

Abstract
Recovery from stroke is limited, in part, by an inhibitory environment in the postischemic brain, but factors preventing successful remodeling are not well known. Using cultured cortical neurons from mice, brain endothelial cells, and a mouse model of ischemic stroke, we show that signaling from the axon guidance molecule Sema3A via eicosanoid second messengers can contribute to this inhibitory environment. Either 90 nM recombinant Sema3A, or the 12/15-lipoxygenase (12/15-LOX) metabolites 12-HETE and 12-HPETE at 300 nM, block axon extension in neurons compared to solvent controls, and decrease tube formation in endothelial cells. The Sema3A effect is reversed by inhibiting 12/15-LOX, and neurons derived from 12/15-LOX-knockout mice are insensitive to Sema3A. Following middle cerebral artery occlusion to induce stroke in mice, immunohistochemistry shows both Sema3A and 12/15-LOX are increased in the cortex up to 2 wk. To determine whether a Sema3A-dependent damage pathway is activated following ischemia, we injected recombinant Sema3A into the striatum. Sema3A alone did not cause injury in normal brains. But when injected into postischemic brains, Sema3A increased cortical damage by 79%, and again, this effect was reversed by 12/15-LOX inhibition. Our findings suggest that blocking the semaphorin pathway should be investigated as a therapeutic strategy to improve stroke recovery.—Pekcec, A., Yigitkanli, K., Jung, J. E., Pallast, S., Xing, C., Antipenko, A., Minchenko, M., Nikolov, D. B., Holman, T. R., Lo, E. H.,van Leyen, K. Following experimental stroke, the recovering brain is vulnerable to lipoxygenase-dependent semaphorin signaling.
Keywords: eicosanoid, 12-HPETE, cortex, axon growth, second messenger
Stroke, a leading cause of death and disability in the United States, is a major public health issue. The only acute stroke treatment with U.S. Food and Drug Administration approval, tissue plasminogen activator (tPA) is only given to a minor percentage of patients because of side effects, including hemorrhage (1–3). Many other neuroprotective strategies have failed (4), leading to a renewed emphasis of research efforts toward improving stroke recovery (5–7). For this, a better understanding of the factors both promoting and limiting neural connectivity is needed.
The brain is an intricate assembly of neural connections and blood vessels, which is initially organized during brain development, and matures as the organ grows. Guidance signals, including the semaphorins, netrins, ephrins, and others, direct the formation of axons, but also affect vascular development. Among the semaphorins, the secretory protein semaphorin 3A (Sema3A) is an important soluble repulsive signal, which leads to growth cone collapse, limiting axonal growth (8, 9). Major injuries, such as stroke, damage the entire neurovascular unit, leading to disruptions of the neural network as well as the cerebrovasculature (10–12). These events, accompanied by massive cell death, occur during the first hours to days following the stroke. Eventually, the situation stabilizes, and the brain enters a lengthy phase of recovery (13). A successful recovery requires the reestablishment of both neural connections and the microvasculature. These are hindered by an inhibitory environment, which may consist of repulsive and growth-inhibitory proteins like Sema3A, Nogo, and myelin-associated glycoprotein (14–16). Sema3A itself has mostly been studied in the developing brain but may be increased following stroke and contribute to brain damage in the acute phase (17). A possible functional role during stroke recovery, however, has not been studied to date.
A related question of Sema3A function concerns elements of the Sema3A pathway that may be amenable to therapeutic intervention. While Sema3A is known to signal through its receptor neuropilin-1 (NRP-1; refs. 18, 19), with plexins as coreceptors (20, 21), downstream effectors are less well studied. Factors purportedly mediating the Sema3A signal include eicosanoids (22, 23) generated by phospholipases (24), the kinases Rho and Rac (25–27), and Mical interacting with the actin cytoskeleton (28), as well as integrin-based adhesion (29), but few specific inhibitors have been shown to be effective in blocking Sema3A signaling (30–32). We initiated the current study to determine the role of eicosanoids produced by 12/15-lipoxygenase (12/15-LOX) in mediating the Sema3A signal. Our results show that these pathways may be viable targets to block detrimental effects of increased Sema3A in the damaged brain.
MATERIALS AND METHODS
Reagents
Growth factor-reduced Matrigel matrix and NuSerum were purchased from BD Biosciences (San Jose, CA, USA). All other cell culture media were from Invitrogen (Carlsbad, CA, USA). The human brain microvascular endothelial cell line was kindly provided by Dr. M. Stins (Johns Hopkins University, Baltimore, MD, USA). 12-(S)-hydroperoxyeicosatetraenoic acid (12-HPETE), 12-(S)-hydroxyeicosatetraenoic acid (12-HETE), and 5-(S)-hydroxyeicosatetraenoic acid (5-HETE) were from Cayman Chemicals (Ann Arbor, MI, USA). Phalloidin-fluorescein isothiocyanate (FITC) was from Sigma (St. Louis, MO, USA). LOXBlock-1 (ID no. 5680672) was from Chembridge (San Diego, CA, USA). Recombinant Sema3A was generated as described previously (33). For cell culture experiments, full-length Sema3A (aa 26-737) was used (see Fig. 1A). For injection experiments, we used a shorter Sema3A (aa 26-572), comprising the Sema domain and the PSI domain, which was available at a higher concentration (10 mg/ml). Both of these Sema3A constructs have been shown to be biologically active (33).
Figure 1.
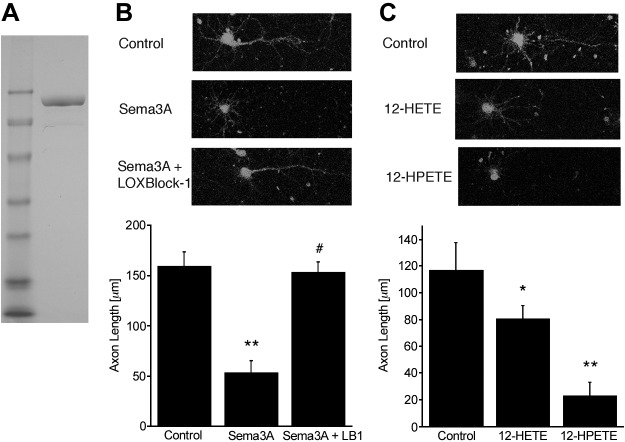
A) Recombinant full-length (90 kDa) Sema3A was used in the cell culture experiments. B) Phalloidin-FITC staining documents the reduced axon growth in Sema3A-treated cultured cortical neurons. The effect is reversed by cotreatment with 5 μM LOXBlock-1, an inhibitor of 12/15-LOX. C) The 12/15-LOX metabolites 12-HPETE and 12-HETE, added at 300 nM, both inhibit axon extension, bypassing the requirement for Sema3A *P < 0.05, **P < 0.01 vs. control; #P < 0.05 vs. Sema3A alone.
Primary neuronal culture
Primary cortical neurons were established following standard protocol. Briefly, neurons from wild-type C57BL6 or ALOX15−/− mice were isolated from E15 embryonic cortices and plated onto poly-d-lysine-coated 24-well plates (BD Biosciences, San Jose, CA, USA) with Dulbecco's minimum essential medium (DMEM) containing 10% fetal bovine serum (FBS) and 100 U/ml penicillin and streptomycin. The next day, medium was switched to NeuroBasal containing 2% B27, 0.5 mM l-glutamine, 100 U/ml penicillin, and streptomycin. Half of the medium was changed every 3 d using the same medium with antioxidant-free B27. Cells were treated as indicated on d 2 of culture, and phase-contrast pictures were randomly taken from cells on d 8 of culture. Alternately, neurons were stained using phalloidin-FITC (Sigma), used according to the manufacturer's suggestion, and imaged using a fluorescent microscope (Nikon Eclipse Ti; Nikon, Tokyo, Japan). In each experiment, neuronal axon length was evaluated in ≥3 pictures for each treatment group, with the longest neuritic extensions of 10 cells from each picture measured, using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA). Three independent experiments were averaged, and the investigator was blinded as to treatment conditions.
Endothelial cell line culture and in vitro angiogenesis assay
A human brain microvascular endothelial cell line (kind gift from Monique Stins, Johns Hopkins University) was cultured as described previously (34, 35). The standard Matrigel assay was used to assess the spontaneous formation of capillary-like structures by the endothelial cells. Cells (6×104 cells/well) were seeded in 24-well plates 30 min after coating with growth factor reduced Matrigel, treated as indicated, and incubated at 37°C for 16 h. Tube formation was assessed by counting the number of tubes in 4 random fields from each well as described before (36).
Mouse transient focal ischemia
All experiments were performed following protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Transient focal cerebral ischemia was induced in male CD-1 mice (Charles River Laboratories, Raleigh, NC, USA) by a standard intraluminal middle cerebral artery occlusion (MCAO) method (37, 38). In all in vivo experiments, investigators were blinded to the experimental group. Mice were kept under anesthesia with 1–1.5% isoflurane via facemask. Body temperature was monitored and maintained at 36.5–37.5°C with a feedback heating pad. Laser-Doppler flowmetry was used to confirm adequate induction of focal ischemia for 60 min, and successful reperfusion (3 mm lateral to bregma). At 7 d after MCAO, mice were randomly assigned to 3 groups. Group I received an intrastriatal injection of 20 μg (300 pmol) recombinant Sema3A, group II received equal volumes of buffer to the striatum, and group III received an intraperitoneal injection of 50 mg/kg LOXBlock-1, 5 min before injection of Sema3A into the striatum.
Intrastriatal Sema3A injections
Under isoflurane anesthesia (1.5%) with spontaneous respiration in a nitrous oxide/oxygen mixture, a small borehole was drilled via the following coordinates from the bregma: 0.0 mm anterior, 2 mm lateral. A 32-gauge 5-μl microinjection needle (Hamilton 7000; Hamilton, Reno, NV, USA) was lowered 3.5 mm deep into the borehole with a stereotactic frame (39). Over 5 min, 2 μl of Sema3A or buffer was injected into the right striatum. The needle was left in place for 10 min and then slowly removed over 5 min. Body temperature was monitored and maintained at 36.5–37.5°C with a feedback heating pad. Afterward, the borehole was sealed with bone wax, the scalp was sutured closed, and the mice were allowed to recover. The whole surgical procedure lasted ∼35 min.
Measurement of injury volumes
Following transcardial perfusion with phosphate-buffered saline (PBS) to remove blood, mice were euthanized 14 d after MCAO. Coronal sections (20 μm), taken at 1-mm intervals, were stained with hematoxylin and eosin. Infarct and injury volumes were quantitated using ImageJ. Because of the large cavities seen in the cortex of Sema3A-treated mice, we used the indirect method [100 × (contralateral hemispheric volume − uninjured ipsilateral volume)/contralateral hemispheric volume] to calculate infarct sizes as a percentage of the uninjured hemisphere. The ventricle, which was highly variable in size both ipsilaterally and contralaterally, was excluded from these measurements.
Immunohistochemistry
For immunohistology, anesthetized CD1 mice were perfused transcardially with ice-cold PBS (pH 7.4), followed with ice-cold 4% paraformaldehyde in PBS (pH 7.4). Brains were removed, immersed in the same fixative overnight at 4°C, and cryoprotected in 15 and 30% sucrose solutions in PBS at 4°C. Frozen coronal sections (20 μm thick) were prepared; blocked with PBS, 0.2% Triton X-100, and 3% bovine serum albumin (BSA); and incubated overnight at 4°C with primary antibodies to 12/15-LOX (affinity-purified rabbit polyclonal antibody directed against the C terminus of mouse and human 12/15-LOX; 1:100 dilution), Sema3A (1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), NeuN (MAB377; Chemicon, Temecula, CA, USA), CD31 (BD Pharmingen, San Diego, CA, USA), or GFAP (Santa Cruz Biotechnology), in PBS, 0.2% Triton X-100, and 2% BSA. Sema3A and 12/15-LOX were imaged in adjacent sections; colocalization studies were carried out with both primary antibodies incubated simultaneously in the same section. The sections were washed with PBS and incubated with secondary antibodies (1:1000; Invitrogen, Carlsbad, CA, USA) and 1 μM To-Pro-3 iodide (Invitrogen) for 30 min. Brain sections were imaged using a Zeiss LSM5 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
Data are expressed as means ± se, and multigroup comparisons were performed by analysis of variation (ANOVA) followed by Tukey's test between individual groups, or by unpaired Student's t test when only 2 groups were involved. Differences with values of P < 0.05 were considered significant.
RESULTS
Sema3A inhibition of axonal growth depends on 12/15-LOX activity, and is relayed by 12-HETE and 12-HPETE as second messengers.
To study effectors affecting Sema3A signaling in cultured cortical neurons, we treated murine neurons with recombinant Sema3A (Fig. 1A). In Fig. 1B, 90 nM of Sema3A is shown to significantly reduce axonal outgrowth. Because 12-HETE was reported to mediate Sema3A-induced growth cone collapse in neurons derived from dorsal root ganglion (22), we tested whether inhibition of 12/15-LOX, which can generate 12-HETE, could also reverse the Sema3A effect on axonal outgrowth. Western blotting showed that Sema3A treatment increased protein levels of 12/15-LOX (see Supplemental Fig. S1A). The 12/15-LOX inhibitor LOXBlock-1 (5 μM; refs. 38, 40) completely blocked the effect of Sema3A (Fig. 1B). LOXBlock-1 alone did not increase axon length (Supplemental Fig. S2C). In these experiments, Sema3A was not toxic (see Supplemental Fig. S3), in contrast to some previous reports (41). Axon growth inhibition by Sema3A was also reversed by a peptide that disrupts the interaction of Sema3A with its receptor NRP-1 (17, 31) but not a scrambled control peptide, demonstrating that Sema3A signals via NRP-1 in these cells (Supplemental Fig. S2A, B).
Previous studies reported that 12-HETE is the obligate lipoxygenase-derived eicosanoid needed for Sema3A-induced growth cone collapse (22). The murine 12/15-LOX initially oxidizes arachidonic acid to the hydroperoxide 12-HPETE, which is then reduced to form the hydroxide 12-HETE. 12-HPETE has several known second messenger activities in its own right: it inhibits Ca2+/calmodulin-dependent protein kinase II (CaM-kinase II; ref. 42), acts as a second messenger by increasing K+ channel activity (43), and is known to activate soluble guanylate cyclase, regulating intracellular levels of cGMP (16). These latter findings led to the observation that 12-HPETE converts a normally attractant netrin signal for growth cone turning to a repulsive one (44). On the basis of these findings, we investigated whether 12-HPETE might also inhibit axonal outgrowth. Strikingly, when we applied physiologically relevant doses (300 nM) of either 12-HETE or 12-HPETE to cortical neurons, 12-HPETE was more active than 12-HETE in mimicking the effect of Sema3A (Fig. 1C). In contrast to 12-HETE and 12-HPETE, the 5-LOX metabolite 5-HETE did not appear to affect axon extension (Supplemental Fig. S2D). Some cell death occurred through the action of 12-HPETE (∼15%), whereas 12-HETE did not affect viability (Supplemental Fig. S3B). Nevertheless, both 12/15-LOX metabolites can replicate the growth inhibitory action of Sema3A.
12/15-LOX is the lipoxygenase isoform required for Sema3A signaling
Several lipoxygenase isoforms can generate 12-HPETE and 12-HETE. Of these, 12/15-LOX is the most prominent in the rodent brain (45, 46). To test whether murine 12/15-LOX gives rise to the 12-HETE and 12-HPETE needed for Sema3A signaling, we derived neurons from 12/15-LOX-knockout mice (ALOX15−/− genotype). Knockout neurons were no longer responsive to Sema3A, nor to the combination of Sema3A and LOXBlock-1, indicating a requirement for 12/15-LOX (Fig. 2A, B). Nonetheless, the machinery to relay the Sema3A signal downstream of 12/15-LOX is still intact, because the 12/15-LOX-knockout neurons were still susceptible to axon growth inhibition by 12-HPETE and 12-HETE (Fig. 2C).
Figure 2.
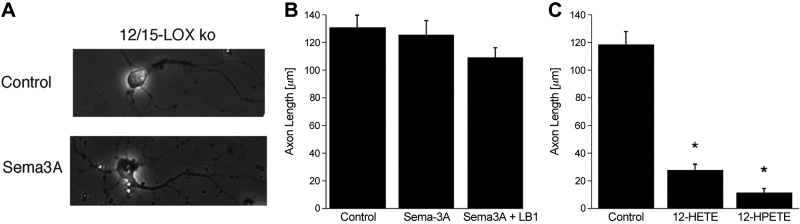
A) Phase-contrast images show that cortical neurons from 12/15-LOX-knockout mice are not susceptible to Sema3A treatment. B) This is confirmed by quantitation of images from 3 independent experiments. C) The knockout neurons do respond to 12-HPETE and 12-HETE treatment (300 nM each), suggesting that the effectors downstream of 12/15-LOX are still active in the 12/15-LOX knockout. *P < 0.05 vs. control.
Sema3A inhibits tube formation by brain endothelial cells in a 12/15-LOX-dependent process
In addition to repulsive effects of Sema3A on axonal growth cones (47), antiangiogenic effects of Sema3A have been reported (48–50). In part, this may occur by counteracting the growth-promoting effects of VEGF, which also signals through the NRP-1 receptor (49, 51). Thus, we studied an in vitro model of angiogenesis in a human brain microvascular endothelial cell line, to determine whether the same signaling pathway operates in the endothelial cells. Sema3A significantly suppressed tube formation in a standard Matrigel assay (Fig. 3A, B). These cells express 12/15-LOX (see Supplemental Fig. S1B), and again, the effect of Sema3A was abrogated by the lipoxygenase inhibitor LOXBlock-1 (Fig. 3B). Similar to the situation in growing axons, 12-HPETE, and to a lesser extent 12-HETE, were able to mimic the Sema3A effect (Fig. 3C). Neither 12-HETE nor 12-HPETE affected viability in these experiments (see Supplemental Fig. S3C). Together, these results indicate that Sema3A inhibits both axonal outgrowth and angiogenesis through a 12/15-LOX-dependent mechanism.
Figure 3.
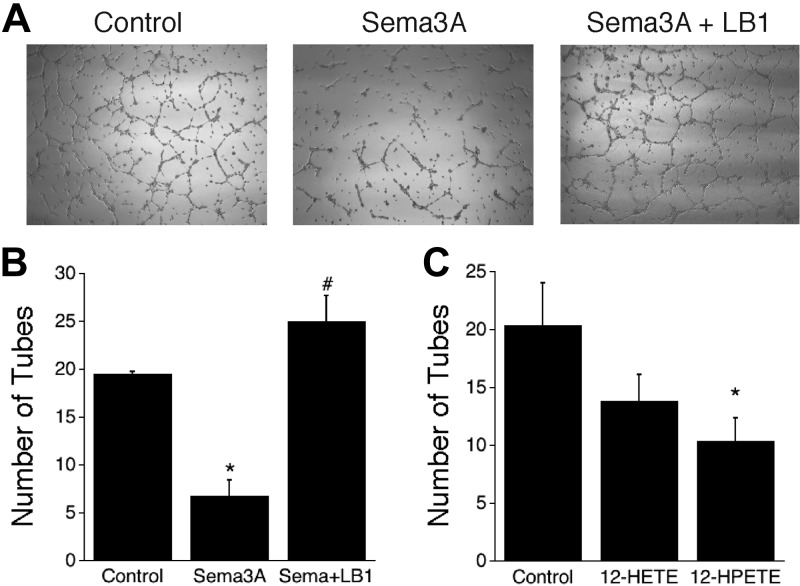
Effects of Sema3A on in vitro angiogenesis. A) Phase-contrast images show that tube formation in endothelial cells is reduced in the presence of Sema3A. B) Tube formation in endothelial cells is reduced by Sema3A and protected by LOXBlock-1. C) 12-HPETE is more effective than 12-HETE at reducing tube formation *P < 0.05 vs. control; #P < 0.05 vs. Sema3A alone.
Sema3A/12/15-LOX system is active in the recovering brain following experimental stroke
To study whether growth-inhibitory signaling of Sema3A is activated in the recovery phase after stroke, we used a mouse model of transient focal ischemia. In a preliminary experiment, we studied the expression of Sema3A, and of its downstream mediator 12/15-LOX, in the cortex over time after 60 min of unilateral MCAO. Both Sema3A and 12/15-LOX were increased in the ipsilateral cortex for up to 14 d, suggesting that the Sema3A/12/15-LOX pathway may be active throughout the recovery phase (Fig. 4 and refs. 6, 52). As control for specificity of the antibody against 12/15-LOX used, no cell-specific staining was detected when we stained a brain section from an ALOX15−/− mouse subjected to MCAO (see Supplemental Fig. S4A). To determine which cells give rise to the Sema3A signal, we carried out double-labeling experiments with marker proteins for neurons (NeuN), endothelial cells (CD31), and astrocytic cells (GFAP), in brain sections taken at d 7 after MCAO. The Sema3A signal was clearly separated from strong GFAP immunoreactivity, suggesting that the increased Sema3A likely is not a result of the glial response to ischemic injury (ref. 53 and Fig. 5). Because Sema3A appeared to be in proximity of neuronal and endothelial cells, we investigated images at higher resolution (Fig. 6). Here, the contralateral Sema3A, present at relatively low levels, appeared mostly vascular. A cellular localization for the ipsilateral Sema3A could still not readily be determined from these images, suggesting that high levels of this soluble protein are secreted into the extracellular milieu. These experiments suggest that the Sema3A signaling pathway may be up-regulated in the recovering brain.
Figure 4.
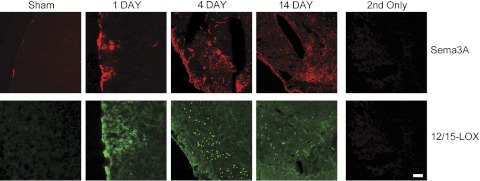
Long-term up-regulation of Sema3A. Both Sema3A and 12/15-LOX are increased up until 14 d following ischemia. Little staining is seen in sham-operated controls, and staining is abolished when only secondary antibodies are used. Scale bar = 100 μm.
Figure 5.

Colocalization of Sema3A with marker proteins 7 d after focal ischemia. No significant colocalization was seen in the cortex with the astrocytic marker GFAP, suggesting that Sema3A is not primarily a component of the glial response. Colocalization with the endothelial marker CD31 and the neuronal marker NeuN cannot be clearly confirmed from these images. Scale bar = 100 μm.
Figure 6.

Higher magnification suggests a partial colocalization of Sema3A with endothelial and neuronal cells, but the diffuse staining for Sema3A suggests mostly extracellular localization in the ipsilateral cortex. In contrast, the Sema3A signal in the contralateral cortex is present at lower levels than ipsilateral, but appears to be restricted to the vasculature. Scale bar = 20 μm.
Intrastriatal injection of Sema3A disrupts the recovery process and increases brain injury, but can be blocked by 12/15-LOX inhibition in vivo
Measuring direct effects of the endogenous Sema3A is difficult, but to investigate whether adding exogenous Sema3A is beneficial or detrimental for the recovery process, we injected 20 μg (300 pmol) recombinant Sema3A into the ipsilateral striatum on d 7 after MCAO. We chose this time point for injections because, by this time, acute injury is fully developed, and regenerative activities are known to be active (6). There was no immediate toxicity, nor was any behavioral effect detectable after injection. But when the mice were euthanized at d 14, significant additional damage was detected in the ipsilateral hemisphere (compare Fig. 7B, C, black arrows). As a control, we injected Sema3A into the striatum of nonischemic brains, which did not cause visible damage when the mice were euthanized 7 d later (Fig. 7A), aside from some minor injection site injury (Fig. 7A, white arrow). Because of the in vitro efficacy of LOXBlock-1 in blocking the effects of Sema3A, we studied whether coadministration of LOXBlock-1 could reverse the Sema3A-induced damage. When LOXBlock-1 (50 mg/kg) was given intraperitoneally at the time of Sema3A injection into the striatum, this reversed the additional damage caused by Sema3A (Fig. 7D). These results were reproducible and statistically significant (Fig. 7E). Strikingly, the greatest effect was not close to the injection site, but instead in the cortex, which exhibited massive tissue loss (Fig. 7C, F).
Figure 7.
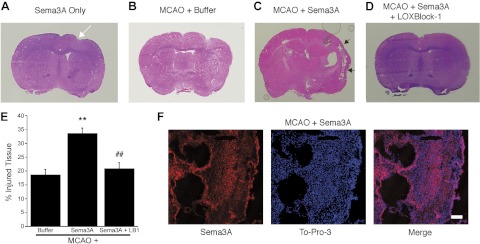
Brain damage after experimental stroke is increased by intrastriatal injection of Sema3A, but reduced by cotreatment with the 12/15-LOX inhibitor LOXBlock-1. A) Besides some injection site injury (white arrow), Sema3A injection into the nonischemic brain does not cause noticeable brain injury 7 d later, as judged by hematoxylin and eosin staining. B, C) When injected into the ischemic striatum 7 d after experimental stroke, recombinant Sema3A dramatically increases damage especially in the cortex (C, black arrows), compared to injection of buffer (B). D) The increased tissue injury is reversed by intraperitoneal administration of LOXBlock-1. E) The increased damage through Sema3A is reflected in the injury volume (expressed as percentage of contralateral side, ventricle excluded). **P < 0.01 vs. MCAO only control; ##P < 0.01 vs. MCAO + Sema3A. F) Immunohistochemistry shows Sema3A distribution and tissue loss (large cavities) in the cortex, detected by the nuclear stain, To-Pro-3. Strong Sema3A staining appears in cells surrounding the cavities. Scale bar = 100 μm.
DISCUSSION
We have previously demonstrated that inhibition of 12/15-LOX protects the ischemic brain in the acute phase of stroke (40, 46, 54). We show here that 12/15-LOX inhibition may also be beneficial during the recovery phase. The two major findings in this study are that the anti-axon growth and antiangiogenic effects of Sema3A act through the metabolites of 12/15-lipoxygenase, 12-HETE, and 12-HPETE; and that a damaging effect of Sema3A specific to the brain recovering from experimental stroke can be blocked by inhibiting 12/15-LOX in vivo.
Eicosanoids 12-HETE and 12-HPETE act as second messengers for Sema3A signaling.
The 12-lipoxygenase-derived eicosanoid 12-HPETE was first reported as a second messenger molecule in 1987, to mediate the signal of the neuroactive peptide FRMFamide in Aplysia neurons (43). 12-HETE, which was not active in Aplysia, nonetheless appears to function here in a manner very similar to 12-HPETE. The reduced activity of 12-HETE compared to 12-HPETE has been noted before in vasoconstrictive signaling of these eicosanoids (55). The characteristics that qualify 12-HPETE and 12-HETE as second messenger molecules are as follows (43). 1) Synthesis: the enzyme is present in neurons. 2) Receptor mediation: 12-HETE production (and by inference, that of the precursor 12-HPETE) was stimulated by Sema3A application in DRG neurons (22, 24), and the Sema3A effect on axonal growth is receptor dependent, as shown by blockade through the blocking peptide (Supplemental Fig. S1D). 3) Simulation: 12-HETE and 12-HPETE replicate the effects of Sema3A on axonal growth. 4) Selective blockade: inhibition of 12/15-LOX blocks the effects of Sema3A, as does the gene knockout. 5) Termination: this can easily occur by further metabolization of 12-HETE and 12-HPETE, as well as by reincorporation into phospholipid.
Involvement of 12-HETE in the Sema3A signaling cascade was demonstrated several years ago by Pfenninger and colleagues (22). We extend those findings here by demonstrating that besides 12-HETE, 12-HPETE is also active in mediating the Sema3A signal. Furthermore, neurons derived from 12/15-LOX-knockout mice do not respond to Sema3A, but can still respond to the 12/15-LOX metabolites 12-HETE and 12-HPETE when they are furnished exogenously. This documents a requirement for the 12/15-LOX isoform to mediate Sema3A signaling.
Damaging effects of Sema3A injected into the postischemic brain
We found that both Sema3A and 12/15-LOX increased in the mouse cortex up to 2 wk after 60 min MCAO. This suggests that the Sema3A pathway is up-regulated at this late time point after stroke (see Fig. 4). These findings are in line with previous reports of long-term up-regulation of Sema3A mRNA (56) and protein (57). Our colocalization studies did not allow for a specific assignment of originating cells for the Sema3A signal in the postischemic cortex. However, Sema3A appeared in proximity to both neurons and endothelial cells, presumably reflecting the fact that Sema3A is a soluble secreted molecule (8, 9).
Injecting recombinant Sema3A into the postischemic striatum had a striking long-range effect 7 d later, increasing injury not at the site of injection, but instead leading to tissue loss in the cortex (see Fig. 7C, F). Notably, this effect was specific for the poststroke brain, as injecting Sema3A into the uninjured mouse brain did not cause significant damage (Fig. 7A). The massive cortical damage observed when recombinant Sema3A is injected into the postischemic striatum is somewhat reminiscent of studies by the Rothwell group, who found increased cortical injury when interleukin-1β was injected into the striatum immediately following MCAO (58), presumably caused by excitotoxic or proinflammatory effects (59, 60). One decisive difference between the two studies concerns the timing of administration; whereas the interleukin 1β injections were carried out in the acute phase of stroke, we are here injecting the recombinant Sema3A later, during the recovery phase. This suggests the mechanisms of increased injury may be different. Nonetheless, both studies demonstrate long-range effects of injected proteins, which in the case of Sema3A may be caused by the inhibition of axonal connectivity, and/or neovascularization proceeding from the striatum to the cortex. The important finding here is that we can prevent this excessive tissue damage from occurring, by intraperitoneal injection of the 12/15-LOX inhibitor LOXBlock-1. This demonstrates that 12/15-LOX activity is required in vivo to mediate Sema3A-induced damage after stroke, and suggests a treatment strategy to prevent detrimental effects of Sema3A signaling during the recovery phase after stroke.
Dual role of 12/15-LOX after stroke
We have previously shown that 12/15-LOX has a damaging effect in the acute phase of stroke, most likely through an organelle damage pathway (37, 46, 54). The findings here document a second, separate pathway by which 12/15-LOX is also detrimental to stroke recovery. Conversely, metabolites of 12/15-LOX derived from docosahexaenoic acid and other ω-3 fatty acids have been shown to have a strong neuroprotective effect (61–63). This means that optimizing the treatment schedule with a 12/15-LOX inhibitor like LOXBlock-1 will be important, and this may both minimize the initial injury, and enhance stroke recovery. Some previous studies have also reported proangiogenic effects of several HETEs, including both S and R stereoisomers of 12-HETE (64, 65). These generally involved cells derived from outside the CNS, some of them cancerous, suggesting they may differ in their signaling properties compared to the brain endothelial cells used here. Both our cellular studies, and the protective effects of LOX inhibition in vivo, would seem to indicate an antiangiogenic role of LOX metabolites.
Several important caveats to the study presented here need to be made. First, besides 12-HETE and 12-HPETE, it is possible that secondary 12/15-LOX metabolites, such as hepoxilin A3 (66), are involved in transducing the Sema3A signal. Indeed, 12-HETE and 12-HPETE might be converted to these secondary metabolites intracellularly. Future studies should focus on the details of the Sema3A signaling pathway, including identifying the exact role played by various LOX metabolites. Second, although we found little indication of cell death caused by Sema3A in our cell culture experiments, cell death effects of Sema3A might play a bigger role in the injection experiments. If so, this does not seem to lead to acute toxicity, because we detected no change in mouse behavior immediately after injection, and the mice easily survived for another 7 d thereafter. Furthermore, we did not detect any obvious tissue loss in the ischemic striatum, into which we had injected the Sema3A. Third, we cannot rule out that the protective effect of LOXBlock-1 is independent of effects on Sema3A signal transduction. However, LOXBlock-1 itself did not enhance axon extension in our cell culture assays (see Supplemental Fig. S2), suggesting it only affects neurite outgrowth in the context of Sema3A signaling. Finally, the injection of recombinant Sema3A is, of course, a fairly artificial situation, and the endogenous Sema3A may have somewhat different effects. Overall, we see these experiments as a proof of principle, demonstrating both the disruptive effect of elevated Sema3A, and the possibility of its prevention by inhibiting 12/15-LOX.
In summary, we demonstrate that Sema3A growth-inhibitory signaling is mediated by the 12/15-LOX metabolites 12-HETE and 12-HPETE in both neurons and endothelial cells. Furthermore, signaling through the Sema3A pathway can be damaging in the aftermath of an experimental stroke, and blocking the 12/15-LOX activity prevents this damage. Future studies are needed to determine whether blocking the Sema3A pathway through 12/15-LOX inhibition during the recovery phase will enhance recovery and improve long-term outcome after stroke.
Supplementary Material
Acknowledgments
The authors thank Dr. Emiri Mandeville for help with the injection experiments.
This work was supported by U.S. National Institutes of Health grants NS049430 and NS069939 to K.V.L., NS048372 to D.B.N., and GM056062 to T.R.H.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 5-HETE
- 5-(S)-hydroxyeicosatetraenoic acid
- 12-HETE
- 12-(S)-hydroxyeicosatetraenoic acid
- 12-HPETE
- 12-(S)-hydroperoxyeicosatetraenoic acid
- 12/15-LOX
- 12/15-lipoxygenase
- BSA
- bovine serum albumin
- FITC
- fluorescein isothiocyanate
- MCAO
- middle cerebral artery occlusion
- NRP-1
- neuropilin-1
- Sema3A
- semaphorin 3A
- tPA
- tissue plasminogen activator
REFERENCES
- 1. Wang X., Tsuji K., Lee S. R., Ning M., Furie K. L., Buchan A. M., Lo E. H. (2004) Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke 35, 2726–2730 [DOI] [PubMed] [Google Scholar]
- 2. Mostany R., Chowdhury T. G., Johnston D. G., Portonovo S. A., Carmichael S. T., Portera-Cailliau C. (2010) Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J. Neurosci. 30, 14116–14126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dawson T. M., Dawson V. L. (2006) Taming the clot-buster tPA. Nat. Med. 12, 993–994 [DOI] [PubMed] [Google Scholar]
- 4. Willmot M., Gray L., Gibson C., Murphy S., Bath P. M. (2005) A systematic review of nitric oxide donors and L-arginine in experimental stroke; effects on infarct size and cerebral blood flow. Nitric Oxide 12, 141–149 [DOI] [PubMed] [Google Scholar]
- 5. Moskowitz M. A., Lo E. H., Iadecola C. (2010) The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiong Y., Mahmood A., Chopp M. (2010) Angiogenesis, neurogenesis and brain recovery of function following injury. Curr. Opin. Investig. Drugs 11, 298–308 [PMC free article] [PubMed] [Google Scholar]
- 7. Hoang S., Liauw J., Choi M., Guzman R. G., Steinberg G. K. (2009) Netrin-4 enhances angiogenesis and neurologic outcome after cerebral ischemia. J. Cereb. Blood Flow Metab. 29, 385–397 [DOI] [PubMed] [Google Scholar]
- 8. Kolodkin A. L., Matthes D. J., Goodman C. S. (1993) The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 75, 1389–1399 [DOI] [PubMed] [Google Scholar]
- 9. Luo Y., Raible D., Raper J. A. (1993) Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 75, 217–227 [DOI] [PubMed] [Google Scholar]
- 10. Zlokovic B. V. (2010) Neurodegeneration and the neurovascular unit. Nat. Med. 16, 1370–1371 [DOI] [PubMed] [Google Scholar]
- 11. Lo E. H., Rosenberg G. A. (2009) The neurovascular unit in health and disease: introduction. Stroke 40, S2–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hara M. R., Thomas B., Cascio M. B., Bae B. I., Hester L. D., Dawson V. L., Dawson T. M., Sawa A., Snyder S. H. (2006) Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. U. S. A. 103, 3887–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayashi T., Noshita N., Sugawara T., Chan P. H. (2003) Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J. Cereb. Blood. Flow Metab. 23, 166–180 [DOI] [PubMed] [Google Scholar]
- 14. He Z., Koprivica V. (2004) The Nogo signaling pathway for regeneration block. Annu. Rev. Neurosci. 27, 341–368 [DOI] [PubMed] [Google Scholar]
- 15. Lee J. K., Kim J. E., Sivula M., Strittmatter S. M. (2004) Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J. Neurosci. 24, 6209–6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wiessner C., Bareyre F. M., Allegrini P. R., Mir A. K., Frentzel S., Zurini M., Schnell L., Oertle T., Schwab M. E. (2003) Anti-Nogo-A antibody infusion 24 hours after experimental stroke improved behavioral outcome and corticospinal plasticity in normotensive and spontaneously hypertensive rats. J. Cereb. Blood Flow Metab. 23, 154–165 [DOI] [PubMed] [Google Scholar]
- 17. Jiang S. X., Whitehead S., Aylsworth A., Slinn J., Zurakowski B., Chan K., Li J., Hou S. T. (2010) Neuropilin 1 directly interacts with Fer kinase to mediate semaphorin 3A-induced death of cortical neurons. J. Biol. Chem. 285, 9908–9918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Z., Tessier-Lavigne M. (1997) Neuropilin is a receptor for the axonal chemorepellent semaphorin III. Cell 90, 739–751 [DOI] [PubMed] [Google Scholar]
- 19. Kolodkin A. L., Levengood D. V., Rowe E. G., Tai Y. T., Giger R. J., Ginty D. D. (1997) Neuropilin is a semaphorin III receptor. Cell 90, 753–762 [DOI] [PubMed] [Google Scholar]
- 20. Tamagnone L., Artigiani S., Chen H., He Z., Ming G. I., Song H., Chedotal A., Winberg M. L., Goodman C. S., Poo M., Tessier-Lavigne M., Comoglio P. M. (1999) Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99, 71–80 [DOI] [PubMed] [Google Scholar]
- 21. Takahashi T., Fournier A., Nakamura F., Wang L. H., Murakami Y., Kalb R. G., Fujisawa H., Strittmatter S. M. (1999) Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell 99, 59–69 [DOI] [PubMed] [Google Scholar]
- 22. Mikule K., Gatlin J. C., de la Houssaye B. A., Pfenninger K. H. (2002) Growth cone collapse induced by semaphorin 3A requires 12/15-lipoxygenase. J. Neurosci. 22, 4932–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mikule K., Sunpaweravong S., Gatlin J. C., Pfenninger K. H. (2003) Eicosanoid activation of protein kinase C epsilon: involvement in growth cone repellent signaling. J. Biol. Chem. 278, 21168–21177 [DOI] [PubMed] [Google Scholar]
- 24. Sanford S. D., Yun B. G., Leslie C. C., Murphy R. C., Pfenninger K. H. (2012) Group IVA phospholipase A(2) is necessary for growth cone repulsion and collapse. J. Neurochem. 120, 974–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kruger R. P., Aurandt J., Guan K. L. (2005) Semaphorins command cells to move. Nat. Rev. Mol. Cell Biol. 6, 789–800 [DOI] [PubMed] [Google Scholar]
- 26. Liu B. P., Strittmatter S. M. (2001) Semaphorin-mediated axonal guidance via Rho-related G proteins. Curr. Opin. Cell Biol. 13, 619–626 [DOI] [PubMed] [Google Scholar]
- 27. Jin Z., Strittmatter S. M. (1997) Rac1 mediates collapsin-1-induced growth cone collapse. J. Neurosci. 17, 6256–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hung R. J., Yazdani U., Yoon J., Wu H., Yang T., Gupta N., Huang Z., van Berkel W. J., Terman J. R. (2010) Mical links semaphorins to F-actin disassembly. Nature 463, 823–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barberis D., Artigiani S., Casazza A., Corso S., Giordano S., Love C. A., Jones E. Y., Comoglio P. M., Tamagnone L. (2004) Plexin signaling hampers integrin-based adhesion, leading to Rho-kinase independent cell rounding, and inhibiting lamellipodia extension and cell motility. FASEB J. 18, 592–594 [DOI] [PubMed] [Google Scholar]
- 30. Kaneko S., Iwanami A., Nakamura M., Kishino A., Kikuchi K., Shibata S., Okano H. J., Ikegami T., Moriya A., Konishi O., Nakayama C., Kumagai K., Kimura T., Sato Y., Goshima Y., Taniguchi M., Ito M., He Z., Toyama Y., Okano H. (2006) A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat. Med. 12, 1380–1389 [DOI] [PubMed] [Google Scholar]
- 31. Williams G., Eickholt B. J., Maison P., Prinjha R., Walsh F. S., Doherty P. (2005) A complementary peptide approach applied to the design of novel semaphorin/neuropilin antagonists. J. Neurochem. 92, 1180–1190 [DOI] [PubMed] [Google Scholar]
- 32. Montolio M., Messeguer J., Masip I., Guijarro P., Gavin R., Antonio Del Rio J., Messeguer A., Soriano E. (2009) A semaphorin 3A inhibitor blocks axonal chemorepulsion and enhances axon regeneration. Chem. Biol. 16, 691–701 [DOI] [PubMed] [Google Scholar]
- 33. Antipenko A., Himanen J. P., van Leyen K., Nardi-Dei V., Lesniak J., Barton W. A., Rajashankar K. R., Lu M., Hoemme C., Puschel A. W., Nikolov D. B. (2003) Structure of the semaphorin-3A receptor binding module. Neuron 39, 589–598 [DOI] [PubMed] [Google Scholar]
- 34. Willmot M., Gibson C., Gray L., Murphy S., Bath P. (2005) Nitric oxide synthase inhibitors in experimental ischemic stroke and their effects on infarct size and cerebral blood flow: a systematic review. Free Radic. Biol. Med. 39, 412–425 [DOI] [PubMed] [Google Scholar]
- 35. Simao F., Pagnussat A. S., Seo J. H., Navaratna D., Leung W., Lok J., Guo S., Waeber C., Salbego C. G., Lo E. H. (2012) Pro-angiogenic effects of resveratrol in brain endothelial cells: nitric oxide-mediated regulation of vascular endothelial growth factor and metalloproteinases. J Cereb Blood Flow Metab 32, 884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xing C., Lee S., Kim W. J., Wang H., Yang Y. G., Ning M., Wang X., Lo E. H. (2009) Neurovascular effects of CD47 signaling: promotion of cell death, inflammation, and suppression of angiogenesis in brain endothelial cells in vitro. J. Neurosci. Res. 87, 2571–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pallast S., Arai K., Pekcec A., Yigitkanli K., Yu Z., Wang X., Lo E. H., van Leyen K. (2010) Increased nuclear apoptosis-inducing factor after transient focal ischemia: a 12/15-lipoxygenase-dependent organelle damage pathway. J. Cereb. Blood Flow Metab. 30, 1157–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Leyen K., Arai K., Jin G., Kenyon V., Gerstner B., Rosenberg P. A., Holman T. R., Lo E. H. (2008) Novel lipoxygenase inhibitors as neuroprotective reagents. J. Neurosci. Res. 86, 904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Foerch C., Arai K., Jin G., Park K. P., Pallast S., van Leyen K., Lo E. H. (2008) Experimental model of warfarin-associated intracerebral hemorrhage. Stroke 39, 3397–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yigitkanli K., Pekcec A., Karatas H., Pallast S., Mandeville E., Joshi N., Smirnova N., Gazaryan I., Ratan R. R., Witztum J. L., Montaner J., Holman T. R., Lo E. H., van Leyen K. (2012) Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann. Neurol. In press; doi: 10.1002/ana.23734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shirvan A., Ziv I., Fleminger G., Shina R., He Z., Brudo I., Melamed E., Barzilai A. (1999) Semaphorins as mediators of neuronal apoptosis. J. Neurochem. 73, 961–971 [DOI] [PubMed] [Google Scholar]
- 42. Piomelli D., Wang J. K., Sihra T. S., Nairn A. C., Czernik A. J., Greengard P. (1989) Inhibition of Ca2+/calmodulin-dependent protein kinase II by arachidonic acid and its metabolites. Proc. Natl. Acad. Sci. U. S. A. 86, 8550–8554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Piomelli D., Volterra A., Dale N., Siegelbaum S. A., Kandel E. R., Schwartz J. H., Belardetti F. (1987) Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature 328, 38–43 [DOI] [PubMed] [Google Scholar]
- 44. Nishiyama M., Hoshino A., Tsai L., Henley J. R., Goshima Y., Tessier-Lavigne M., Poo M. M., Hong K. (2003) Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 423, 990–995 [DOI] [PubMed] [Google Scholar]
- 45. Clarkson A. N., Carmichael S. T. (2009) Cortical excitability and post-stroke recovery. Biochem. Soc. Trans. 37, 1412–1414 [DOI] [PubMed] [Google Scholar]
- 46. van Leyen K., Kim H. Y., Lee S. R., Jin G., Arai K., Lo E. H. (2006) Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke 37, 3014–3018 [DOI] [PubMed] [Google Scholar]
- 47. Goodman C. S. (1996) Mechanisms and molecules that control growth cone guidance. Annu. Rev. Neurosci. 19, 341–377 [DOI] [PubMed] [Google Scholar]
- 48. Serini G., Valdembri D., Zanivan S., Morterra G., Burkhardt C., Caccavari F., Zammataro L., Primo L., Tamagnone L., Logan M., Tessier-Lavigne M., Taniguchi M., Puschel A. W., Bussolino F. (2003) Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424, 391–397 [DOI] [PubMed] [Google Scholar]
- 49. Neufeld G., Cohen T., Shraga N., Lange T., Kessler O., Herzog Y. (2002) The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc. Med. 12, 13–19 [DOI] [PubMed] [Google Scholar]
- 50. Neufeld G., Kessler O. (2008) The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer 8, 632–645 [DOI] [PubMed] [Google Scholar]
- 51. Soker S., Takashima S., Miao H. Q., Neufeld G., Klagsbrun M. (1998) Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92, 735–745 [DOI] [PubMed] [Google Scholar]
- 52. Zhang Z. G., Tsang W., Zhang L., Powers C., Chopp M. (2001) Up-regulation of neuropilin-1 in neovasculature after focal cerebral ischemia in the adult rat. J. Cereb. Blood Flow Metab. 21, 541–549 [DOI] [PubMed] [Google Scholar]
- 53. Pasterkamp R. J., Giger R. J., Ruitenberg M. J., Holtmaat A. J., De Wit J., De Winter F., Verhaagen J. (1999) Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol. Cell. Neurosci. 13, 143–166 [DOI] [PubMed] [Google Scholar]
- 54. Jin G., Arai K., Murata Y., Wang S., Stins M. F., Lo E. H., van Leyen K. (2008) Protecting against cerebrovascular injury: contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 39, 2538–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nishiyama M., Okamoto H., Watanabe T., Hori T., Sasaki T., Kirino T., Shimizu T. (1998) Endothelium is required for 12-hydroperoxyeicosatetraenoic acid-induced vasoconstriction. Eur. J. Pharmacol. 341, 57–63 [DOI] [PubMed] [Google Scholar]
- 56. Carmichael S. T., Archibeque I., Luke L., Nolan T., Momiy J., Li S. (2005) Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp. Neurol. 193, 291–311 [DOI] [PubMed] [Google Scholar]
- 57. Hou S. T., Keklikian A., Slinn J., O'Hare M., Jiang S. X., Aylsworth A. (2008) Sustained up-regulation of semaphorin 3A, Neuropilin1, and doublecortin expression in ischemic mouse brain during long-term recovery. Biochem. Biophys. Res. Commun. 367, 109–115 [DOI] [PubMed] [Google Scholar]
- 58. Stroemer R. P., Rothwell N. J. (1998) Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1β in the rat. J. Cereb. Blood Flow Metab. 18, 833–839 [DOI] [PubMed] [Google Scholar]
- 59. Grundy R. I., Rothwell N. J., Allan S. M. (2002) Site-specific actions of interleukin-1 on excitotoxic cell death in the rat striatum. Brain Res. 926, 142–148 [DOI] [PubMed] [Google Scholar]
- 60. Kunz A., Abe T., Hochrainer K., Shimamura M., Anrather J., Racchumi G., Zhou P., Iadecola C. (2008) Nuclear factor-κB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J. Neurosci. 28, 1649–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bazan N. G., Marcheselli V. L., Cole-Edwards K. (2005) Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Ann. N. Y. Acad. Sci. 1053, 137–147 [DOI] [PubMed] [Google Scholar]
- 62. Belayev L., Khoutorova L., Atkins K. D., Bazan N. G. (2009) Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke 40, 3121–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marcheselli V. L., Hong S., Lukiw W. J., Tian X. H., Gronert K., Musto A., Hardy M., Gimenez J. M., Chiang N., Serhan C. N., Bazan N. G. (2003) Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 278, 43807–43817 [DOI] [PubMed] [Google Scholar]
- 64. Nie D., Tang K., Diglio C., Honn K. V. (2000) Eicosanoid regulation of angiogenesis: role of endothelial arachidonate 12-lipoxygenase. Blood 95, 2304–2311 [PubMed] [Google Scholar]
- 65. Nie D., Krishnamoorthy S., Jin R., Tang K., Chen Y., Qiao Y., Zacharek A., Guo Y., Milanini J., Pages G., Honn K. V. (2006) Mechanisms regulating tumor angiogenesis by 12-lipoxygenase in prostate cancer cells. J. Biol. Chem. 281, 18601–18609 [DOI] [PubMed] [Google Scholar]
- 66. Zafiriou M. P., Deva R., Ciccoli R., Siafaka-Kapadai A., Nigam S. (2007) Biological role of hepoxilins: upregulation of phospholipid hydroperoxide glutathione peroxidase as a cellular response to oxidative stress? Prostaglandins Leukot. Essent. Fatty Acids 77, 209–215 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.