

Abstract
Pasteurella multocida is the causative agent of a number of epizootic and zoonotic diseases. Its major virulence factor associated with atrophic rhinitis in animals and dermonecrosis in bite wounds is P. multocida toxin (PMT). PMT stimulates signal transduction pathways downstream of heterotrimeric G proteins, leading to effects such as mitogenicity, blockade of apoptosis, or inhibition of osteoblast differentiation. On the basis of Gαi2, it was demonstrated that the toxin deamidates an essential glutamine residue of the Gαi2 subunit, leading to constitutive activation of the G protein. Here, we studied the specificity of PMT for its G-protein targets by mass spectrometric analyses and by utilizing a monoclonal antibody, which recognizes specifically G proteins deamidated by PMT. The studies revealed deamidation of 3 of 4 families of heterotrimeric G proteins (Gαq/11, Gαi1,2,3, and Gα12/13 of mouse or human origin) by PMT but not by a catalytic inactive toxin mutant. With the use of G-protein fragments and chimeras of responsive or unresponsive G proteins, the structural basis for the discrimination of heterotrimeric G proteins was studied. Our results elucidate substrate specificity of PMT on the molecular level and provide evidence for the underlying structural reasons of substrate discrimination.—Orth, J. H. C., Fester, I., Siegert, P., Weise, M., Lanner, U., Kamitani, S., Tachibana, T, Wilson, B. A., Schlosser, A., Horiguchi, Y., Aktories, K. Substrate specificity of Pasteurella multocida toxin for α subunits of heterotrimeric G proteins.
Keywords: PMT, GTPase domain, helical domain, deamidation, oncogene
The 146-kDa toxin from Pasteurella multocida (PMT) is a major virulence factor responsible for a number of the severe symptoms associated with various zoonotic and epizootic diseases in wild and domestic animals, including pasteurellosis and bite-wound dermonecrosis. In swine and rabbits, PMT exposure leads to atrophic rhinitis, which is characterized by destruction of the nasal turbinates (1–3). Intoxication of mammalian cells by PMT leads to increased total inositol phosphate levels due to activation of phospholipase Cβ (PLCβ; ref. 4). PMT exhibits strong mitogenic (5, 6) and antiapoptotic effects in various cell lines (7) and alters gene expression by activation of calcium (8), mitogen-activated protein (MAP) kinase (9, 10), and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) (11, 12) signaling pathways. A number of these pathways are involved in tumorigenesis, in particular those leading to sustained proliferative signaling or impaired apoptosis (13). These special features of PMT action led to the hypothesis of a link between bacterial toxins and cancer (14, 15). The cellular effects of PMT are induced by the activation of heterotrimeric G proteins of different families. Increased PLCβ activity is caused by PMT-induced activation of Gαq (4). Initial gene deletion studies and reconstitution experiments, using Gαq/11-deficient mouse embryonic fibroblasts (MEFs), showed PMT-induced signaling via Gαq, whereas the toxin did not activate PLCβ via the closely related Gα11 (10, 16). In addition to activation of Gαq, PMT can inhibit adenylyl cyclase activity through activation of Gαi (17) and can induce RhoA-mediated actin stress fiber formation through activation of Gα13 of the G12/13 family (18); however, it was not determined whether PMT could discriminate between the G12 and G13 proteins.
PMT is an AB toxin, consisting of a receptor binding and translocation domain at the N terminus and a biological active domain at the C terminus (19, 20). The crystal structure of a C-terminal fragment of PMT (aa 569–1285) revealed 3 domains (C1–C3) (21). Whereas the function of domain C2 is still enigmatic, domain C1 is involved in membrane targeting (22), and domain C3 is responsible for the biological activity (21, 23). The latter domain contains an active site catalytic triad, consisting of the essential amino acids Cys-1165 (19, 24), His-1205 (25, 26), and Asp-1220. Recently, we identified the molecular mechanism of PMT as the deamidation of a specific Gln residue, which is essential for catalyzing GTP hydrolysis in the α subunits of targeted G proteins (27). The resulting deamidation by PMT constitutively activates the α subunit of heterotrimeric G proteins.
The family of heterotrimeric G proteins consists of 4 major families: Gαs, Gαi, Gαq, and Gα12/13 (28). So far, the substrate specificity of PMT was deduced indirectly from the effects of PMT on G-protein-dependent signal transduction, e.g., PMT-induced activation of PLCβ1 (4), activation of RhoA (18), or inhibition of adenylyl cyclase (17). To unambiguously clarify the substrate specificity of PMT, we have now analyzed the PMT-induced deamidation of Gα subunits directly by a combination of mass spectrometry (MS) and immunoblot analysis using a monoclonal antibody, which selectively recognizes the deamidated Gln in the switch II region of Gα proteins (29). To further refine substrate specificity of PMT, we studied the effects of PMT on Gα-protein chimeras and on fragments of Gαi2, consisting of the Ras-like GTPase domain only.
MATERIALS AND METHODS
Materials
PCR primers were from Apara (Denzlingen, Germany). All other reagents were of analytical grade and purchased from commercial sources.
Cell culture, transfection, virus production, and transduction
HEK-293 cells were transfected using PEI as described previously (30). MEFs derived from wild-type (WT) or Gαq/11-gene-deficient mice were cultured as described previously (31, 32). The retroviral vector was produced as described previously (18). In brief, HEK-293T cells were cotransfected with pMD-G, pMD-g/p, and the retroviral transfer vector, using the calcium phosphate method. The supernatant was collected after 3 d and centrifuged to spin down cellular debris. The virus-containing medium was filtered. Cells were infected in the presence of polybrene. Protein expression was monitored by immunoblot analysis. Caco-2 cells were cultured in DMEM supplemented with 10% FCS, 1% nonessential amino acids, and 1% sodium pyruvate.
Plasmids and retroviral vector construction
cDNA of mouse Gα11 was from Dr. B. Nürnberg (Universität Tübingen, Tübingen, Germany), and cDNA of bovine Gαs was from Dr. C. Kleuss (Freie Universität Berlin, Berlin, Germany). The plasmids pMD-G and pMD-g/p (33) were kindly provided by Dr. R. Mulligan (Harvard Medical School, Boston, MA, USA). The plasmid pLNCX2 was purchased from Clontech (Heidelberg, Germany). The pcDNA3.1 mammalian expression vectors, encoding human Gαi1, Gαi2, Gαi3, Gαs, Gα11, Gα12, and Gα13, were obtained from Missouri S&T cDNA Resource Center (http://www.cDNA.org).
The plasmid encoding the chimera Gαq-Gαs (Gαq1–216-Gαs221–380; human Gαq; and bovine Gαs) was generated by splicing using overlap extension and cloned into pcDNA3.1 without the His tag. Only one of the two complementary overlapping primers used for the chimera is listed: Gαq1–216-Gαs221–380, 5′-TCAGAGAGAAGAAAATGGATCCAATGCTTCAATGAT-3′.
Gαi1 and Gαi3 were cloned into the bacterial expression vector pET41a by standard cloning techniques. The mutations in the previously described Gαi2-pET41a vector (27) were introduced by site-directed mutagenesis. Only one of the complementary primers used is listed: Gαi2S207D, 5′-GGTGGTCAGCGGGATGAGCGGAAGAAGTGG-3′; Gαi2E208Q, 5′-GGTCAGCGGTCTCAACGGAAGAGGTGGATC-3′.
Fragments of Gαi2, deletion of the helical domain (HD; Gαi2ΔHD) and the switch II region of Gαi2 (Gαi2swII), were cloned into the pET41a vector by standard PCR techniques. Gαi2ΔHD consists of the GTPase domain, whereas the HD (residues 61–175) was replaced by a spacer coding for Ser-Ala-Gly-Ala. Gαi2swII comprises the region directly around the switch II region (residues 184–218).
An internal flag tag was introduced at position 139 of Gα13 by replacing Thr-139 or at position 121 of Gαi1 by replacing Thr-121 with the sequence SGGGGYPDYKDDDDKGGGGS, using standard PCR cloning procedures.
MS analysis
For in-gel digestion, the excised gel bands were destained with 30% acetonitrile, shrunk with 100% acetonitrile, and dried in a vacuum concentrator (Concentrator 5301; Eppendorf, Hamburg, Germany). Digests with trypsin were performed overnight at 37°C in 0.05 M NH4HCO3 (pH 8). About 0.1 μg of protease was used for each gel band. Peptides were extracted from the gel slices with 5% formic acid. All LC-MS/MS analyses were performed with the 1200 Agilent Chip-HPLC system (Agilent Technologies, Böblingen, Germany), either coupled to a quadrupole time-of-flight (Q-TOF; Agilent 6520) or an ion trap (Agilent 6340) mass spectrometer. Peptides were separated on the HPLC-Chip with an analytical column of 75 μm inner diamter and 150 mm length and a 40 nl trap column, both packed with Zorbax 300SB C-18 (5 μm particle size; Agilent). Peptides were eluted with a linear acetonitrile gradient with 1%/min at a flow rate of 300 nl/min (starting with 3% acetonitrile). The Q-TOF spectrometer was operated in the 2 GHz extended dynamic range mode. MS/MS analyses were performed using data-dependent acquisition mode. After a MS scan (2 spectra/s), a maximum of 3 peptides were selected for MS/MS (2 spectra/s). Singly charged precursor ions were excluded from selection. Internal calibration was applied using one reference mass. LC-MS/MS analyses on the ion trap were performed using data-dependent acquisition mode. After a MS scan (standard enhanced mode), a maximum of 3 peptides were selected for MS/MS (collision-induced dissociation, standard enhanced mode). The automated gain control was set to 350,000. The maximum accumulation time was set to 300 ms.
Protein expression
Expression of WT or catalytically inactive mutants (C1165S) of PMT or PMT fragment (PMT-C) or coexpression with Gαi1, Gαi3, or Gαi2 mutants and deletions was performed as described previously (19, 27).
Immunoprecipitation of Gα proteins
Cells were washed twice with cold PBS and lysed in 50 mM Na-HEPES (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and complete protease inhibitor cocktail (Roche, Mannheim, Germany) at 4°C for 1 h. After centrifugation at 3500 g for 10 min at 4°C, the supernatant was used for immunoprecipitation. To remove proteins that bind nonspecifically to protein G-Sepharose, lysates were incubated with protein A/G-Sepharose (sc-2003; Santa Cruz Biotechnology, Heidelberg, Germany) for 30 min at 4°C. After centrifugation at 1000 g for 1 min, the supernatant was used for immunoprecipitation by incubation overnight at 4°C with the indicated antibody and protein G-Sepharose 4B (096K1347; Sigma-Aldrich, Taufkirchen, Germany). Sepharose beads were collected and washed with lysis buffer. Bound proteins were released by treating the Sepharose beads with 15 μl 2× Laemmli-buffer for 30 min at 50°C and followed by 5 min at 85°C. Sepharose beads were separated from eluted proteins by centrifugation, and the proteins were further analyzed by SDS-PAGE and immunoblotting.
Immunoblot analysis
For immunoblotting, samples were subjected to SDS-PAGE and transferred onto polyvinylidene difluoride membrane. Gαq, Gα12, Gαs, and Gαq/11 antibodies were purchased from Santa Cruz Biotechnology. Gαi and Gαpan antibodies were obtained from Cell Signaling Technology (Frankfurt, Germany). Flag-tag (M2), and tubulin antibodies were purchased from Sigma-Aldrich. Deamidation-specific antibody anti-Gαq Q209E (3G3) was used as described previously (29). Binding of the appropriate horseradish peroxidase-coupled secondary antibody was detected by using enhanced chemiluminescent detection reagent and the LAS-3000 imaging system (Fujifilm, Düsseldorf, Germany).
RESULTS
PMT and Gαi family members
The Gαi family of heterotrimeric G proteins consists of Gαi1, Gαi2, and Gαi3, inter alia (28). Since previous studies showed that PMT deamidates Gαi2, leading to an inhibition of adenylyl cyclase (17, 27), we asked whether PMT also activates Gαi1 and Gαi3 by deamidation. To this end, an active fragment of PMT (PMT-C) was coexpressed in Escherichia coli with Gαi1 or Gαi3. The G-protein α subunits were tested for functionality by a nucleotide-binding assay and then subjected to MS analysis (Fig. 1 and Supplemental Fig. S1). MS analysis identified a tryptic peptide of Gαi1 corresponding to aa 198–205 (m/z 455.2162+). This peptide included the Gln residue (Gln-204) crucial for GTP hydrolysis. A second peptide was also identified with a mass shift of 1 Da (m/z 455.7082+). Tandem MS analysis identified a Glu residue at position 204, indicating that Gln-204 had been deamidated by PMT. Similar results were obtained for Gαi3 (Fig. 1B). No relevant deamidation product was identified when Gαi1 or Gαi3 was expressed without PMT (Fig. 1A, B).
Figure 1.
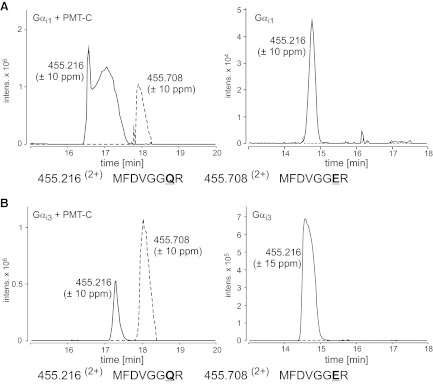
Gαi family members are substrates of PMT. Gαi proteins were coexpressed with an active PMT fragment and subjected to MS analysis. Left panels: combined extracted ion chromatograms (Q-TOF data) for m/z 455.216 (±10 ppm; solid line) and 455.708 (±10 ppm; dashed line), corresponding to the tryptic peptides MFDVGGQR and MFDVGGER (aa 198–205) of Gαi1 (A, left panel) and Gαi3 (B, left panel). The presence of the tryptic peptide MFDVGGER indicates deamidation of Gαi1 and Gαi3 at the essential Gln-204 by PMT. Right panels: no relevant deamidation product was detectable when Gαi1 (A) and Gαi3 (B) were expressed without PMT.
In addition, we utilized a recently described monoclonal antibody (29), which specifically recognizes the deamidated state of the Gln residue in position 204 of Gαi. This anti-QE antibody was engineered to detect the switch II region of Gαq after PMT-induced deamidation of the crucial Gln residue. Due to the high sequence identity in the switch II region of Gα proteins, the anti-QE antibody also recognizes other deamidated forms of G-protein family members, such as Gαi or Gαs. The specificity of the antibody was verified by using mutants of Gαi (e.g., Gαi1Q204E), which mimic the toxin-induced deamidation (29). To study the substrate specificity of PMT toward Gαi family members, Gαi1, Gαi2, and Gαi3 were ectopically expressed in HEK-293 cells. Gα-expressing cells were intoxicated with PMT, and cell lysates were analyzed by immunoblotting for deamidated G proteins. Overexpression of Gαi1-3 largely increased the immunoblot signal by the QE-specific antibody, indicating a PMT-induced deamidation of the ectopically expressed G proteins (Fig. 2A). Thus, the data indicate that PMT accepts all Gαi family members tested as substrates. Additionally, we performed a time course of PMT-induced deamidation. We treated Caco-2 cells for 1 h with the toxin and, subsequently, incubated the cells for up to 7 d. We detected deamidation of G proteins up to 6 d after initial PMT treatment (Fig. 2B).
Figure 2.

Determination of the substrate specificity of PMT using a monoclonal anti-QE antibody that detects toxin-deamidated Gα proteins. A) HEK-293 cells were transfected with pcDNA3-based plasmids expressing the indicated Gα subunits. After 24 h of incubation, the cells were treated with or without PMT (1 nM) for a further 24 h. Cells were lysed and subjected to SDS-PAGE, followed by immunoblotting with monoclonal rat anti-Gαq Q209E (3G3; anti-QE), polyclonal rabbit anti-Gαi, polyclonal rabbit anti-Gαs, and monoclonal mouse anti-tubulin antibody as described in Materials and Methods. Note that control HEK-293 cells (pcDNA) and recombinant Gαs-expressing cells contain small amounts of endogenous G proteins, which are deamidated by PMT and labeled with anti-QE (double bands in top panel, lanes 2 and 10). The double bands labeled by anti-Gαs indicate the two isoforms of Gαs. B) CaCo-2 cells were treated with PMT (1 nM) for 1 h. Toxin-containing medium was discarded, and cells were washed with medium and incubated for indicated times. Cells were lysed and subjected to SDS-PAGE, followed by immunoblotting with monoclonal rat anti-Gαq Q209E (3G3; anti-QE) and polyclonal rabbit anti-Gαq/11 antibody.
PMT acts on the Gαq/11 family
Activation of Gαq-dependent signaling pathways is a consequence of PMT intoxication (4, 16). Therefore, we wanted to analyze the direct action of the toxin on the G proteins involved. For this purpose, Gαq was immunoprecipitated from PMT-treated cells by a specific antibody (Fig. 3C), and then subjected to MS analysis of tryptic peptides, as described previously (27). As shown in Fig. 3A, Gln-209 of Gαq was deamidated by PMT intoxication. The deamidation of Gln-209 to Glu was verified by a 1-Da mass shift of the peptide and a retention time shift of ∼1 min (m/z 431.22+ 203-MVDVGGQR-210; m/z 431.72+ 203-MVDVGGER-210). In line with these findings are the corresponding MS/MS spectra (Supplemental Fig. S2).
Figure 3.
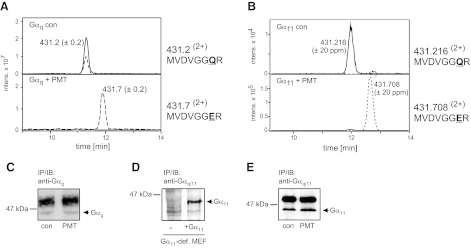
PMT substrate specificity with respect to Gαq and Gα11. A) Combined extracted ion chromatograms (ion trap data) for m/z 431.2 ± 0.2 (solid line) and 431.7 ± 0.2 (dashed line), corresponding to the doubly protonated precursors of the tryptic peptides MVDVGGQR and MFDVGGER (aa 203–210) of Gαq. Top panel: endogenous Gαq from untreated WT-MEFs. Bottom panel: endogenous Gαq from PMT-treated WT-MEFs. B) Combined extracted ion chromatograms (Q-TOF data) for m/z 431.216 ± 20 ppm (solid line) and 431.708 ± 20 ppm (dashed line), corresponding to the doubly protonated precursors of the tryptic peptides MVDVGGQR and MFDVGGER (aa 203–210) of Gα11. Gα11 was precipitated from Gαq/11-deficient MEFs, which were transduced with retrovirus encoding for Gα11.Top panel: Gα11 from untreated cells. Bottom panel: Gα11 from PMT-treated cells. Deamidation of Gln-209 of Gαq and Gα11 is complete, and MVDVGGQR is no longer detectable after PMT treatment. C) Immunoblot of endogenous Gαq after immunoprecipitation with a specific Gαq antibody. Precipitations were performed from untreated MEF (con) or from PMT-treated MEF (1 nM, 18 h). D) Detection of Gα11 in Gαq/11-deficient MEFs (−) or in Gαq/11-deficient MEFs transduced with Gα11-encoding retrovirus (+Gα11). E) Immunoblot of Gα11 after immunoprecipitation with anti-Gαq/11 antibody from Gαq/11-deficient MEFs transduced with Gα11-encoding retrovirus.
Gα11 also belongs to the Gαq family and shares 89% amino acid sequence identity with Gαq. To avoid cross contamination of Gα11 with Gαq during immunoprecipitation, we utilized MEF cells deficient for Gαq and Gα11. In Gαq/11-deficient MEFs, Gα11 was expressed by retroviral transfection (Fig. 3D). Immunoprecipitation of Gα11 from PMT-treated cells was performed (Fig. 3E). As was observed for Gαq, MS analysis revealed deamidation of Gln-209 of Gα11 (Fig. 3B and Supplemental Fig. S2), indicating that both Gαq and Gα11 are targeted by PMT. These data are in contrast to previous results, suggesting that only Gαq- but not Gα11-mediated signaling is affected by PMT (10, 16).
Gα12/13 are substrates of PMT
RhoA is activated by Gα12/13- and Gαq/11-dependent signaling pathways (32, 34). The Gα subunits cause activation of specific RhoA guanine nucleotide exchange factors (GEFs), such as p115RhoGEF or p63RhoGEF, which are specific for Gα12/13 or Gαq/11, respectively (35, 36). Previously, we reported the involvement of Gα12/13 in PMT-induced activation of RhoA (18). However, these studies did not permit distinction between Gα12 and Gα13 as substrates of PMT. To determine whether Gα12 and/or Gα13 are targets of PMT, Gα13 was ectopically expressed with an internal flag tag. Gα13 was precipitated from PMT-treated or untreated cells by using an anti-flag antibody (Fig. 4B). MS analysis of the immunoprecipitated flag-tagged Gα13 subunits identified a 1-Da shift of 2 tryptic peptides, encompassing the amino acid residues 220 to 227 that harbor the essential Gln residue (control peptide precursor with m/z 431.2162+ (220-MVDVGGQR-227) and toxin-modified peptide precursor with m/z 431.7082+ (220-MVDVGGER-227). Tandem MS analysis confirmed deamidation of Gln-226 in the switch II region of Gα13 to Glu (Fig. 4 and Supplemental Fig. S3).
Figure 4.
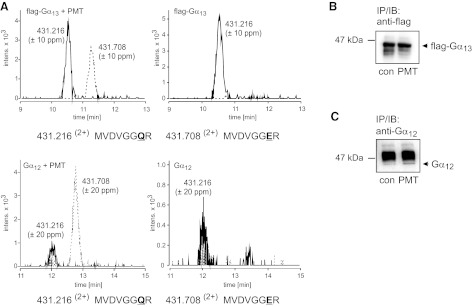
PMT substrate specificity with respect to Gα12 and Gα13. A) Combined extracted ion chromatograms for m/z 431.216 ± 10 ppm (solid line) and 431.708 ± 10 ppm (dashed line), corresponding to the doubly protonated precursors of the tryptic peptides MVDVGGQR and MFDVGGER (Gα13: aa 220–227, Gα12: aa 225–232) of Gα13 and Gα12. Top panel: Gα13 from PMT-treated (left) or untreated cells (right; Q-TOF data). Bottom panel: Gα12 from PMT-treated (left) or untreated cells (right; Q-TOF data). The 1-Da shift demonstrates the deamidation of Gln-226 (Gα13) and Gln-231 (Gα12) to Glu. The deamidated form of the tryptic peptide (MVDVGGER) is only detectable when Gα13 or Gα12 is precipitated from PMT-treated cells. In addition, unmodified peptide (MVDVGGQR) is present in precipitates from PMT-treated cells. B) Gα13 cDNA was engineered to harbor an internal flag tag. The construct was ectopically expressed in HEK-293 cells. After treatment with or without PMT (1 nM, 18 h), cells were lysed, and flag-tagged protein was immunoprecipitated, isolated, and subjected to MS analysis. Shown is an immunoblot of Gα13 after immunoprecipitation with anti-flag-tag antibody from PMT-treated or untreated (con) cells. C) Gα12 was ectopically expressed in HEK-293 cells. After treatment with or without PMT (1 nM, 18 h), cells were lysed, and Gα12 protein was immunoprecipitated, using a specific anti-Gα12 antibody, isolated, and subjected to MS analysis. Immunoblot shows Gα12 after immunoprecipitation with anti-Gα12 antibody from PMT-treated or untreated (con) cells. The strong band above Gα12 is part of the antibody used for immunoprecipitation.
In addition, we overexpressed Gα12 (Fig. 4C) to clarify whether both members of the Gα12/13 family are substrates of PMT. MS analysis of Gα12 from PMT-intoxicated or untreated cells again revealed two tryptic peptides, encompassing amino acid residue 225 to 232, that harbor the crucial Gln-231 of the switch II region of Gα12 (m/z 431.2162+ 225-MVDVGGQR-232; m/z 431.7082+ 225-MVDVGGER-232). Tandem MS analysis indicated deamidation of Gln-231 (Fig. 4 and Supplemental Fig. S3).
G-protein mutants and fragments
The switch II region of α subunits of heterotrimeric G proteins is highly conserved throughout all G-protein family members, with only minor sequence differences (Fig. 5A). We tested whether changes in this region might define the substrate specificity of PMT. Gαs, which is not a target for PMT (see below), possesses an Asp two residues upstream of the crucial Gln, while the known PMT substrates have Ser at this position. We changed the corresponding Ser-207 in Gαi2 to Asp (Gααi2S207D). In addition, Gα12, which is a target of PMT, has a unique Gln three residues upstream of the crucial Gln, whereas all other PMT substrates have Glu at the equivalent position. To detect any unspecific effect of mutations within the switch II region, we also replaced the corresponding Glu of Gαi2 to Gln (Gααi2E208Q). The respective Gαi2 mutants were coexpressed with the active C-terminal fragment of PMT (PMT-Cwt) in E. coli and were tested for functionality by a nucleotide-binding assay (Supplemental Fig. S4A). Tryptic peptides of the isolated G proteins were analyzed by HPLC and MS/MS. As shown in Fig. 5B, C (left panels), two peptides, 199-MFDVGGER-206 (m/z 455.7082+) and 199-MFDVGGQR-206 (m/z 455.2162+), were obtained by MS analysis from both mutants. The peptides differed by 1 Da, indicating the specific deamidation of Gln-205 of Gαi2 by PMT (Supplemental Fig. S4). No relevant deamidation product was identified when Gααi2S207D or Gααi2E208Q was expressed without PMT (Fig. 5B, C; right panels). These results showed that both of the switch II mutant Gαi2 proteins (Gααi2S207D and Gααi2E208Q) were still substrates of PMT, indicating that the residues changed have no major effect on substrate recognition by PMT.
Figure 5.
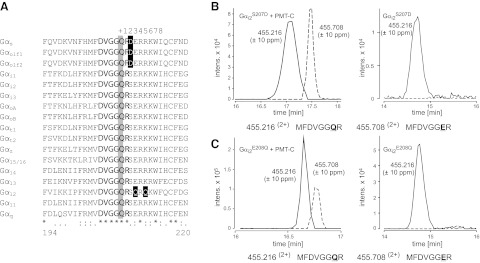
Switch II of G-protein α subunits is not involved in PMT substrate specificity. A) Alignment of amino acid sequences of the switch II region in the α subunits of mouse heterotrimeric GTPases. Nucleotide sequences are obtained from the U.S. National Center for Biotechnology Information (NCBI): Gαs (P63094), Gαolf1 (NP_034437), Gαolf2 (NP_796111), Gαi1 (NP_034435), Gαi2 (AAH65159), Gαi3 (NP_034436), GαoA (NP_034438), GαoB (P18873.3), Gαt1 (NP_032166), Gαt2 (NP_032167), Gαz (NP_034441), Gα15 (NP_034434), Gα14 (NP_032163), Gα13 (NP_034433), Gα12 (NP_034432), Gα11 (NP_034431), and Gαq (NP_032165). Numbers below the alignment correspond to the amino acid positions of Gαq. The active site Gln (at position 209 in Gαq) is indicated by a gray box, highly conserved flanking residues by boldface type, and flanking residues that result in notable charge differences by are indicated in reverse type on a black box. Asterisks denote identical amino acid residues; colons denote highly conserved residues; periods denote conserved residues. Numbers in the top row indicate positions downstream of the essential Gln. B, C) Combined extracted ion chromatograms (Q-TOF data) for m/z 455.216 ± 10 and 455.708 ± 10 ppm, corresponding to the tryptic peptides MFDVGGQR and MFDVGGER (aa 199–206) of Gαi2S207D (B) and Gαi2E208Q (C). Gαi2 mutants were coexpressed with an active PMT fragment and subjected to MS analysis. The presence of the tryptic peptide MFDVGGER indicates deamidation of Gαi2S207D and Gαi2E208Q at Gln-205 by PMT. No relevant deamidation product was detectable when Gαi2S207D and Gαi2E208Q were expressed without PMT (right panels).
Because substrate specificity of PMT is dependent on the N terminus of Gα subunits, including their HDs, but not on the C terminus (37), which regulates the interaction with effectors, we next addressed whether the interaction of G proteins with specific effectors influences the action of PMT. To this end, we constructed a chimera of the PMT target Gαq with Gαs, which is not a target of PMT (Fig. 6). The chimeric protein consisted of the N terminus of Gαq, including the switch I and switch II regions of GTPase domain and the full HD, fused to the C terminus of the GTPase domain of Gαs (Gαq1–216-Gαs221–380; Fig. 6A). This chimera is still able to activate adenylyl cyclase via Gs-coupled receptors (38). PMT treatment of HEK-293 cells overexpressing the Gαq-Gαs chimera but not Gαq nor Gαs increased cAMP production in a dose-dependent manner (Fig. 6B), indicating stimulation of adenylyl cyclase by PMT via activation of the Gαq-Gαs chimera. We also observed that in pcDNA control transfectants, basal adenylyl cyclase activity was inhibited by PMT. This is in line with our previous findings, showing that PMT activates Gαi to inhibit adenylyl cyclase activity (17, 27). The data obtained with the Gαq-Gαs chimera suggest that PMT activation overrode the inhibition of adenylyl cyclase caused by toxin-induced activation of Gαi. To verify that the observed activation of adenylyl cyclase was indeed due to PMT-induced deamidation of the Gαq-Gαs chimera, the chimera was immunoprecipitated from toxin-treated HEK-293 cells (Fig. 6C) and analyzed by LC-MS/MS, which revealed that PMT catalyzed the deamidation of Gln-209 of the Gαq-Gαs chimera (Fig. 6D and Supplemental Fig. S5). Two peptides were identified, corresponding to the unaffected chimera (m/z 431.2162+) and the toxin-deamidated form (m/z 431.7082+). Altogether, these results confirmed the involvement of the N terminus of Gα subunits in substrate recognition by PMT. We further tested whether Gαs could serve as a target of PMT. To this end, Gαs was immunoprecipitated from PMT-treated MEFs and subjected to LC-MS/MS analysis, but no deamidation product was detectable (Fig. 6E, F and Supplemental Fig. S5). In line with this finding, we did not detect deamidation of Gαs overexpressed in HEK-293 cells by using the QE-antibody (Fig. 2A). Both results confirm previous findings that Gαs is not a substrate of PMT.
Figure 6.
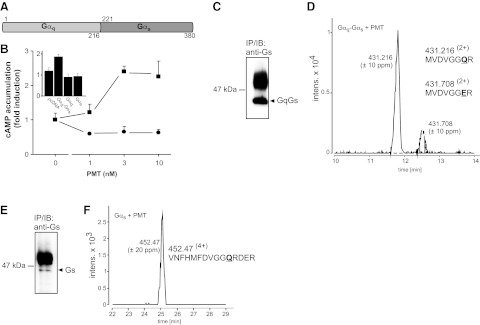
PMT-induced activation of Gαs-containing chimera. A) Scheme of Gαq-Gαs chimera. N-terminal part including the HD and switch II of Gαq (aa 1–216) were fused to the C-terminal effector-binding domain of Gαs (aa 221–380). B) cAMP accumulation was measured after transfection of Gαq-Gαs chimera into HEK-293-cells (squares) or after transfection with an empty pcDNA vector (circles). PMT was added at indicated concentrations for 3 h. Inset: cAMP accumulation of PMT-treated (10 nM, 3 h) cells transfected as indicated. Gαq-and Gαs-overexpressing cells show no increase in cAMP levels after PMT incubation. C) Chimera Gαq-Gαs was immunoprecipitated from PMT-treated cells (1 nM, 18 h) with a Gs antibody against the C-terminal region. Immunoblot shows Gαq-Gαs chimera after immunoprecipitation with anti-Gs antibody. D) Combined extracted ion chromatograms (Q-TOF data) for m/z 431.216 ± 10 and 431.708 ± 10 ppm, corresponding to the doubly protonated precursors of the tryptic peptides MVDVGGQR and MFDVGGER (Gαq-Gαs: aa 203–210) of Gαq-Gαs. The 1-Da shift demonstrates the deamidation of Gln-209 to Glu. E) Endogenous Gαs was immunoprecipitated from PMT-treated MEF (1 nM, 18 h) with a Gs antibody against the C-terminal region. Immunoblot shows Gαs after immunoprecipitation with anti-Gs antibody. F) Combined extracted ion chromatogram (Q-TOF data) for m/z 452.464 ± 20 ppm corresponding to the quadruple protonated precursors of the tryptic peptide VNFHMFDVGGQRDER (Gαs: aa 203–217) of Gαs. No deamidation of Gln-213 was detectable.
To further define the minimal region of the G proteins that can serve as a substrate for PMT, fragments of Gαi2 were constructed (Fig. 7). In Gαi2ΔHD, the HD insert was deleted and consists therefore only of the GTPase domain of Gαi2. Gαi2swII comprises the amino acid residues of the switch II region. Gαi2 and both constructs were coexpressed in E. coli with the active C-terminal fragment of PMT (PMT-Cwt) or the catalytically inactive PMT fragment (PMT-CC1165S). Purified Gαi2 mutant proteins were subjected to immunoblot analysis to detect deamidation of the essential Gln by PMT (Fig. 7B). Gαi2, Gαi2ΔHD, and Gαi2swII were detected by the anti-Gαpan antibody. Gαi2 and Gαi2ΔHD coexpressed with active PMT-Cwt, but not with PMT-CC1165S, were recognized by the anti-QE antibody. Gαi2swII was not detected by the QE-specific antibody under either condition. Gαi2ΔHD or Gαi2swII coexpressed with WT or inactive PMT were also examined by mass spectral analysis (Fig. 7C and Supplemental Fig. S6). Tryptic digestion and LC-MS/MS analysis confirmed PMT-mediated deamidation of the target Gln in Gαi2ΔHD, as evidenced by detection of the deamidated peptide (199-MFDVGGER-206; m/z 455.7082+) vs. the unaffected peptide (199-MFDVGGQR-206; m/z 455.2162+). In the case of Gαi2swII, only the unaffected peptide (199-MFDVGGQR-206; m/z 455.2162+) was detected; no deamidation product was detected.
Figure 7.
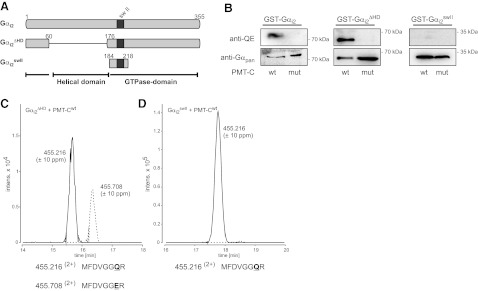
PMT effect on Gαi2 fragments. A) Scheme of Gαi2 fragments. Gαi2 consists of the GTPase domain and an HD insert. In the case of Gαi2ΔHD, the HD (residues 61–175) was deleted and replaced by a short linker (Ser-Ala-Gly-Ala). The construct Gαi2swII comprises only the region around the switch II (residues 184–218). B) GST-fusion proteins of Gαi2, Gαi2ΔHD, and Gαi2swII were coexpressed with active or inactive PMT-C and subjected to immunoblot analysis with monoclonal rat anti-Gαq Q209E (3G3; anti-QE), Gαpan antibody as described in Materials and Methods. Deamidation of Gαi2 and Gαi2ΔHD was detectable. C) Combined extracted ion chromatograms (Q-TOF data) for m/z 455.216 ± 10 ppm (solid line) and 455.708 ± 10 ppm (dashed line), corresponding to the tryptic peptides MFDVGGQR and MFDVGGER (aa 198–205) of Gαi2ΔHD coexpressed with active PMT-Cwt. D) Combined extracted ion chromatograms (Q-TOF data) for m/z 455.216 ± 10 ppm (solid line) and 455.708 ± 10 ppm (dashed line) of Gαi2swII coexpressed with active PMT-Cwt. Only the tryptic peptide MFDVGGQR (m/z 455.216), but no MFDVGGER (m/z 455.708), was identified.
DISCUSSION
Here, we studied the substrate specificity of PMT for α subunits of heterotrimeric G proteins. To analyze which of the Gi family members Gαi1/2/3 serve as substrate of PMT, we utilized the previously described coexpression system of the G-protein α subunit and PMT in E. coli (27). In addition, toxin-catalyzed deamidation of the Gαi family members Gαi1, Gαi2, or Gαi3 was observed after overexpression in HEK-293 cells.
MS analysis data indicate that Gαq and Gα11 are substrates of PMT. With the use of Gq/11 gene-deletion models, it has been suggested that PMT activates Gαq but not Gα11 (10, 16). Recent studies by the laboratory of Horiguchi (29) presented the first evidence that Gα11 might also be a target of PMT. In this study, an antibody specific for the deamidated form of Glu-209 of Gαq/11 was used, but no MS analysis of Gα11 was offered. Previous studies, which identified Gαq but not Gα11 as a target of PMT, were mainly based on activation of PLCβ by the G proteins. One possible explanation for the observed discrepancy could be a difference in the interaction potential of the deamidated/activated forms of Gαq vs. Gα11 with the downstream effector PLCβ. In line with this model is the finding of Kamitani et al. (29) that PMT-induced Gα11-dependent activation of PLCβ was markedly lower than the Gαq-dependent activation of PLCβ.
In intact cells, PMT is a potent activator of Rho proteins (10, 18). It is well known that the activity of the small GTPase RhoA is regulated by heterotrimeric G proteins of the G12/13 family (39). In the case of G12, LARG is stimulated to catalyze the GDP-GTP exchange of RhoA (40). G13 binds p115RhoGEF, which then activates RhoA (41). Studies with Gα12/13-deficient MEFs showed the involvement of Gq/11 in RhoA activation (32) and that p63RhoGEF activates RhoA exclusively via Gq/11 (36).
PMT induces RhoA activation via both families of heterotrimeric G proteins. However, it was not clear whether PMT activated both members of the Gα12/13 family. Here we show by immunoprecipitation of ectopically expressed Gα12 or flag-tagged Gα13 and subsequent LC-MS/MS analysis that PMT induced deamidation of the essential Gln residue in Gα12 (Glu-231) and Gα13 (Glu-226). Therefore, PMT targets both members of the Gα12/13 family.
The observed substrate specificity of PMT prompted us to study additional structural determinants of PMT-targeted Gα subunits. An alignment of responsive (Gi1/2/3, Gq, Gα11, G13, and G12), nonresponsive (Gs), and other G-protein α subunits (Fig. 5A) revealed high amino acid sequence identity within the switch II regions, with only minor changes present in some of the targets. The region upstream of the essential Gln (209 in Gαq) that is modified by the toxin is nearly identical among the α subunits. In contrast, differences occur at some downstream positions; most notably, the conserved Ser at position +2 is an Asp in the unresponsive Gαs/olf family, and a conserved Glu at position +3 is a Gln in the responsive Gα12. However, MS data of the respective mutants revealed that these residues are not sufficient to define substrate recognition by PMT.
Moreover, we studied whether parts of G proteins, which are responsible for direct interaction with their specific effectors, are crucial for PMT activation. We observed that a chimera, consisting of the first 216 amino acid residues of Gαq fused to the C terminus of Gαs (residues 221–380), was activated by PMT. These results indicate that the G-protein effector-binding region is not crucial for PMT action. The intensity of PMT-deamidated Gαq-Gαs in MS analysis is remarkably lower compared with other tested PMT substrates. However, utilized MS techniques do not allow quantification. Therefore, no conclusion can be drawn regarding the effectiveness of deamidation. Based on the interaction sites of Gs with adenylyl cyclase, which mainly encompass the α3-β5 loop of Gs and the switch II region (42), our data suggest that the effector interaction site downstream of the switch II region is not essentially involved in defining substrate specificity of PMT. These findings might offer the possibility to engineer G proteins of specific functions (e.g., for substitution of genetic malfunction), which are deamidated and constitutively activated by PMT.
To clarify the involvement of the HD in substrate recognition and/or specificity, we deleted this domain from Gαi2 (Gαi2ΔHD). The construct was created in analogy to studies with Gαs on the structure/function relationship of the Ras-like GTPase domain and the HD (43). The remaining Ras-like GTPase domain of Gαi2 still harbors the essential Gln residue in the switch II region. Coexpression with active PMT, followed by immunoblot and LC-MS/MS analysis, revealed that the toxin also accepts this deletion mutant as a substrate. In contrast, another mutant of Gαi2, consisting of a small region around switch II itself (Gαi2swII, residues 184–218), was not deamidated by PMT. These results indicate that the HD is not essential for substrate recognition.
Recently, the structure of the active state ternary complex comprised of agonist occupied β-adrenoceptor and Gs was reported (44). Interestingly, this nucleotide-free structure showed a strong displacement of the HD relative to the Ras-like GTPase domain. Westfield et al. (45) proposed that the reorientation of the HD of Gαs facilitates accessibility to the catalytic Arg-201 for ADP-ribosylation by cholera toxin. In addition, the catalytic Gln residue of Gαs is particularly exposed in the nucleotide-free Gαs. There is reasonable evidence (46) that other family members of the heterotrimeric G proteins, including the PMT targets Gαi and Gαq/11, show similar structural changes during nucleotide exchange. The nucleotide-free state with a reoriented HD could present a PMT-accessible switch II Gln residue. Further studies will be needed to clarify whether this nucleotide-free structure of the heterotrimeric complex is the preferred target structure for PMT action.
The relevance of the HD for PMT deamidation should also be reinvestigated with respect to the new structural findings. Alignments of the α subunits of heterotrimeric G proteins show a high similarity in their GTPase domains yet striking differences in the HDs (47). The result that the HD of the PMT target Gαi2 is not necessary for toxin recognition does not exclude a possible negative effect of the HD in the nonresponsive Gαs. In future studies, we will test whether the HD of Gαs is an inhibitory element in the Gαs subunit for PMT-induced deamidation.
Taken together, the studies reported herein extend our knowledge of the substrate specificity of PMT. The toxin deamidates the Gi family members Gαi1/2/3. Furthermore, Gαq and Gα11 and both members of the G12/13 family are deamidated. All these deamidation reactions, catalyzed by PMT, cause persistent activation of the G proteins. Mutations of the crucial Gln residue of Gα subunits, which is involved in GTP hydrolysis, are known to have transforming capability. This is true for Gαi, presumably through activation of the mitogen-activated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK) signaling pathway (48). Because this same Gln of Gαi is a target of PMT and the same pathway is stimulated by PMT (9), it is possible that this contributes to the transforming potential of PMT. Recently, somatic mutations of GNAQ or GNA11, the genes encoding for Gαq and Gα11, were found in uveal melanoma and blue naevi (49, 50). Again, the identical Gln-209 is affected, which is deamidated by PMT. Moreover, GTPase-deficient mutants of Gα12 or Gα13 were described as most powerful transforming Gα subunits (51).
Usually, all these mutants occur at the crucial sites of the switch II Gln, but also at the essential Arg in the switch I region (48, 51). In contrast to G-protein-coupled receptor-induced effects, which occur fast and transiently, GTPase-deficient mutants signal continuously. Therefore, tightly regulated cell functions become deregulated and can contribute to the hallmarks of cancer, such as sustained proliferative signaling, resisting cell death or metastasis (13, 52).
It is fascinating that PMT affects heterotrimeric G proteins at the identical site where oncogenic mutations were identified. Although PMT does not induce gene mutations, deamidation of the crucial Gln in the switch II is long lasting and is apparently terminated only by G-protein degradation (Fig. 2B). Consequently, PMT is already discussed as a potential carcinogen (15).
Supplementary Material
Acknowledgments
This study was financially supported by the Deutsche Forschungsgemeinschaft, SFB 746 (to J.O. and K.A.), and U.S. National Institutes of Health grant R01-AI-038936 (to B.A.W.).
The authors thank Dr. S. Offermanns (Max-Planck-Institut für Herz- und Lungenforschung, Bad Nauheim, Germany) for the kind gift of Gαq/11-deficient cells and Silke Fieber and Petra Bartholome for excellent technical assistance. The authors thank Dr. Mengfei Ho and Dr. Shuhong Luo (University of Illinois at Urbana-Champaign, Urbana, IL, USA) for construction of the plasmids containing flag-tagged Gα proteins.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- GEF
- guanine nucleotide exchange factor
- HD
- helical domain
- MEF
- mouse embryonic fibroblast
- MS
- mass spectrometry
- PLCβ
- phospholipase Cβ
- PMT
- Pasteurella multocida toxin
- Q-TOF
- quadrupole time-of-flight
- WT
- wild type
REFERENCES
- 1. Orth J. H. C., Aktories K. (2010) Pasteurella multocida toxin activates various heterotrimeric G proteins by deamidation. Toxins 2, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilson B. A., Ho M. (2010) Recent insights into Pasteurella multocida toxin and other G-protein-modulating bacterial toxins. Future Microbiol. 5, 1185–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lax A. J., Grigoriadis A. E. (2001) Pasteurella multocida toxin: the mitogenic toxin that stimulates signalling cascades to regulate growth and differentiation. Int. J. Med. Microbiol. 291, 261–268 [DOI] [PubMed] [Google Scholar]
- 4. Wilson B. A., Zhu X., Ho M., Lu L. (1997) Pasteurella multocida toxin activates the inositol triphosphate signaling pathway in Xenopus oocytes via Gqα-coupled phospholipase C-β1. J. Biol. Chem. 272, 1268–1275 [DOI] [PubMed] [Google Scholar]
- 5. Rozengurt E., Higgins T., Chanter N., Lax A. J., Staddon J. M. (1990) Pasteurella multocida toxin: potent mitogen for cultured fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 87, 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilson B. A., Aminova L. R., Ponferrada V. G., Ho M. (2000) Differential modulation and subsequent blockade of mitogenic signaling and cell cycle progression by Pasteurella multocida toxin. Infect. Immun. 68, 4531–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Preuss I., Hildebrand D., Orth J. H., Aktories K., Kubatzky K. F. (2010) Pasteurella multocida toxin is a potent activator of anti-apoptotic signalling pathways. Cell. Microbiol. 12, 1174–1185 [DOI] [PubMed] [Google Scholar]
- 8. Aminova L. R., Wilson B. A. (2007) Calcineurin-independent inhibition of 3T3-L1 adipogenesis by Pasteurella multocida toxin: suppression of Notch1, stabilization of beta-catenin and pre-adipocyte factor 1. Cell. Microbiol. 9, 2485–2496 [DOI] [PubMed] [Google Scholar]
- 9. Seo B., Choy E. W., Maudsley W. E., Miller W. E., Wilson B. A., Luttrell L. M. (2000) Pasteurella multocida toxin stimulates mitogen-activated protein kinase via Gq/11-dependent transactivation of the epidermal growth factor receptor. J. Biol. Chem. 275, 2239–2245 [DOI] [PubMed] [Google Scholar]
- 10. Zywietz A., Gohla A., Schmelz M., Schultz G., Offermanns S. (2001) Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. Involvement of Gq but not G11. J. Biol. Chem. 276, 3840–3845 [DOI] [PubMed] [Google Scholar]
- 11. Orth J. H., Aktories K., Kubatzky K. F. (2007) Modulation of host cell gene expression through activation of STAT transcription factors by Pasteurella multocida toxin. J. Biol. Chem. 282, 3050–3057 [DOI] [PubMed] [Google Scholar]
- 12. Hildebrand D., Walker P., Dalpke A., Heeg K., Kubatzky K. F. (2010) Pasteurella multocida toxin-induced Pim-1 expression disrupts suppressor of cytokine signalling (SOCS)-1 activity. Cell. Microbiol. 12, 1732–1745 [DOI] [PubMed] [Google Scholar]
- 13. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 14. Lax A. J., Thomas W. (2002) How bacteria could cause cancer: one step at a time. Trends Microbiol. 10, 293–299 [DOI] [PubMed] [Google Scholar]
- 15. Lax A. J. (2005) Bacterial toxins and cancer - a case to answer? Nat. Rev. Microbiol. 3, 343–349 [DOI] [PubMed] [Google Scholar]
- 16. Orth J. H., Lang S., Aktories K. (2004) Action of Pasteurella multocida toxin depends on the helical domain of Galphaq. J. Biol. Chem. 279, 34150–34155 [DOI] [PubMed] [Google Scholar]
- 17. Orth J. H., Fester I., Preuss I., Agnoletto L., Wilson B. A., Aktories K. (2008) Activation of Galphai and subsequent uncoupling of receptor-Galphai signaling by Pasteurella multocida toxin. J. Biol. Chem. 283, 23288–23294 [DOI] [PubMed] [Google Scholar]
- 18. Orth J. H., Lang S., Taniguchi M., Aktories K. (2005) Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of Gα proteins, Gαq and Gα12/13. J. Biol. Chem. 280, 36701–36707 [DOI] [PubMed] [Google Scholar]
- 19. Busch C., Orth J., Djouder N., Aktories K. (2001) Biological activity of a C-terminal fragment of Pasteurella multocida toxin. Infect. Immun. 69, 3628–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pullinger G. D., Sowdhamini R., Lax A. J. (2001) Localization of functional domains of the mitogenic toxin of Pasteurella multocida. Infect. Immun. 69, 7839–7850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kitadokoro K., Kamitani S., Miyazawa M., Hanajima-Ozawa M., Fukui A., Miyake M., Horiguchi Y. (2007) Crystal structures reveal a thiol protease-like catalytic triad in the C-terminal region of Pasteurella multocida toxin. Proc. Natl. Acad. Sci. U. S. A. 104, 5139–5144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamitani S., Kitadokoro K., Miyazawa M., Toshima H., Fukui A., Abe H., Miyake M., Horiguchi Y. (2010) Characterization of the membrane-targeting C1 domain in Pasteurella multocida toxin. J. Biol. Chem. 285, 25467–25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aminova L. R., Luo S., Bannai Y., Ho M., Wilson B. A. (2008) The C3 domain of Pasteurella multocida toxin is the minimal domain responsible for activation of Gq-dependent calcium and mitogenic signaling. Protein Sci. 17, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ward P. N., Miles A. J., Sumner I. G., Thomas L. H., Lax A. J. (1998) Activity of the mitogenic Pasteurella multocida toxin requires an essential C-terminal residue. Infect. Immun. 66, 5636–5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orth J. H., Blöcker D., Aktories K. (2003) His1205 and His 1223 are essential for the activity of the mitogenic Pasteurella multocida toxin. Biochemistry 42, 4971–4977 [DOI] [PubMed] [Google Scholar]
- 26. Pullinger G. D., Lax A. J. (2007) Histidine residues at the active site of the Pasteurella multocida toxin. Open Biochem. J. 1, 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orth J. H., Preuss I., Fester I., Schlosser A., Wilson B. A., Aktories K. (2009) Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc. Natl. Acad. Sci. U. S. A. 106, 7179–7184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hepler J. R., Gilman A. G. (1992) G proteins. Trends Biochem. Sci. 17, 383–387 [DOI] [PubMed] [Google Scholar]
- 29. Kamitani S., Ao S., Toshima H., Tachibana T., Hashimoto M., Kitadokoro K., Fukui-Miyazaki A., Abe H., Horiguchi Y. (2011) Enzymatic actions of Pasteurella multocida toxin detected by monoclonal antibodies recognizing the deamidated alpha subunit of the heterotrimeric GTPase G(q). FEBS J. 278, 2702–2712 [DOI] [PubMed] [Google Scholar]
- 30. Ehrhardt C., Schmolke M., Matzke A., Knoblauch A., Will C., Wixler V., Ludwig S. (2006) Polyethylenimine, a cost-effective transfection reagent. Signal Transduct. 6, 179–184 [Google Scholar]
- 31. Offermanns S., Zhao L. P., Gohla A., Sarosi I., Simon M. I., Wilkie T. M. (1998) Embryonic cardiomyocyte hypoplasia and craniofacial defects in G alpha q/G alpha 11-mutant mice. EMBO J. 17, 4304–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vogt S., Grosse R., Schultz G., Offermanns S. (2003) Receptor-dependent RhoA activation in G12/G13-deficient cells. J. Biol. Chem. 278, 28743–28749 [DOI] [PubMed] [Google Scholar]
- 33. Ory D. S., Neugeboren B. A., Mulligan R. C. (1996) A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. U. S. A. 93, 11400–11406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buhl A. M., Johnson N. L., Dhanasekaran N., Johnson G. L. (1995) Gα12 and Gα13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 270, 24631–24634 [DOI] [PubMed] [Google Scholar]
- 35. Sah V. P., Seasholtz T. M., Sagi S. A., Brown J. H. (2000) The role of Rho in G protein-coupled receptor signal transduction. Annu. Rev. Pharmacol. Toxicol. 40, 459–489 [DOI] [PubMed] [Google Scholar]
- 36. Lutz S., Shankaranarayanan A., Coco C., Ridilla M., Nance M. R., Vettel C., Baltus D., Evelyn C. R., Neubig R. R., Wieland T., Tesmer J. J. (2007) Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923–1927 [DOI] [PubMed] [Google Scholar]
- 37. Orth J. H., Lang S., Preuss I., Milligan G., Aktories K. (2007) Action of Pasteurella multocida toxin on Galpha(q) is persistent and independent of interaction with G-protein-coupled receptors. Cell. Signal. 19, 2174–2182 [DOI] [PubMed] [Google Scholar]
- 38. Venkatakrishnan G., Exton J. H. (1996) Identification of determinants in the alpha-subunit of Gq required for phospholipase C activation. J. Biol. Chem. 271, 5066–5072 [DOI] [PubMed] [Google Scholar]
- 39. Kurose H. (2003) Gα12 and Gα13 as key regulatory mediator in signal transduction. Life Sci. 74, 155–161 [DOI] [PubMed] [Google Scholar]
- 40. Suzuki N., Nakamura S., Mano H., Kozasa T. (2003) Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl. Acad. Sci. U. S. A. 100, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kreutz B., Hajicek N., Yau D. M., Nakamura S., Kozasa T. (2007) Distinct regions of Galpha13 participate in its regulatory interactions with RGS homology domain-containing RhoGEFs. Cell. Signal. 19, 1681–1689 [DOI] [PubMed] [Google Scholar]
- 42. Tesmer J. J., Sunahara R. K., Gilman A. G., Sprang S. R. (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science 278, 1907–1916 [DOI] [PubMed] [Google Scholar]
- 43. Markby D. W., Onrust R., Bourne H. R. (1993) Separate GTP binding and GTPase activating domains of a Gα subunit. Science 262, 1895–1901 [DOI] [PubMed] [Google Scholar]
- 44. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Westfield G. H., Rasmussen S. G., Su M., Dutta S., DeVree B. T., Chung K. Y., Calinski D., Velez-Ruiz G., Oleskie A. N., Pardon E., Chae P. S., Liu T., Li S., Woods V. L., Jr., Steyaert J., Kobilka B. K., Sunahara R. K., Skiniotis G. (2011) Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc. Natl. Acad. Sci. U. S. A. 108, 16086–16091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van E. N., Preininger A. M., Alexander N., Kaya A. I., Meier S., Meiler J., Hamm H. E., Hubbell W. L. (2011) Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc. Natl. Acad. Sci. U. S. A. 108, 9420–9424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dohlman H. G., Jones J. C. (2012) Signal activation and inactivation by the galpha helical domain: a long-neglected partner in g protein signaling. Sci. Signal. 5, re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Radhika V., Dhanasekaran N. (2001) Transforming G proteins. Oncogene 20, 1607–1614 [DOI] [PubMed] [Google Scholar]
- 49. Van Raamsdonk C. D., Bezrookove V., Green G., Bauer J., Gaugler L., O'Brien J. M., Simpson E. M., Barsh G. S., Bastian B. C. (2008) Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Van Raamsdonk C. D., Griewank K. G., Crosby M. B., Garrido M. C., Vemula S., Wiesner T., Obenauf A. C., Wackernagel W., Green G., Bouvier N., Sozen M. M., Baimukanova G., Roy R., Heguy A., Dolgalev I., Khanin R., Busam K., Speicher M. R., O'Brien J., Bastian B. C. (2010) Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 363, 2191–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gudermann T., Grosse R., Schultz G. (2000) Contribution of receptor/G protein signaling to cell growth and transformation. Naunyn Schmiedebergs Arch. Pharmacol. 361, 345–362 [DOI] [PubMed] [Google Scholar]
- 52. Entschladen F., Zanker K. S., Powe D. G. (2011) Heterotrimeric G protein signaling in cancer cells with regard to metastasis formation. Cell Cycle 10, 1086–1091 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.