

Abstract
211At is a promising radionuclide for α-particle therapy of cancers. Its physical characteristics make this radionuclide particularly interesting to consider when bound to cancer-targeting biomolecules for the treatment of microscopic tumors. 211At is produced by cyclotron irradiation of 209Bi with α-particles accelerated at ∼28 MeV and can be obtained in high radionuclidic purity after isolation from the target. Its chemistry resembles iodine, but there is also a tendency to behave as a metalloid. However, the chemical behavior of astatine has not yet been clearly established, primarily due to the lack of any stable isotopes of this element, which precludes the use of conventional analytical techniques for its characterization. There are also only a limited number of research centers that have been able to produce this element in sufficient amounts to carry out extensive investigations. Despite these difficulties, chemical reactions typically used with iodine can be performed, and a number of biomolecules of interest have been labeled with 211At. However, most of these compounds exhibit unacceptable instability in vivo due to the weakness of the astatine–biomolecule bond. Nonetheless, several compounds have shown high potential for the treatment of cancers in vitro and in several animal models, thus providing a promising basis that has allowed initiation of the first two clinical studies.
Key words: α-immunotherapy, Astatine-211, radiolabeling
Introduction
While β−-emitters have proven their usefulness in the therapy of localized macroscopic cancerous tumors, limited successes have been obtained in the case of microscopic and disseminated cancers. The disappointing results are explained by the relatively long path length of the β−-particles in tissue (several millimeters), leading to the loss of most of the deposited energy in healthy tissues when the tumor being treated is too small. In contrast, α-particle emitters are promising radionuclides for the treatment of such small tumors. When such a radionuclide is conjugated to a suitable targeting agent, the short path length of the α-particles (∼25–100 μm), in association with their high energy (∼4–8.5 MeV), makes them highly efficient at eradicating small clusters or isolated cancerous cells with a reduced dose deposited to the surrounding healthy tissues. Such radiopharmaceuticals are of high interest for the treatment of disseminated micrometastasis, diseases consisting of monocellular cancer cells such as lymphoma and leukemia, or in the case of residual disease after surgical debulking. Among the hundred α-particle emitters known, a limited number of radionuclides exhibit characteristics that have been considered as potentially suitable for therapeutic applications (225Ac, 211At, 212Bi, 213Bi, 223Ra, 149Tb, 227Th, and 212Pb as an in vivo generator of 212Bi, see Table 1). All have been the object of increasing attention in recent years, and several have reached different stages of clinical evaluation while production and availability of the radionuclides and their labeling chemistry issues are being overcome with varying levels of success.1
Table 1.
Characteristics of α-Emitting Radionuclides of Potential Interest for the Therapy of Cancers
| Nuclide | Half-life | Decays | Energy α (MeV) | Production | Clinical trial status |
|---|---|---|---|---|---|
| 225Ac | 10 days | 4 α, 2 β− | 5.1–8.4 | 233U decay/cyclotron | First phase I ongoing |
| 211At | 7.2 hours | 1 α, 1 EC | 5.9 or 7.4 | Cyclotron | Two phase I published |
| 212Bi | 61 minutes | 1 α, 1 β− | 6.1/7.8 | 228Th decay/224Ra generator | Preclinical |
| 213Bi | 46 minutes | 1 α, 2 β− | 6.0/8.4 | 225Ac generator | Phase I/II |
| 223Ra | 11.4 days | 4 α, 2 β− | 5.7–7.5 | 227Ac generator | Phase I/II/III |
| 149Tb | 4.1 hours | 1 α, EC | 4.0 | Accelerator | Preclinical |
| 227Th | 18.7 days | 5 α, 2 β− | 5.7–7.5 | 227Ac generator | Preclinical |
| 212Pb/212Bia | 10.6 hours | 1 α, 2 β− | 6.1/7.8 | 224Ra generator | First phase I ongoing |
Although 212Pb is not an α-particle emitter, it is included in this table for its considerations in several studies as an in vivo generator of the α-particle emitter 212Bi. See Yong and Brechbiel2 for a definition of the concept.
Among these radionuclides, 211At exhibits particularly favorable characteristics, with a 7.2-hour half-life suitable to the kinetics of full antibodies or other biomolecules that require several hours to reach an optimal tumor-to-blood dose ratio. Furthermore, with 100% of its decay leading to the production of an α-particle, high efficiency of the treatments and limited toxicity are expected at relatively low doses. However, the introduction of 211At-based radiopharmaceuticals into clinics has been slowed by difficulties related to the production and availability of the radionuclide, the limited knowledge of the chemistry of elemental astatine, the lack of stability of the astatine–biomolecule bond, and concerns about the potential toxicity of α-radiation on humans.
In this review, the characteristics of 211At and its methods of production and purification for radiolabeling purposes are presented. The different aspects of its chemistry as well as the strategies for the radiolabeling of biomolecules are also described. Finally, the latest advances in the use of 211At-based radiopharmaceuticals in radiotherapy of cancers from the in vitro studies to the very first clinical trials that have been published recently are presented.
211At: Characteristics and Production
The 85th element in the periodic table classification was first produced in 1940 when Corson irradiated 209Bi with a beam of α-particles accelerated to 32 MeV.3 Expected by Mendeleïev to be below the element iodine (and named eka-iodine at that time), it is the only halogen without a stable isotope, which is why its discoverers named it astatine, from the Greek word άστατοζ (astatos=unstable).
Astatine exists on the Earth in very small quantities (estimated to be a few milligrams in the whole Earth's crust) by some decay branches of 235U, 238U, and 232Th, which actually makes it the rarest natural element.4 There are 32 astatine isotopes known: 191At and all the rest from 193At to 223At. Their half-lives range from 125 nanoseconds for 213At up to 8.1 hours for 210At, and most of them are α-particle emitters. Among these isotopes, only 211At exhibits physical characteristics that are reasonably adapted for targeted radiotherapy.
The most relevant characteristics of the radionuclide are summarized below, and the reader is referred to the Gmelin Handbook of Inorganic Chemistry on Astatine for an exhaustive review of the topic.5
Physical properties
The disintegration of 211At follows a branched decay scheme with a 7.21 hours half-life (Fig. 1). One branch leads to 207Bi by emission of an α-particle. 207Bi, with a 33.9 year half-life, decays to 207Pb via electron capture. The other decay branch occurs via electron capture and leads to the 516 milliseconds half-life radionuclide 211Po, which in turn decays to stable 207Pb by emission of an α-particle. The result of these decay pathways is 100% α-particle emission during the decay of 211At (5.87 and 7.45 MeV in 42% and 58% of the decays, respectively). Concerns have been raised regarding the fate of the 211Po decay during targeted radiotherapy. Palm et al. investigated the influence of the diffusion of 211Po on the dose absorbed by isolated cells in suspension.6 They estimated that the diffusion distance from the initial 211At nucleus bound to the cell reduces the absorbed dose by a factor of 2 in this model. Part of the deposited dose may indeed be lost from the target in case of dispersed cells, but this phenomenon might be reduced in cell cluster environments, where the dose could be deposited toward a neighboring malignant cell. Also, as noted by the authors of the study, cellular localization may reduce the diffusion of 211Po depending upon the viscosity of the tissue. Another concern is the fate of 207Bi with its long half-life (32.9 years), and the potential issues due to its uptake in the bone, liver, and kidneys. However, 347 MBq of 211At, which is the highest dose that has been administered to a human, leads to only 310 kBq of 207Bi, making its potential toxicity negligible.
FIG. 1.
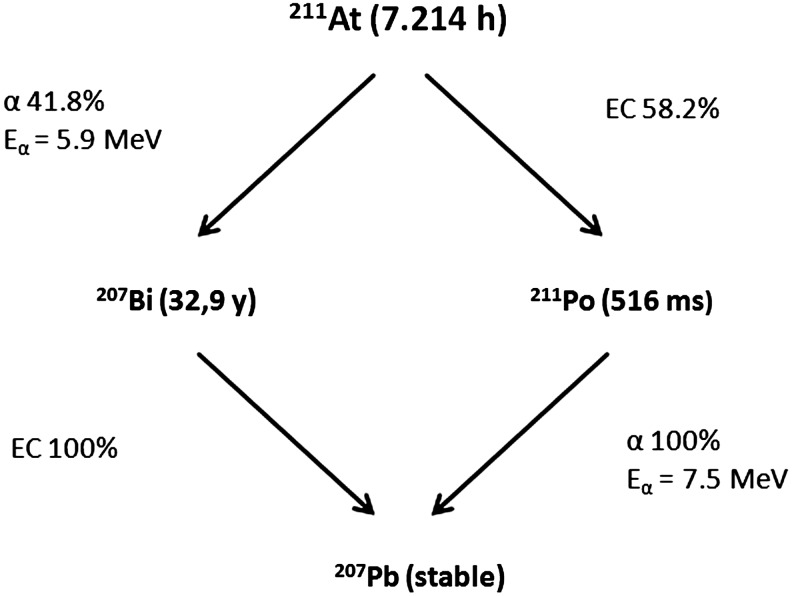
Simplified decay scheme of 211At.
Also, another interesting characteristic is the emission of X-rays during the decay to 211Po. With energies between 77 and 92 keV, these X-rays can be easily monitored by γ-detectors classically used in laboratory or clinical centers. This combination of emission and instrumentation makes detection relatively easy and useful for in vivo monitoring by SPECT imaging.7
Production
211At is produced by irradiation of natural bismuth (209Bi) following the nuclear reaction: 209Bi(α, 2n)211At. Other production methods have been investigated, such as the production of a 211Rn/211At generator by irradiation of 209Bi with 7Li,8 or by fusion of 9Li with 208Pb.9 However, these alternative methods remain anecdotal with respect to the production of medical research quantities of the radionuclide.
211At production is possible for α-beam energies ranging from 21 to more than 40 MeV, with a maximum cross-section observed for 31 MeV (Fig. 2). However, at this level of energy, 210At is also produced. This 8.1-hour half-life isotope exhibits characteristics that are not compatible with pharmaceutical applications because of its decay into the long half-life 210Po (138.2 days), which is highly toxic, especially to the bone marrow.10 Production of 210Po is also observed from 26.7 MeV beam energies. Nonetheless, the presence of this isotope after irradiation is generally irrelevant due to an efficient isolation of 211At from the target and impurities during the purification process. For these reasons, the beam energy is generally controlled to 28–29 MeV to limit formation of 210At while maintaining acceptable yields of 211At. However, only a limited number of cyclotrons in the world are able to generate α-beam energies beyond 25 MeV, which strongly limits the availability of this radionuclide.11
FIG. 2.
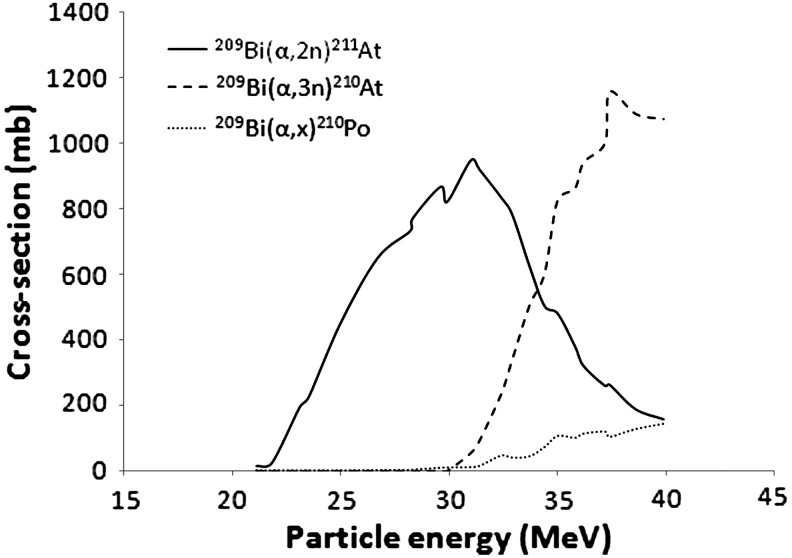
Cross-section of the irradiation of 209Bi as a function of the alpha particles energy. Plotted from the experimental data from Hermanne et al.12
Another limitation to consider is the low melting point of the irradiated material (Bismuth mp=272°C) and its low thermal conductivity (k=7.9 W m−1 K−1). Together, with the relatively high volatility of astatine (337°C), this potentially leads to vaporization of the produced activity due to overheating of the target during the irradiation. Solutions have been developed to limit this overheating issue, such as cooling systems using cold gas and water during irradiation, or the use of an irradiation angle that spreads the beam over a larger impact surface on the bismuth. Furthermore, the bismuth is deposited as a thin layer on a material with a better thermal conductivity to enhance cooling. Aluminum has frequently been used as backing material (k=250 W m−1 K−1), although some reports make reference to the use of copper (k=390 W m−1 K−1).
Purification
After irradiation, the target contains the initial 209Bi, traces of 210Po, and the desired 211At activity, which must be obtained in solution with the highest purity. Two methods are described to harvest 211At: dry distillation that is probably the most widely used or liquid extraction that requires dissolution of the target in an acidic solution.
Dry distillation
In this method, the target is placed in a furnace and heated above the boiling point of astatine. The boiling points of bismuth and polonium being 1564°C and 962°C, respectively, the furnace temperature is generally set to 650°C–900°C. During the distillation, while the bismuth and the polonium melt and stay on the support, the volatile astatine is carried away by a stream of gas (generally nitrogen or argon) and trapped at the outlet. The astatine activity can be obtained by bubbling that stream of gas directly into the solvent of choice, or it can be captured in capillary tubing cooled in dry ice/ethanol placed at the outlet.13 In the latter case, the activity is subsequently dissolved in the solvent of choice (Fig. 3). With this kind of system, the astatine activity is collected in the trap in 20–30 minutes with recovery yields up to 80%.
FIG. 3.

Example of a typical furnace used for the distillation of 211At. (A) Gas inlet with desiccant. (B) Furnace heated to 650°C–900°C. (C) Capillary trap cooled in (D) a dry ice/ethanol bath. It can be replaced by a cooled bubbler containing the solvent of interest. (E) Gas-wash bottle containing a reducing agent such as Na2S2O5 to trap traces of astatine that would escape from the capillary. Reprinted by permission from Lindegren et al13.
Wet extraction
This alternative procedure consists of dissolution of the target in concentrated nitric acid. After evaporation of the nitric acid, the residue containing the bismuth and the astatine is dissolved in dilute nitric acid and extracted with di-isopropyl ether, which is the most efficient and practical solvent for this purpose.14,15 With this method, the astatine activity is obtained in high yields (∼90%) in ∼1 hour with high radionuclidic purity. The advantage of this technique over dry distillation is that it makes use of simple and cheap materials. Moreover, harvesting yields are obtained with an excellent reproducibility. However, the choice of extraction solvent is reduced to di-isopropyl ether or analogous solvents that can limit radiochemistry work. Furthermore, some nitric acid is extracted into the organic phase in concentrations that are not negligible, and that can cause side reactions during the radiolabeling chemistry. Nonetheless, it has been demonstrated that biomolecules can be radiolabeled efficiently using standard procedures with 211At recovered with this method.16
Chemical Properties of Astatine
Despite more than 70 years of research on astatine, its chemistry is still not clearly established. The main reason is the fact that no stable isotope exists, the most stable isotope being 210At with a half-life of only 8.1 hours. The highest reported activity produced is 6.59 GBq (which corresponds to 0.087 μg of 211At).17 These amounts preclude the use of standard analysis techniques such as nuclear magnetic resonance, mass spectrometry (although some experiments have been reported18), UV, IR, or X-rays. Furthermore, with the astatine being obtained in highly dilute solutions, traces of unavoidable contaminants in solution can become competitive species and cause side reactions. Another impediment to the understanding of astatine chemistry is the lack of cyclotrons to produce this element leading to a reduced number of research laboratories able to run experiments.11 Despite these obstacles, a relatively clear idea of some of the chemical properties of astatine has been established by analogy with its closest element in the periodic table of the elements, iodine. However, in its positive oxidation states, astatine exhibits many properties specific to metal ions. While almost identical to iodine in the At(−I) state, it is, in some respects, also comparable to silver in the At(+I) state, and close to polonium in its higher oxidation states.
In this section, the inorganic and organic chemistry of astatine is covered. Radiolabeling approaches for biomolecules of interest are also discussed as well as the issue of the limited in vivo stability of some 211At-radiolabeled compounds.
Inorganic chemistry of astatine
Six oxidation states have been identified or hypothesized with varying degrees of certainty: −I, 0, +I, +III, +V, and +VII. These forms of astatine were determined at low concentration with chemistry techniques that can be used at a tracer level such as co-precipitation, liquid extraction, or electromigration experiments, with the knowledge of iodine chemistry as a guide. Most of the descriptions of the oxidation states presented below rely on works by Johnson et al.,19 Appelman,20 Visser and Diemer,21,22 and Dreyer et al.23–25 Also, more exhaustive information can be found in the DOE-Sponsored Nuclear Science Series monogram by Ruth et al.26
At(−I)
The (−I) oxidation state is probably the most clearly established form of astatine with a strong similarity with iodide. It is obtained in the presence of reducing agents such as SO32−, Zn0, or cysteine, which form the astatide species (At−). It is characterized by electromigration at the anode or by co-precipitation with AgI, TlI, or PbI2, and is stable over a wide pH range. The astatide anion exhibits soft base characteristics, similar to iodide. It forms stable complexes with soft cations such as Hg2+, Rh3+, or Ir3+. Pruszyński et al.27 investigated the complexation of astatide in the presence of mercury nitrate in aqueous solution. Interestingly, the complex formed, assumed to be Hg(OH)At, exhibited a higher stability than the iodinated analog.27 Similarly, complexes have been prepared with rhodium (III) and iridium (III), and chelated in a macrocyclic agent bearing soft donors sulfur atoms (Fig. 4).28 These compounds could lead to an interesting approach for the radiolabeling of biomolecules if they exhibit acceptable in vivo stability.
FIG. 4.
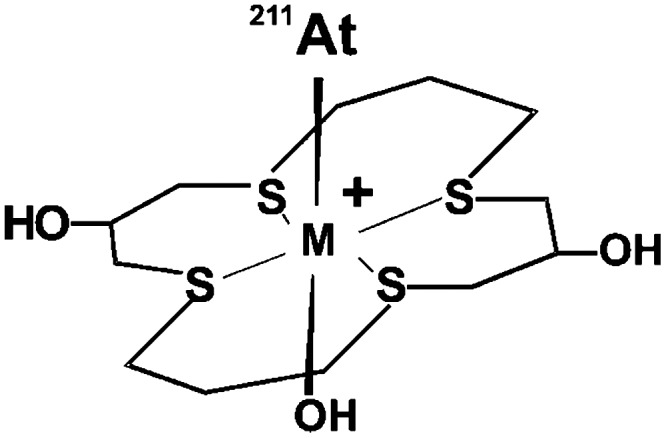
211At-M(16-S4-diol) complex, with M=Rh(III) or Ir(III).
At(0)
This oxidation state of astatine, also denoted At0, remains unresolved, mainly because of its highly irreproducible behavior. It is hypothesized that astatine is obtained as At2 in the gas phase (such as during the distillation process), but in solution, At2 or the radical At• is highly unlikely at this level of dilution. Recombination with impurities or other species of the solvent is supposed to occur very rapidly. At0 has often been referred to as the species that does not exhibit mobility in an electric field. It is supposed to be in the AtX form (with X=HSO4, NO3, OAc, etc.) and extracted from an aqueous medium by organic solvents as AtXLn (n depending on the solvent L used). Also, At0 was observed to co-precipitate with elemental Ag or Tl.
At(+I)
This oxidation state can be obtained under moderately oxidizing conditions (i.e., dilute HClO4 or H2Cr2O7) in the At+ form in acidic medium, or AtO− in alkaline medium. In the cationic form, it exhibits a relatively weak electrophilic behavior similarly to I+. Characteristics similar to metals are also observed, with a cationic form stable toward hydrolysis in acidic medium, unlike the other halogens. Denoted At(θ)+, this cationic form of astatine exhibits the characteristics of a soft acid with an affinity for soft-base ligands (I−, CN−, and SCN−) similar to Ag+, as demonstrated by the stability of the interhalogen complexes according to the following sequence29:
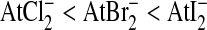 |
Several pseudohalogen ligands such as SCN−, N3− or C(CN3)−, with lower stability than AtI2−, have also been investigated.30 Better results were obtained with selenoureas and thioureas with complexes assumed to be in the At(L2) form (Fig. 5).31,32 Soft-donor ligands containing phosphorus have also been considered. However, their use seems unlikely for medical applications because of the reductive properties of these ligands at pH>2, leading to the reduction of At+ to At− (Fig. 5).33
FIG. 5.
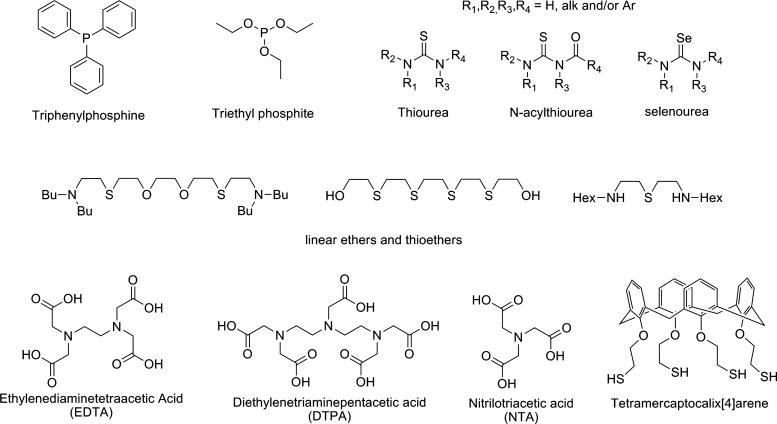
Organic ligands investigated for the complexation of 211At discussed in the text.
The metalloid-like behavior of astatine led Milesz et al. to study well-known chelating agents for the sequestration of metals such as EDTA,34 DTPA,35 or NTA36 (Fig. 5). The feasibility of radiolabeling an antibody with 211At complexed to DTPA has been demonstrated, but the in vivo stability was too low for biomedical applications.37 While the proof of concept of astatine as a metal for radiolabeling an antibody has been demonstrated, polyaminocarboxylates do not appear to be appropriate for the soft-cation At+ due to the presence of only hard-donor atoms in their structure (nitrogen and negatively changed oxygen). According to the cited studies, soft donors should be more appropriate for complexation of At+. This was confirmed by a comparative study of linear nitrogen-, oxygen-, and sulfur-containing ligands, with better results being obtained with the sulfur-containing compounds (Fig. 5).38 However, no chelating agents stable enough for biomedical applications have been reported yet, the most recent example of the use of 211At in the At+ form being a sulfur-containing calixarene, which exhibited a weak in vivo stability (Fig. 5).39
At(III)
This oxidation state is obtained in the presence of stronger oxidizing agents such as S2O82− or H2O2. However, it is difficult to obtain in a pure form, and is often associated with At+ in proportions dependent upon experimental conditions. It has been described as a component in anionic (AtOX2− and AtX4−), neutral [XAtO, At(OH)3, At(OH)X2, or AtX3], and cationic forms (AtSO4+ and AtCrO4+). This form of astatine behaves similarly to At+, however, as a harder acid with higher affinity for harder bases such as Cl−, NO3−, or SO42−. Recent examples of the investigation of this species are limited to a comparative study of the complexation of the At(I) and the At(III) species with thiocyanate and calixarene ligands.40
At(V)
Obtained as AtO3− in strong oxidizing conditions (CeIV, NaBiO3, hot S2O82−, or IO4−), it is characterized by co-precipitation with AgIO3, Ba(IO3)2, or Pb(IO3)2. No complexation chemistry has been reported yet, but a relatively hard acid behavior can be expected.
At(VII)
Many studies have reported the inability to demonstrate the formation of this high oxidation state of astatine, even under the strongest oxidizing conditions. Although preparation of AtO4− by use of xenon fluoride in hot NaOH has been reported,41 its existence remains highly hypothetical.
In conclusion for this section, it must be kept in mind that none of the forms of astatine presented above have actually been definitely established, but rather that some of them seem to be in good agreement with the observed reactivity. Furthermore, extreme caution must be used in the experiments to obtain reproducible results. The purity of the solutions used is a primary factor influencing the results, but clearly, elapsed time also plays an important role in the behavior of astatine as demonstrated by studies by Pozzi and Zalutsky.42 Indeed, evolution of the oxidation state of astatine in solution has been observed over time. This evolution has been attributed to the high ionizing energy emitted by the α-particles in the medium. As a consequence, oxidizing species such as peroxides can be generated, leading to evolution of astatine to higher oxidation states. On the other hand, it was also hypothesized that solvents such has methanol could be radiolyzed into reducing species (such as hydrogen or formaldehyde) that can reduce At+ into At−. These phenomena can highly influence the results of the complexation chemistry of astatine as well as the organic chemistry discussed that follows below.
The organic chemistry of astatine
While the inorganic chemistry of astatine is difficult to comprehend, in part because of its dual behavior between halogen and metalloid, its organic chemistry appears to be closer to halogens, and reactions typically used with iodine are generally applicable to the formation of C–At bonds.43 However, given the tendency of the carbon–halogen bond energy to decrease from the lighter to the heavier halogens, the bonding of At has been mostly limited to sp2 carbons (preferentially aromatic carbons) rather than sp3 carbons, whose resulting bond energies are too weak to provide sufficient stability for biomedical applications (see Table 2).
Table 2.
Phenyl- and Alkyl-Halogen Bond Energies (kJ/mol)
| X | Phenyl-X | Alkyl-X |
|---|---|---|
| F | 523 | 444 |
| Cl | 398 | 339 |
| Br | 335 | 285 |
| I | 268 | 222±12 |
| At | 197±20 | 163±12 |
Data from Coenen et al.44
The reactions employed are similar to the ones used for labeling with radioiodine. They are generally optimized to accelerated procedures required by the relatively short half-life of 211At. Synthetic approaches have been considered with nucleophilic At− in halogen exchange or dediazoniation processes, but also with electrophilic At+ in direct aromatic electrophilic substitution (EAS) or in demetallation reactions (Fig. 6). Alternatively, boron–astatine bonds in boron cages with improved stability have been the subject of several investigations recently.
FIG. 6.
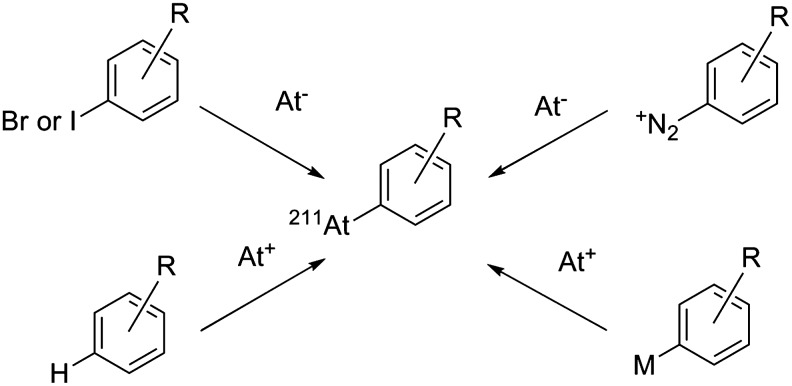
Main reaction pathways used for the formation of a C–At bond.
In most cases, At− is formed in the presence of sodium sulfite, while At+ is often obtained with hydrogen peroxide, peracetic acid, or N-chlorosuccinimide, the latter being the most commonly employed. Advantages and drawbacks of each synthetic approach are discussed in the following sections.
Halogen exchange
Because of the higher nucleofugal character of iodine over the lighter halogens, this reaction is generally performed on iodinated derivatives. Examples are described for substitution on alkyl carbons, but they are unusual because of the weakness of the Calkyl–At bond and the resulting low interest in these compounds for biomedical applications. Aromatic nucleophilic substitution is generally preferred, and while iodinated compounds with which the reaction is facilitated are often the starting material, bromoaryls can be advantageously used to facilitate the chromatographic purification; thanks to the higher difference in polarity between them and the astatinated compounds.
The advantages of this approach are the rapidity of the reaction and good radiochemical yields that can be obtained. However, because of the high temperatures required, its use is restrained to substrates tolerant of relatively harsh conditions. Catalysts such as crown ethers or Cu(I) can be used to promote the reaction (see examples in Table 3). However, the effective specific activity that may be achieved through this route remains low.
Table 3.
Examples of Astatinated Compounds Obtained Via a Halogen Exchange Reaction
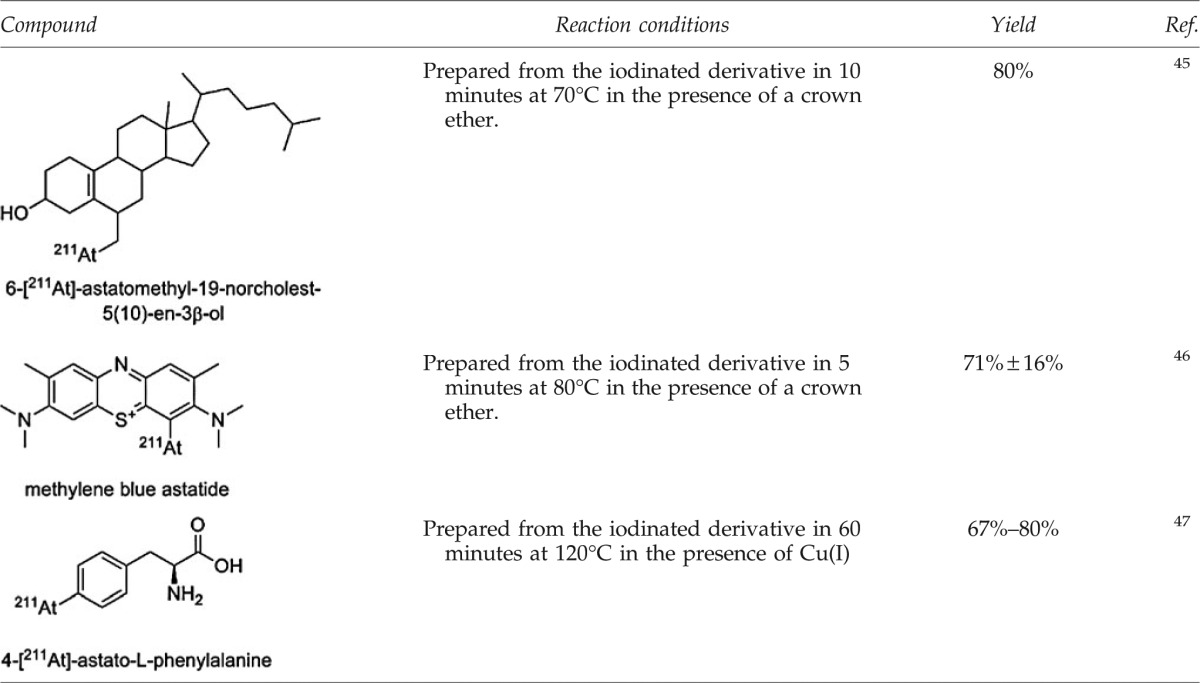 |
Dediazoniation
Diazonium salts are widely used in organic synthesis for the derivatization of aromatic rings, including halogenations. In standard organic synthesis, this reaction is performed with an excess of halogen to limit the competing reaction of water with the diazonium leading to the phenol derivative. Because of the high dilution of astatine involved, it seems unlikely to obtain a good radiochemical yield by this method. Indeed, reactions with radioiodine have been shown to work, but with low yields (max 15%). However, much better results have been obtained on identical compounds with 211At (up to 90%).48 This difference in reactivity can be explained by looking at the dediazoniation mechanisms. Indeed, two modes of cleavage of the C–N bond can occur: heterolytic cleavage leading to the formation of an aryl cation, or homolytic cleavage leading to an aryl radical (Fig. 7). As suggested by Meyer et al.,49 the homolytic pathway seems more likely. Because of its higher polarizability, astatine has a higher propensity to form a stable complex with the diazonium than iodine. For the same reason, astatine also has a higher propensity to cede an electron to form a radical that is able to recombine with the aryl radical according to the following sequence:
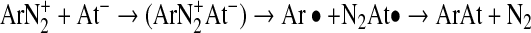 |
FIG. 7.
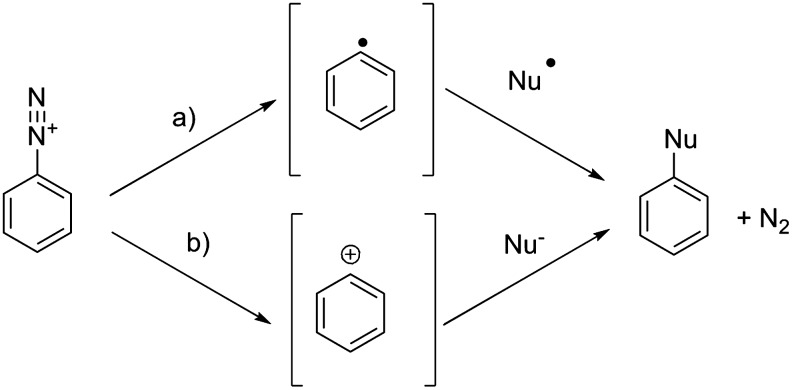
Two possible dediazoniation pathways: (a) homolytic cleavage and (b) heterolytic cleavage.
The use of this approach is marginal due to the harsh conditions required (oxidative and acidic media) for the preparation of the diazonium, thus limiting the method to less-sensitive substrates. Furthermore, a high proportion of side-products, often precipitated in the medium, makes purification and isolation of the product difficult. Examples of astatinated compounds obtained by this approach are uncommon (see Table 4).
Table 4.
Examples of Astatinated Compounds Obtained Via Dediazoniation
 |
Direct EAS
Hydrogen substitution on aromatic rings has been investigated by several research groups to understand the mechanisms of introduction of astatine on protein (which differ from iodine, see next section). Thus, it was demonstrated that the astatination of tyrosine requires conditions that are denaturing for proteins (T=160°C in the presence of an oxidizing agent),51 highlighting the fact that direct radiolabeling of antibodies on tyrosine residues as it is performed with radioiodine is unlikely with astatine.
Only a limited number of biomolecules were obtained using direct EAS because of the harsh conditions required for this chemistry. One of the most interesting examples of such procedure is the preparation of methylene blue astatide (which was shown to accumulate in melanoma). It was obtained in 15 minutes at 100°C in the presence of sodium persulfate as oxidizing agent, with a 68% radiolabeling yield.52 However, because of the high amounts of starting material required, the specific activity is very low, and purification is necessary.
Demetallation
Electrophilic substitution of At+ on an organometallic precursor is currently the most widely used reaction. It exhibits many advantages over direct EAS because of the high reactivity of the carbon–metal bond leading to high yields in mild conditions, thereby allowing its use on a wide variety of substrates. Furthermore, high specific activities can be obtained.
Organomercuric compounds were the first precursors to be studied that allowed the introduction of 211At with high yields on various compounds.53,54 However, the presence of highly toxic traces of mercury in the final compound, even after purification, hampered their use for pharmaceutical purposes. Metals from group IV of the periodic table have been preferred, especially Boron, Silicon, and Tin. Organotin compounds are the most interesting because of the weakness of the carbon–metal bond, making the tin group an excellent leaving group (see Table 5). Furthermore, tin precursors are easily introduced on a large variety of compounds by well-established synthetic approaches.
Table 5.
Carbon–Metal Bond Characteristics (Hydrogen Included for Comparison)
| Element M | Electronegativity | Covalent radius (Å) | Energy of the C–M bond (kJ/mol) | % Ionicity of the C–M bond |
|---|---|---|---|---|
| H | 2.20 | 0.37 | 418.8 | 2 |
| B | 2.01 | 0.79 | 372.7 | 6 |
| Si | 1.74 | 1.18 | 301.5 | 16 |
| Sn | 1.72 | 1.40 | 226.1 | 16 |
| Hg | 2.00 | 1.50 | 113.1 | 6 |
Data from Coenen et al.44
Even with toxicity, several orders of magnitude lower than mercury compounds, organotin compounds remain highly toxic molecules that must be separated efficiently from the desired astatinated compound. While high-performance liquid chromatography can be used to remove the tin precursor with a relatively high efficiency, other methods have been developed that achieve a very low level of tin in the final compounds, such as the use of a precursor grafted on a polymer support. This method simplifies the purification process and highly limits the amount of liberated tin. For example, meta-[211At]astatobenzylguanidine (MABG), a molecule with potential clinical applications in the treatment of neuroblastoma, was synthesized via a solid-supported tin precursor with good yields and low levels of tin in the final product ([Sn]<1 ppm) (Fig. 8).55
FIG. 8.
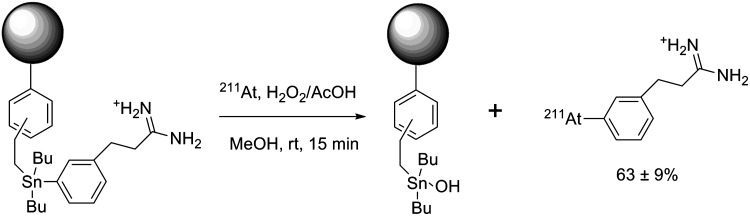
Synthesis of meta-[211At]astatobenzylguanidine (MABG) via a supported tin precursor.55
Organotin precursors have become the most widely used method for the introduction of 211At on biomolecules. Many examples describe the radiolabeling of proteins with N-succinimidyl astatobenzoate (SAB), a prosthetic group prepared from the tin precursor, as well as a variety of small molecules (see Table 6). Because of its lower toxicity, the tributyltin derivatives seem more appropriate for pharmaceutical applications than the trimethyltin, especially since no significant differences in the reactivity are detectable.56
Table 6.
Examples of Tin Precursors Used for the Radiolabeling with 211At
 |
The B–At bond in boron clusters
Because of the higher dissociation energy of the B–I bond compared to C–I (381±21 kJ/mol and 222±12 kJ/mol, respectively), the use of boron clusters such as decaborate, dodecaborate and carborane has been considered recently to improve the stability of antibodies labeled with 211At. Carboranes, containing both boron and carbon atoms, are particularly interesting for the functionalization of biomolecules because of their orthogonal reactivity. While the CHs are weak acids (pKa ranging from 22 to 27, depending on position) that can be deprotonated to generate a nucleophile, the borons are reactants toward electrophiles, and many reactions characteristic to aromatic carbons can be considered.60 These characteristics were used to prepare various structures radiolabeled with 211At with excellent radiolabeling efficiency (yields up to 90% in<10 minutes, see Fig. 9).61 Their advantages and drawbacks for the radiolabeling of proteins are discussed in the next section.
FIG. 9.
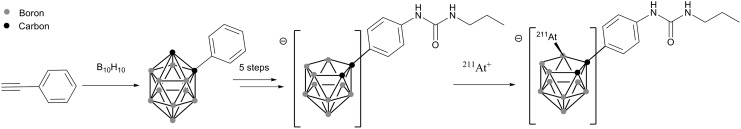
Preparation of a nido-carborane precursor and its radiolabeling with 211At+.61
Astatination and stability of biomolecules of interest
Since the first studies of astatinated biomolecules, concerns about the lack of in vivo stability of the At–biomolecule bond have been raised.62 Release of free astatine in vivo (assumed to be in the form of At−) leading to irradiation of nontargeted tissues can occur dramatically depending on the nature of the molecules considered and on the radiolabeling method employed. As defined in several animal studies, astatide has a similar behavior to iodide with a high uptake in the thyroid and stomach. However, unlike iodide, significant uptake is also observed in the spleen and lungs (Fig. 10). This phenomenon could be attributed to the in vivo oxidation of At− into At+, as it was observed that preinjection of thiocyanide (known to form complexes with At+) decreased astatine uptake in these organs, and that preinjection of periodate, known to oxidize At− to At+, increased the uptake in the lung and spleen.63
FIG. 10.
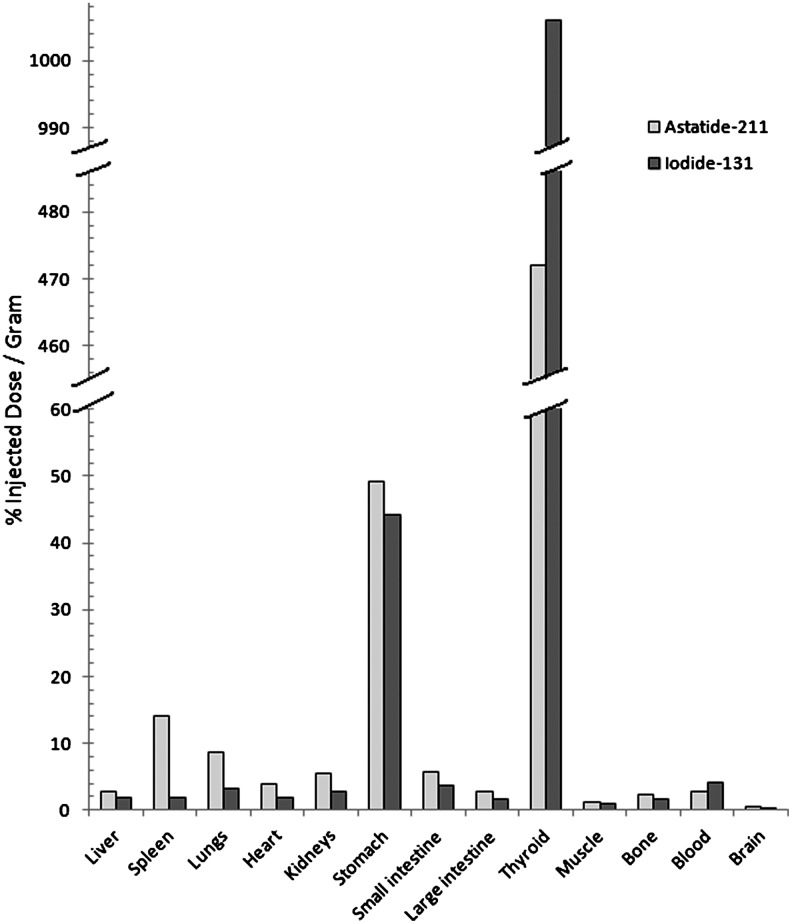
Uptake of 211At and 131I in normal mice 1 hour postdual injection of [211At]astatide and [131I]iodide. Chart plotted with data from Larsen et al.63
Several animal studies have highlighted the toxicity of free 211At−, with damage observed to various organs, especially the thyroid and the ovaries, and development of cancerous tumors due to the ionizing radiation, and to the disturbance of the endocrine system.64,65 In a long-term study by McLendon et al.,66 the LD10 of [211At]-astatide was found to be 0.56 MBq (15.1 μCi) in B6C3F1 mice with 37.8% weight difference versus saline control and 0.28 MBq (7.7 μCi) in BALB/c (nu/nu) mice with 9.44% weight difference versus controls at the 0.37 MBq (10 μCi) dose 1 year after injection. Histological analyses revealed that damage to the bone marrow, testes, heart, spleen, and stomach increased with the injected dose.66
To avoid toxicity issues, a highly stable radiolabeled compound must be achieved. Studies with promising astatinated compounds have been hampered because of their insufficient stability in vivo. Depending on the class of biomolecule considered, different levels of stability are observed. There is a tendency for the smaller molecules, which are more rapidly metabolized, to release free astatine more extensively.
Protein-based carriers
Thanks to the increasing number of successful applications of radioimmunotherapy (RIT) in the treatment of cancers over the last 30 years; antibodies and their derivatives have quickly become the vectors of choice for 211At.67 The half-life of 211At seems reasonably long enough to fit the kinetics of antibodies or their fragments. The first astatination studies using well-known methods for radioiodination of proteins were disappointing. In 1975, Aaij et al. demonstrated that it was possible to directly astatinate by using electrophilic 211At (using chloramine-T or H2O2 as oxidizing agent), without denaturation of the protein.68 The products of this direct labeling procedure were later demonstrated to be unstable in vivo, as free 211At was released.69 This instability was first attributed to the weakness of the At–C bond, with astatination assumed to occur on the same sites as for the radioiodination, that is, tyrosine and histidine residues (this kind of bonding having shown to be unstable in previous studies70), but later, Visser et al. demonstrated in a series of reports that the conditions employed did not allow the introduction of 211At on tyrosine and histidine.53,71 Based on their observations, they hypothesized that astatine reacted with the cysteine residues of the proteins when using the direct radiolabeling approach.72 Consequently, the weak At–S bond formed is easily hydrolyzed in vivo, resulting in release of free astatide. These early investigations led to the conclusion that unlike iodine, astatine could not be used in direct radiolabeling procedures of proteins, and that development of prosthetic groups for indirect radiolabeling analogous to those developed for radioiodination would be necessary.
The first stably astatinated proteins were obtained via three-step procedures by production of astatobenzoic acid in a first step, followed by activation of the carboxyl moiety to a mixed anhydride. This activated astatinated compound was then conjugated to the protein in the last step. Animal studies of bovine serum albumin labeled with this method indicated a low uptake of 211At in the stomach, thyroid, spleen, and intestine compared to free 211At in mice.73 This methodology was improved and simplified by Zalutsky and Narula74 with the development of an aromatic organotin precursor bearing an activated ester as the conjugation moiety. With a precursor activated for conjugation, this time-saving and more efficient procedure became a standard for radiolabeling proteins with radiolabeling of various antibodies in ∼2 hours in sufficient yields to support production of clinical trial doses (Fig. 11).
FIG. 11.
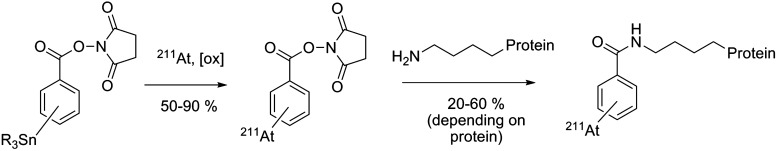
Standard indirect labeling procedure for the astatination of proteins via the preparation of succinimidyl astatobenzoate (SAB).
While initially performed in chloroform in the presence of N-chlorosuccinimide with good radiochemical yields (up to 90%), radiolabeling the organotin precursor exhibited decreasing yields at higher activities. In a study by Pozzi and Zalutsky,75 this phenomenon was attributed to radiolysis of the chloroform when exposed to high radiation doses of 211At, leading to free-radical chlorination reaction capable of competing with astatine during the electrophilic substitution. A comparison study of different solvents determined methanol as the optimal solvent among chloroform, benzene, and methanol for the production of SAB at high levels of activity.75
With the improved stability provided by SAB, clinical translation became feasible as demonstrated by the first clinical study with astatinated ch81C6 antitenascin antibody for the treatment of glioma published recently by Zalutsky et al.76 Despite the notable in vivo stability using of SAB for most of the full antibodies studied, several reports have highlighted the lack of stability for more rapidly metabolized proteins. This phenomenon is observed with rapidly internalized antibodies that are exposed to accelerated metabolization due to additional catabolic processes occurring in the cells, such as the lysosomal proteolysis. To resolve this issue, positively charged prosthetic groups such as pyridine carboxylate or guanidinomethylbenzoate derivatives were developed with the purpose of retaining the positively charged astatinated catabolites inside the tumor cell.77,78 The conjugation of N-succinimidyl 3-[211At]-4-astato-guanidinomethyl benzoate to monoclonal antibody L8A4 known to be internalized in cells expressing the anti-epidermal growth factor (EGF) receptor variant III interestingly resulted with uptake in the spleen, lungs, and thyroid similar to the radioiodinated counterpart.
The main concerns have been for antibody fragments such as Fab or F(ab′)2, which are extensively deastatinated in vivo when radiolabeled with SAB, as demonstrated by the significant uptake of activity in the thyroid, stomach, spleen, and lung compared to their radioiodinated counterparts (Fig. 12).79,80 Several approaches, mostly based on structural modifications of the SAB, have been considered to improve the stability. They have consisted either of increasing the electronic density of the aromatic ring of the prosthetic group to strengthen the C–At bond (e.g., addition of electron donor groups and suppression of electron withdrawing group56,81,82), or by the addition of hindering groups such as a methyl in the vicinity of the 211At atom, which was proposed to sterically hinder dehalogenation mechanisms83 (see examples in Fig. 13). However, limited successes were obtained, except in the case of methyl-SAPS, which appears to improve the in vivo characteristics of the antibodies investigated. However, none of these compounds seems to be a real final solution to the stability issue with antibody fragments.
FIG. 12.
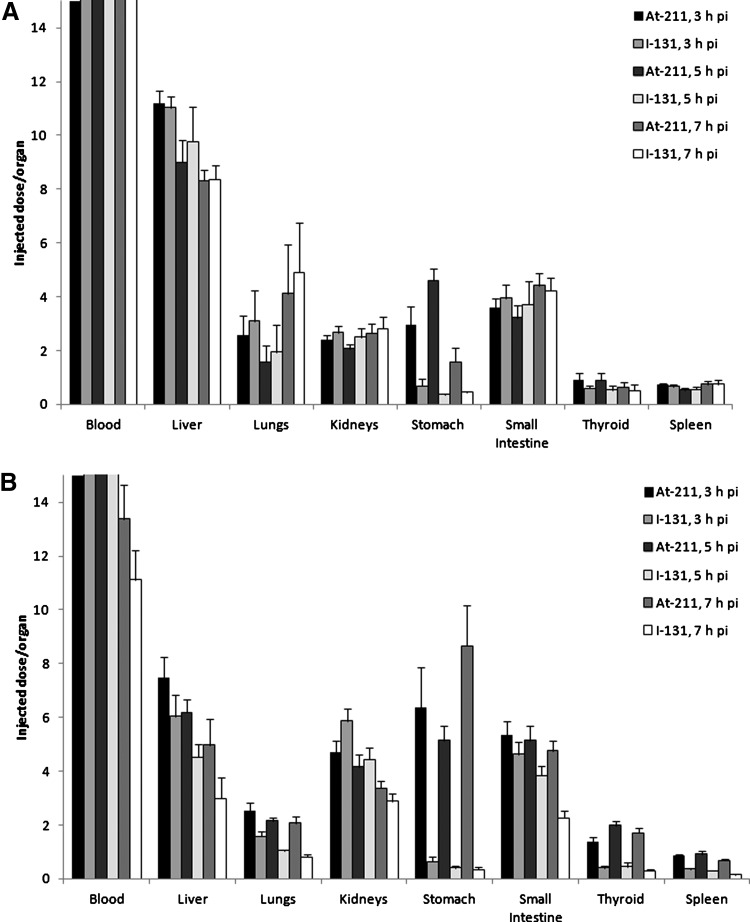
Comparative biodistribution of [211At]-astatinated and [131I]-iodinated full antibody C110 IgG (A) with its F(ab′)2 fragment (B) in mice. Charts plotted from data by Garg et al.80
FIG. 13.
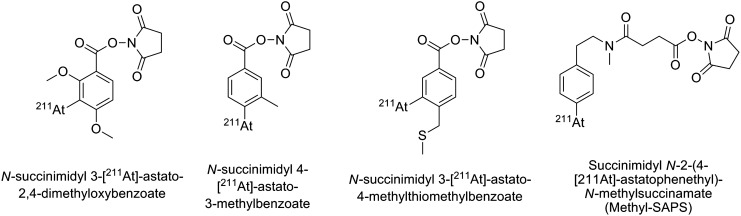
Modified prosthetic groups based on the SAB structure.56,81–83
Alternatives to astatoaryl derivatives, boron cages, were proposed more recently by Wilbur et al.61 with promising results (see structure in Fig. 9—previous section). In a series of reports, it was first demonstrated with small molecules that nido-carboranyls are much more stable than aryls toward deastatination.61 These interesting results led the authors to investigate the radiolabeling of a Fab′ antibody fragment with selected nido-carboranes and closo-decaborate derivatives and to compare them to the classic radiolabeling results obtained from using SAB.84 It was demonstrated that the first advantage of the boron derivatives over the SAB was the possibility of direct astatination of the antibody fragment. With a modification of the Fab′ by conjugation to the boron clusters before radiolabeling, it was possible to reach astatination yields up to 75% in only one step, in 10 minutes. Then, biodistribution studies in mice indicated the superiority of the radiolabeling via the boron cages with a significantly lower uptake of 211At in the thyroid, stomach, spleen, and lungs. The best results were obtained with the closo-decaborate derivatives, but significant uptake in the liver and kidneys were observed, suggesting that modifications to the boron cage or to the linkers are necessary. Studies by this research group are ongoing to solve this issue, such as the investigation of cleavable linkers to increase the clearance rate from these organs.85,86
Peptide/biotin carriers
Because of the relatively short half-life of 211At, smaller biomolecules have been considered as better carriers for targets that are not quickly accessible to full antibodies. This includes peptides that exhibit fast kinetics, or biotin derivatives for pretargeting strategies. Octreotide has been a peptide of interest investigated by Vaidyanathan et al.87 for targeting somatostatin receptors. In an initial article, they demonstrated that it was possible to radiolabel octreotide via a two-step procedure by conjugation to SAB. Further studies were performed with an octreotide premodified with guadininomethyl trimethylstannylbenzoate for direct radiohalogenation of the peptide. Assays indicated an excellent binding to the somatostatin receptor in vitro. No biodistribution study was performed with the astatinated octreotide, but the results obtained with the radioiodinated counterpart, although good in tumor-binding properties, unfortunately demonstrated an elevated uptake of the peptide in the liver and in the kidneys. These results precluded the use of this compound for i.v. administration, although its use in locoregional administration for targeting of medulloblastoma was suggested.88 Later improvements were attempted by glycosylation of the octreotide that met with limited success on improving binding properties and pharmacokinetics.89 Nevertheless, while astatination of the octreotide derivatives cited above was demonstrated, no data on the in vivo stability are available, and instability of the SAB-like compound would be expected for this kind of rapidly internalized compound.
Several reports relate the use of astatinated biotin for pretargeting (for general details on pretargeting, see Lesch et al.90). Initial studies by Foulon et al.91 described direct astatination of biotin preconjugated to a trialkylstannylaniline moiety. However, the resulting astatinated compound was unstable when incubated in murine serum.91 The same instability was observed with the radioiodinated counterpart, and analysis of the catabolites led the authors to conclude that cleavage of the amide bond occurred in vitro due to a biotinidase enzyme. This issue was resolved by reversing the position of the nitrogen and the carbonyl of the amide bond to avoid the action of the biotinidase (see Fig. 14), and provided high stability of the astatinated compound in murine serum.92 Unfortunately, biodistribution studies indicated release of the astatine with significant uptake in the lungs, stomach, and thyroid. Thereafter, modifications using the promising boron cages developed by Wilbur et al.93 provided a higher in vivo stability with a nido-carborane compared to the aryl compounds. While biodistribution studies indicated that deastatination was still occurring (but at a lower level than with aryl derivatives), latest results suggest that closo-decaborate(2−) is the most promising group for a strong stabilization of the astatination of biotin derivatives (see Fig. 14).94
FIG. 14.
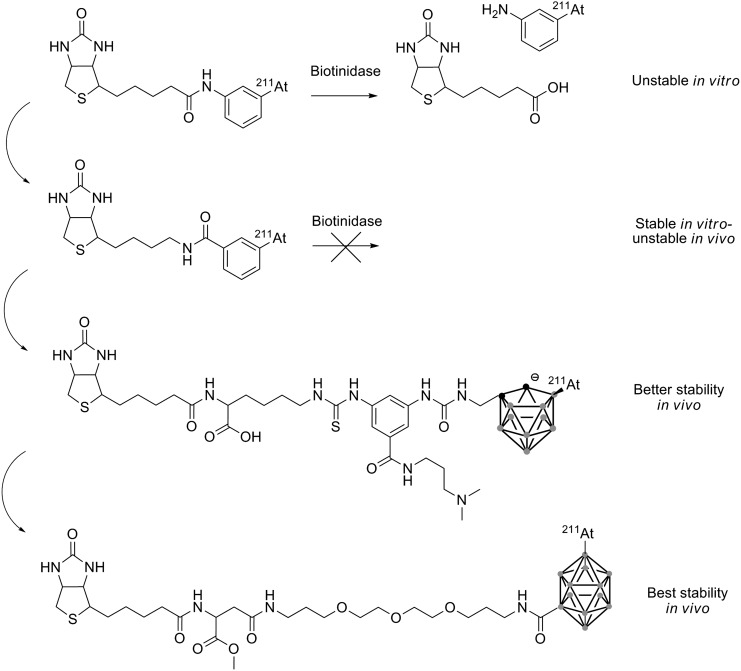
Successive improvements to the biotin to increase the astatination stability.91–94
Alternatively, Lindegren's group investigated biotinylated polylysines conjugated to SAB in a pretargeting strategy. In the first study, the poly-l-lysines had molecular weights of 13, 38, and 363 kDa with excellent avidin binding in vitro. As expected, biodistribution studies indicated that the smaller compound was rapidly excreted through the kidneys while the larger compounds had an increased liver excretion pathway. Uptake in the thyroid, stomach, lungs, and spleen indicated that release of astatine also increased with the size of the polymer.95 In a second study, both l- and d-isomers of the polylysine were compared for their biodistribution properties. Interestingly, the poly-d-lysine, probably more resistant to metabolism, exhibited a significantly lower release of astatine as indicated by a decreased uptake in the lungs, spleen, stomach, and thyroid. However, because of an elevated uptake in kidneys for this isomer, the poly-l-lysine was preferred.96 Recently, this astatinated biotinylated poly-l-lysine was investigated for use with mAb MX35 conjugated to avidin and administered intraperitonealy to tumor-free mice. High uptake of astatine was observed in the lungs, spleen, stomach, and throat, which could be reduced in a second experiment by preadministration of sodium perchlorate as a blocking agent.97 However, while more studies are necessary to assess the efficiency of this poly-l-lysine derivative for the treatment of tumors, it remains equally clear that improvement of the astatine stability is required.
Other small molecules
Several other small astatinated molecules have been investigated for their propensity to be incorporated quickly to some tumor cells. However, because of their small size, many of these compounds have a natural tendency to be rapidly metabolized, resulting in marked deastatination being observed in vivo. This is the case of the majority of such investigated compounds, for example, 6-[211At]-astatomethyl-19-norcholest-5(10)-en-3β-ol, which targets the adrenal gland,45 4-[211At]-astato-N-piperidinoethyl benzamide considered for the treatment of melanoma and glioma,57 or 5-[211At]-astato-2′-deoxyuridine for DNA-targeting strategy58 (structures in Fig. 15).
FIG. 15.
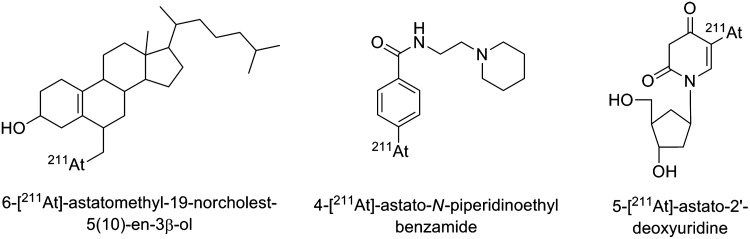
Examples of small astatinated molecules unstable in vivo.45,57,58
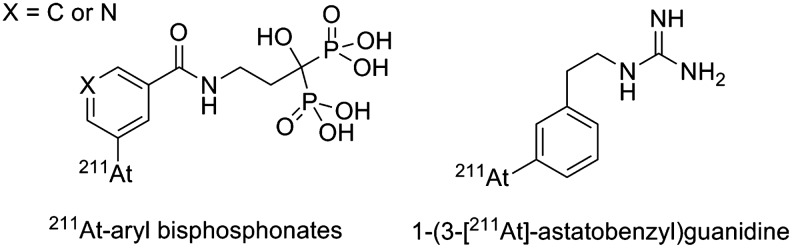
Other examples, however, show that small molecules can be very stable with minimal release of 211At. This is the case of two 211At-arylbisphosphonates studied for applications in bone metastasis pain palliation98 and 1-(3-[211At]-astatobenzyl)guanidine ([211At]MABG) with potential applications in the treatment of glioma99 (see structures in Fig. 16). These compounds could be interesting to consider as prosthetic groups for radiolabeling substrates that have demonstrated instabilities cited above, although it is not clear if the stability comes from the molecular structures themselves or from biodistribution pathways circumventing deastatination mechanisms.
FIG. 16.
Nanoparticles and microspheres
Although devoid of active targeting properties, nanoparticles and larger particles have been investigated with potential applications for locoregional administration. Larsen et al.100 prepared 211At-microspheres by conjugation of aminated polymer particles to SAB. The resulting particles with a size of 1.8 μm were stable in various media in vitro.100 They were compared to free 211At− and astatinated antibodies by i.p. administration to mice inoculated with the K13 hybridoma cell line.101 Biodistribution studies indicated that free 211At− spread rapidly in the whole body, whereas the microsphere and the antibodies were mostly retained in the peritoneal cavity, highlighting a good stability. However, the authors concluded that the efficiency of the astatinated antibodies was superior to the microspheres due to a better diffusion in the peritoneal area.
Another approach developed by Kucka et al.102 is the preparation of astatinated silver nanoparticles coated with poly(ethylene oxide) (PEO). The authors suggested that the high affinity of silver for astatine could result in a stable radiolabeling. They incubated 211At with nanoparticles made of a silver proteinate core coated with PEO (size of 2000 and 5000 g/mol) in the presence of a reducing agent (NaBH4). Quantitative yields were obtained, and in vitro measurements indicated a good stability, even in the presence of high concentration of chloride ions as a competitive agent. However, no data on in vivo behavior are available.
More recently, use of carbon nanotubes was proposed by Hartman et al.103 The free carbon nanotubes, with a length in the 20–50 nm range, were incubated with [211At]-astatide. The astatine trapped inside the nanotubes was then oxidized into the AtCl form in the presence of chloramine-T or N-chlorosuccinimide. The stability of the labeling was assessed by successive washing with water or with human serum. Even though moderate, a loss of activity was observed in both cases. This phenomenon was attributed to either a release of the AtCl portion close to the ends of the nanotubes, or by leakage through damage in the walls of the nanotubes caused by the high-energy α-particles from 211At decay.
Alpha-Therapy of Cancers with 211At-Labeled Targeting Agents
In vitro studies
A number of in vitro studies on astatinated bioconjugates have focused on the determination of the cytotoxicity and microdosimetric data with various carriers (mostly antibodies) and cell lines. Cell conditions have varied from suspended isolated cells to monolayers to spheroid cell clusters, thereby modeling different possible configurations occurring in vivo, but then also making difficult any actual direct comparison between the studies. While it is not intended to make an exhaustive description of all in vitro studies, a selection of reports covering different aspects of 211At effects on in vitro cells is described below.
Larsen et al. investigated the cytotoxicity of astatinated mAb 81C6 (targeting tenascin in the extracellular matrix of the cell), mAb Me1-14 (targeting proteoglycan chondroitin sulfate on the cell membrane), and TPS3.2 (nonspecific) on microcolonies of D-247 MG glioma cells and SK-MEL-28 melanoma cells.104 After 1-hour incubation with 18 kBq/mL activity concentration, results indicated two-to-five times more activity bound to the cells of the specific antibodies compared to the nonspecific mAb. A survival fraction of 0.37 (D0) was obtained with one to two α-decays hitting the cell nucleus with only little differences between both specific mAbs and both cell lines. In another study, the cytotoxicity of 211At-trastuzumab was investigated against three breast carcinoma cell lines expressing the HER2 receptor (SKBr-3, BT-474, and MCF7/HER2-18).105 Cell survival studies indicated a relative biological effectiveness (RBE) of 211At-trastuzumab 10-fold higher than external irradiation. At the low amount of antibody used, the toxicity was attributed exclusively to the α-decay of 211At, with no contribution from trastuzumab. To estimate absorbed dose and toxicity in vivo on nonspecific tissues, the anti-CD20 mAb, rituximab, labeled with 211At was investigated on cultured RAEL lymphoma cells and bone marrow cells.106 The absorbed dose when using a 10 kBq/mL activity concentration was 0.645 and 0.021 Gy for lymphoma cells and bone marrow cells, respectively, after 1-hour exposure. However, a higher radiosensitivity to α-particles was observed for bone marrow (D0=0.34 Gy) compared to lymphoma cells (D0=0.55 Gy), whereas reversed sensitivity was observed from exposure to 60Co γ-irradiation (D0=1.21 and 0.72 Gy for bone marrow and lymphoma, respectively). Despite these observations, the high specificity of RAEL cell binding of 211At-rituximab over bone marrow cells made these results interesting in the perspective of the treatment of non-Hodgkin's lymphoma. Recently, Petrich et al. investigated 211At-anti-CD33 antibodies to overcome the cellular resistance observed in a number of patients to gemtuzumab ozogamicin (GO), the recently withdrawn toxin–mAb conjugate Mylotarg, used in the treatment of acute myeloid lymphoma.107 The better results observed for 211At-anti-CD33 over GO were promising in the perspective of treatment of GO-resistant cases of acute myeloid lymphoma.
Within the realm of preclinical studies, cetuximab was investigated in combination with astatinated cMab U36 on cultured squamous cell carcinomas. The results indicated a higher growth inhibition for the combination therapy compared to RIT alone or cetuximab alone. However, a radioprotective effect was observed in the presence of cetuximab, as well as a decrease of the internalization of cMab U36.108 Although these phenomena have not been explained yet, the results suggest that RIT after treatment with cetuximab would be more efficient than combination of both treatments at the same time to avoid this radioprotective effect.
Alternatively, a limited number of nonantibody carriers have also been studied in vitro, including 211At-EGF, a small protein (MW≈6 kDa) targeting the EGF receptor, which is overexpressed in a number of cancerous tumors. Internalization of 211At-EGF in A-431 carcinoma cells was compared to the [125I]iodinated counterpart. Interestingly, a longer retention for the astatinated compound was observed, resulting in an increased biological half-life from 1.5 hours for 125I-EGF to 3.5 hours for 211At-EGF.109 These results suggested that 211At conjugated to EGF would be a radionuclide of choice due to its favorable biological and physical characteristics. In another study, 5-[211At]-astato-2′-deoxyuridine (211AtdU) was investigated for its DNA incorporation properties in Chinese hamster V79 lung fibroblasts. It was shown that<1% of free 211At− was incorporated compared to 211AtdU. The survival study indicated a D0 of 1.3 decays/cell, and that unlabeled cells were killed by neighboring labeled cells. Furthermore, DNA double-strand break (DSB) levels were 10-fold higher with 211AtdU compared to the [125I]iodinated counterpart.58
A limited number of studies have specifically focused on the radiobiology of 211At and the biological consequences of the α-decay to the cell material. The most recent reports were published by Claesson et al.,110 who investigated the RBE for induction of DSB for 211At on human fibroblasts compared to 60Co γ-irradiation and X-rays. In a first article, it was observed that two to three times more DSB occurred with 211At compared to 60Co γ-rays and X-rays. Furthermore, the study of the size of the DNA fragments induced by 211At revealed a nonrandom distribution compared to γ-rays and X-rays, which resulted in a random distribution.110 In a second study, the RBE of DSB for 211At and X-rays was investigated at different cell cycle phases (V79-379A fibroblasts synchronized with mimosine in G1, early, mid-, and late S phase). As expected, 211At exhibited a higher efficiency in generating DSB, with DSB varying in the cell cycle phases according to the following sequence: G1<S early<S late<S mid<mitosis. It was however observed that X-rays were more efficient in inducing clustered damage than 211At.111
Preclinical therapy studies
A number of preclinical therapy animal studies have been performed to assess the targeting properties of the astatinated biomolecules and their efficacy and toxicity, sometimes in comparison with other radionuclides of interest. Therapy studies were conducted mostly with antibodies or their fragments as delivery vectors as described below. Interestingly, models involving locoregional administration of the radiopharmaceutical have been more frequently investigated over systemic injection. Indeed, the biological and physical half-lives of the astatinated proteins seem particularly appropriate to localized compartmental tumors such as in i.p. implanted ovarian tumor models. Furthermore, concerns on the deastatination of the proteins as a result of accelerated metabolism when administered intravenously may have been a limiting factor with some carriers.
Nonetheless, the feasibility of therapy using i.v. injected astatinated antibodies has been demonstrated in several studies, and promising results have been obtained for the treatment of disseminated cancers. The astatinated anti-CD25 antibody 7G7/B6 was evaluated in a murine model of leukemia. Therapy with i.v. injection of 0.55 MBq (15 μCi) 211At-7G7/B6 in karpas299 leukemia-bearing mice resulted in 70% survival at 46 days and 33% survival after 5 months, whereas untreated mice and mice treated with nonspecific antibody (211At-11F11) all died after 46 days.112 These promising results led the authors to investigate 211At-7G7/B6 in combination with mAb daclizumab for therapy in mice inoculated with MET-1 human T-cell leukemia. Excellent results were obtained with improved survival for combination therapy (91% survival at 94 days) compared to RIT with 0.44 MBq (12 μCi) 211At-7G7/B6 (32% survival at 94 days) or daclizumab treatment alone (47% at 94 days) while all mice died after 70 days when treated with nonspecific 211At-11F11.113 The same authors also investigated the anti-CD30 antibody 211At-HeFi-1 in a model of karpas299 leukemia-bearing mice. Again, the combination of RIT with cold HeFi-1 significantly prolonged the survival of the animals (33% and 84% survival at 120 days for RIT alone and combination therapy, respectively).114 In another study, Cheng et al.115 evaluated the astatinated mAb U36 for the treatment of head and neck squamous cell carcinoma (HNSCC). Intravenous injection of 200 kBq radiolabeled antibody in mice inoculated with HNSCC resulted in the reduction or the stabilization of the tumor volume, while control group (treated with nonlabeled U36) had its tumor volume steadily increasing after treatment.115 Alternatively to full antibodies, Robinson et al.116 investigated the smaller C6.5 diabody (targeting HER2 receptors) that exhibits faster targeting properties compared to antibodies. Mice bearing HER2/neu-positive MDA-MB-361/DYT2 tumors were treated by i.v. injection of 0.74 MBq (20 μCi) to 1.67 MBq (45 μCi) 211At-C6.5. Compared to the control group, the tumor volume doubling time was delayed by 30 days for the 20 μCi group and by 57 days with 60% of the mice being tumor free after 1 year for the 45 μCi group.116 Unfortunately, no biodistribution data were reported for the 211At-C6.5 diabody. The in vivo stability of the 211At-diabody bond of this engineered antibody fragment, which can be expected to be rapidly metabolized, would be interesting to study.
In the case of locoregional injections, the most documented astatinated compound is probably MX35 in its F(ab′)2 form. MX35 targets an antigen (Ley) expressed in 90% of human epithelial ovarian cancers. It has been extensively studied for α-RIT in a murine model of ovarian NIH:OVCAR-3 cancer cells inoculated intraperitoneally at the Sahlgrenska academy at Göteborg University. Early investigations on 211At-MX35 F(ab′)2 allowed for the determination of the minimum required dose by i.p. administration of doses ranging from 25 to 200 kBq 4 weeks after inoculation of NIH:OVCAR-3 cells in mice. With an increase from 22% to 50% of animals presenting no sign of tumor with doses of 50 and 100 kBq, respectively, it was concluded that 100 kBq of 211At-MX35 F(ab′)2 was the minimal required dose to give clear evidence of the therapeutic efficacy of this treatment.117 The authors also investigated the efficacy of the treatment against different tumor sizes. In this study, groups of mice were treated with 400 kBq 211At-MX35 F(ab′)2 at different times (1, 3, 4, 5, or 7 weeks) after inoculation of the NIH:OVCAR-3 cells. Eight weeks after treatment, the mice were sacrificed, and the proportions of tumor- free mice were found to be 95%, 68%, 58%, 47%, and 26% for 1–7 weeks post-treatment, respectively, thus confirming the suitability of α-radiation for the treatment of smaller tumors.118 Fractionated administration of the treatment was also compared to single injection. Four weeks after inoculation of the NIH:OVCAR-3 cells, groups of mice received a total of 800, 400, or 50 kBq 211At-MX35 F(ab′)2 in a single injection or in three equal injections separated by 4 days. However, no advantages were found in fractionated treatment with the best results being obtained for the 800 kBq single-injection group (56% tumor-free animals 8 weeks post-treatment compared to 41% for the corresponding fractionated-injection group).119 However, a subsequent investigation showed that weekly repeated injections of a 400 kBq dose of 211At-MX35 F(ab′)2 (from one to six injections) led to a significantly higher therapeutic efficacy after three or more injection with 17% free tumor animals after one injection, 39% after three injection, and up to 67% after six injections. Furthermore, no ascites were detected in the group receiving five or six injections, while 15 out of 18 animals exhibited ascites when receiving one injection.120 Recently, the 211At-MX35 F(ab′)2 efficacy was compared to its 213Bi counterpart. Groups of mice were treated with either 2.7 MBq of 213Bi-MX35 F(ab′)2 or 440 kBq of 211At-MX35 F(ab′)2 2 or 4 weeks after inoculation of NIH:OVCAR-3 cells. No significant superiority of either radionuclide was observed.121 Finally, with the promising results obtained for 211At-MX35 F(ab′)2 in preclinical studies and the absence of significant toxicity of these treatments at the doses employed, one of the first clinical trial with a 211At radiopharmaceutical was recently initiated with this antibody (see next section).
Another antibody that deserves attention is ch81C6, as it was used in the first phase I 211At-mAb clinical trial (see next section). It was initially used in the murine form for the treatment of neoplastic meningitis in preclinical studies. In a rat model, animals were inoculated with TE-671 human rhabdomyosarcoma cells by intrathecal injection into the subarachnoid space. In a series of experiments, dose escalation was first performed by intrathecal administration of 0.15–0.48 MBq (4–13 μCi) of 211At-81C6 mAb to determine the dose inducing a therapeutic effect. Increases in median survival of 30%, 29%, and 51% were observed with 4, 7, and 13 μCi, respectively, compared to control (saline) with 20% of the animals still alive after 190 days in the 13 μCi group. In a second experiment, animals were treated with 0.44 MBq (12 μCi) or 0.67 MBq (18 μCi) of 211At-81C6 and compared with nonspecific 211At-45.6 mAb and saline. No significant increase in the median survival was observed for the nonspecific mAb compared to saline while 113% and 357% survival prolongation was observed for the 12 μCi and the 18 μCi 211At-81C6 groups, respectively.122 To reduce the potential immunogenicity of the treatment from the perspective of the clinical use of the 81C6 mAb, the human/mouse chimeric version of the antibody (ch81C6) was assessed. Biodistribution, dosimetry, and toxicity studies were performed and confirmed the superior characteristics of the ch81C6 mAb over its murine form.123
Trastuzumab, an anti-HER2, labeled with 211At is another antibody that has shown promising results in recent studies. In a radioresistant SKOV-3 ovarian tumor model implanted in mice intraperitoneally, 211At-trastuzumab was investigated in a series of experiments. First, a dose–escalation study from 0 to 800 kBq of the radiolabeled antibody injected i.p. was performed. A dose-dependent reduction of the tumor size was observed from 0 to 400 kBq without better results at 800 kBq compared to the 400 kBq dose. In a second experiment, the mice were treated with 400 kBq 211At-trastuzumab in combination with increasing doses of cold trastuzumab (5–500 μg). Increased efficacy was observed while increasing the dose of cold trastuzumab with the best result obtained at 500 μg cold trastuzumab with a total eradication of the tumors. Finally, fractionated administration at various doses of 211At-trastuzumab combined with various doses of cold trastuzumab did not exhibit any advantages.124 In another study, 211At-trastuzumab was also investigated for the treatment of breast carcinomatous meningitis by intrathecal injection with significantly improved survival compared to the control groups (saline or nonspecific astatinated mAb).125
Clinical studies
To date, only two phase-I clinical trials have been reported. The first one was published in 2008 by the Zalutsky group at the Duke University. Astatinated ch81C6 was used for the treatment of residual disease after surgical removal of the glioma tumor in the brain. The classical treatment for this pathology is surgery followed by external radiotherapy and chemotherapy. In this study, 18 patients with glioblastoma multiforme or anaplastic oligodendroglioma were enrolled and received an injection of 211At-ch81C6 (71–347 MBq) in the cavity resulting from the surgery. The biodistribution was monitored directly by a γ-camera and demonstrated that 96.7%±3.6% of the 211At decays occurred within the cavity. Blood counts indicated minimal leakage from the cavity with<0.05% of injected dose measured. These data highlighted the low catabolism of the antibody when injected into the cavity. Final results were promising with a median survival time that increased from 31 weeks with the classical treatment of GMB to 54 weeks with additional 211At-RIT without dose-limiting toxicity being observed.76
The second clinical trial was reported in 2009 for the treatment of ovarian cancer with 211At-MX35 F(ab′)2. The classical treatment for ovarian carcinoma is surgery in combination with chemotherapy, but in many cases, relapse occurs with a low chance of survival. The purpose of this study was to demonstrate the potential of α-RIT after a clinical trial has failed as a phase-III study with a β−-emitter (HMFG1 mAb radiolabeled with 90Y). The disappointing results obtained with 90Y-HMFG1 were attributed to the nature of the radionuclide and its associated β−-particles being unsuitable for the treatment of microscopic tumors. Nine female patients in total remission after chemotherapy were enrolled in the study. After laparoscopy to check the absence of macroscopic tumor, the patients received 20–100 MBq 211At-MX35 via a catheter. The biodistribution was monitored with a γ-camera and indicated that most of the radioactivity was retained in the abdomen. A noticeable uptake in the thyroid was observed for the patients who did not receive a blocking agent before the treatment. No significant uptake was observed in the other organs. Analyses of the dosimetric data indicated that this treatment allowed for the deposition of the necessary dose for the destruction of micrometastases without noticeable toxicity to the bone marrow and to the other healthy organs.126 No conclusions on the therapeutic efficacy of this treatment are available as yet, but further investigations and reports can be expected in the near future.
Conclusions
Shortly after the discovery of 211At in 1940, the therapeutic potential of this radionuclide was investigated, with applications considered for the treatment of thyroid disorders.127 However, because of the high toxicity of α-radiation to healthy tissue, it appeared obvious that 211At could not be used alone, and needed to be conjugated to an appropriate targeting carrier, especially for potential applications in the treatment of cancers.
With the exploration of the chemical properties of astatine, methodologies for the radiolabeling of various molecules of interest have been proposed. However, it is still difficult today to work with this radionuclide because of the unpredictable aspects of its reactivity and its halogen/metalloid duality. Fortunately, simple, fast, and reproducible radiolabeling methods applicable to a number of molecules of interest have been developed. Particularly, antibodies and their engineered fragments promising for RIT of cancers can now be astatinated efficiently using astatobenzoyl-based prosthetic groups. The high potential of 211At for the treatment of residual disease has been recently highlighted by the first clinical trials realized with a chimeric antibody (ch81C6) and a F(ab′)2 fragment (MX35) using such radiolabeling methods. Promising results were obtained with a real therapeutic gain and/or limited toxicity observed.
However, many compounds of interest labeled with 211At have been eliminated from the process of development of new radiopharmaceuticals because of the lack of stability on radiolabeling with astatine. From the review of the recent literature, it is clear that an effort in the understanding of the chemistry of astatine for the development of new radiolabeling approaches is required to provide new astatinated radiopharmaceuticals. Recent reports show that several research groups are working on alternative chemistries to reach that goal.40,128,129 Furthermore, implementation of new high-energy cyclotrons capable of producing 211At should improve the availability of this radionuclide in the near future and accelerate the development of new astatinated radiopharmaceuticals.11,130
Acknowledgment
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Disclosure Statement
No competing financial interests exist.
References
- 1.Kim Y-S. Brechbiel MW. An overview of targeted alpha therapy. Tumor Biol. 2012;33:573. doi: 10.1007/s13277-011-0286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yong K. Brechbiel MW. Towards translation of 212Pb as a clinical therapeutic; getting the lead in! Dalton Trans. 2011;40:6068. doi: 10.1039/c0dt01387k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corson DR. MacKenzie KR. Segrè E. Possible production of radioactive isotopes of element 85. Phys Rev. 1940;57:1087. [Google Scholar]
- 4.Hyde EK. The present status of elements 85 and 87. J Phys Chem. 1954;58:21. [Google Scholar]
- 5.Gmelin L. Berei K. Kugler HK, et al. Gmelin Handbook of Inorganic Chemistry: At, Astatine. Berlin, New York: Springer-Verlagt; 1985. [Google Scholar]
- 6.Palm S. Humm JL. Rundqvist R, et al. Microdosimetry of astatine-211 single-cell irradiation: Role of daughter polonium-211 diffusion. Med Phys. 2004;31:218. doi: 10.1118/1.1640951. [DOI] [PubMed] [Google Scholar]
- 7.Turkington TG. Zalutsky MR. Jaszczak RJ, et al. Measuring astatine-211 distributions with SPECT. Phys Med Biol. 1993;38:1121. doi: 10.1088/0031-9155/38/8/010. [DOI] [PubMed] [Google Scholar]
- 8.Meyer GJ. Lambrecht RM. Excitation function for the 209-Bi(7-Li,5n)211-Rn nuclear reaction as a route to the 211-Rn-211-At generator. J Label Compd Radiopharm. 1981;28:233. [Google Scholar]
- 9.Vinodkumar AM. Loveland W. Sprunger PH, et al. Fusion of 9Li with 208Pb. Phys Rev C. 2009;80:054609. [Google Scholar]
- 10.Harrison J. Leggett R. Lloyd D, et al. Polonium-210 as a poison. J Radiol Prot. 2007;27:17. doi: 10.1088/0952-4746/27/1/001. [DOI] [PubMed] [Google Scholar]
- 11.Zalutsky MR. Pruszynski M. Astatine-211: Production and availability. Current Radiopharm. 2011;4:177. doi: 10.2174/1874471011104030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hermanne A. Tárkányi F. Takács S, et al. Experimental study of the cross-sections of [alpha]-particle induced reactions on 209Bi. Appl Radiat Isot. 2005;63:1. doi: 10.1016/j.apradiso.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Lindegren S. Bäck T. Jensen HJ. Dry-distillation of astatine-211 from irradiated bismuth targets: A time-saving procedure with high recovery yields. Appl Radiat Isot. 2001;55:157. doi: 10.1016/s0969-8043(01)00044-6. [DOI] [PubMed] [Google Scholar]
- 14.Yordanov A. Pozzi O. Carlin S, et al. Wet harvesting of no-carrier-added 211At from an irradiated 209Bi target for radiopharmaceutical applications. J Radioanal Nucl Chem. 2005;262:593. [Google Scholar]
- 15.Alliot C. Cherel M. Barbet J, et al. Extraction of astatine-211 in diisopropylether (DIPE) Radiochim Acta. 2009;97:161. [Google Scholar]
- 16.Bourgeois M. Guerard F. Alliot C, et al. Feasibility of the radioastatination of a monoclonal antibody with astatine-211 purified by wet extraction. J Label Compd Radiopharm. 2008;51:379. doi: 10.1002/jlcr.1543. [DOI] [PubMed] [Google Scholar]
- 17.Zalutsky MR. Zhao XG. Alston KL, et al. High-level production of alpha-particle-emitting (211)At and preparation of (211)At-labeled antibodies for clinical use. J Nucl Med. 2001;42:1508. [PubMed] [Google Scholar]
- 18.Appelman EH. Sloth EN. Studier MH. Observation of astatine compounds by time-of-flight mass spectrometry. Inorg Chem. 1966;5:766. [Google Scholar]
- 19.Johnson GL. Leininger RF. Segrè E. Chemical properties of astatine. I. J Chem Phys. 1949;17:1. [Google Scholar]
- 20.Appelman EH. The oxidation states of astatine in aqueous solution. J Am Chem Soc. 1961;83:805. [Google Scholar]
- 21.Visser GWM. Diemer EL. Inorganic astatine chemistry: Formation of complexes of astatine. Radiochim Acta. 1983;33:145. [Google Scholar]
- 22.Visser GWM. Inorganic astatine chemistry part II: The chameleon behaviour and electrophilicity of At-species. Radiochim Acta. 1989;47:97. [Google Scholar]
- 23.Dreyer I. Dreyer R. Chalkin VA. Investigation of the movement of astatine in an electric field. Radiochem Radioanal Lett. 1978;35:257. [Google Scholar]
- 24.Dreyer I. Dreyer R. Chalkin VA. Cations of astatine in water: illustration and properties. Radiochem Radioanal Lett. 1978;36:389. [Google Scholar]
- 25.Dreyer R. Dreyer I. Rosch F, et al. Studies of polyhalogenide ions of astatine. Radiochem Radioanal Lett. 1982;54:165. [Google Scholar]
- 26.Ruth TJ. D'Auria JM. Dombsky M, et al. The Radiochemistry of Astatine. DOE sponsored Nuclear Science Series monogram (NAS-NS-3064; DE88015386) 1988. www.ntis.gov/search/product.aspx?ABBR=DE88015386. [Jul 16;2012 ]. www.ntis.gov/search/product.aspx?ABBR=DE88015386
- 27.Pruszyński M. Bilewicz A. Wąs B, et al. Formation and stability of astatide-mercury complexes. J Radioanal Nucl Chem. 2006;268:91. [Google Scholar]
- 28.Pruszyński M. Bilewicz A. Zalutsky MR. Preparation of Rh[16aneS4-diol]211At and Ir[16aneS4-diol]211At complexes as potential precursors for astatine radiopharmaceuticals. Part I: Synthesis. Bioconjug Chem. 2008;19:958. doi: 10.1021/bc700413r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appelman EH. Solvent extraction studies of interhalogen compounds of astatine. J Phys Chem. 1961;65:325. [Google Scholar]
- 30.Fischer S. Dreyer R. Albrecht S. Pseudohalogen compounds of astatine: Synthesis and characterization of At/I/-tricyanomethanide-and At/I/-azide-compounds. J Radioanal Nucl Chem. 1987;117:275. [Google Scholar]
- 31.Dreyer R. Dreyer I. Fischer S, et al. Synthesis and characterization of cationic astatine compounds with sulphur-containing ligands stable in aqueous solutions. J Radioanal Nucl Chem. 1985;96:333. [Google Scholar]
- 32.Fischer S. Dreyer R. Hussein H, et al. Synthesis and first characterization of cationic At/I/-compounds with selenium-containing neutral ligands. J Radioanal Nucl Chem. 1987;119:181. [Google Scholar]
- 33.Ludwig R. Dreyer R. Fisher S. First investigation of complex formation of At(I) with phosphorous organic compounds. Radiochim Acta. 1989;47:129. [Google Scholar]
- 34.Milesz S. Jovchev M. Schumann D, et al. The EDTA complexes of astatine. J Radioanal Nucl Chem. 1988;127:193. [Google Scholar]
- 35.Milesz S. Norseev YV. Szücs Z, et al. Characterization of DTPA complexes and conjugated antibodies of astatine. J Radioanal Nucl Chem. 1989;137:365. [Google Scholar]
- 36.Schumann D. Milesz S. Jovchev M, et al. Nitrilotriacetate complex of univalent astatine. Radiochim Acta. 1992;56:173. [Google Scholar]
- 37.Ning L. Jiannan J. Shangwu M, et al. Preparation and preliminary evaluation of astatine-211 labeled IgG via DTPA anhydride. J Radioanal Nucl Chem. 1998;227:187. [Google Scholar]
- 38.Ludwig R. Fischer S. Dreyer R, et al. Complex formation equilibria between astatine(I) and sulphur-containing chelating ligands. Polyhedron. 1991;10:11. [Google Scholar]
- 39.Yordanov AT. Deal K. Garmestani K, et al. Synthesis and biodistribution study of a new 211-At-calix[4]arene complex. J Label Compd Radiopharm. 2000;43:1219. [Google Scholar]
- 40.Champion J. Alliot C. Huclier S, et al. Determination of stability constants between complexing agents and At(I) and At(III) species present at ultra-trace concentrations. Inorg Chim Acta. 2009;362:2654. [Google Scholar]
- 41.Greenwood N. Earnshaw A. Chemistry of the Elements. 2nd. Elsevier, Oxford, UK: Butterworth-Heinemann; 1997. Chapter 17: The halogens: Fluorine, Chlorine, Bromine, Iodine and Astatine; p. 789. [Google Scholar]
- 42.Pozzi OR. Zalutsky MR. Radiopharmaceutical chemistry of targeted radiotherapeutics, part 3: Alpha-particle-induced radiolytic effects on the chemical behavior of (211)At. J Nucl Med. 2007;48:1190. doi: 10.2967/jnumed.106.038505. [DOI] [PubMed] [Google Scholar]
- 43.Berei K. Vasáros L. PATAI'S Chemistry of Functional Groups. John Wiley and Sons, Ltd; 2009. [Jul 16;2012 ]. Recent Advances in the Organic Chemistry of Astatine. [Google Scholar]
- 44.Coenen HH. Moerlein SM. Stöcklin G. No-carrier added radiohalogenation methods with heavy halogens. Radiochim Acta. 1983;34:47. [Google Scholar]
- 45.Liu BL. Jin YT. Liu ZH, et al. Halogen exchanges using crown ethers: Synthesis and preliminary biodistribution of 6-[211At]astatomethyl-19-norcholest-5(10)-en-3 beta-ol. Int J Appl Radiat Isot. 1985;36:561. doi: 10.1016/0020-708x(85)90110-3. [DOI] [PubMed] [Google Scholar]
- 46.Brown I. Carpenter RN. Link E, et al. Potential diagnostic and therapeutic agents for malignant melanoma: Synthesis of heavy radiohalogenated derivatives of methylene blue by electrophilic and nucleophilic methods. J Radioanal Nucl Chem Lett. 1986;107:337. [Google Scholar]
- 47.Meyer GJ. Walte A. Sriyapureddy SR, et al. Synthesis and analysis of 2-[211At]-l-phenylalanine and 4-[211At]-l-phenylalanine and their uptake in human glioma cell cultures in-vitro. Appl Radiat Isot. 2010;68:1060. doi: 10.1016/j.apradiso.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 48.Visser GWM. Diemer EL. The reaction of astatine with aromatic diazonium compounds. Radiochem Radioanal Lett. 1982;51:135. [Google Scholar]
- 49.Meyer GJ. Rössler K. Stöcklin G. Reaction of aromatic diazonium salts with carrier-free radioiodine and astatine. Evidence for complex formation. J Am Chem Soc. 1979;101:3121. [Google Scholar]
- 50.Wunderlich G. Fischer S. Dreyer R, et al. A simple method for labelling proteins with 211At via diazotized aromatic diamine. J Radioanal Nucl Chem Lett. 1987;117:197. [Google Scholar]
- 51.Norseyev YV. Nhan DD. Khalkin VA, et al. The preparation of astatine labelled tyrosine using an electrophilic reaction. J Radioanal Nucl Chem. 1985;94:185. [Google Scholar]
- 52.Norseev YV. Synthesis of astatine-tagged methylene blue, a compound for fighting micrometastases and individual cells of melanoma. J Radioanal Nucl Chem. 1998;237:155. [Google Scholar]
- 53.Visser GWM. Diemer EL. Kaspersen FM. The preparation and stability of astatotyrosine and astato-iodotyrosine. Int J Appl Radiat Isot. 1979;30:749. [Google Scholar]
- 54.Visser GWM. Diemer E. Kaspersen FM. The preparation of aromatic astatine compounds through aromatic mercury compounds part II: Astatination of pyrimidines and steroids. J Label Compd Radiopharm. 1981;18:799. [Google Scholar]
- 55.Vaidyanathan G. Affleck DJ. Alston KL, et al. A kit method for the high level synthesis of [211At]MABG. Bioorg Med Chem. 2007;15:3430. doi: 10.1016/j.bmc.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vaidyanathan G. Affleck DJ. Zalutsky MR. Monoclonal antibody F(ab′)2 fragment labeled with N-succinimidyl 2,4-dimethoxy-3-halobenzoates: In vivo comparison of iodinated and astatinated fragments. Nucl Med Biol. 1994;21:105. doi: 10.1016/0969-8051(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 57.Garg PK. John CS. Zalutsky MR. Preparation and preliminary evaluation of 4-[211At]astato-N-piperidinoethyl benzamide. Nucl Med Biol. 1995;22:467. doi: 10.1016/0969-8051(94)00134-6. [DOI] [PubMed] [Google Scholar]
- 58.Vaidyanathan G. Larsen RH. Zalutsky MR. 5-[211At]Astato-2’-deoxyuridine, an {alpha}-particle-emitting endoradiotherapeutic agent undergoing DNA incorporation. Cancer Res. 1996;56:1204. [PubMed] [Google Scholar]
- 59.Narula AS. Zalutsky MR. No-carrier added astatination of N-succinimidyl-3-(tri-n-butylstannyl) benzoate (ATE) via electrophilic destannylation. Radiochim Acta. 1989;47:131. [Google Scholar]
- 60.Valliant JF. Guenther KJ. King AS, et al. The medicinal chemistry of carboranes. Coord Chem Rev. 2002;232:173. [Google Scholar]
- 61.Wilbur DS. Zalutsky MR. Wedge TJ, et al. Reagents for astatination of biomolecules: Comparison of the in vivo distribution and stability of some radioiodinated/astatinated benzamidyl and nido-carboranyl compounds. Bioconjug Chem. 2004;15:203. doi: 10.1021/bc034175k. [DOI] [PubMed] [Google Scholar]
- 62.Wilbur DS. [211At]Astatine-labeled compound stability: Issues with released [211At]astatide and development of labeling reagents to increase stability. Current Radiopharm. 2008;1:144. [Google Scholar]
- 63.Larsen RH. Slade S. Zalutsky MR. Blocking [211At]astatide accumulation in normal tissues: Preliminary evaluation of seven potential compounds. Nucl Med Biol. 1998;25:351. doi: 10.1016/s0969-8051(97)00230-8. [DOI] [PubMed] [Google Scholar]
- 64.Hamilton JG. Asling CW. Garrison WM, et al. Destructive action of astatine 211 (element 85) on the thyroid gland of the rat. Proc Soc Exp Biol Med. 1950;73:51. doi: 10.3181/00379727-73-17572. [DOI] [PubMed] [Google Scholar]
- 65.Durbin PW. Asling CW. Johnston ME, et al. The induction of tumors in the rat by astatine-211. Radiat Res. 1958;9:378. [PubMed] [Google Scholar]
- 66.McLendon RE. Archer GE. Garg PK, et al. Radiotoxicity of systematically administered [211At]astatide in B6C3F1 and BALB/c (nu/nu) mice: A long-term survival study with histologic analysis. Int J Radiat Oncol Biol Phys. 1996;35:69. doi: 10.1016/s0360-3016(96)85013-9. [DOI] [PubMed] [Google Scholar]
- 67.Zalutsky MR. Reardon DA. Pozzi OR, et al. Targeted alpha-particle radiotherapy with 211At-labeled monoclonal antibodies. Nucl Med Biol. 2007;34:779. doi: 10.1016/j.nucmedbio.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aaij C. Tschroots WRJM. Lindner L, et al. The preparation of astatine labelled proteins. Int J Appl Radiat Isot. 1975;26:25. doi: 10.1016/0020-708x(75)90006-x. [DOI] [PubMed] [Google Scholar]
- 69.Vaughan AT. Fremlin JH. The preparation of astatine labelled proteins using an electrophilic reaction. Int J Nucl Med Biol. 1978;5:229. doi: 10.1016/0047-0740(78)90145-6. [DOI] [PubMed] [Google Scholar]
- 70.Vaughan AT. Fremlin JH. The preparation of astatotyrosine. Int J Appl Radiat Isot. 1977;28:595. [Google Scholar]
- 71.Visser GWM. Diemer EL. Kaspersen FM. The preparation and stability of 211At-astato-imidazoles. Int J Appl Radiat Isot. 1980;31:275. [Google Scholar]
- 72.Visser GWM. Diemer EL. Kaspersen FM. The nature of the astatine-protein bond. Int J Appl Radiat Isot. 1981;32:905. doi: 10.1016/0020-708x(81)90078-8. [DOI] [PubMed] [Google Scholar]
- 73.Friedman AM. Zalutsky MR. Fitch FW, et al. Preparation of a biologically stable and immunogenically competent astatinated protein. Int J Nucl Med Biol. 1977;4:219. doi: 10.1016/0047-0740(77)90146-2. [DOI] [PubMed] [Google Scholar]
- 74.Zalutsky MR. Narula AS. Astatination of proteins using an N-succinimidyl tri-n-butylstannyl benzoate intermediate. Int J Rad Appl Instrum A. 1988;39:227. doi: 10.1016/0883-2889(88)90176-1. [DOI] [PubMed] [Google Scholar]
- 75.Pozzi OR. Zalutsky MR. Radiopharmaceutical chemistry of targeted radiotherapeutics, part 1: Effects of solvent on the degradation of radiohalogenation precursors by 211At alpha-particles. J Nucl Med. 2005;46:700. [PubMed] [Google Scholar]
- 76.Zalutsky MR. Reardon DA. Akabani G, et al. Clinical experience with alpha-particle emitting 211At: Treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med. 2008;49:30. doi: 10.2967/jnumed.107.046938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reist CJ. Foulon CF. Alston K, et al. Astatine-211 labeling of internalizing anti-EGFRvIII monoclonal antibody using N-succinimidyl 5-[211At]astato-3-pyridinecarboxylate. Nucl Med Biol. 1999;26:405. doi: 10.1016/s0969-8051(98)00120-6. [DOI] [PubMed] [Google Scholar]
- 78.Vaidyanathan G. Affleck DJ. Bigner DD, et al. N-succinimidyl 3-[211At]astato-4-guanidinomethylbenzoate: An acylation agent for labeling internalizing antibodies with alpha-particle emitting 211At. Nucl Med Biol. 2003;30:351. doi: 10.1016/s0969-8051(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 79.Hadley SW. Wilbur DS. Gray MA, et al. Astatine-211 labeling of an antimelanoma antibody and its Fab fragment using N-succinimidyl p-astatobenzoate: Comparisons in vivo with the p-[125I]iodobenzoyl conjugate. Bioconjug Chem. 1991;2:171. doi: 10.1021/bc00009a006. [DOI] [PubMed] [Google Scholar]
- 80.Garg PK. Harrison CL. Zalutsky MR. Comparative tissue distribution in mice of the alpha-emitter 211At and 131I as labels of a monoclonal antibody and F(ab′)2 fragment. Cancer Res. 1990;50:3514. [PubMed] [Google Scholar]
- 81.Talanov VS. Yordanov AT. Garmestani K, et al. Preparation and in vivo evaluation of novel linkers for 211At labeling of proteins. Nucl Med Biol. 2004;31:1061. doi: 10.1016/j.nucmedbio.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 82.Talanov VS. Garmestani K. Regino CAS, et al. Preparation and in vivo evaluation of a novel stabilized linker for 211At labeling of protein. Nucl Med Biol. 2006;33:469. doi: 10.1016/j.nucmedbio.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 83.Yordanov AT. Garmestani K. Zhang M, et al. Preparation and in vivo evaluation of linkers for 211At labeling of humanized anti-Tac. Nucl Med Biol. 2001;28:845. doi: 10.1016/s0969-8051(01)00257-8. [DOI] [PubMed] [Google Scholar]
- 84.Wilbur DS. Chyan M-K. Hamlin DK, et al. Reagents for astatination of biomolecules. 2. Conjugation of anionic boron cage pendant groups to a protein provides a method for direct labeling that is stable to in vivo deastatination. Bioconjug Chem. 2007;18:1226. doi: 10.1021/bc060345s. [DOI] [PubMed] [Google Scholar]
- 85.Wilbur DS. Chyan M-K. Hamlin DK, et al. Preparation and in vivo evaluation of radioiodinated closo-decaborate(2-) derivatives to identify structural components that provide low retention in tissues. Nucl Med Biol. 2010;37:167. doi: 10.1016/j.nucmedbio.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilbur DS. Chyan M-K. Hamlin DK, et al. Reagents for astatination of biomolecules. 5. Evaluation of hydrazone linkers in 211At- and 125I-labeled closo-decaborate(2-) conjugates of fab′ as a means of decreasing kidney retention. Bioconjug Chem. 2011;22:1089. doi: 10.1021/bc1005625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vaidyanathan G. Affleck D. Welsh P, et al. Radioiodination and astatination of octreotide by conjugation labeling. Nucl Med Biol. 2000;27:329. doi: 10.1016/s0969-8051(00)00098-6. [DOI] [PubMed] [Google Scholar]
- 88.Vaidyanathan G. Boskovitz A. Shankar S, et al. Radioiodine and 211At-labeled guanidinomethyl halobenzoyl octreotate conjugates: Potential peptide radiotherapeutics for somatostatin receptor-positive cancers. Peptides. 2004;25:2087. doi: 10.1016/j.peptides.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 89.Vaidyanathan G. Affleck DJ. Schottelius M, et al. Synthesis and evaluation of glycosylated octreotate analogues labeled with radioiodine and 211At via a tin precursor. Bioconjug Chem. 2006;17:195. doi: 10.1021/bc0502560. [DOI] [PubMed] [Google Scholar]
- 90.Lesch HP. Kaikkonen MU. Pikkarainen JT, et al. Avidin-biotin technology in targeted therapy. Expert Opin Drug Deliv. 2010;7:551. doi: 10.1517/17425241003677749. [DOI] [PubMed] [Google Scholar]
- 91.Foulon CF. Schoultz BW. Zalutsky MR. Preparation and biological evaluation of an astatine-211 labeled biotin conjugate: Biotinyl-3-[211At]astatoanilide. Nucl Med Biol. 1997;24:135. doi: 10.1016/s0969-8051(96)00185-0. [DOI] [PubMed] [Google Scholar]
- 92.Foulon CF. Alston KL. Zalutsky MR. Astatine-211-labeled biotin conjugates resistant to biotinidase for use in pretargeted radioimmunotherapy. Nucl Med Biol. 1998;25:81. doi: 10.1016/s0969-8051(97)00166-2. [DOI] [PubMed] [Google Scholar]
- 93.Wilbur DS. Hamlin DK. Chyan M-K, et al. Biotin reagents in antibody pretargeting. 6. Synthesis and in vivo evaluation of astatinated and radioiodinated aryl- and nido-carboranyl-biotin derivatives. Bioconjug Chem. 2004;15:601. doi: 10.1021/bc034229q. [DOI] [PubMed] [Google Scholar]
- 94.Wilbur DS. Chyan M-K. Hamlin DK, et al. Reagents for astatination of biomolecules. 3. Comparison of closo-decaborate(2-) and closo-dodecaborate(2-) moieties as reactive groups for labeling with astatine-211. Bioconjug Chem. 2009;20:591. doi: 10.1021/bc800515d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lindegren S. Andersson H. Jacobsson L, et al. Synthesis and biodistribution of 211At-labeled, biotinylated, and charge-modified poly-L-lysine: Evaluation for use as an effector molecule in pretargeted intraperitoneal tumor therapy. Bioconjug Chem. 2002;13:502. doi: 10.1021/bc010054d. [DOI] [PubMed] [Google Scholar]
- 96.Lindegren S. Karlsson B. Jacobsson L, et al. (211)At-labeled and biotinylated effector molecules for pretargeted radioimmunotherapy using poly-L- and poly-D-lysine as multicarriers. Clin Cancer Res. 2003;9:3873S. [PubMed] [Google Scholar]
- 97.Frost SHL. Bäck T. Chouin N, et al. In vivo distribution of avidin-conjugated MX35 and 211At-labeled, biotinylated poly-L-lysine for pretargeted intraperitoneal α-radioimmunotherapy. Cancer Biother Radiopharm. 2011;26:727. doi: 10.1089/cbr.2011.1046. [DOI] [PubMed] [Google Scholar]
- 98.Larsen RH. Murud KM. Akabani G, et al. 211At- and 131I-labeled bisphosphonates with high in vivo stability and bone accumulation. J Nucl Med. 1999;40:1197. [PubMed] [Google Scholar]
- 99.Vaidyanathan G. Zalutsky MR. 1-(m-[211At]astatobenzyl)guanidine: Synthesis via astato demetalation and preliminary in vitro and in vivo evaluation. Bioconjug Chem. 1992;3:499. doi: 10.1021/bc00018a006. [DOI] [PubMed] [Google Scholar]
- 100.Larsen RH. Hassfjell SP. Hoff P, et al. 211-At-labelling of polymer particles for radiotherapy: Synthesis, purification and stability. J Label Compd Radiopharm. 1993;33:977. [Google Scholar]
- 101.Larsen RH. Hoff P. Vergote IB, et al. Alpha-particle radiotherapy with 211At-labeled monodisperse polymer particles, 211At-labeled IgG proteins, and free 211At in a murine intraperitoneal tumor model. Gynecol Oncol. 1995;57:9. doi: 10.1006/gyno.1995.1093. [DOI] [PubMed] [Google Scholar]
- 102.Kucka J. Hrubý M. Konák C, et al. Astatination of nanoparticles containing silver as possible carriers of 211At. Appl Radiat Isot. 2006;64:201. doi: 10.1016/j.apradiso.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 103.Hartman KB. Hamlin DK. Wilbur DS, et al. 211AtCl@US-tube nanocapsules: A new concept in radiotherapeutic-agent design. Small. 2007;3:1496. doi: 10.1002/smll.200700153. [DOI] [PubMed] [Google Scholar]
- 104.Larsen RH. Akabani G. Welsh P, et al. The cytotoxicity and microdosimetry of astatine-211-labeled chimeric monoclonal antibodies in human glioma and melanoma cells in vitro. Radiat Res. 1998;149:155. [PubMed] [Google Scholar]
- 105.Akabani G. Carlin S. Welsh P, et al. In vitro cytotoxicity of 211At-labeled trastuzumab in human breast cancer cell lines: Effect of specific activity and HER2 receptor heterogeneity on survival fraction. Nucl Med Biol. 2006;33:333. doi: 10.1016/j.nucmedbio.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 106.Aurlien E. Kvinnsland Y. Larsen RH, et al. Radiation doses to non-Hodgkin's lymphoma cells and normal bone marrow exposed in vitro. Comparison of an alpha-emitting radioimmunoconjugate and external gamma-irradiation. Int J Radiat Biol. 2002;78:133. doi: 10.1080/09553000110094788. [DOI] [PubMed] [Google Scholar]
- 107.Petrich T. Korkmaz Z. Krull D, et al. In vitro experimental 211At-anti-CD33 antibody therapy of leukaemia cells overcomes cellular resistance seen in vivo against gemtuzumab ozogamicin. Eur J Nucl Med Mol Imaging. 2010;37:851. doi: 10.1007/s00259-009-1356-x. [DOI] [PubMed] [Google Scholar]
- 108.Nestor M. Sundström M. Anniko M, et al. Effect of cetuximab in combination with alpha-radioimmunotherapy in cultured squamous cell carcinomas. Nucl Med Biol. 2011;38:103. doi: 10.1016/j.nucmedbio.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 109.Orlova A. Sjöstrom A. Lebeda O, et al. Targeting against epidermal growth factor receptors. Cellular processing of astatinated EGF after binding to cultured carcinoma cells. Anticancer Res. 2004;24:4035. [PubMed] [Google Scholar]
- 110.Claesson AK. Stenerlöw B. Jacobsson L, et al. Relative biological effectiveness of the alpha-particle emitter (211)At for double-strand break induction in human fibroblasts. Radiat Res. 2007;167:312. doi: 10.1667/RR0668.1. [DOI] [PubMed] [Google Scholar]
- 111.Claesson K. Magnander K. Kahu H, et al. RBE of α-particles from 211At for complex DNA damage and cell survival in relation to cell cycle position. Int J Rad Biol. 2011;87:372. doi: 10.3109/09553002.2011.538127. [DOI] [PubMed] [Google Scholar]
- 112.Zhang M. Yao Z. Zhang Z, et al. The anti-CD25 monoclonal antibody 7G7/B6, armed with the α-emitter 211At, provides effective radioimmunotherapy for a murine model of leukemia. Cancer Res. 2006;66:8227. doi: 10.1158/0008-5472.CAN-06-1189. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Z. Zhang M. Garmestani K, et al. Effective treatment of a murine model of adult T-cell leukemia using 211At-7G7/B6 and its combination with unmodified anti-Tac (daclizumab) directed toward CD25. Blood. 2006;108:1007. doi: 10.1182/blood-2005-11-4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang M. Yao Z. Patel H, et al. Effective therapy of murine models of human leukemia and lymphoma with radiolabeled anti-CD30 antibody, HeFi-1. Proc Natl Acad Sci U S A. 2007;104:8444. doi: 10.1073/pnas.0702496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng J. Ekberg T. Engström M, et al. Radioimmunotherapy with astatine-211 using chimeric monoclonal antibody U36 in head and neck squamous cell carcinoma. Laryngoscope. 2007;117:1013. doi: 10.1097/MLG.0b013e31804b1a6d. [DOI] [PubMed] [Google Scholar]
- 116.Robinson MK. Shaller C. Garmestani K, et al. Effective treatment of established human breast tumor xenografts in immunodeficient mice with a single dose of the α-emitting radioisotope astatine-211 conjugated to anti-HER2/neu diabodies. Clin Cancer Res. 2008;14:875. doi: 10.1158/1078-0432.CCR-07-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Elgqvist J. Andersson H. Bernhardt P, et al. Administered activity and metastatic cure probability during radioimmunotherapy of ovarian cancer in nude mice with 211At-MX35 F(ab′)2. Int J Radiat Oncol Biol Phys. 2006;66:1228. doi: 10.1016/j.ijrobp.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 118.Elgqvist J. Andersson H. Back T, et al. {alpha}-radioimmunotherapy of intraperitoneally growing OVCAR-3 tumors of variable dimensions: Outcome related to measured tumor size and mean absorbed dose. J Nucl Med. 2006;47:1342. [PubMed] [Google Scholar]
- 119.Elgqvist J. Andersson H. Bäck T, et al. Fractionated radioimmunotherapy of intraperitoneally growing ovarian cancer in nude mice with 211At-MX35 F(ab′)2: Therapeutic efficacy and myelotoxicity. Nucl Med Biol. 2006;33:1065. doi: 10.1016/j.nucmedbio.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 120.Elgqvist J. Andersson H. Jensen H, et al. Repeated intraperitoneal α-radioimmunotherapy of ovarian cancer in mice. J Oncol. 2010;2010:1. doi: 10.1155/2010/394913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gustafsson AME. Bäck T. Elgqvist J, et al. Comparison of therapeutic efficacy and biodistribution of 213Bi- and 211At-labeled monoclonal antibody MX35 in an ovarian cancer model. Nucl Med Biol. 2012;39:15. doi: 10.1016/j.nucmedbio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 122.Zalutsky MR. McLendon RE. Garg PK, et al. Radioimmunotherapy of neoplastic meningitis in rats using an alpha-particle-emitting immunoconjugate. Cancer Res. 1994;54:4719. [PubMed] [Google Scholar]
- 123.Zalutsky MR. Stabin MG. Larsen RH, et al. Tissue distribution and radiation dosimetry of astatine-211-labeled chimeric 81C6, an α-particle-emitting immunoconjugate. Nucl Med Biol. 1997;24:255. doi: 10.1016/s0969-8051(97)00060-7. [DOI] [PubMed] [Google Scholar]
- 124.Palm S. Back T. Claesson I, et al. Therapeutic efficacy of astatine-211–labeled trastuzumab on radioresistant SKOV-3 tumors in nude mice. Int J Radiat Oncol Biol Phys. 2007;69:572. doi: 10.1016/j.ijrobp.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 125.Boskovitz A. McLendon RE. Okamura T, et al. Treatment of HER2-positive breast carcinomatous meningitis with intrathecal administration of [alpha]-particle-emitting 211At-labeled trastuzumab. Nucl Med Biol. 2009;36:659. doi: 10.1016/j.nucmedbio.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Andersson H. Cederkrantz E. Bäck T, et al. Intraperitoneal {alpha}-particle radioimmunotherapy of ovarian cancer patients: Pharmacokinetics and dosimetry of 211At-MX35 F(ab′)2-a phase I study. J Nucl Med. 2009;50:1153. doi: 10.2967/jnumed.109.062604. [DOI] [PubMed] [Google Scholar]
- 127.Hamilton JG. Soley MH. A comparison of the metabolism of iodine and of element 85 (eka-iodine) Proc Natl Acad Sci U S A. 1940;26:483. doi: 10.1073/pnas.26.8.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wilbur DS. Thakar MS. Hamlin DK, et al. Reagents for astatination of biomolecules. 4. Comparison of maleimido-closo-decaborate(2-) and meta-[211At]astatobenzoate conjugates for labeling anti-CD45 antibodies with [211At]astatine. Bioconjug Chem. 2009;20:1983. doi: 10.1021/bc9000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guerard F. Rajerison H. Faivre-Chauvet A, et al. Radiolabelling of proteins with stabilised hypervalent astatine-211: Feasability and stability. J Nucl Med. 2011;52:1486. [Google Scholar]
- 130.Haddad F. Barbet J. Chatal JF. The ARRONAX project. Current Radiopharm. 2011;4:186. doi: 10.2174/1874471011104030186. [DOI] [PubMed] [Google Scholar]