

Abstract
Triplex-forming oligonucleotides (TFOs) can bind to the major groove of homopurine-homopyrimidine stretches of double-stranded DNA in a sequence-specific manner through Hoogsteen hydrogen bonding to form DNA triplexes. TFOs by themselves or conjugated to reactive molecules can be used to direct sequence-specific DNA damage, which in turn results in the induction of several DNA metabolic activities. Triplex technology is highly utilized as a tool to study gene regulation, molecular mechanisms of DNA repair, recombination, and mutagenesis. In addition, TFO targeting of specific genes has been exploited in the development of therapeutic strategies to modulate DNA structure and function. In this review, we discuss advances made in studies of DNA damage, DNA repair, recombination, and mutagenesis by using triplex technology to target specific DNA sequences.
Keywords: Triplex-forming oligonucleotides, DNA damage, DNA repair, triplex technology, recombination, mutagenesis, DNA structure
Introduction
In 1953 Watson and Crick described the double-helical structure of DNA [1]. Four years later it was shown by Felsenfeld and Rich (1957) that nucleic acids can also interact to form triple helical (or triplex) structures. Their RNA diffraction studies revealed that a stable complex of poly(U) (polyuridylate) and poly(A) (polyadenylate) strands was formed in a 2:1 ratio in the presence of Mg2+ ions [2]. However, the advent of “triplex technology” occurred nearly thirty years later once the synthesis of mixed sequence oligonucleotides was more practical. In the late 1980s, it was demonstrated that short (typically 15 to 30 bp), synthetic, triplex-forming oligonucleotides (TFOs) could bind through the major groove of duplex DNA to form stable triple helical structures [3, 4]. The interaction between a TFO and its target duplex DNA is sequence specific in nature [5] and is derived from the ability of polypyrimidine TFOs to bind to polypurine stretches of B-DNA (C+•GC or T•AT base triplets, where C+ indicates a protonated cytosine) in a parallel orientation (5′→3′ direction with respect to the purine-rich strand of the underlying duplex) through Hoogsteen hydrogen bonding [6]. Similarly, polypurine TFOs can bind to polypurine stretches of B-DNA (G•GC or A•AT base triplets) through reverse Hoogsteen hydrogen bonding in an anti-parallel orientation (3′→5′ direction with respect to the purine-rich strand of the underlying duplex) (Fig. 1). The Hoogsteen hydrogen bonds afford specificity and affinity of the TFO for its target duplex. Thermodynamically, TFO base pairing with duplex DNA is similar to the binding energy of Watson-Crick base pairing (∼0.22-0.64 kcal/mole less per base triplet for Hoogsteen hydrogen bonds compared to a Watson-Crick base pair). TFO binding is dependent on a number of factors, including ionic conditions, base composition, and the sequence context of the TFO-binding site [7]. For example, melting temperature studies indicated that the presence of GC repeats flanking the TFO-binding site increased triplex stability [8]. Physical studies of triplex structures revealed that binding of TFOs to target DNA induced significant structural distortions in the underlying duplex [9-15]. X-ray diffraction studies [16], nuclear magnetic resonance studies [17] and studies using chemical probing of triplex structure [5] provided evidence of helical distortions induced in the DNA upon triplex formation [18]. X-ray crystallographic studies on triplex structure revealed changes in the phosphate backbone torsion angles on the purine-rich strand. It was also shown that the duplex-triplex junction had lower twists and higher openings, indicating partial unwinding of the DNA, and that the presence of protonated cytosine (C+) caused an additional narrowing of the purine-rich and Hoogsteen strands [15]. Such changes in the helical parameters of DNA after formation of triplexes in vivo likely mediate induction of several cellular DNA metabolic events, such as repair or recombination. These features, along with facile chemistries to conjugate DNA damaging agents to TFOs, make triplexes useful probes to study the structural basis of DNA damage recognition and processing. In addition to their use as probes to study the structural basis of damage recognition, the repair processing of triplexes is of interest because of the expanding use of TFOs to modify mammalian genomes in vivo.
Figure 1. Model of a psoralen-modified TFO bound to duplex DNA.
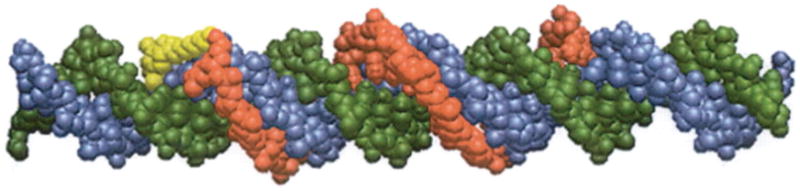
Space filling model of a 19-mer psoralen-modified TFO bound to its target 38 bp duplex. The TFO is shown in red, the psoralen derivative is in yellow, the pyrimidine-rich strand of the target duplex is in green, and the purine-rich strand in blue. The TFO is bound to the target purine-rich strand (blue) in an anti-parallel fashion via reverse Hoogsteen hydrogen bonding in the major groove. Reprinted with permission from Vasquez et al., Biochemistry, 35, 10712-10719, Copyright 1996 American Chemical Society.
Because TFOs bind to their target duplex DNA sequences specifically and with high affinity, they make useful tools for targeted genome modifications. Some of the initial demonstrations of sequence-specific modification of DNA by chemically-modified TFOs were shown through site-specific photo-crosslinking with azidoproflavine derivatives attached to TFOs [19], and targeted cleavage of duplex DNA via an artificial photo-endonuclease attached to the 3′-phosphate of a TFO [20]. Subsequently, it was demonstrated that TFOs covalently linked to a photoactivatble crosslinking agent, psoralen, can be used in a sequence-specific manner to induce DNA damage through the formation of DNA interstrand crosslinks (ICLs) in mammalian genes [21, 22]. Through the use of TFO-directed ICLs, DNA damage recognition and repair related processes and targeted mutagenesis have been extensively studied (reviewed in [23, 24]). Antigene strategies based on triplex formation via inhibition of transcription was one of the earliest [4], and continues to be an important biological application of TFOs. In the past two decades triplex technology has been used to study mechanisms of DNA damage and repair, mutation, recombination, and gene targeting. This review will focus primarily on the involvement of triplex technology in the advancement of our understanding of the mechanisms of DNA repair, recombination, and mutagenesis in mammalian cells.
TFO target sequences are abundant in mammalian genomes
In order to maximize the utility of triplex technology for either mechanistic studies or for therapeutic application, purine-rich stretches in the DNA target sites must be available to allow for stable triplex formation. Further, to allow for TFO binding to unique sites, a purine-rich stretch of 15 bases or more is desired. To predict the number of such TFO target sequences (TTS) in the human and mouse genomes, Gaddis et al. [25] developed an algorithm to search both strands of the entire human (NCBI Human Genome; build 35.1; 2004) and mouse genomes (NCBI Mouse Genome; build 33; 2004) for potential TFO target sites. Potential TTS were defined as purine stretches of a minimum of 15 bases, with at least 50% G with one pyrimidine interruption allowed within the purine-rich site. Sites with more than 50% simple repeat sequences (e.g. G, A, GA, GAA, GGA) or sites within known repeat sequence elements in the RepBase update database (Volume 10) were eliminated, as were sites that were found more than 10 times in the dataset. This search engine is available at http://spi.mdanderson.org/tfo, where the user can insert sequences of interest to identify potential TTS. The results predicted that ∼98% of annotated human genes have at least one TTS within the promoter/transcribed region, and ∼87% of the identified TTS are unique to one gene. Similarly, ∼93% of the known protein coding genes of the mouse genome have at least one potential TTS with ∼83% of the identified TTS unique to one gene. When two human and twelve mouse genes predicted to have TTS by this search engine were tested for their binding affinities to synthesized target duplexes in vitro, specific, high affinity binding was detected with apparent dissociation constants between 1 × 10−9 and 1 × 10−7 M, as predicted [26].
In 2009, Jenjaroenpun and Kuznetsov [27] developed a database to predict and annotate probable TTS, G-quadruplex motifs, and other regulatory DNA elements in the human genome. This search engine is available for public access at http://ggeda.bii.a-star.edu.sg/∼piroonj/TTS_mapping/TTS_mapping.php. Existing TTS models predict from the human genome sequence that TTS are frequently present in gene regulatory regions [25, 27-29]. In an attempt to understand why TTS are overrepresented in mammalian genomes, Goñi et al. (2006) predicted that the curved and rigid structure of TTS may confer unique physical properties to the promoter region that allow for enhanced DNA-protein interactions [29]. Nevertheless, these preliminary predictions suggest tremendous potential for modulating gene function for a large number of genes in the human genome through the proper utilization of triplex technology. TFO binding in the major groove of the duplex DNA causes local distortions in the helix, which can initiate DNA repair events at the TFO targeted site. Similarly, TFOs associated with reactive molecules can initiate DNA repair activities once bound to the duplex DNA. However, there are several barriers to successful association of TFOs to their cognate binding sites particularly in vivo. Much research has been done to chemically modify TFOs to overcome these barriers. Some of these limitations and potential solutions to expand the utility of triplex technology via TFO modification are discussed in the following section.
Modifications of TFOs to increase triplex formation and stability
There are several potential limitations to the efficacy of triplex technology, particularly in a cellular environment. Efficient uptake of TFOs into the nucleus, stability once inside the cell, and specific, high-affinity binding in a chromosomal context must be achieved for successful triplex formation in vivo. Pyrimidine TFOs which form triplexes in the parallel motif require a protonated cytosine (C+:GC) for Hoogsteen hydrogen bond formation, and are therefore pH sensitive [30]. Purine TFOs form stable triplexes in the anti-parallel motif in a pH-independent fashion; however, these G-rich oligonucleotides tend to form alternative structures, such as G-quartets under physiological K+ concentrations [31-34]. Stable triplex formation also requires a contiguous homopurine run, as pyrimidine interruptions within the purine stretch can inhibit triplex formation [35-37]. Moreover, in an intracellular environment, charge repulsion between the three polyanionic strands and accessibility of binding sites in a chromosomal context can create obstacles to stable triplex formation [38].
Significant progress has been made in developing chemically modified TFOs to improve triplex stability (for reviews see Jain et al, Biochimie, 2008 [39]; and Fox & Brown, Quart. Rev. Biophys., 2005 [40]). For example, replacement of cytosine by 5-methylcytosine abolishes the pH dependency of pyrimidine-rich TFOs [41, 42]. The use of 2′-deoxy-6-thioguanosine or 7-deaza-2′-deoxyxanthosine in G-rich TFOs inhibits secondary structure formation [31, 33, 34]. Introduction of W-shaped nucleoside analogs (WNA) decreases steric repulsion and therefore increases the stability of purine-rich TFOs, particularly at sites of pyrimidine interruptions in the polypurine target site [43-45]. Similarly, modifications of thymidine to 2′ deoxyuridine (dU), 5-propargylamino-dU and 2′-aminoethoxy, 5-propargylamino-dU bis-substituted derivatives have been shown to increase triplex stability [8, 46-49]. Recently, Semenyuk et al. [50] characterized several pyrmidine motif TFOs with various analogues for binding to a target sequence containing a CG inversion. They found that N4-alkyl-5-methylcytosine derivatives improved binding at the target site, and that a TFO containing an N4-(2-guanidoethyl)-5-methylcytosine at the inversion site was active in a cell-based gene targeting assay [50]. Such studies using base analogues to improve the binding and bioactivity of TFOs at base interruptions will contribute toward the successful expansion of available triplex targets in cells.
TFOs modified with a 3′ propylamine group show reduced nuclease mediated degradation in vivo [51]. Backbone modifications such as replacement of the phosphodiester linkages of natural DNA with positively charged, RNAase H-insensitive phosphoramidate linkages or morpholino oligonucleotides can increase triplex stability under conditions of neutral pH [52-55]. Though these morpholino oligonucleotides are nuclease resistant and form triplexes under physiological K+ concentrations, there are some limitations associated with their application; e.g., triplex formation with morpholino backbone-modified TFOs is preferred in the pyrimidine motif [56]. Triplex-forming peptide nucleic acids (PNAs) represent a backbone modification in which the bases are attached to a polyamide backbone. PNAs can be used to form highly stable triplexes [57, 58] with nuclease and protease resistance [59]. Important sugar modifications include 2′-O-methyl ribonucleotides [60, 61] and 2′-O-aminoethylribose [62-64], which form RNAase H-resistant, stable triplexes with lower levels of structural distortion in the duplex DNA compared to unmodified TFO-derived triplexes [65]. Locked nucleic acids (LNA) contain 2′-O, 4′-C-methylene-linked ribonucleotides, which result in a locked C3′-endo sugar conformation. This modification reduces the flexibility of the ribose and allows for stable triplex formation (reviewed by Peterson and Wengel, 2003 [66]). Importantly, extensive sugar modification has been shown to decrease the biological activity of TFOs in vivo [67]. Thus, chemical modifications of TFOs should be considered based upon the specific application. Table 1 summarizes some of the improvements in TFO binding through the use of such modifications. Clearly, continued advances in chemical modifications to oligonucleotides are essential for the successful application of TFOs in vivo, but comprehensive treatment of this subject is beyond the scope of this review. For recent and more comprehensive reviews on chemical modifications of oligonucleotides, please see the following [68, 69].
Table 1. Chemical modifications of TFOs.
| Position | Modifications | Improvements | Reference number |
|---|---|---|---|
| Nucleobase | Replacement of C1 by 5-MeC2 | Alleviates pH dependency of Pyrich3 TFOs | [41] |
| WNA4 | Decrease steric repulsion of Purich5 TFOs, stabilization against TA/GC interruptions | [43-45] | |
| 2′-dU6 | Enhance thermal stability and binding affinity | [46] | |
| 5-AdU7 | Enhance stability and binding affinity at neutral pH | [47] | |
| 2′-AE7,8 | Enhance thermal stability | [48] | |
|
| |||
| Sugar | 2′-O-Me-RNA | Enhance thermal stability | [60] |
| 2′-AE ribose | Reduce charge repulsion, increase in association rate at physiological pH, nuclease resistant | [63, 64] | |
| LNA9 | Enhance thermal stability at neutral pH, decrease in dissociation rate | [66, 156] | |
|
| |||
| Backbone | Phosphoramidate linkages | Triplex stability in the absence of Mg2+ ions at neutral pH, decrease in dissociation rate, RNase H resistant | [52, 54] |
| (Morpholino oligonucleotides) | Higher melting temperature, increase in association rate, RNase H resistant | [53] | |
| Phosphorothioates | Nuclease resistant, increased half-life in cells | [55, 157, 158] | |
| PNA | Higher stability, nuclease and protease resistant | [57, 59, 159] | |
Cytosine;
5 methyl-cytosine;
Pyrimidine-rich;
W-shaped nucleoside analogs;
Purine-rich;
C-5 propyne analogs of 2′-deoxyuridine;
5-aminopropargyl-dU;
aminoethoxy;
Locked nucleic acid;
Peptide Nucleic Acid
Triplex technology in studies of DNA damage and repair
TFOs conjugated to DNA damaging agents (as well as unmodified TFOs) have been successfully used as tools to study mechanisms of DNA repair. An early example of such a TFO conjugate was provided by Povsic and Dervan [70] where TFOs were conjugated at the 5′ end to an electrophile, which was shown to alkylate a guanine base adjacent to the TFO target site. Another example of triplex-directed DNA alkylation damage includes TFOs conjugated to aryl nitrogen mustards which can specifically alkylate strands of the target duplex DNA [71]. Other well studied TFO-DNA damaging agent conjugates include phenylsulfoxide derivatives of 2-amino-6-vinylpurine [72], and DNA crosslinking agents such as psoralen [21, 22, 73]. Psoralen is a photoreactive DNA crosslinking agent that has been extensively employed in triplex technology to induce site-specific DNA damage. The repair of psoralen DNA ICLs has been recently reviewed by Vasquez (2010) [23]. TFOs conjugated to reactive molecules that introduce DNA strand breaks are also of great interest. Panyutin and Neumann (1994) showed that the decay of 125I labeled TFOs caused DNA double-strand breaks (DSBs) within 10 bp of the position of the 125I deoxycytosine residue in the TFOs [74]. In a subsequent study, it was shown that their repair was mutagenic in a cellular context [75]. TFOs attached to semi-synthetic nucleases [76], to EDTA•Fe [3], and to restriction endonucleases, which function in a Mg2+-dependent manner [77] efficiently induce DSBs but lack efficient control over the timing of DNA damage induction. This can be overcome by covalently linking photoactivatable derivatives to TFOs, so that the timing of damage can be controlled. For example, ellipticine, quinacridine, and pyrene derivatives linked to TFOs can be activated upon irradiation, resulting in cleavage on both strands of the target duplex [20, 78, 79]. In the sections below we discuss the use of unmodified TFOs to induce DNA damage and repair, followed by studies of DNA damage and repair induced by TFOs covalently linked to specific DNA damaging agents.
Induction of DNA repair by TFOs
The ability of TFOs to bind to undamaged DNA and stimulate DNA repair was demonstrated by initiation of repair synthesis on a TFO-bound plasmid in HeLa cell-free extracts [80]. Following incubation of the TFOs with the plasmid in HeLa cell-free extracts, increased α-32P dCTP incorporation was detected compared to a control plasmid (that did not contain a TFO binding site), indicating TFO-induced DNA repair synthesis [80]. Higher rates of TFO-induced repair synthesis correlated with higher levels of mutagenesis, which indicated that the processing of triplexes was, at least in part, error-prone. In order to determine which DNA repair pathway(s) was involved, the experiment was repeated using XPA-deficient cells, which are deficient in the nucleotide excision repair (NER) mechanism. The results indicated no significant increase in mutagenesis in the absence of functional NER, suggesting involvement of the NER pathway in the mutagenic or error-prone repair of the triplex structure [80]. The ability of TFOs to induce mutations at specific sites has important implications for triplex-mediated therapeutic approaches. These findings are also consistent with the ability of endogenous intra-molecular triplex structures (i.e. H-DNA) to induce mutagenesis in vivo [81-85].
The role of transcription-coupled NER (TC-NER) in triplex processing was studied on plasmids in HeLa nuclear extracts, where the triplex structure was used as a transcription inhibitor [86]. The results indicated a strong correlation between the presence of a promoter and the level of induction of transcription-related repair in the presence of triplex structures. Subsequently, when individual transcription components TFIIH, TFIID, and RNA Pol II were immuno-depleted in HeLa nuclear extracts, inhibition of the repair was evident, suggesting that the triplex-induced repair synthesis was indeed related to transcription. The ability to repair triplex structures was altered in nuclear extracts of Cockayne's syndrome A cells (which are defective in TC-NER), XPC cells (defective in global genome-NER (GG-NER)), and XPA cells (defective in both GG-NER and TC-NER), indicating that GG-NER and TC-NER are both involved in the repair of triplex structures [86]. These studies, and others highlight the potential of triplex technology, in the absence of covalent modifications to the genome, to stimulate DNA repair in cells for use in gene therapeutic applications.
DNA repair stimulated by TFOs conjugated to DNA damaging agents
The conjugation of TFOs to psoralen derivatives presents a powerful approach to modify DNA in vitro and in cells. When irradiated with UVA light (365 nm), psoralen forms monoadducts and covalent ICLs preferably at 5′-TpA-3′ sites. Such ICLs are processed by the UvrABC nuclease in E. coli. This endonuclease system also identifies and processes triplex structures that are associated with DNA ICLs. There is evidence to suggest that psoralen ICLs directed by pyrimidine TFOs are processed differently by UvrABC compared to those directed by purine TFOs. Duval-Valentin et al. (1998), reported that a 14-mer pyrimidine, 3′-psoralen and a 5′-acridine conjugated TFO when photo-crosslinked to its target sites was identified by the UvrABC excinuclease, but in vitro excision experiments indicated that the presence of the triplex structure was inhibitory to the excision process [87]. On the other hand, Christensen et al., (2008) demonstrated that psoralen ICLs directed by purine-rich TFOs were efficiently processed by the UvrABC excinuclease. The purine TFO was displaced by the UvrA and UvrB subunits, and the psoralen ICLs were incised efficiently in a UvrA-concentration dependent manner [88]. The apparent differences in the processing of ICLs targeted by pyrimidine-rich TFOs versus purine-rich TFOs should be further explored to establish the biological relevance of these observations.
Plasmid-based studies of TFO-directed psoralen adducts in S. cerevisiae indicated that efficient repair depends on the nature of the lesion formed; monoadducts were repaired more efficiently than ICLs, as expected [89]. In addition, the introduction of ICLs resulted in frequent mutations, with A→T, T→A and T→G transversions and multiple insertions at the targeted site. Analysis of mutations in NER, error-prone repair (EPR) or recombinational repair-deficient yeast strains indicated that either NER and EPR together or EPR alone could be responsible for generating the mutations. When the psoralen-modified TFO was bound to a target site in the yeast URA3 gene, the furan-sided adducts and the pyrone-sided adducts produced by psoralen plus UVA were not processed in a similar manner [90]. Bypass of these adducts and/or error-generating repair led to T→G substitutions or G insertions for furan-sided adducts, while processing of pyrone-sided adducts resulted in T→A or T→C substitutions or insertions of A, indicating that the enzymes responsible for lesion bypass may recognize structural differences of the lesions.
To study the effect of the third strand in the processing of TFO-directed psoralen ICLs, plasmid substrates were designed to contain a single psoralen ICL in the presence or absence of an adjacent TFO [91]. When substrates containing psoralen ICLs were incubated in HeLa cell extracts an ∼5-fold increase in DNA synthesis (i.e. repair synthesis) was observed compared to the background level. However, in the presence of a triplex structure adjacent to the crosslink, the repair synthesis was decreased by 30-40%, suggesting that repair of psoralen ICLs is modulated in the presence of an adjacent triple-stranded DNA structure [91]. Similar repair studies in human HEC59 cancer cell extracts deficient in the DNA mismatch repair (MMR) protein MSH2 resulted in ∼50% reduction of repair synthesis, indicating that MSH2 is an important component in the efficient processing of TFO-induced psoralen ICLs in mammalian cells [92]. TFO-directed ICL-induced mutagenesis studies in the same MSH2 proficient and deficient human cells indicated that MMR was not required for the mutagenic repair of ICLs, and thus is likely involved in error-free processing of these lesions, perhaps through a pathway involving homologous recombination during S-phase [92].
A psoralen-conjugated pyrimidine TFO containing 2′-O-(2-aminoethyl) residues was used to target mutations to a site in the hamster HPRT in Chinese hamster ovary (CHO) cells [93]. The TFO-directed ICLs led to mutations, predominantly in the form of base substitutions and deletions at the targeted site. Studies in repair-deficient mammalian cell lines indicated that TC-NER was not essential for the base substitutions or deletions, nor was MMR [94], consistent with observations from studies in MSH2-deficient human cells described above [92]. The NER complex, ERCC1-XPF, was involved in generating base substitutions, but was not required for the deletion events. Cells that were engineered to block polymerase ζ expression showed ∼3-fold fewer base substitutions compared to wild-type cells, indicating that these base substitutions could result from lesion bypass by the error-prone polymerase ζ [94].
Using a PNA clamp to form a PNA:DNA:PNA triplex to stimulate DNA repair in HeLa cell-free extracts, Rogers et al. (2002) found that the repair synthesis induced by the PNA triplex was significantly higher than that of a DNA triplex at the same site [95]. The increased repair stimulated by the PNA triplex relative to a DNA triplex may be due to greater helical distortions created by PNAs and/or the differences in the chemical composition of the two types of triplex structures [95]. The repair synthesis stimulated by the PNA-generated triplex was substantially reduced in XPA immune-depleted HeLa cell extracts, implicating NER in the processing of PNA triplex structures [95].
Recombinational repair stimulated by triplex formation
Intermolecular recombination events
Homologous recombination (HR) has been used successfully to modify mammalian genomes for a variety of applications [96-98]. However, there are several limitations associated with the targeted modification of mammalian genomes via HR. One is the low frequency of HR in mammalian cells, and the high frequency of non-targeted integration of transfected DNA [99]. In an effort to overcome these barriers and to enhance targeted recombination frequencies, several strategies have been developed (summarized in Table 2 [100-105]). Among these strategies, the sequence-specific DNA binding by TFOs has provided a tool through which the frequency of targeted HR can be increased. For example, in 1995 Sandor and Bredberg studied the role of HR in the repair of TFO-directed psoralen ICLs and reported that HR can be stimulated by such treatment in human cells [106]. Their studies were carried out using a supF-based mutation/recombination reporter system, which contained a TFO binding sequence at the 3′ end of a mutant supF gene. These shuttle vectors, which can replicate in mammalian and bacterial cells, were treated with psoralen-conjugated TFOs and UVA irradiated to produce site-specific ICLs and were subsequently co-transfected into Jurkat human lymphoblastoid T-cells. A replication-defective plasmid carrying the homologous wild-type supF gene, the donor plasmid, was also transfected into the cells. Thus, correction of mutations in the supF gene was possible through a successful intermolecular recombination event [106]. The results indicated that TFO-directed ICLs induced a relatively low frequency of intermolecular recombination, at a frequency of ∼0.05% compared to a 0.003% spontaneous reversion mutation frequency. The induction of HR by the TFO-directed ICL was ∼40 times lower than that induced by DSBs, an efficient HR substrate [106]. Studies from our laboratory on the role of TFO-directed ICLs in inducing intermolecular HR indicated an ∼6-fold increase in HR frequencies above the spontaneous background levels in mammalian cells, which was relatively high when compared to the ∼12-fold increase in HR caused by the DSBs in this system [107]. The differences in levels of HR stimulation seen in various studies may be due in part to the systems used to analyze the effects of TFO-directed ICLs on HR. For example, Liu et al. (2009) used plasmids containing one copy of the supF reporter gene with either a 4 bp or a 24 bp deletion (as compared to point mutations in the supF gene used in studies by Sandor and Bredberg, 1995) in an SV40 shuttle vector system along with a replication-proficient donor plasmid containing a wild-type supF gene. The recombination spectra consisted largely of gene conversion (GC) events (∼70%), with the frequency of such events being highest with TFO-directed ICL treatment, whereas treatments with the TFO only or a DSB resulted in many illegitimate recombination events [107]. The recombination frequencies were found to be dependent on the size of the deletion in the supF gene, and the polarity of the TFO-directed ICL relative to the target gene [107].
Table 2. Strategies to stimulate targeted HR.
| Strategies | Mechanisms | Limitations | Reference number |
|---|---|---|---|
| Microinjection into nuclei | Homology-directed integration | Cell damage | [105] |
| AAV1 vectors | Homology-directed integration | Significant random integration | [101, 102] |
| Rare cuttingendonucleases(e.g. I-SceI) | Site-directed DNA double-strand breaks | Prior introduction of recognition site | [103] |
| ZFN2 | Site-directed HR | Off target effects not fully characterized | [100] |
| TFO | Site-specific distortion of DNA | Nuclear delivery and accessibility of binding sites in genome | [104] |
| TFO-conjugated to a DNA damaging agent | Site-specific distortion and damage of DNA | In vivo activation of the reactive molecule required | [104] |
| PNA with donor DNA | Site-directed HR | Cellular uptake | [95, 108] |
Adeno-associated virus;
Zinc finger nucleases
PNA triplexes have also been shown to induce site-directed HR. Rogers et al. (2002) demonstrated that a bis-PNA conjugated to a donor DNA molecule induced mutation frequencies at the targeted supF gene ∼5-fold (0.82%) above background, and was found to be more active than its TFO-DNA counterpart [95]. Similar studies using PNA triplexes to correct a splice site mutation, IVS2-1, in the beta-globin gene in mammalian cells demonstrated recombination frequencies ∼3-fold above the background levels [108].
Intramolecular recombination events
Intramolecular recombination can also be stimulated by psoralen-conjugated TFOs. Faruqi et al. (1996) investigated targeted recombination induced by TFO-directed ICL formation in episomal substrates within mammalian COS-7 cells [109]. These studies were carried out using SV40 shuttle vectors with two tandem supF genes containing different point mutations and a TFO-binding site between the gene copies. Correction of the mutations through a successful intramolecular recombination event between the two gene copies was detected as a wild-type blue bacterial colony phenotype. TFO-directed ICLs stimulated recombination to a greater extent than ICLs alone, and a variety of products were generated from gene conversions events and point mutations, suggesting that the damage induced by TFO-directed psoralen ICLs was processed via several mechanisms [109].
A number of parameters involved in triplex-induced intramolecular recombination events, such as the distance between the reporter genes and the orientation of the triplex structure relative to the reporter genes, indicated that the distance between the two mutant reporter genes influenced the intramolecular recombination events, largely via single-strand annealing (SSA), while the position of the reporter genes relative to the ICL did not have a substantial effect on SSA [110]. Using an SV40 shuttle vector that harbored two tandem mutant supF genes, one with small (8 bp) deletion and the other with a larger (24 bp) deletion, Liu et al. (2008) found that when the distance between the two supF genes was less than 60 bp, the recombination events were mostly gene conversions (GC). Interestingly, these GC events had a bias in that nearly every GC event occurred in the supF gene containing the smaller 8 bp deletion [110]. Further experimentation is needed to understand the events that lead to such a preference in copy choice. However, the location of the ICL on the intervening sequence between the two mutant supF genes did not have a significant effect on the HR spectra [110]. In the presence of ICLs, recombination between the two mutant supF genes was dependent on the distance between the two genes. In the presence of TFO-directed psoralen ICLs a distance of 1.4 kb between the two reporter genes stimulated recombination ∼4-fold above the background levels. When this distance was reduced to ∼60 bp, the resulting induction of recombination was reduced to 2-fold above background. The nature of this distance-dependent increase in TFO-directed ICL stimulated recombination was not observed with the triplex structure only (in the absence of the ICL), while the ICL only (in the absence of the TFO) was able to stimulate recombination albeit to a lesser extent than that of the TFO-directed ICL [110].
Triplex formation alone can stimulate recombination; for example, it was found that a TFO targeted to a region between tandem copies of mutated supF genes induced an intramolecular recombination frequency 5-fold above background in COS-7 cells [111]. The TFO-induced recombination was dependent on functional NER, while the recombination stimulated by TFO-directed psoralen ICLs was only partially dependent on the NER pathway [111].
Recombination in a chromosomal context
TFOs have also been used to stimulate recombination in a chromosomal context in cells. TFOs alone or conjugated to psoralen have been designed to target sites within the CHO adenine phosphoribosyltranasferase gene (APRT) specifically and with high affinity [22, 112]. Such high affinity interactions of a specific psoralen-conjugated TFO with the APRT gene stimulated HR ∼5-fold in CHO cells [104]. Knauert et al. (2006) demonstrated that TFOs attached to short single-stranded oligonucleotides (donor molecules) can induce HR in CHO-derived cells at a chromosomal locus [113]. In this study, a mutant firefly luciferase (Fluc) gene containing a TFO-binding site was cloned into a plasmid and transfected into CHO cells. When the cells were treated with TFOs conjugated to donor wild-type DNA, the correction of the luciferase gene was 5 to 9-fold higher (recombination frequencies up to 0.1%) compared to the donor DNA alone, indicating that TFOs can stimulate intermolecular recombination within mammalian cells [113]. The authors extended these studies to examine the ability of phosphoramidate-modified TFOs and 2′-O, 4′-C-methylene LNA-modified TFOs conjugated to homologous donor DNA to induce HR. Previous structural studies by Asensio et al. revealed that upon binding, DNA-based TFOs induce more distortion of the underlying DNA duplex compared to RNA-based TFOs [114]. Consistent with this observation, in this particular study the phosphoramidate-modified TFOs stimulated HR, while the LNA-modified TFOs did not, suggesting that the helical distortion induced by the phosphoramidate TFO was important in stimulating HR in these assays [113].
The ability of TFOs to induce a high frequency of recombination within a chromosomal locus via gene conversion events was demonstrated by Luo et al. (2000) by using microinjection to deliver TFOs and psoralen-conjugated TFOs into the nucleus of mouse derived LTK cell lines [115]. Using the herpes simplex virus thymidine kinase (tk) gene as a marker, two tandem mutant copies of the tk gene were integrated into a single locus in the mouse genome containing a supF reporter gene and a TFO binding site between the copies. Microinjection of the TFOs generated recombinants at a frequency of ∼1%, ∼2,500-fold over the background frequency [115]. Subsequent analysis of the recombinants indicated that they were generated exclusively through gene conversion events. Microinjection of psoralen-conjugated TFOs led to similar recombination frequencies, which were comparable with I-SceI induced DSB-mediated recombination frequencies in mammalian cells [116, 117].
Taken together, these studies indicate that TFOs and TFO-directed psoralen ICLs can be used to stimulate recombination events at specific genomic sites. However, this approach is currently limited because psoralen requires UVA irradiation to form ICLs, and UVA does not penetrate internal organs. Thus, approaches such as the use of laser-induced two-photon excitation, where irradiation at 765 nm is used to photoactivate psoralen, may provide advances for clinical applications [118]. A summary of triplex-mediated induction of recombination in mammalian cells is provided in Table 3.
Table 3. TFO-induced HR in mammalian cells.
| TFO | Type of recombination | Cell line | Reporter gene | Recombination frequency at target site | Mechanism | Ref number |
|---|---|---|---|---|---|---|
| Pso-AG301 | Intra-molecular, extra-chromosomal | COS-7 | supF2 | 0.58% (∼29-fold)3 | GC4 | [109] |
| AG305 | Intra-molecular, extra-chromosomal | COS-7 cells | supF | 0.37% (∼5-fold) | ND6 | [111] |
| AG30, pso-AG30 | Intra-molecular, extra-chromosomal | XP2OS7 | supF | 0.03% | NER-dependent | [111] |
| Pso-AG30 | Intra-chromosomal | LTK− | TK8 | 1.2% (∼3000-fold) | GC | [115] |
| AG30 | Intra-chromosomal | LTK− | TK | 1.0% (∼2500-fold) | GC | [115] |
| AG30 + 51-mer donor DNA | Inter-molecular, chromosomal | CHO-derived | Fluc9 | 0.05% (∼6-fold) | ND | [113] |
| aTC1810 +51-mer donor DNA | Inter-molecular, chromosomal | CHO-derived | Fluc | 0.07 % (∼ 6-fold) | ND | [113] |
| TFO11 | Inter-molecular, extra-chromosomal | Jurkat T lymphoblastoid cells | supF | ∼0.05% (∼ 15-fold) | ND | [106] |
| TFO1 | Intra-chromosomal | CHO-derived | APRT12 | 3-5 fold | ND | [104] |
| pTFO113 | Intra-molecular, extra-chromosomal | HeLa | supF | 0.002% (∼4-fold) | GC | [110] |
| pTFO1 | Inter-molecular, extra-chromosomal | HeLa | supF | 0.0027% (∼6-fold) | ND | [107] |
Psoralen-conjugated, purine TFO;
Bacterial suppressor tRNA;
Fold over background;
Gene conversion;
Purine-rich TFO;
Not determined;
Human XPA deficient cells;
Thymidine kinase;
Firefly luciferase gene;
18-mer, pyrimidine TFO;
22-mer, purine-rich TFO;
Adenine phosphoribosyltransferase;
Psoralen-conjugated, 19-mer, purine-rich TFO conjugated to psoralen
Triplex-stimulated mutagenesis
Mutagenesis in mammalian cells using plasmid-based assays
Another well-studied application of triplex technology is the use of TFOs to induce site-specific mutations to modify or inactivate targeted genes. Initial studies using psoralen-modified TFOs to target a site in a double stranded λ-phage genome indicated that mutagenesis could be stimulated ∼100-fold above the background level by TFO-directed psoralen ICLs [119]. Approximately 56% of the mutations characterized were T:A→A:T transversions at the target site, as expected from error-prone repair of psoralen ICLs. In order to study TFO-directed mutagenesis in mammalian cells, this system was modified such that the supF mutation-reporter gene was incorporated into an SV40-based shuttle vector. This vector was treated with a psoralen-modified TFO and UVA irradiation, and was then transfected into monkey COS-7 cells. After 48 hours of replication and repair the plasmids were isolated and grown in bacterial cells to identify mutations that had occurred in the supF gene. The results indicated that targeted mutagenesis occurred efficiently in mammalian cells, where ∼6% of the plasmids were mutated compared to 0.2% in the bacterial cells [120]. Characterization of the mutants revealed ∼55% T:A→A:T transversions, ∼15% other point mutations, and ∼30% small deletions at the targeted site [120]. Triplex formation in the absence of psoralen ICLs or other DNA damaging agents has also been shown to be mutagenic. For example, using a system similar to the supF-shuttle vector described above, Wang et al. (1996) demonstrated an ∼13-fold increase in TFO-targeted mutagenesis in COS-7 cells above the background, which was dependent on TC-NER [80].
Mutagenesis in mammalian cells in a chromosomal context
To study triplex-mediated mutagenesis in mammalian chromosomes, transgenic mice with chromosomally integrated copies of a recoverable lambda vector containing a 30-bp triplex target site within the supF mutation-reporter gene were developed, and transformed cell lines from the mice were established. When these cells were treated with psoralen-modified TFOs in the presence or absence of UVA irradiation, mutations in the supF target gene were stimulated 10-fold above background levels, suggesting that the triplex structure alone was mutagenic [121]. Results from these experiments and other similar TFO targeting experiments [122] led to the conclusion that TFOs were capable of inducing targeted mutagenesis on chromosomal targets in mammalian cells.
Homopurine-homopyrimidine mirror-repeat sequences in the genome have the capacity to adopt intramolecular triplex structures or H-DNA [123, 124]. Naturally occurring intramolecular triplex-forming sequences have been shown to stimulate mutagenesis in cells and in mice, providing further evidence for the mutagenic potential of triplex-forming structures in vivo. The predominant form of mutation generated by H-DNA in mammalian cells and mice are deletions resulting from DSBs [82] [81]. Bacolla et al. [83] found that breakpoints of large-scale deletions coincide with non-canonical DNA structures such as H-DNA, and such structure-forming sequences have been found to correlate with chromosomal breakpoints in tumors, implicating these structures in chromosomal rearrangements and translocation-related cancer etiology. For example, an H-DNA-forming sequence from the human c-MYC gene, which maps to translocation breakpoints in cancer, was found to stimulate mutations ∼20-fold above background levels in mammalian cells [82]. The majority of these mutants contained large-scale deletions surrounding the triplex site [82]. This same H-DNA-forming sequence was shown to be mutagenic in a chromosomal context when incorporated on chromosomes of transgenic mutation reporter mice, where ∼8% of the F1 offspring suffered deletions and/or rearrangements near the H-DNA region [81]. The expansion of GAA repeats in the frataxin gene is associated with the autosomal recessive neurological disorder, Freidreich's ataxia, and these GAA repeats have the capacity to adopt H-DNA triplex structures [125, 126]. Studies in S. cerevisiae demonstrated that GAA H-DNA-forming repeats led to chromosome fragility that was dependent on the repeat length and orientation [84]. These sequences have also been shown to lead to large-scale expansions, consistent with the early onset of Friedreich's ataxia [85]. Together, these findings suggest that naturally occurring triplex-forming sequences in the genome can be mutagenic and involved in genomic instability, disease etiology, and genome evolution.
The efficiency of TFO targeting appears to be cell-cycle dependent in mammalian cells, with most mutations occurring in S-phase, likely due to a chromatin structure that allows for greater efficiency of TFO binding [127]. For example, TFO-induced mutagenesis was more efficient in CHO cells in S-phase (mutation frequencies of 0.15 – 0.22%) compared to cells at quiescence (mutation frequencies 0.02 – 0.03%). A triplex target site in the HPRT gene in CHO cells was used to measure the efficiency of TFO binding, and it was estimated that up to 30% of the HPRT targets were crosslinked with the psoralen-modified TFOs in S phase [127]. Similar experiments with free psoralen resulted in mutation frequencies ∼5-fold above the background frequencies, but were not cell-cycle dependent [127].
Targeted mutagenesis in animal models
An important “proof of concept” study supporting therapeutic applications of triplex technology was reported in 2000 [128]. TFOs were successfully used to direct specific mutations on chromosomal targets in somatic cells of mice. To induce mutagenesis, transgenic mice containing chromosomally integrated copies of a recoverable lambda vector containing a 30-bp TFO binding site within the supF gene were treated with specific TFOs or scrambled control oligonucleotides. The 3′-propanolamine-modified oligonucleotides were delivered through intraperitoneal injections of 1 mg per day for five days, and DNA was isolated from various tissues 10 days post-injection for mutation screening. Mutation frequencies in the targeted supF gene were ∼5-fold higher than background levels in most tissues screened. However, there was no increase in mutations in the brain, consistent with the inability of the oligonucleotides to cross the blood-brain barrier [51]. As expected, the majority (∼85%) of the mutations were located within the 30-bp target site, and 40% of those mutations were single-base insertions or deletions, which were speculated to occur through template misalignment at the triplex site, perhaps via error-prone repair events provoked by the triplex structure.
Cellular proteins that recognize triplex DNA structures
Naturally occurring non-canonical DNA structures such as intramolecular triplexes (H-DNA) can cause genome instability [81, 82]. Proteins that interact with such structures have been of great interest as they may have roles in maintaining genome integrity. Triplex technology has been extensively used to identify and characterize such cellular proteins. In 1991 Kiyama and Camerini-Otero identified a 55 kDa protein from HeLa cells that interacted with a T(34).A(34).T(34) pyrimidine motif triplex [129]. This was the first demonstration of human cellular proteins interacting specifically with triplex structures with high affinity, and suggested that triplex structures are indeed formed in vivo. However, the biological functions of these proteins were not clear. Six years later Guleysse et al. (1997) demonstrated a similar phenomenon with a pyrimidine motif triplex formed by a single-stranded 55-bp oligonucleotide that could fold to form an intramolecular triplex structure and isolated a ∼55 KDa protein, which interacted efficiently with and stabilized the triplex structure [130].
Using several purine motif intermolecular triplexes as probes, Musso et al. (1998) isolated three different triplex-specific polypeptides with apparent molecular masses of ∼100 KDa, ∼60 KDa and ∼15 KDa from HeLa cell extracts [131]. Interestingly, these proteins did not interact with a variety of pyrimidine motif triplexes tested, but interacted with purine triplexes that contained either phosphodiester or phosphorothioate backbones. A similar approach was used to identify and characterize triplex binding proteins in S. cerevisiae. A purine triplex-specific binding protein of ∼35 KDa was isolated and identified as STM1, a protein involved in transcriptional repression and mitosis in yeast [132]. Further characterization of this protein established it as a ribosome-associated protein, which also interacts with telomere-proximal Y element DNA sequences [133]. This 80S ribosome associated protein was shown to facilitate protein synthesis under nutrient starved conditions and may be involved in the mTOR signaling pathway [134]. Similar purine motif triplex probes and southwestern library screening identified another protein that binds triplex structures, a product of the yeast CPD1 gene, which is involved in chromosome segregation [135].
In addition to using triplexes as probes to identify proteins that bind to such structures, triplex technology has been used to study a critical step in DNA repair mechanisms; the recognition of DNA damage and structural distortions. The DNA damage recognition complexes, XPA-RPA and XPC-RAD23B, from the NER mechanism have been shown to bind to TFO-directed psoralen ICLs independently and together as a complex [136, 137]. Recognition of the TFO-directed psoralen ICL by XPC-RAD23B was not affected by the presence of XPA. However, RPA and XPC-RAD23B influenced each other, with a biphasic dependence on RPA concentrations. The MMR protein complex, MutSβ, also formed a high affinity complex with TFO-directed ICLs in vitro [138]. Chromatin immunoprecipitation experiments demonstrated that MutSβ interacted with TFO-directed psoralen ICLs on a plasmid transfected into human 293T cells [138]. Moreover, when the NER damage recognition complexes, XPA-RPA and XPC-RAD23B, were incubated with MutSβ in the presence of TFO-directed ICLs, stable complex formation occurred between the proteins on the lesions [138]. These results suggest that proteins from NER and MMR interact to recognize TFO-directed ICLs, and underscore the possibility of crosstalk between the MMR and NER pathways in resolving genome destabilizing DNA lesions.
Triplex technology has also been used to study helicases that unwind mutagenic non-B DNA structures. RPA, though not a helicase, when present in equimolar concentration with triplex substrates has been reported to dissolve triplex structures, but not G4 tetraplex structures, which suggests that the high in vivo concentration of RPA may function as a negative regulator of triplex formation [139]. RecQ helicases, such as the Bloom and Werner helicases unwind triplex structures with a 3′→5′ polarity and require a 3′ single stranded tail [140]. The FANCJ helicase can efficiently unwind purine and pyrimidine motif triplex substrates in an ATP-dependent manner, and requires a 5′ overhang on the third strand for efficient unwinding [141]. Recently it was demonstrated that the DHX9 helicase immunoprecipitated with intermolecular triplex substrates in mammalian cell extracts [142]. Further characterization of this helicase indicated that it can unwind a triplex substrate with a 3′→5′ polarity with respect to the third strand in the presence of ATP and a 3′-overhang on the third strand [142]. The ability of these helicases to unwind intermolecular triplexes implicates them in genome maintenance through resolving mutagenic DNA structures.
Emerging roles of architectural DNA binding proteins in triplex-mediated DNA repair
It is becoming increasingly evident that architectural DNA binding proteins are involved in triplex-associated DNA damage recognition and repair. One of the most abundant non-histone chromosomal DNA binding proteins is the high mobility group protein 1, HMGB1. HMGB1 is known to bind non-canonical DNA structures through interactions in the minor groove of the DNA duplex [143-146]. HMGB1 has also been shown to bind to certain types of DNA lesions, in particular, those that are substrates for NER. Examples include UV-induced DNA lesions [147], cisplatin-DNA lesions [148], and TFO-directed psoralen ICLs [149]. In agreement with its high affinity binding to non-canonical DNA structures, HMGB1 and some family members (e.g. HMGB2 and SRY) have been shown to enhance triplex formation on plasmid DNA containing (GGA/TCC)11 repeats with d(GGA)11 oligonucleotides [150]. Due to its preference for distorted DNA, it is thought that HMGB1 may interfere with the binding and processing of DNA lesions by NER proteins, a phenomenon known as “repair shielding” [151]. However, subsequent investigation by Reddy et al. (2005) demonstrated that the architectural protein HMGB1 forms stable ternary complexes with TFO-directed psoralen ICLs with the NER DNA damage recognition proteins, XPA and RPA, in a cooperative fashion [149]. This association suggests that HMGB1 may modulate DNA repair by recruiting NER damage recognition factors to lesion-containing sites. Later it was also demonstrated that the NER damage recognition complex, XPC-RAD23B, could form stable ternary complexes with HMGB1 on TFO-directed psoralen DNA ICLs. Further, association of XPA and XPC-RAD23B on TFO-directed psoralen ICLs in the presence of HMGB1 was also demonstrated [152]. These interactions of HMGB1 with NER proteins on triplex substrates indicate that HMGB1 may function to enhance interactions between DNA damage recognition proteins and their substrates. Further assessment of processing of TFO-directed ICLs on plasmids using a supF mutation-reporter system transfected into mouse embryonic fibroblasts lacking the HMGB1 protein demonstrated that HMGB1 is indeed an NER co-factor in the sense that it promotes repair of TFO-directed psoralen ICLs in mammalian cells [153].
Conclusion
Triplex technology continues to provide an important tool to study targeted DNA damage and repair. While the mechanisms involved in processing TFO-directed triplex structures and associated lesions are not fully understood, it is known that proteins from several repair pathways are involved. This repair processing can be error-prone as evidenced by TFO-targeted induction of mutations. Advances in synthesizing chemically modified TFOs have been essential to improve our understanding of the complexity of the cellular DNA repair machinery, where proteins from various DNA repair pathways can interact in concert or compete with each other in processing DNA lesions directed by TFOs. From the existing literature it appears that NER is the predominant pathway involved in triplex-associated lesion repair. Through the use of triplex technology, induction of unscheduled DNA repair synthesis (as occurs during NER) could be used as a strategy to improve the efficacy of antimetabolite chemotherapeutics [154]. The estimated high frequency of TTS in the human genome provides feasibility to the use of triplex technology as a therapeutic targeting approach with broad application. Interestingly, these TTS display a high frequency of single nucleotide polymorphisms (SNPs) [28]. Using a SNP prediction software, PupasView, which can map SNPs in the genome provided by Ensemble, 364,314 SNPs were mapped on the 5.4 million putative TTS (∼7%) within the entire human genome [155], which in turn opens the possibility of individualized treatment options.
Limitations to triplex technology still exist, particularly for in vivo applications, such as uptake of the TFOs into cells, affinity and specificity for the binding site, and in vivo stability. Thus, contemporary research is focused on improving the effectiveness of this approach to genome modification primarily through chemical modifications of TFOs and improved cellular delivery systems. Though restrictions still exist, triplex technology has been a key strategy in moving the field of targeted genome manipulation forward.
Figure 2. Schematic of TFO-induced recombination detection through a blue-/white-screening assay.
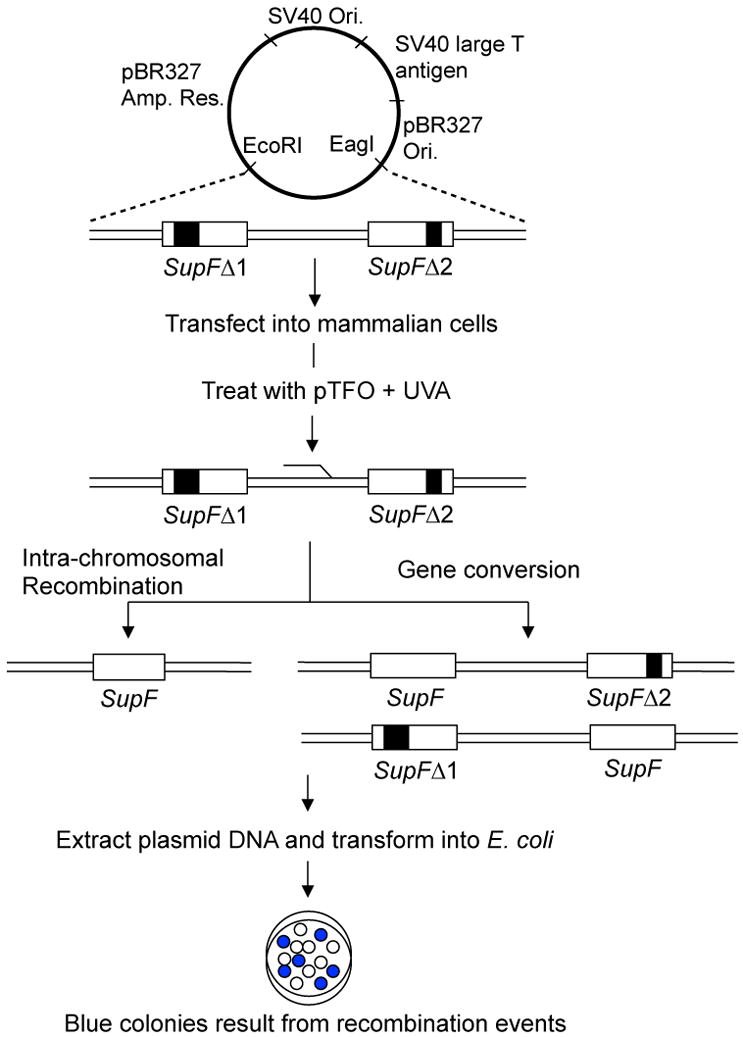
‘Gain of gene function’ or ‘loss of gene function’ is studied through formation of blue and white bacterial colonies. The production of a blue colony on Isopropyl β-D-1-thiogalactopyranoside (IPTG) and 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-Gal) containing agar plates requires a functional supF gene. The supF gene encodes for a suppressor tRNA for an amber mutation in the LacZ gene present in the genomic DNA of E. coli MBM7070 indicator bacterial cells. Functional beta-galactosidase oxidizes the X-Gal present in the media to indole blue, which gives rise to the blue colony phenotype. In the schematic representation, two copies of the supF gene, which have deletions at different positions (SupFΔ1 and SupFΔ2) are inserted into a replication-proficient shuttle vector. When E. coli MBM7070 cells are transformed with plasmid and grown on IPTG/X-Gal agar plates, the resulting colonies are white due to non-functional supF genes. Recombination events in the mammalian cells can produce a functional supF gene, so that when these recombinants are transformed into E. coli MBM7070 cells, they form blue colonies on IPTG/X-Gal plates. The recombination frequency is calculated by dividing the number of recombinant (blue) colonies by the total number of colonies (blue and white). Plasmid map adapted from (Liu et al., NAR, 2008).
Acknowledgments
We thank members of the Vasquez Lab for helpful discussions and Dr. Rick A. Finch for critical review of the manuscript. We also thank Ms. Sarah Henninger for manuscript preparation. This work is supported by an NIH grant, NIH/NCI CA093729 (to KMV).
Abbreviations
- APRT
adenine phosphoribosyltransferase
- bp
base pair
- CHO
Chinese hamster ovary
- DSB
DNA double-strand break
- EPR
error-prone repair
- GC
gene conversion
- GG-NER
global- genome nucleotide excision repair
- HPRT
hypoxanthine-guanine phosphoribosyltransferase
- HR
homologous recombination
- ICL
DNA interstrand crosslink
- LNA
locked nucleic acid
- MMR
mismatch repair
- NER
nucleotide excision repair
- PNA
peptide nucleic acid
- SSA
single-strand annealing
- SNP
single nucleotide polymorphism
- TC-NER
transcription-coupled nucleotide excision repair
- TFO
triplex-forming oligonucleotide
- TFO-ICL
TFO-directed psoralen DNA interstrand crosslink
- tk
thymidine kinase
- TTS
TFO target sequence
- UVA
ultraviolet radiation A (320 – 400 nm)
- WNA
W-shaped nucleoside analog
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 2.Felsenfeld G, Rich A. Studies on the formation of two- and three-stranded polyribonucleotides. Biochim Biophys Acta. 1957;26:457–468. doi: 10.1016/0006-3002(57)90091-4. [DOI] [PubMed] [Google Scholar]
- 3.Moser HE, Dervan PB. Sequence-specific cleavage of double helical DNA by triple helix formation. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 4.Cooney M, Czernuszewicz G, Postel EH, Flint SJ, Hogan ME. Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science. 1988;241:456–459. doi: 10.1126/science.3293213. [DOI] [PubMed] [Google Scholar]
- 5.Francois JC, Saison-Behmoaras T, Helene C. Sequence-specific recognition of the major groove of DNA by oligodeoxynucleotides via triple helix formation. Footprinting studies. Nucleic Acids Res. 1988;16:11431–11440. doi: 10.1093/nar/16.24.11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoogsteen K. The structure of crystals containing a hydrogen-bonded complex of 1-methylthymine and 9-methyladenine. Acta Cryst. 1959;12:822–823. [Google Scholar]
- 7.Pilch DS, Brousseau R, Shafer RH. Thermodynamics of triple helix formation: spectrophotometric studies on the d(A)10.2d(T)10 and d(C+3T4C+3).d(G3A4G3).d(C3T4C3) triple helices. Nucleic Acids Res. 1990;18:5743–5750. doi: 10.1093/nar/18.19.5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rusling DA, Rachwal PA, Brown T, Fox KR. The stability of triplex DNA is affected by the stability of the underlying duplex. Biophys Chem. 2009;145:105–110. doi: 10.1016/j.bpc.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Sun XG, Cao EH, He YJ, Qin JF. Spectroscopic comparison of different DNA structures formed by oligonucleotides. J Biomol Struct Dyn. 1999;16:863–872. doi: 10.1080/07391102.1999.10508298. [DOI] [PubMed] [Google Scholar]
- 10.Asensio JL, Dosanjh HS, Jenkins TC, Lane AN. Thermodynamic, kinetic, and conformational properties of a parallel intermolecular DNA triplex containing 5′ and 3′ junctions. Biochemistry. 1998;37:15188–15198. doi: 10.1021/bi980057m. [DOI] [PubMed] [Google Scholar]
- 11.Han ZJ, Rhee S, Liu K, Miles HT, Davies DR. Crystallization and preliminary crystallographic study of triple-helical DNA. Acta Crystallogr D Biol Crystallogr. 2000;56:104–105. doi: 10.1107/s0907444999012895. [DOI] [PubMed] [Google Scholar]
- 12.He Y, Scaria PV, Shafer RH. Studies on formation and stability of the d[G(AG)5]* d[G(AG)5]. d[C(TC)5] and d[G(TG)5]* d[G(AG)5]. d[C(TC)5] triple helices. Biopolymers. 1997;41:431–441. doi: 10.1002/(SICI)1097-0282(19970405)41:4<431::AID-BIP7>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 13.Johnson KH, Durland RH, Hogan ME. The vacuum UV CD spectra of G.G.C triplexes. Nucleic Acids Res. 1992;20:3859–3864. doi: 10.1093/nar/20.15.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kan LS, Callahan DE, Trapane TL, Miller PS, Ts′o PO, Huang DH. Proton NMR and optical spectroscopic studies on the DNA triplex formed by d-A-(G-A)7-G and d-C-(T-C)7-T. J Biomol Struct Dyn. 1991;8:911–933. doi: 10.1080/07391102.1991.10507857. [DOI] [PubMed] [Google Scholar]
- 15.Rhee S, Han Z, Liu K, Miles HT, Davies DR. Structure of a triple helical DNA with a triplex-duplex junction. Biochemistry. 1999;38:16810–16815. doi: 10.1021/bi991811m. [DOI] [PubMed] [Google Scholar]
- 16.Arnott S, Selsing E. Structures for the polynucleotide complexes poly(dA) with poly (dT) and poly(dT) with poly(dA) with poly (dT) J Mol Biol. 1974;88:509–521. doi: 10.1016/0022-2836(74)90498-7. [DOI] [PubMed] [Google Scholar]
- 17.Radhakrishnan I, de los Santos C, Patel DJ. Nuclear magnetic resonance structural studies of intramolecular purine.purine.pyrimidine DNA triplexes in solution. Base triple pairing alignments and strand direction. J Mol Biol. 1991;221:1403–1418. [PubMed] [Google Scholar]
- 18.Hartman DA, Kuo SR, Broker TR, Chow LT, Wells RD. Intermolecular triplex formation distorts the DNA duplex in the regulatory region of human papillomavirus type-11. J Biol Chem. 1992;267:5488–5494. [PubMed] [Google Scholar]
- 19.Le Doan T, Perrouault L, Praseuth D, Habhoub N, Decout JL, Thuong NT, Lhomme J, Helene C. Sequence-specific recognition photocrosslinking and cleavage of the DNA double helix by an oligo-[alpha]-thymidylate covalently linked to an azidoproflavine derivative. Nucleic Acids Res. 1987;15:7749–7760. doi: 10.1093/nar/15.19.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrouault L, Asseline U, Rivalle C, Thuong NT, Bisagni E, Giovannangeli C, Le Doan T, Helene C. Sequence-specific artificial photo-induced endonucleases based on triple helix-forming oligonucleotides. Nature. 1990;344:358–360. doi: 10.1038/344358a0. [DOI] [PubMed] [Google Scholar]
- 21.Takasugi M, Guendouz A, Chassignol M, Decout JL, Lhomme J, Thuong NT, Helene C. Sequence-specific photo-induced cross-linking of the two strands of double-helical DNA by a psoralen covalently linked to a triple helix-forming oligonucleotide. Proc Natl Acad Sci U S A. 1991;88:5602–5606. doi: 10.1073/pnas.88.13.5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasquez KM, Wensel TG, Hogan ME, Wilson JH. High-efficiency triple-helix-mediated photo-cross-linking at a targeted site within a selectable mammalian gene. Biochemistry. 1996;35:10712–10719. doi: 10.1021/bi960881f. [DOI] [PubMed] [Google Scholar]
- 23.Vasquez KM. Targeting and processing of site-specific DNA interstrand crosslinks. Environ Mol Mutagen. 2010 doi: 10.1002/em.20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chin JY, Glazer PM. Repair of DNA lesions associated with triplex-forming oligonucleotides. Mol Carcinog. 2009;48:389–399. doi: 10.1002/mc.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaddis SS, Wu Q, Thames HD, DiGiovanni J, Walborg EF, MacLeod MC, Vasquez KM. A web-based search engine for triplex-forming oligonucleotide target sequences. Oligonucleotides. 2006;16:196–201. doi: 10.1089/oli.2006.16.196. [DOI] [PubMed] [Google Scholar]
- 26.Wu Q, Gaddis SS, MacLeod MC, Walborg EF, Thames HD, DiGiovanni J, Vasquez KM. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol Carcinog. 2007;46:15–23. doi: 10.1002/mc.20261. [DOI] [PubMed] [Google Scholar]
- 27.Jenjaroenpun P, Kuznetsov VA. TTS mapping: integrative WEB tool for analysis of triplex formation target DNA sequences, G-quadruplets and non-protein coding regulatory DNA elements in the human genome. BMC Genomics. 2009;10(3):S9. doi: 10.1186/1471-2164-10-S3-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goni JR, de la Cruz X, Orozco M. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res. 2004;32:354–360. doi: 10.1093/nar/gkh188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goni JR, Vaquerizas JM, Dopazo J, Orozco M. Exploring the reasons for the large density of triplex-forming oligonucleotide target sequences in the human regulatory regions. BMC Genomics. 2006;7:63. doi: 10.1186/1471-2164-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JS, Johnson DA, Morgan AR. Complexes formed by (pyrimidine)n . (purine)n DNAs on lowering the pH are three-stranded. Nucleic Acids Res. 1979;6:3073–3091. doi: 10.1093/nar/6.9.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milligan JF, Krawczyk SH, Wadwani S, Matteucci MD. An anti-parallel triple helix motif with oligodeoxynucleotides containing 2′-deoxyguanosine and 7-deaza-2′-deoxyxanthosine. Nucleic Acids Res. 1993;21:327–333. doi: 10.1093/nar/21.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng AJ, Van Dyke MW. Monovalent cation effects on intermolecular purine-purine-pyrimidine triple-helix formation. Nucleic Acids Res. 1993;21:5630–5635. doi: 10.1093/nar/21.24.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olivas WM, Maher LJ., 3rd Overcoming potassium-mediated triplex inhibition. Nucleic Acids Res. 1995;23:1936–1941. doi: 10.1093/nar/23.11.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao TS, Durland RH, Seth DM, Myrick MA, Bodepudi V, Revankar GR. Incorporation of 2′-deoxy-6-thioguanosine into G-rich oligodeoxyribonucleotides inhibits G-tetrad formation and facilitates triplex formation. Biochemistry. 1995;34:765–772. doi: 10.1021/bi00003a009. [DOI] [PubMed] [Google Scholar]
- 35.Cheng AJ, Van Dyke MW. Oligodeoxyribonucleotide length and sequence effects on intermolecular purine-purine-pyrimidine triple-helix formation. Nucleic Acids Res. 1994;22:4742–4747. doi: 10.1093/nar/22.22.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gowers DM, Fox KR. DNA triple helix formation at oligopurine sites containing multiple contiguous pyrimidines. Nucleic Acids Res. 1997;25:3787–3794. doi: 10.1093/nar/25.19.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orson FM, Klysik J, Bergstrom DE, Ward B, Glass GA, Hua P, Kinsey BM. Triple helix formation: binding avidity of acridine-conjugated AG motif third strands containing natural, modified and surrogate bases opposed to pyrimidine interruptions in a polypurine target. Nucleic Acids Res. 1999;27:810–816. doi: 10.1093/nar/27.3.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown PM, Fox KR. Nucleosome core particles inhibit DNA triple helix formation. Biochem J. 1996;319(Pt 2):607–611. doi: 10.1042/bj3190607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jain A, Wang G, Vasquez KM. DNA triple helices: biological consequences and therapeutic potential. Biochimie. 2008;90:1117–1130. doi: 10.1016/j.biochi.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fox KR, Brown T. An extra dimension in nucleic acid sequence recognition. Q Rev Biophys. 2005;38:311–320. doi: 10.1017/S0033583506004197. [DOI] [PubMed] [Google Scholar]
- 41.Lee JS, Woodsworth ML, Latimer LJ, Morgan AR. Poly(pyrimidine) . poly(purine) synthetic DNAs containing 5-methylcytosine form stable triplexes at neutral pH. Nucleic Acids Res. 1984;12:6603–6614. doi: 10.1093/nar/12.16.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singleton SF, Dervan PB. Influence of pH on the equilibrium association constants for oligodeoxyribonucleotide-directed triple helix formation at single DNA sites. Biochemistry. 1992;31:10995–11003. doi: 10.1021/bi00160a008. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki S, Yamauchi H, Takahasi R, Taniguchi Y, Maeda M. New base analogs for the formation of non-natural triplexes. Nucleic Acids Res Suppl. 2001:23–24. doi: 10.1093/nass/1.1.23. [DOI] [PubMed] [Google Scholar]
- 44.Sasaki S, Taniguchi Y, Takahashi R, Senko Y, Kodama K, Nagatsugi F, Maeda M. Selective formation of stable triplexes including a TA or a CG interrupting site with new bicyclic nucleoside analogues (WNA) J Am Chem Soc. 2004;126:516–528. doi: 10.1021/ja037211z. [DOI] [PubMed] [Google Scholar]
- 45.Taniguchi Y, Uchida Y, Takaki T, Aoki E, Sasaki S. Recognition of CG interrupting site by W-shaped nucleoside analogs (WNA) having the pyrazole ring in an anti-parallel triplex DNA. Bioorg Med Chem. 2009;17:6803–6810. doi: 10.1016/j.bmc.2009.08.040. [DOI] [PubMed] [Google Scholar]
- 46.Froehler BC, W S, Terhorst TJ, Gerrard SR. Oligodeoxynucleotides containing C-5 propyne analogs of 2′-deoxyuridine and 2′-deoxycytidine. Tetrahedron Letters. 1992;33:5307–5310. [Google Scholar]
- 47.Bijapur J, Keppler MD, Bergqvist S, Brown T, Fox KR. 5-(1-propargylamino)-2′-deoxyuridine (UP): a novel thymidine analogue for generating DNA triplexes with increased stability. Nucleic Acids Res. 1999;27:1802–1809. doi: 10.1093/nar/27.8.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sollogoub M, Darby RA, Cuenoud B, Brown T, Fox KR. Stable DNA triple helix formation using oligonucleotides containing 2′-aminoethoxy,5-propargylamino-U. Biochemistry. 2002;41:7224–7231. doi: 10.1021/bi020164n. [DOI] [PubMed] [Google Scholar]
- 49.Osborne SD, Powers VE, Rusling DA, Lack O, Fox KR, Brown T. Selectivity and affinity of triplex-forming oligonucleotides containing 2′-aminoethoxy-5-(3-aminoprop-1-ynyl)uridine for recognizing AT base pairs in duplex DNA. Nucleic Acids Res. 2004;32:4439–4447. doi: 10.1093/nar/gkh776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Semenyuk A, Darian E, Liu J, Majumdar A, Cuenoud B, Miller PS, Mackerell AD, Jr, Seidman MM. Targeting of an interrupted polypurine:polypyrimidine sequence in mammalian cells by a triplex-forming oligonucleotide containing a novel base analogue. Biochemistry. 2010;49:7867–7878. doi: 10.1021/bi100797z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zendegui JG, Vasquez KM, Tinsley JH, Kessler DJ, Hogan ME. In vivo stability and kinetics of absorption and disposition of 3′ phosphopropyl amine oligonucleotides. Nucleic Acids Res. 1992;20:307–314. doi: 10.1093/nar/20.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Escude C, Giovannangeli C, Sun JS, Lloyd DH, Chen JK, Gryaznov SM, Garestier T, Helene C. Stable triple helices formed by oligonucleotide N3′-->P5′ phosphoramidates inhibit transcription elongation. Proc Natl Acad Sci U S A. 1996;93:4365–4369. doi: 10.1073/pnas.93.9.4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lacroix L, Arimondo PB, Takasugi M, Helene C, Mergny JL. Pyrimidine morpholino oligonucleotides form a stable triple helix in the absence of magnesium ions. Biochem Biophys Res Commun. 2000;270:363–369. doi: 10.1006/bbrc.2000.2438. [DOI] [PubMed] [Google Scholar]
- 54.Michel T, Debart F, Vasseur JJ, Geinguenaud F, Taillandier E. FTIR and UV spectroscopy studies of triplex formation between alpha-oligonucleotides with non-ionic phoshoramidate linkages and DNA targets. J Biomol Struct Dyn. 2003;21:435–445. doi: 10.1080/07391102.2003.10506938. [DOI] [PubMed] [Google Scholar]
- 55.Xodo L, Alunni-Fabbroni M, Manzini G, Quadrifoglio F. Pyrimidine phosphorothioate oligonucleotides form triple-stranded helices and promote transcription inhibition. Nucleic Acids Res. 1994;22:3322–3330. doi: 10.1093/nar/22.16.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basye J, Trent JO, Gao D, Ebbinghaus SW. Triplex formation by morpholino oligodeoxyribonucleotides in the HER-2/neu promoter requires the pyrimidine motif. Nucleic Acids Res. 2001;29:4873–4880. doi: 10.1093/nar/29.23.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 58.Nielsen PE. Targeted gene repair facilitated by peptide nucleic acids (PNA) Chembiochem. 2010;11:2073–2076. doi: 10.1002/cbic.201000346. [DOI] [PubMed] [Google Scholar]
- 59.Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, Nielsen PE. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol. 1994;48:1310–1313. doi: 10.1016/0006-2952(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 60.Shimizu M, Konishi A, Shimada Y, Inoue H, Ohtsuka E. Oligo(2′-O-methyl)ribonucleotides. Effective probes for duplex DNA. FEBS Lett. 1992;302:155–158. doi: 10.1016/0014-5793(92)80428-j. [DOI] [PubMed] [Google Scholar]
- 61.Inoue H, Hayase Y, Imura A, Iwai S, Miura K, Ohtsuka E. Synthesis and hybridization studies on two complementary nona(2′-O-methyl)ribonucleotides. Nucleic Acids Res. 1987;15:6131–6148. doi: 10.1093/nar/15.15.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puri N, Majumdar A, Cuenoud B, Natt F, Martin P, Boyd A, Miller PS, Seidman MM. Targeted gene knockout by 2′-O-aminoethyl modified triplex forming oligonucleotides. J Biol Chem. 2001;276:28991–28998. doi: 10.1074/jbc.M103409200. [DOI] [PubMed] [Google Scholar]
- 63.Blommers MJ, Natt F, Jahnke W, Cuenoud B. Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides: the solution structure of an intramolecular triplex obtained by NMR spectroscopy. Biochemistry. 1998;37:17714–17725. doi: 10.1021/bi9816352. [DOI] [PubMed] [Google Scholar]
- 64.C B, Cuenoud F, Husken D, Natt F, Wolf RM, Altman RM, Martin P, Moser HE. Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides. Angew Chem Intl Ed Engl. 1998;37:1288–1291. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1288::AID-ANIE1288>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 65.Asensio JL, Brown T, Lane AN. Solution conformation of a parallel DNA triple helix with 5′ and 3′ triplex-duplex junctions. Structure. 1999;7:1–11. doi: 10.1016/s0969-2126(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 66.Petersen M, Wengel J. LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol. 2003;21:74–81. doi: 10.1016/S0167-7799(02)00038-0. [DOI] [PubMed] [Google Scholar]
- 67.Alam MR, Majumdar A, Thazhathveetil AK, Liu ST, Liu JL, Puri N, Cuenoud B, Sasaki S, Miller PS, Seidman MM. Extensive sugar modification improves triple helix forming oligonucleotide activity in vitro but reduces activity in vivo. Biochemistry. 2007;46:10222–10233. doi: 10.1021/bi7003153. [DOI] [PubMed] [Google Scholar]
- 68.Simon P, Cannata F, Concordet JP, Giovannangeli C. Targeting DNA with triplex-forming oligonucleotides to modify gene sequence. Biochimie. 2008;90:1109–1116. doi: 10.1016/j.biochi.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 69.Kuwahara M, Sugimoto N. Molecular evolution of functional nucleic acids with chemical modifications. Molecules. 2010;15:5423–5444. doi: 10.3390/molecules15085423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Povsic TJ, Dervan PB. Sequence-specific alkylation of double-helical DNA by oligonucleotide-directed triple-helix formation. Journal of the American Chemical Society; (United States) 1990;112:25. Medium: X; Size: Pages: 9428-9430. [Google Scholar]
- 71.Reed MW, Lukhtanov EA, Gorn V, Kutyavin I, Gall A, Wald A, Meyer RB. Synthesis and reactivity of aryl nitrogen mustard-oligodeoxyribonucleotide conjugates. Bioconjug Chem. 1998;9:64–71. doi: 10.1021/bc970134a. [DOI] [PubMed] [Google Scholar]
- 72.Nagatsugi F, Kawasaki T, Tokuda N, Maeda M, Sasaki S. Site-directed alkylation to cytidine within duplex by the oligonucleotides containing functional nucleobases. Nucleosides Nucleotides Nucleic Acids. 2001;20:915–919. doi: 10.1081/NCN-100002458. [DOI] [PubMed] [Google Scholar]
- 73.Perkins BD, Wensel TG, Vasquez KM, Wilson JH. Psoralen photo-cross-linking by triplex-forming oligonucleotides at multiple sites in the human rhodopsin gene. Biochemistry. 1999;38:12850–12859. doi: 10.1021/bi9902743. [DOI] [PubMed] [Google Scholar]
- 74.Panyutin IG, Neumann RD. Sequence-specific DNA double-strand breaks induced by triplex forming 125I labeled oligonucleotides. Nucleic Acids Res. 1994;22:4979–4982. doi: 10.1093/nar/22.23.4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mezhevaya K, Winters TA, Neumann RD. Gene targeted DNA double-strand break induction by (125)I-labeled triplex-forming oligonucleotides is highly mutagenic following repair in human cells. Nucleic Acids Res. 1999;27:4282–4290. doi: 10.1093/nar/27.21.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pei D, Corey DR, Schultz PG. Site-specific cleavage of duplex DNA by a semisynthetic nuclease via triple-helix formation. Proc Natl Acad Sci U S A. 1990;87:9858–9862. doi: 10.1073/pnas.87.24.9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eisenschmidt K, Lanio T, Simoncsits A, Jeltsch A, Pingoud V, Wende W, Pingoud A. Developing a programmed restriction endonuclease for highly specific DNA cleavage. Nucleic Acids Res. 2005;33:7039–7047. doi: 10.1093/nar/gki1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Teulade-Fichou MP, Perrin D, Boutorine A, Polverari D, Vigneron JP, Lehn JM, Sun JS, Garestier T, Helene C. Direct photocleavage of HIV-DNA by quinacridine derivatives triggered by triplex formation. J Am Chem Soc. 2001;123:9283–9292. doi: 10.1021/ja0109040. [DOI] [PubMed] [Google Scholar]
- 79.Benfield AP, Macleod MC, Liu Y, Wu Q, Wensel TG, Vasquez KM. Targeted generation of DNA strand breaks using pyrene-conjugated triplex-forming oligonucleotides. Biochemistry. 2008;47:6279–6288. doi: 10.1021/bi7024029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang G, Seidman MM, Glazer PM. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science. 1996;271:802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- 81.Wang G, Carbajal S, Vijg J, DiGiovanni J, Vasquez KM. DNA structure-induced genomic instability in vivo. J Natl Cancer Inst. 2008;100:1815–1817. doi: 10.1093/jnci/djn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bacolla A, Jaworski A, Larson JE, Jakupciak JP, Chuzhanova N, Abeysinghe SS, O'Connell CD, Cooper DN, Wells RD. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc Natl Acad Sci U S A. 2004;101:14162–14167. doi: 10.1073/pnas.0405974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim HM, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, Lobachev KS. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. Embo J. 2008;27:2896–2906. doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, Narayanan V, Lobachev KS, Mirkin SM. Large-scale expansions of Friedreich's ataxia GAA repeats in yeast. Mol Cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang G, Chen Z, Zhang S, Wilson GL, Jing K. Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclear extracts. Nucleic Acids Res. 2001;29:1801–1807. doi: 10.1093/nar/29.8.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Duval-Valentin G, Takasugi M, Helene C, Sage E. Triple helix-directed psoralen crosslinks are recognized by Uvr(A)BC excinuclease. J Mol Biol. 1998;278:815–825. doi: 10.1006/jmbi.1998.1728. [DOI] [PubMed] [Google Scholar]
- 88.Christensen LA, Wang H, Van Houten B, Vasquez KM. Efficient processing of TFO-directed psoralen DNA interstrand crosslinks by the UvrABC nuclease. Nucleic Acids Res. 2008;36:7136–7145. doi: 10.1093/nar/gkn880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barre FX, Giovannangeli C, Helene C, Harel-Bellan A. Covalent crosslinks introduced via a triple helix-forming oligonucleotide coupled to psoralen are inefficiently repaired. Nucleic Acids Res. 1999;27:743–749. doi: 10.1093/nar/27.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barre FX, Asseline U, Harel-Bellan A. Asymmetric recognition of psoralen interstrand crosslinks by the nucleotide excision repair and the error-prone repair pathways. J Mol Biol. 1999;286:1379–1387. doi: 10.1006/jmbi.1999.2550. [DOI] [PubMed] [Google Scholar]
- 91.Guillonneau F, Guieysse AL, Nocentini S, Giovannangeli C, Praseuth D. Psoralen interstrand cross-link repair is specifically altered by an adjacent triple-stranded structure. Nucleic Acids Res. 2004;32:1143–1153. doi: 10.1093/nar/gkh267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu Q, Christensen LA, Legerski RJ, Vasquez KM. Mismatch repair participates in error-free processing of DNA interstrand crosslinks in human cells. EMBO Rep. 2005;6:551–557. doi: 10.1038/sj.embor.7400418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Puri N, Majumdar A, Cuenoud B, Natt F, Martin P, Boyd A, Miller PS, Seidman MM. Minimum number of 2′-O-(2-aminoethyl) residues required for gene knockout activity by triple helix forming oligonucleotides. Biochemistry. 2002;41:7716–7724. doi: 10.1021/bi025734y. [DOI] [PubMed] [Google Scholar]
- 94.Richards S, Liu ST, Majumdar A, Liu JL, Nairn RS, Bernier M, Maher V, Seidman MM. Triplex targeted genomic crosslinks enter separable deletion and base substitution pathways. Nucleic Acids Res. 2005;33:5382–5393. doi: 10.1093/nar/gki851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rogers FA, Vasquez KM, Egholm M, Glazer PM. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc Natl Acad Sci U S A. 2002;99:16695–16700. doi: 10.1073/pnas.262556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Babinet C, Cohen-Tannoudji M. Genome engineering via homologous recombination in mouse embryonic stem (ES) cells: an amazingly versatile tool for the study of mammalian biology. An Acad Bras Cienc. 2001;73:365–383. doi: 10.1590/s0001-37652001000300007. [DOI] [PubMed] [Google Scholar]
- 97.Mortensen R. Overview of gene targeting by homologous recombination. Curr Protoc Neurosci. 2007 doi: 10.1002/0471142301.ns0429s40. Chapter 4. Unit 4 29. [DOI] [PubMed] [Google Scholar]
- 98.Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6:507–512. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- 99.Bollag RJ, Waldman AS, Liskay RM. Homologous recombination in mammalian cells. Annu Rev Genet. 1989;23:199–225. doi: 10.1146/annurev.ge.23.120189.001215. [DOI] [PubMed] [Google Scholar]
- 100.Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- 101.Hendrie PC, Hirata RK, Russell DW. Chromosomal integration and homologous gene targeting by replication-incompetent vectors based on the autonomous parvovirus minute virus of mice. J Virol. 2003;77:13136–13145. doi: 10.1128/JVI.77.24.13136-13145.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chamberlain JR, Schwarze U, Wang PR, Hirata RK, Hankenson KD, Pace JM, Underwood RA, Song KM, Sussman M, Byers PH, Russell DW. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science. 2004;303:1198–1201. doi: 10.1126/science.1088757. [DOI] [PubMed] [Google Scholar]
- 103.Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- 104.Vasquez KM, Marburger K, Intody Z, Wilson JH. Manipulating the mammalian genome by homologous recombination. Proc Natl Acad Sci U S A. 2001;98:8403–8410. doi: 10.1073/pnas.111009698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Folger KR, Wong EA, Wahl G, Capecchi MR. Patterns of integration of DNA microinjected into cultured mammalian cells: evidence for homologous recombination between injected plasmid DNA molecules. Mol Cell Biol. 1982;2:1372–1387. doi: 10.1128/mcb.2.11.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sandor Z, Bredberg A. Triple helix directed psoralen adducts induce a low frequency of recombination in an SV40 shuttle vector. Biochim Biophys Acta. 1995;1263:235–240. doi: 10.1016/0167-4781(95)00109-t. [DOI] [PubMed] [Google Scholar]
- 107.Liu Y, Nairn RS, Vasquez KM. Targeted gene conversion induced by triplex-directed psoralen interstrand crosslinks in mammalian cells. Nucleic Acids Res. 2009;37:6378–6388. doi: 10.1093/nar/gkp678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chin JY, Kuan JY, Lonkar PS, Krause DS, Seidman MM, Peterson KR, Nielsen PE, Kole R, Glazer PM. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc Natl Acad Sci U S A. 2008;105:13514–13519. doi: 10.1073/pnas.0711793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Faruqi AF, Seidman MM, Segal DJ, Carroll D, Glazer PM. Recombination induced by triple-helix-targeted DNA damage in mammalian cells. Mol Cell Biol. 1996;16:6820–6828. doi: 10.1128/mcb.16.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu Y, Nairn RS, Vasquez KM. Processing of triplex-directed psoralen DNA interstrand crosslinks by recombination mechanisms. Nucleic Acids Res. 2008;36:4680–4688. doi: 10.1093/nar/gkn438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Faruqi AF, Datta HJ, Carroll D, Seidman MM, Glazer PM. Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol Cell Biol. 2000;20:990–1000. doi: 10.1128/mcb.20.3.990-1000.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vasquez KM, Wensel TG, Hogan ME, Wilson JH. High-affinity triple helix formation by synthetic oligonucleotides at a site within a selectable mammalian gene. Biochemistry. 1995;34:7243–7251. doi: 10.1021/bi00021a040. [DOI] [PubMed] [Google Scholar]
- 113.Knauert MP, Kalish JM, Hegan DC, Glazer PM. Triplex-stimulated intermolecular recombination at a single-copy genomic target. Mol Ther. 2006;14:392–400. doi: 10.1016/j.ymthe.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 114.Asensio JL, C R, Brown T, Lane AN. Conformational and thermodynamic properties of parallel intramolecular triple helices containing a DNA, RNA, or 2′-OMeDNA third strand. J Am Chem Soc. 1999;121:11063–11070. [Google Scholar]
- 115.Luo Z, Macris MA, Faruqi AF, Glazer PM. High-frequency intrachromosomal gene conversion induced by triplex-forming oligonucleotides microinjected into mouse cells. Proc Natl Acad Sci U S A. 2000;97:9003–9008. doi: 10.1073/pnas.160004997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taghian DG, Nickoloff JA. Chromosomal double-strand breaks induce gene conversion at high frequency in mammalian cells. Mol Cell Biol. 1997;17:6386–6393. doi: 10.1128/mcb.17.11.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Donoho G, Jasin M, Berg P. Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol Cell Biol. 1998;18:4070–4078. doi: 10.1128/mcb.18.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Oh DH, King BA, Boxer SG, Hanawalt PC. Spatially localized generation of nucleotide sequence-specific DNA damage. Proc Natl Acad Sci U S A. 2001;98:11271–11276. doi: 10.1073/pnas.201409698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Havre PA, Gunther EJ, Gasparro FP, Glazer PM. Targeted mutagenesis of DNA using triple helix-forming oligonucleotides linked to psoralen. Proc Natl Acad Sci U S A. 1993;90:7879–7883. doi: 10.1073/pnas.90.16.7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Havre PA, Glazer PM. Targeted mutagenesis of simian virus 40 DNA mediated by a triple helix-forming oligonucleotide. J Virol. 1993;67:7324–7331. doi: 10.1128/jvi.67.12.7324-7331.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vasquez KM, Wang G, Havre PA, Glazer PM. Chromosomal mutations induced by triplex-forming oligonucleotides in mammalian cells. Nucleic Acids Res. 1999;27:1176–1181. doi: 10.1093/nar/27.4.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Majumdar A, Khorlin A, Dyatkina N, Lin FL, Powell J, Liu J, Fei Z, Khripine Y, Watanabe KA, George J, Glazer PM, Seidman MM. Targeted gene knockout mediated by triple helix forming oligonucleotides. Nat Genet. 1998;20:212–214. doi: 10.1038/2530. [DOI] [PubMed] [Google Scholar]
- 123.Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, Frank-Kamenetskii MD. DNA H form requires a homopurine-homopyrimidine mirror repeat. Nature. 1987;330:495–497. doi: 10.1038/330495a0. [DOI] [PubMed] [Google Scholar]
- 124.Belotserkovskii BP, Veselkov AG, Filippov SA, Dobrynin VN, Mirkin SM, Frank-Kamenetskii MD. Formation of intramolecular triplex in homopurine-homopyrimidine mirror repeats with point substitutions. Nucleic acids research. 1990;18:6621–6624. doi: 10.1093/nar/18.22.6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wells RD. DNA triplexes and Friedreich ataxia. Faseb J. 2008;22:1625–1634. doi: 10.1096/fj.07-097857. [DOI] [PubMed] [Google Scholar]
- 126.Koeppen AH. Friedreich's ataxia: Pathology, pathogenesis, and molecular genetics. J Neurol Sci. 2011 doi: 10.1016/j.jns.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Majumdar A, Puri N, Cuenoud B, Natt F, Martin P, Khorlin A, Dyatkina N, George AJ, Miller PS, Seidman MM. Cell cycle modulation of gene targeting by a triple helix-forming oligonucleotide. J Biol Chem. 2003;278:11072–11077. doi: 10.1074/jbc.M211837200. [DOI] [PubMed] [Google Scholar]
- 128.Vasquez KM, Narayanan L, Glazer PM. Specific mutations induced by triplex-forming oligonucleotides in mice. Science. 2000;290:530–533. doi: 10.1126/science.290.5491.530. [DOI] [PubMed] [Google Scholar]
- 129.Kiyama R, Camerini-Otero RD. A triplex DNA-binding protein from human cells: purification and characterization. Proc Natl Acad Sci U S A. 1991;88:10450–10454. doi: 10.1073/pnas.88.23.10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guieysse AL, Praseuth D, Helene C. Identification of a triplex DNA-binding protein from human cells. J Mol Biol. 1997;267:289–298. doi: 10.1006/jmbi.1997.0884. [DOI] [PubMed] [Google Scholar]
- 131.Musso M, Nelson LD, Van Dyke MW. Characterization of purine-motif triplex DNA-binding proteins in HeLa extracts. Biochemistry. 1998;37:3086–3095. doi: 10.1021/bi9717486. [DOI] [PubMed] [Google Scholar]
- 132.Nelson LD, Musso M, Van Dyke MW. The yeast STM1 gene encodes a purine motif triple helical DNA-binding protein. J Biol Chem. 2000;275:5573–5581. doi: 10.1074/jbc.275.8.5573. [DOI] [PubMed] [Google Scholar]
- 133.Van Dyke MW, Nelson LD, Weilbaecher RG, Mehta DV. Stm1p, a G4 quadruplex and purine motif triplex nucleic acid-binding protein, interacts with ribosomes and subtelomeric Y' DNA in Saccharomyces cerevisiae. J Biol Chem. 2004;279:24323–24333. doi: 10.1074/jbc.M401981200. [DOI] [PubMed] [Google Scholar]
- 134.Van Dyke N, Baby J, Van Dyke MW. Stm1p, a ribosome-associated protein, is important for protein synthesis in Saccharomyces cerevisiae under nutritional stress conditions. J Mol Biol. 2006;358:1023–1031. doi: 10.1016/j.jmb.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 135.Musso M, Bianchi-Scarra G, Van Dyke MW. The yeast CDP1 gene encodes a triple-helical DNA-binding protein. Nucleic Acids Res. 2000;28:4090–4096. doi: 10.1093/nar/28.21.4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vasquez KM, Christensen J, Li L, Finch RA, Glazer PM. Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc Natl Acad Sci U S A. 2002;99:5848–5853. doi: 10.1073/pnas.082193799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thoma BS, Wakasugi M, Christensen J, Reddy MC, Vasquez KM. Human XPC-hHR23B interacts with XPA-RPA in the recognition of triplex-directed psoralen DNA interstrand crosslinks. Nucleic Acids Res. 2005;33:2993–3001. doi: 10.1093/nar/gki610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhao J, Jain A, Iyer RR, Modrich PL, Vasquez KM. Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks. Nucleic Acids Res. 2009;37:4420–4429. doi: 10.1093/nar/gkp399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu Y, Rawtani N, Thazhathveetil AK, Kenny MK, Seidman MM, Brosh RM., Jr Human replication protein A melts a DNA triple helix structure in a potent and specific manner. Biochemistry. 2008;47:5068–5077. doi: 10.1021/bi702102d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brosh RM, Jr, Majumdar A, Desai S, Hickson ID, Bohr VA, Seidman MM. Unwinding of a DNA triple helix by the Werner and Bloom syndrome helicases. J Biol Chem. 2001;276:3024–3030. doi: 10.1074/jbc.M006784200. [DOI] [PubMed] [Google Scholar]
- 141.Sommers JA, Rawtani N, Gupta R, Bugreev DV, Mazin AV, Cantor SB, Brosh RM., Jr FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem. 2009;284:7505–7517. doi: 10.1074/jbc.M809019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jain A, Bacolla A, Chakraborty P, Grosse F, Vasquez KM. Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry. 49:6992–6999. doi: 10.1021/bi100795m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem Sci. 2001;26:167–174. doi: 10.1016/s0968-0004(01)01801-1. [DOI] [PubMed] [Google Scholar]
- 144.Hamada H, Bustin M. Hierarchy of binding sites for chromosomal proteins HMG 1 and 2 in supercoiled deoxyribonucleic acid. Biochemistry. 1985;24:1428–1433. doi: 10.1021/bi00327a022. [DOI] [PubMed] [Google Scholar]
- 145.Bianchi ME, Beltrame M, Paonessa G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science. 1989;243:1056–1059. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- 146.Lange SS, Vasquez KM. HMGB1: the jack-of-all-trades protein is a master DNA repair mechanic. Mol Carcinog. 2009;48:571–580. doi: 10.1002/mc.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pasheva EA, Pashev IG, Favre A. Preferential binding of high mobility group 1 protein to UV-damaged DNA. Role of the COOH-terminal domain. J Biol Chem. 1998;273:24730–24736. doi: 10.1074/jbc.273.38.24730. [DOI] [PubMed] [Google Scholar]
- 148.Pil PM, Lippard SJ. Specific binding of chromosomal protein HMG1 to DNA damaged by the anticancer drug cisplatin. Science. 1992;256:234–237. doi: 10.1126/science.1566071. [DOI] [PubMed] [Google Scholar]
- 149.Reddy MC, Christensen J, Vasquez KM. Interplay between human high mobility group protein 1 and replication protein A on psoralen-cross-linked DNA. Biochemistry. 2005;44:4188–4195. doi: 10.1021/bi047902n. [DOI] [PubMed] [Google Scholar]
- 150.Suda T, Mishima Y, Takayanagi K, Asakura H, Odani S, Kominami R. A novel activity of HMG domains: promotion of the triple-stranded complex formation between DNA containing (GGA/TCC)11 and d(GGA)11 oligonucleotides. Nucleic Acids Res. 1996;24:4733–4740. doi: 10.1093/nar/24.23.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc Natl Acad Sci U S A. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lange SS, Reddy MC, Vasquez KM. Human HMGB1 directly facilitates interactions between nucleotide excision repair proteins on triplex-directed psoralen interstrand crosslinks. DNA Repair (Amst) 2009;8:865–872. doi: 10.1016/j.dnarep.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lange SS, Mitchell DL, Vasquez KM. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci U S A. 2008;105:10320–10325. doi: 10.1073/pnas.0803181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Christensen LA, Finch RA, Booker AJ, Vasquez KM. Targeting oncogenes to improve breast cancer chemotherapy. Cancer Res. 2006;66:4089–4094. doi: 10.1158/0008-5472.CAN-05-4288. [DOI] [PubMed] [Google Scholar]
- 155.Conde L, Vaquerizas JM, Ferrer-Costa C, de la Cruz X, Orozco M, Dopazo J. PupasView: a visual tool for selecting suitable SNPs, with putative pathological effect in genes, for genotyping purposes. Nucleic Acids Res. 2005;33:W501–505. doi: 10.1093/nar/gki476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Brunet E, Corgnali M, Perrouault L, Roig V, Asseline U, Sorensen MD, Babu BR, Wengel J, Giovannangeli C. Intercalator conjugates of pyrimidine locked nucleic acid-modified triplex-forming oligonucleotides: improving DNA binding properties and reaching cellular activities. Nucleic Acids Res. 2005;33:4223–4234. doi: 10.1093/nar/gki726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cogoi S, Rapozzi V, Quadrifoglio F, Xodo L. Anti-gene effect in live cells of AG motif triplex-forming oligonucleotides containing an increasing number of phosphorothioate linkages. Biochemistry. 2001;40:1135–1143. doi: 10.1021/bi0012639. [DOI] [PubMed] [Google Scholar]
- 158.McGuffie EM, Pacheco D, Carbone GM, Catapano CV. Antigene and antiproliferative effects of a c-myc-targeting phosphorothioate triple helix-forming oligonucleotide in human leukemia cells. Cancer Res. 2000;60:3790–3799. [PubMed] [Google Scholar]
- 159.Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature. 1993;365:566–568. doi: 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]