JP, Pan-Hammarstrom Q. Cernunnos influences human immunoglobulin class switch recombination and may be associated with B cell lymphomagenesis. J. Exp. Med. 2012;209:291–305. doi: 10.1084/jem.20110325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Casellas R, Nussenzweig A, Wuerffel R, Pelanda R, Reichlin A, Suh H, Qin XF, Besmer E, Kenter A, Rajewsky K, Nussenzweig MC. Ku80 is required for immunoglobulin isotype switching. EMBO J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Manis JP, Gu Y, Lansford R, Sonoda E, Ferrini R, Davidson L, Rajewsky K, Alt FW. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zan H, White CA, Thomas LM, Zhang J, Li G, Yu ES, Mai T, Xu Z, de Wind N, Schimenti JC, Casali P. The translesion DNA synthesis polymerase Rev1 recruits Ung for immunoglobulin class switch DNA recombination. 2012 doi: 10.1016/j.celrep.2012.09.029. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li L, Halaby MJ, Hakem A, Cardoso R, El Ghamrasni S, Harding S, Chan N, Bristow R, Sanchez O, Durocher D, Hakem R. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J. Exp. Med. 2010;207:983–997. doi: 10.1084/jem.20092437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Santos MA, Huen MS, Jankovic M, Chen HT, Lopez-Contreras AJ, Klein IA, Wong N, Barbancho JL, Fernandez-Capetillo O, Nussenzweig MC, Chen J, Nussenzweig A. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J. Exp. Med. 2010;207:973–981. doi: 10.1084/jem.20092308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, Zaheen A, Durocher D, Scharff MD, Martin A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2010;107:809–814. doi: 10.1073/pnas.0913790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Soulas-Sprauel P, Le Guyader G, Rivera-Munoz P, Abramowski V, Olivier-Martin C, Goujet-Zalc C, Charneau P, de Villartay JP. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. J. Exp. Med. 2007;204:1717–1727. doi: 10.1084/jem.20070255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, Alt FW. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.