

Abstract
DYRK kinases are involved in alternative pre-mRNA splicing as well as in neuropathological states such as Alzheimer's disease and Down syndrome. In this study, we present the design, synthesis, and biological evaluation of indirubins as DYRK inhibitors with enhanced selectivity. Modifications of the bis-indole included polar or acidic functionalities at positions 5′ and 6′ and a bromine or a trifluoromethyl group at position 7, affording analogues that possess high activity and pronounced specificity. Compound 6i carrying a 5′-carboxylate moiety demonstrated the best inhibitory profile. A novel inverse binding mode, which forms the basis for the improved selectivity, was suggested by molecular modeling and confirmed by determining the crystal structure of DYRK2 in complex with 6i. Structure–activity relationships were further established, including a thermodynamic analysis of binding site water molecules, offering a structural explanation for the selective DYRK inhibition.
Keywords: DYRK inhibitors, indirubins, kinase selectivity, pre-mRNA splicing, docking calculations, WaterMap
The dual-specificity tyrosine phosphorylation-regulated kinase (DYRK)/minibrain family of protein kinases represents a relatively unexplored portion of the human kinome in terms of inhibitor development. The DYRK1a gene is localized in the Down syndrome (DS) critical region of chromosome 21, and its overexpression has been related to the development of DS features.1 DYRK2 regulates p53 to induce apoptosis in response to DNA damage and serves as a scaffold for an E3 ubiquitin ligase complex.2,3 It is mainly expressed during development, but it has been found to be overexpressed in adenocarcinomas of the esophagus and lung.4 DYRK2 has been recently identified as the priming phosphorylation enzyme for oncogenes c-Jun and c-Myc, although the precise role of DYRK2 in cancer needs further investigation.5 As such, DYRKs can be regarded as emerging targets, and selective inhibitors could assist in elucidating the various biochemical mechanisms in which they are implicated.
A limited number of compounds demonstrate selective inhibition toward DYRK kinases, although binding with cdc2-like kinases (CLKs) is frequently observed.6−10 The bis-indole indirubin has been utilized as an interesting lead for the development of potent kinase inhibitors.11−17 Regardless of the notable selectivity of the various indirubin analogues, they all share a common interaction pattern with their target enzyme active site (for example, see PDB structures 1E9H, 2BHE, 1UV5, and 1UNH). Recently, the structural basis of selective inhibition of CLK3, a closely related kinase, by leucettamine-B derivatives was reported.6 Interestingly, the cocrystal structure revealed a significantly different binding mode from the typical ATP-competitive interaction pattern. A similar binding orientation was subsequently reported as the structural basis for selective DYRK1a inhibition by harmine.7 Those studies prompted us to investigate whether selective indirubin analogues targeting the closely related DYRK2 could be designed.
The initial series of inhibitors had minor modifications on the indirubin core that were designed with the objective of generating a noncanonical binding mode and subsequently an improved selectivity profile toward DYRKs. A bulky modification such as bromine or trifluoromethyl was introduced at position 7 based on previous studies of the analogue 7BIO (7-bromoindirubin-3′ oxime; Table 1) that showed a minor negative influence on affinity due to a putative steric clash between the inhibitor and the kinase hinge.15 In addition, the removal of a hydrogen bond donor from the indirubin core by methylating the lactam nitrogen at position 1 was explored as a more drastic way to perturb the interactions. Such methylated analogues are totally inactive against CDKs and glycogen synthase kinase 3β (GSK3β) and serve as negative control compounds.13 However, the low water solubility of the bis-indole system necessitated the introduction of polar or ionizable functionalities such as carbomethoxy, methylene hydroxy, aldehyde, carboxy, tetrazole, cyano, and oxime groups at positions 5′ and 6′. Moreover, positions 5′ and 6′ represent the least explored sites of this scaffold. Thus, results would contribute toward a systematic exploration of the indirubin SAR landscape. The routes employed for derivative synthesis are summarized in Schemes S1 and S2 in the Supporting Information. All compounds were evaluated against a panel of five kinases (Table 1).
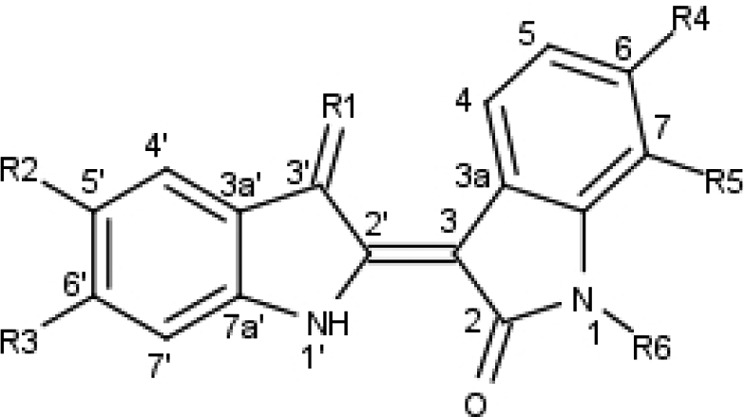
Table 1. IC50 Values (μM) of Indirubin Analogues Binding to Five Kinasesa.
| IC50 (μM) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | R4 | R5 | R6 | CDK5 | GSK3 | CK1 | DYRK1a | DYRK2 |
| 6BIO | NOH | H | H | Br | H | H | 0.083 | 0.005 | 1.2 | 1.7 | 2.1 |
| 7BIO | NOH | H | H | H | Br | H | >10 | >10 | >10 | 1.9 | 1.3 |
| 5a | O | H | H | H | Br | CH3 | >10 | >10 | >10 | >10 | >10 |
| 5b | O | COOCH3 | H | H | H | H | >10 | >10 | 0.58 | >10 | >10 |
| 5c | O | COOH | H | H | H | H | 3.40 | 1.10 | 1.40 | 2.00 | 1.80 |
| 5d | O | COOCH3 | H | H | Br | H | >10 | 25 | >10 | >10 | >10 |
| 5e | O | COOCH3 | H | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 5f | O | CN | H | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 5g | O | tetrazole | H | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 5h | O | COOCH3 | H | H | Br | CH3 | >10 | >10 | >10 | >10 | >10 |
| 5i | O | H | COOH | H | H | H | >10 | >10 | 1.70 | 2.60 | 1.10 |
| 5j | O | H | COOCH3 | H | Br | H | >10 | >10 | >10 | >10 | >10 |
| 5k | O | H | COOCH3 | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 5l | O | H | COOH | H | Br | H | >10 | >10 | 9.30 | 1.30 | 1.30 |
| 5m | O | H | COOH | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 5n | O | CH2OH | H | H | Br | H | >10 | >10 | >10 | >10 | >10 |
| 5o | O | CH=NOH | H | H | Br | H | 0.20 | 0.20 | 0.80 | 1.10 | ND |
| 6a | NOH | H | H | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 6b | NOH | H | H | H | Br | CH3 | >10 | >10 | >10 | >10 | >10 |
| 6c | NOH | COOCH3 | H | H | H | H | 0.52 | 0.40 | 0.30 | >10 | >1 |
| 6d | NOH | COOH | H | H | H | H | 0.53 | 0.07 | 0.42 | 0.31 | 0.35 |
| 6e | NOH | COOCH3 | H | H | Br | H | >10 | >10 | >10 | >10 | >10 |
| 6f | NOH | COOCH3 | H | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 6g | NOH | CN | H | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 6h | NOH | tetrazole | H | H | CF3 | H | >10 | >10 | >10 | >10 | >10 |
| 6i | NOH | COOH | H | H | Br | H | >10 | >10 | >10 | 0.21 | 0.13 |
| 6j | NOH | COOH | H | H | CF3 | H | 1.6 | 1.2 | 4.2 | 0.41 | ND |
| 6k | NOH | COOCH3 | H | H | Br | CH3 | >10 | >10 | >10 | >10 | >10 |
| 6l | NOH | COOH | H | H | Br | CH3 | 0.2 | >10 | >10 | 0.13 | 0.22 |
| 6m | NOH | H | COOH | H | H | H | 3.30 | 6.10 | 0.80 | 0.63 | 1.90 |
| 6n | NOH | H | COOCH3 | H | Br | H | >10 | >10 | >10 | >10 | >10 |
| 6o | NOH | H | COOCH3 | H | CF3 | H | 1.7 | 0.1 | 5.0 | 0.11 | ND |
| 6p | NOH | H | COOH | H | Br | H | >10 | >10 | 7.40 | 0.60 | 1.70 |
| 6q | NOH | H | COOH | H | CF3 | H | 3.1 | >10 | >10 | 7.0 | >10 |
| 6r | NOH | CH2OH | H | H | Br | H | 0.40 | 0.40 | >10 | 1.60 | ND |
| 6s | NOH | CH=NOH | H | H | Br | H | 0.60 | 0.60 | 7.40 | 2.10 | ND |
All measurements were determined in triplicate, and mean values are reported. The standard error of determination in all cases does not exceed 10%. The Km for ATP with DYRK2 is 7.7 μM.
7BIO showed significant gains in selectivity toward DYRKs as compared with 6BIO. Derivatives 5c, 5i, 6d, and 6m lacking a 7-bromo substitution showed a diminished selectivity toward DYRKs. Results also suggested an anionic functionality to maintain efficient inhibition, as protection of the acidic moiety abolished DYRK activity of 7-substituted methylesters 5d, 5e, 5h, 6e, and 6f. The difference of IC50 values between analogues 6d and 6m indicated 5′ rather than 6′ as the preferred position for an acidic substitution to maintain DYRK inhibition. The presence of a 3′-oxime moiety generally improved potency with minor effects on selectivity. Finally, the influence of methyl-capping N1 was unfavorable for DYRK activity (7BIO as compared to 6b). Inspection of the SAR landscape suggested that the combination of the 7-bromo, 3′-oxime, and 5′-carboxy substitutions would be optimal for DYRK selective inhibition. The corresponding 7-bromo-5′-carboxyindirubin-3′ oxime (6i) proved to be a potent inhibitor of DYRK1a and DYRK2 (IC50 values 210 and 130 nM, respectively). DYRK activity was accompanied by a marked selectivity, as 6i was inactive toward cyclin-dependent kinase 5 (CDK5), GSK3β, and casein kinase 1 (CK1). Interestingly, the introduction of the bulky bromine in position 7 enhanced selective DYRK inhibition only when combined with the carboxylate substitution, which was evident comparing 6d and 6i. Quite unexpectedly, the carboxylate bioisostere tetrazole compounds (5g and 6h) were inactive. Moreover, analogues where the carboxylate moiety was replaced by a polar but neutral oxime group (5o and 6s) showed good DYRK inhibition but poor selectivity, despite of the concurrent 7-bromine substitution. The optimal effect of bromine in 6i was denoted by the lack of selectivity of 6j carrying the closely related substituent CF3 at position 7. Surprisingly, the N1-methylated analogue of 6i (compound 6l) retained selectivity and affinity for DYRK kinases with small variations in IC50 values determined for the two family members (IC50 of 130 nM for DYRK1a and 220 nM for DYRK2).
To rationalize the affinity and selectivity profiles, we analyzed poses from previous docking studies to a DYRK1a-derived homology model of DYRK2, which showed that compound 6i may adopt a binding mode that is significantly different from the experimentally established indirubin-kinase binding mode.12 Two distinct modes were observed in the docking studies, distinguished by a 180° flip of the indirubin core with respect to either its primary or secondary axis (Figures S1A and S1B in the Supporting Information, respectively). In both cases, the usually observed hydrogen bond triplet between the indirubin pharmacophore and the kinase hinge was either disrupted (mode I) or inverted (mode II). In both cases, the 5′ carboxylate formed a salt bridge with either the Lys165 (mode I) or the catalytic Lys178 (mode II).
To confirm the inverse binding mode, we determined the crystal structure of 6i bound to DYRK2 at a resolution of 2.28 Å (PDB ID 3KVW). The structure (Figures 1A and S2 in the Supporting Information) revealed that the binding mode was indeed inverted with respect to the geometry of all previously reported indirubin-kinase complexes and consistent with the predicted binding mode II. The predicted salt bridge anchoring the anionic 5′-carboxylate to the side chain of Lys178 was observed in the crystal structure. An additional hydrogen bond was present between the 5′ carboxylate and the NH backbone of Asp295. Furthermore, three water molecules participated in the hydrogen bond network by bridging the inhibitor carboxylate and Lys178 ammonium groups to the side chains of Asp295 and Glu193. At the hinge, two hydrogen bonds were formed between the lactam carbonyl of 6i and the NH backbone of Leu231 (n + 3 from the gatekeeper) and between the lactam nitrogen of the inhibitor and the backbone carbonyl of the same residue. The solvent-accessible surface area buried upon binding was 417 Å2 (229.4 Å2 nonpolar) for the ligand and 254.8 Å2 (222.8 Å2 nonpolar) for the protein, underscoring the hydrophobic nature of this binding site. The bromine at position 7 was almost half-exposed to the solvent. Encouragingly, docking calculations using Glide with the crystal structure of DYRK2 predicted mode II as the best pose of 6i. Interestingly, docking in the crystal structure of DYRK2 showed that 7BIO would also adopt the inverted mode, while 6BIO was predicted to bind in the usual indirubin-kinase orientation, further supporting the putative role of the flipped binding mode in determining selectivity.12
Figure 1.
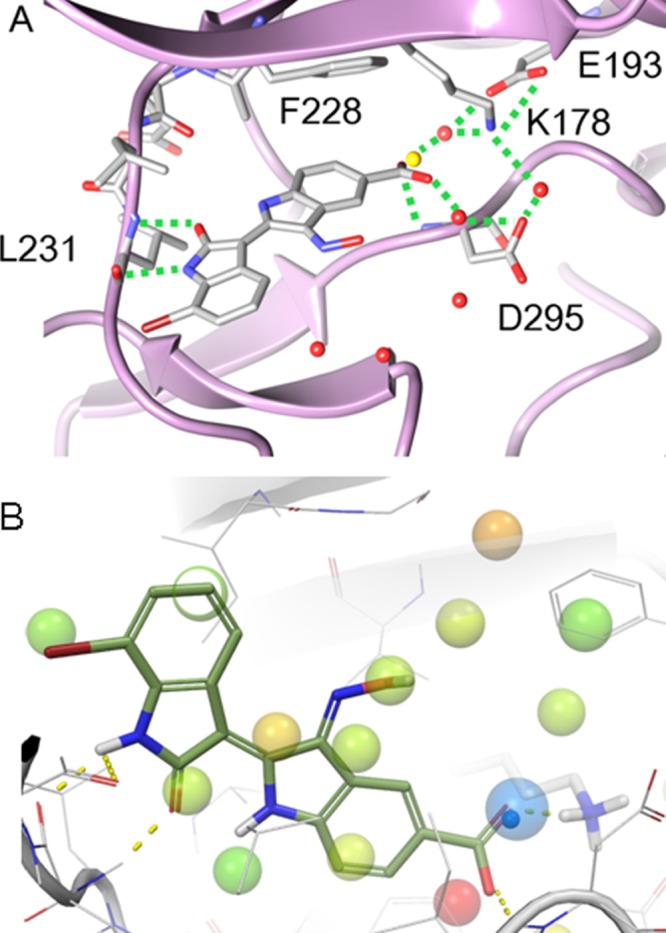
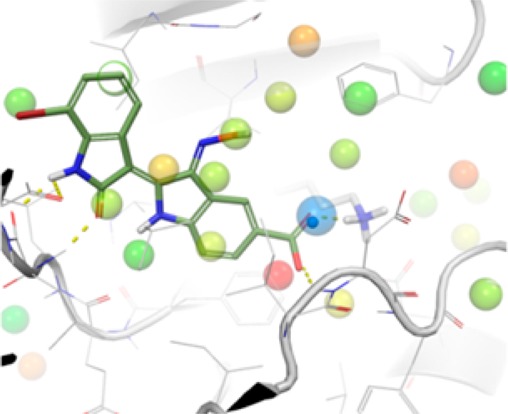
(A) Crystal structure of the 6i-DYRK2 complex revealed a nontypical binding mode, in which the orientation of the indirubin core was in good accordance with the predicted binding mode. The water molecule displaced by the carboxylate group of 6i is depicted as a yellow sphere. (B) Unfavorable DYRK2 hydration sites (>1.4 kcal/mol) from WaterMap calculations on the DYRK2 apo crystal structure. Hydration sites are colored by free energy, with red being the most unfavorable. The hydration site adjacent to Lys178 (ΔG = 1.6 kcal/mol) is shown as a large semitransparent blue sphere, and the crystallographic water is shown as a small opaque blue sphere. Compound 6i is superimposed to show the displacement of the hydration sites. The view has been rotated to improve visibility of the key water.
A superposition of the inhibitor cocrystal structure of DYRK2 with that of the apoenzyme (PDB ID 3K2L, manuscript in preparation) revealed that the 5′-carboxylate moiety of 6i overlaps with a crystallographic water molecule appearing in the active site of the apoenzyme and interacting with Lys178 (Figure 1A). To understand the thermodynamic characteristics of the system and that particular water molecule, calculations were run with the WaterMap program, which combines molecular dynamics, solvent clustering, and statistical thermodynamics to assess the enthalpy, entropy, and free energy of water “hydration sites”.18 WaterMap has been successfully applied to study selectivity in kinases and PDZ domains, as well as several studies of understanding binding affinity and SAR series.19−21 Figure 1B shows the predicted WaterMap hydration sites in the apoenzyme. The site near Lys178 is in near-perfect accordance with the crystallographic water. This hydration site has a thermodynamic profile making the total free energy slightly worse than bulk water (+1.5 kcal/mol). While the site is highly unfavorable entropically (+3.4 kcal/mol) due to the localization around Lys178, it is enthalpically favorable (−1.9 kcal/mol) due to the interactions with Lys178. Displacement of this hydration site by a ligand functional group that also replaces the water interactions is predicted to improve potency, in agreement with the experimental 10-fold affinity difference between 7BIO and its carboxylated analogue 6i. This shows the importance of including water molecules in the analysis and assessment of binding energies.
The selectivity of 6i and 6l toward DYRKs prompted us to investigate their inhibition profile over a broader panel of protein kinases. Compounds 6i and 6l along with 6BIO and 7BIO were assayed in vitro against a panel of 42 kinases (Table S1 in the Supporting Information). Compounds 6i, 6l, and 7BIO were inactive against all assayed kinases. In contrast, 6BIO showed a broad inhibitory profile. Apart from its well-established target, GSK-3β, 6BIO was weakly active toward the receptor tyrosine kinases fibroblast growth factor receptor 3 (FGFR3) and platelet-derived growth factor receptor (PDGFR) and showed a notable inhibition of proto-oncogene tyrosine-protein kinase receptor (RET). Although the crystal structure of 6l-DYRK2 was not determined, the similar specificity profile of 6i and 6l suggests that the presence of the N1-methyl does not induce an alternative binding mode.
To conclude, the combined presence of a bromine substitution at position 7 and an acidic functionality at position 5′ of the indirubin scaffold turns the nonselective bis-indole indirubin into a potent and selective DYRK inhibitor. Structural insights offered by docking, crystallographic studies, and solvent thermodynamic calculations suggest that selective DYRK inhibition can be attributed to a nonstandard kinase binding mode where the indirubin core adopts an inverted pose. Data indicate that the driving force for the inverted binding orientation is the occurrence of a steric clash between the bulky halogen of position 7 and the kinase hinge. The acidic substitution at position 5′ further enhances activity by displacing an unstable water and establishing a salt bridge between the 5′-carboxylate and the Lys178. The need for the simultaneous substitutions at position 5′ and 7 was evident by the fact that neither of the two substitutions alone resulted in the desired activity-selectivity profile. As a result, the desired selectivity profile was achieved with an ATP-competitive but not ATP-mimetic inhibitor using a variety of rational design strategies.22
Glossary
Abbreviations
- FGFR3
fibroblast growth factor receptor 3
- PDGFR
platelet-derived growth factor receptor
- RET
proto-oncogene tyrosine-protein kinase receptor
- CDK5
cyclin-dependent kinase 5
- GSK3β
glycogen synthase kinase 3β
- CK1
casein kinase 1
Supporting Information Available
Detailed information about compound synthesis, computational methodology, protein production, crystallization, data collection, refinement statistics, and biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The 6i-DYRK2 structure has been deposited to the PDB with accession code 3KVW.
Author Contributions
∇ These authors contributed equally.
This research was supported by grants from the “Fonds Unique Interministériel” (FUI) PHARMASEA project (L.M.), the “Association France-Alzheimer (Finistère)” (L.M.), “CRITT-Santé Bretagne” (L.M.), “Fondation Jérôme Lejeune” (L.M.), and EU-FP7REGPOT-2011 project INsPiRE (284460) (V.M.). S.K. and M.S. are supported by the SGC, a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, the Novartis Research Foundation, Takeda, the Ontario Ministry of Research and Innovation, and the Wellcome Trust. P.F. is supported by a Wellcome Trust Career Development Fellowship (095751/Z/11/Z).
The authors declare no competing financial interest.
Supplementary Material
References
- Arron J. R.; Winslow M. M.; Polleri A.; Chang C. P.; Wu H.; Gao X.; Neilson J. R.; Chen L.; Heit J. J.; Kim S. K.; Yamasaki N.; Miyakawa T.; Francke U.; Graef I. A.; Crabtree G. R. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 7093, 595–600. [DOI] [PubMed] [Google Scholar]
- Taira N.; Nihira K.; Yamaguchi T.; Miki Y.; Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 2007, 5, 725–738. [DOI] [PubMed] [Google Scholar]
- Maddika S.; Chen J. Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat. Cell Biol. 2009, 4, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. T.; Aggarwal S.; Lin T. K.; Dagenais S. L.; Contreras J. I.; Orringer M. B.; Glover T. W.; Beer D. G.; Lin L. Amplification and overexpression of the dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 2 (DYRK2) gene in esophageal and lung adenocarcinomas. Cancer Res. 2003, 14, 4136–4143. [PubMed] [Google Scholar]
- Taira N.; Mimoto R.; Kurata M.; Yamaguchi T.; Kitagawa M.; Miki Y.; Yoshida K. DYRK2 priming phosphorylation of c-Jun and c-Myc modulates cell cycle progression in human cancer cells. J. Clin. Invest. 2012, 3, 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debdab M.; Carreaux F.; Renault S.; Soundararajan M.; Fedorov O.; Filippakopoulos P.; Lozach O.; Babault L.; Tahtouh T.; Baratte B.; Ogawa Y.; Hagiwara M.; Eisenreich A.; Rauch U.; Knapp S.; Meijer L.; Bazureau J. P. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 12, 4172–4186. [DOI] [PubMed] [Google Scholar]
- Ogawa Y.; Nonaka Y.; Goto T.; Ohnishi E.; Hiramatsu T.; Kii I.; Yoshida M.; Ikura T.; Onogi H.; Shibuya H.; Hosoya T.; Ito N.; Hagiwara M. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [DOI] [PubMed] [Google Scholar]
- Cuny G. D.; Robin M.; Ulyanova N. P.; Patnaik D.; Pique V.; Casano G.; Liu J. F.; Lin X.; Xian J.; Glicksman M. A.; Stein R. L.; Higgins J. M. Structure-activity relationship study of acridine analogs as haspin and DYRK2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 12, 3491–3494. [PMC free article] [PubMed] [Google Scholar]
- Adayev T.; Wegiel J.; Hwang Y. W. Harmine is an ATP-competitive inhibitor for dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). Arch. Biochem. Biophys. 2011, 2, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal A. S.; Tanega C.; Shen M.; Mott B.; Bougie J. M.; Nguyen D. T.; Misteli T.; Auld D, S.; Maloney D. J.; Thomas C. J. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk). Bioorg. Med. Chem. Lett. 2011, 10, 3152–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoessel R.; Leclerc S.; Endicott J.; Noble M.; Lawrie A.; Tunnah P.; Leost M.; Damiens E.; Marie D.; Marko D.; Niederberger E.; Tang W.; Eisenbrand G.; Meijer L. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Skaltsounis A. L.; Magiatis P.; Polychronopoulos P.; Knockaert M.; Leost M.; Ryan X. P.; Vonica C. A.; Brivanlou A.; Dajani R.; Crovace C.; Tarricone C.; Musacchio A.; Roe S. M.; Pearl L.; Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 2003, 12, 1255–1266. [DOI] [PubMed] [Google Scholar]
- Polychronopoulos P.; Magiatis P.; Skaltsounis A. L.; Myrianthopoulos V.; Mikros E.; Tarricone A.; Musacchio A.; Roe S. M.; Pearl L.; Leost M.; Greengard P.; Meijer L. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem. 2004, 4, 935–946. [DOI] [PubMed] [Google Scholar]
- Sato N.; Meijer L.; Skaltsounis L.; Greengard P.; Brivanlou A. H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3 specific inhibitor. Nat. Med. 2004, 1, 55–63. [DOI] [PubMed] [Google Scholar]
- Myrianthopoulos V.; Magiatis P.; Ferandin Y.; Skaltsounis A. L.; Meijer L.; Mikros E. An integrated computational approach to the phenomenon of potent and selective inhibition of aurora kinases B and C by a series of 7-substituted indirubins. J. Med. Chem. 2007, 17, 4027–4037. [DOI] [PubMed] [Google Scholar]
- Vougogiannopoulou K.; Ferandin Y.; Bettayeb K.; Myrianthopoulos V.; Lozach O.; Fan Y.; Johnson C. H.; Magiatis P.; Skaltsounis A. L.; Mikros E.; Meijer L. Soluble 3′,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase-3 alter circadian period. J. Med. Chem. 2008, 20, 6421–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xingi E.; Smirlis D.; Myrianthopoulos V.; Magiatis P.; Grant K. M.; Meijer L.; Mikros E.; Skaltsounis A. L.; Soteriadou K. 6-Br-5-methylindirubin-3′oxime (5-Me-6-BIO) targeting the leishmanial glycogen synthase kinase-3 (GSK-3) short form affects cell-cycle progression and induces apoptosis-like death: exploitation of GSK-3 for treating leishmaniasis. Int. J. Parasitol. 2009, 12, 1289–1303. [DOI] [PubMed] [Google Scholar]
- Young T.; Abel R.; Byungchan K.; Berne B.; Friesner R. Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc. Natl. Acad. Sci. U.S.A. 2007, 3, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D.; Sherman W.; Farid R. Understanding kinase selectivity through energetic analysis of binding site waters. ChemMedChem 2010, 5, 618–627. [DOI] [PubMed] [Google Scholar]
- Higgs C.; Beuming T.; Sherman W. Hydration site thermodynamics explain SARs for triazolylpurines analogues binding to the A2A receptor. ACS Med. Chem. Lett. 2010, 1, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah F.; Gut J.; Legac J.; Shivakumar D.; Sherman W.; Rosenthal P. J.; Avery M. A. Computer-aided drug design of falcipain inhibitors: virtual screening, structure–activity relationships, hydration site thermodynamics, and reactivity analysis. J. Chem. Inf. Model. 2012, 52, 696–710. [DOI] [PubMed] [Google Scholar]
- Huggins J. D.; Sherman W.; Tidor B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 2012, 55, 1424–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.