

Abstract
Fibroblast activation protein (FAP) is a serine protease selectively expressed on tumor stromal fibroblasts in epithelial carcinomas and is important in cancer growth, adhesion, and metastases. As FAP enzymatic activity is a potent therapeutic target, we aimed to identify inhibitory antibodies. Using a competitive inhibition strategy, we used phage display techniques to identify 53 single-chain variable fragments (scFvs) after three rounds of panning against FAP. These scFvs were expressed and characterized for binding to FAP by surface plasmon resonance and flow cytometry. Functional assessment of these antibodies yielded an inhibitory scFv antibody, named E3, which could attenuate 35% of FAP cleavage of the fluorescent substrate Ala-Pro-7-amido-4-trifluoromethylcoumarin compared with nonfunctional scFv control. Furthermore, a mutant E3 scFv was identified by yeast affinity maturation. It had higher affinity (4-fold) and enhanced inhibitory effect on FAP enzyme activity (3-fold) than E3. The application of both inhibitory anti-FAP scFvs significantly affected the formation of 3-dimensional FAP-positive cell matrix, as demonstrated by reducing the fibronectin fiber orientation from 41.18% (negative antibody control) to 34.06% (E3) and 36.15% (mutant E3), respectively. Thus, we have identified and affinity-maturated the first scFv antibody capable of inhibiting FAP function. This scFv antibody has the potential to disrupt the role of FAP in tumor invasion and metastasis.—Zhang, J., Valianou, M., Simmons, H., Robinson, M. K., Lee, H.-O., Mullins, S. R., Marasco, W. A., Adams, G. P., Weiner, L. M., Cheng, J. D. Identification of inhibitory ScFv antibodies targeting fibroblast activation protein utilizing phage display functional screens.
Keywords: serine protease, single-chain variable fragment
The tumor stroma consists of a heterogeneous mixture of endothelial cells, lymphatic channels, inflammatory cells, supportive connective tissue, and fibroblasts. An increasing body of evidence suggests that the tumor stroma, rather than being a passive bystander in tumor progression, actively participates in cancer invasion and metastasis by providing nutrients, growth factors, and proteolytic enzymes (1, 2). Stromal cells and their cytokines coordinate critical pathways that exert important roles in the ability of tumors to invade and metastasize (3, 4). Development of effective therapeutic interventions against stromal cells might lead to the disruption of these pathways.
Fibroblast activation protein (FAP) is a 97-kDa type II integral membrane glycoprotein that belongs to the serine protease family. It is highly expressed on reactive tumor stromal fibroblasts in >90% of human epithelial carcinomas (e.g., breast, lung, colorectal, ovary), as determined by immunohistochemistry (5–7). Human and murine FAP share an 89% sequence identity and have similar functional homology (8, 9). FAP can cleave N-terminal dipeptides from polypeptides with l-proline or l-alanine in the penultimate position (10) and degrade gelatin or type I collagen (11). Mutation of the catalytic serine residue at position 624 abolishes both the dipeptidyl peptidase and collagenase activity of FAP (11). Thus, FAP is a serine protease with both dipeptidyl peptidase, as well as endopeptidase activity cleaving gelatin and type I collagen. FAP plays an important role in tumor development. In colon cancer patients, higher levels of FAP in tumor stroma have been correlated with aggressive disease progression and decreased survival (12). Preclinical studies by our group (13) and others (14) have shown that overexpression of FAP promotes tumor growth in mouse models, whereas overexpression of catalytically inactive FAP is not protumorgenic (15). Consistent with this, we had previously reported that rabbit sera containing FAP inhibitory polyclonal antibodies attenuated tumor growth in vivo (13). Recently, Kraman et al. (16) reported that depletion of FAP-expressing cells in tumor significantly increased the immunological control of tumor growth in lung and pancreatic cancer models, suggesting that FAP is an immune-suppressive component of the tumor stroma. Using an in vivo-like 3-dimensional (3D) matrix system, we observed that the enrichment of FAP in extracellular matrix (ECM) facilitates the motility of cancer cells and implicates the involvement of FAP in cancer metastasis (17). These findings provide the compelling rationale for developing targeted therapeutics that function by disrupting FAP-driven tumor progression. In the current study, we sought to identify antibodies that could inhibit the enzymatic activity of FAP. We describe here the identification of such FAP inhibitory antibodies utilizing a novel functional screen of a human phage display library.
MATERIALS AND METHODS
Inhibition of FAP dipeptidyl peptidase activity by gelatin
Recombinant murine FAP extracellular domain (ECD) was produced as described previously (13). Briefly, murine FAP ECD (1–761 aa) was cloned into the pSEC/Tag2 expression vector with a (His)6 tag at the 5′ end and a FLAG tag at the 3′ end. FAP-ECD was expressed in HEK293 cells and secreted in the culture supernatant before purified on a nickel resin column (Qiagen, Valencia, CA, USA). The dipeptidyl peptidase (DPP) activity of FAP-ECD was assayed using alanine-proline-7-amido-4-trifluoromethylcoumarin (Ala-Pro-AFC) as a substrate (Bachem, King of Prussia, PA, USA). Gelatin inhibition of FAP DPP activity was performed by incubating serial dilutions of gelatin in reaction buffer consisting of 100 mM NaCl and 100 mM Tris-HCl (pH 7.8) with equal volume of the fluorescent substrate 0.5 mM Ala-Pro-AFC. Serial 10-fold dilutions of gelatin were incubated with 3 nM recombinant murine FAP-ECD at room temperature for 15 min prior to the addition of Ala-Pro-AFC as a competitive substrate. Tests were performed in triplicate after incubation for 1 h at 37°C. Release of free AFC was analyzed in a Cytofluor fluorimeter (Labsystems, Helsinki, Finland) with 395-nm excitation and 490-nm emission to quantitatively measure dipeptidyl peptidase activity.
Panning of mFAP to identify single-chain variable fragment (scFv) inhibitory antibodies
A naive human phage display library containing 1010 unique phages was constructed by Dr. Wayne A. Marasco's group (Dana-Farber Cancer Institute, Boston, MA, USA; ref. 18). Briefly, the 27-billion-member Mehta I/II human scFv phage display library was assembled from rearranged VH and VL genes from 57 healthy donors that were cloned into the pFARBER phagemid vector and rescued using M13K07 helper phage. Nunc immunotubes (Nunc, Rochester, NY, USA) were coated overnight with 200 μg of recombinant murine FAP-ECD. The FAP-coated immunotubes were rinsed 3× in PBS and blocked with 2% milk/PBS at 37°C for 2 h. The naive human phage display library was incubated in the immunotubes in 2% milk/PBS for 60 min prior to washing with PBS/0.1% Tween-20 × 15, and bound phage eluted using 1 ml 100 mM triethylamine (pH 10), and neutralized by adding 0.5 ml 1 M Tris-HCl (pH 7.4). TG1 bacterial culture grown to optical density (OD) 0.4-0.5 was infected with the eluted phage for 30 min at 37°C, centrifuged at 3300 g for 10 min, resuspended, and spread on a 150-mm bioassay dish on antibiotic-resistant 2XYT agar. The bacterial colonies on the bioassay dish were scraped into 2XYT medium with 1% glucose/ampicillin and grown to OD 0.5 prior to infection with M13K07 helper phage for amplification. The culture was incubated at 37°C in 2XYT medium with ampicillin (100 μg/ml) and kanamycin (25 μg/ml) but without glucose. The bacteria were centrifuged at 10,800 g, and the phage supernatant was treated with 20% polyethylene glycol 6000/2.5 M NaCl. This methodology was followed for the first and third round of panning against FAP. The second round of panning used an initial depletion step employing gelatin/FAP complexes that would be expected to have obscured FAP enzymatic sites in order to remove non-FAP inhibitory antibodies from the library. Briefly, gelatin was applied to FAP-coated immunotubes prior to the incubation with the amplified first- round eluted phage containing a mixture of anti-FAP antibodies. Antibodies that did not bind to FAP in the presence of gelatin, i.e., the supernatant from the panning step, was used to infect TG1 bacterial culture, centrifuged, and resuspended in gelatin-free 2XYT medium before repanning against FAP in the absence of gelatin in round 2B. The eluted phages from round 2B that targeted the epitopes previously occupied by gelatin were amplified in the third round, as described previously (19).
Identification of phage display antibodies that bind to FAP
Phages were randomly selected for assessment of binding to FAP by ELISA. Precipitated phages were diluted after the second and third round of panning and plated on 2XYT/ampicillin/glucose/MgCl2 for single-colony selection; 192 colonies from each round, 384 colonies total, were amplified by rescuing with 5 × 108 M13K07 helper phage in 2XYT/ ampicillin/kanamycin, followed by purification using PEG solution. FAP-ECD was coated on a 96-well Nunc Maxisorb plate overnight; blocked with 1% BSA, PBS, and Tween-20; and washed 3× prior to the addition of precipitated phage supernatant clones for 2 h. Anti-M13 (pVIII) antibody conjugated to HRP in a 1:5000 dilution was added as the secondary antibody, and the plate was developed using ABTS substrate. Absorbance >5 times baseline (0.08) was considered positive. PCR fingerprinting analysis was performed on 115 positive clones to eliminate redundant patterns. DNAs of 53 scFvs were isolated from colonies streaked on an ampicillin 2XYT agar plate, followed by digestion with NcoI/NotI restriction enzymes. A 750-bp band was gel extracted and ligated into the pCYN expression vector. TG1 bacteria were transformed with the ligation mixture and plated onto ampicillin/glucose/2XYT agar plates. Colony PCR was performed, and those having the 750-bp insert were selected for DNA preparation and sequencing. The amino acid sequences were deduced from assembled sequence contigs and subjected to cluster analysis using the DNASTAR package (DNASTAR, Inc., Madison, WI, USA) to eliminate redundancy. Out of the initially identified 53 scFvs, 40 clones containing unique open reading frame sequences with a (Gly4Ser1)×3 linker were selected for expression in a pCYN vector using periplasmic secretion and IMAC purification of the 6× His tag, as described previously (20).
Characterization of scFv using flow cytometry
To further characterize the binding of scFvs to FAP, flow cytometry was performed using human embryonic kidney 293 (HEK293) cells transfected with either empty vector (HEK293-Mock) or FAP (HEK293-FAP) vector (13). Cells were incubated with scFv for 30 min on ice, followed by another 30-min incubation with FITC-labeled mouse anti-His antibody. HEK293-Mock or HEK293-FAP incubated only with the secondary antibody was used as negative controls. The samples were analyzed on a BD FACScan (BD Biosciences, San Jose, CA, USA), and the data were processed using FlowJo software (TreeStar Inc., Ashland, OR, USA).
Characterization of scFv by surface plasmon resonance
The specificity of the scFv molecules for FAP and the kinetic analysis were conducted by surface plasmon resonance on the BIAcore 1000 instrument (GE Healthcare, Waukesha, WI, USA), as described previously (21). Briefly, purified murine FAP-ECD was covalently immobilized to the carboxyl groups of carboxymethylated (CM) dextran matrix (CM-5 sensor chip; GE Healthcare) using an amine coupling strategy. For all experiments, an activated and quenched surface served as a control for nonspecific binding to the dextran matrix. Validation of scFv binding was carried out on surfaces containing ∼2000 resonance units (RU) of immobilized ECD. Kinetic and equilibrium binding analyses were carried out by passing serially diluted samples of scFv (62.5 nM to 1 μM) over ∼700 RU of immobilized FAP-ECD. Kinetic rate constants (association/dissociation) and affinities were calculated from double-subtracted data using BIAEvaluation 3.2 software (GE Healthcare).
DPP and endopeptidase inhibition of FAP by scFv
The DPP activity was assayed using Ala-Pro-AFC substrate. To quantitatively measure the inhibition of FAP DPP activity, serial concentrations of 40 distinct recombinant anti-FAP scFv antibodies were incubated with 60 ng FAP-ECD for 30 min at 37°C, followed by an additional 1 h incubation after the addition of 0.5 mM Ala-Pro-AFC substrate in reaction buffer. Free AFC was measured in the Cytofluor fluorimeter at a setting of 395-nm excitation and 490-nm emission, as described previously (13, 15).
The endopeptidase activity of FAP was assayed using a fluorescence resonance energy transfer (FRET) peptide substrate that contained the FAP-sensitive Pro12-Asn13 bond: Arg-Lys (DABCYL)-Thr-Ser-Gly-Pro-Asn-Gln-Glu-Gln-Glu(EDANS)-Arg (kindly provided by Dr. Patrick McKee, University of Oklahoma College of Medicine, Norman, OK, USA). Serial concentrations of parental wild-type (WT) or mutant (Mut) E3 scFvs were incubated with 120 ng FAP-ECD for 30 min at 37°C prior to the addition of the FRET peptide substrate. Hydrolysis of the Pro-Asn bond separates the fluorophore, EDANS, 5-[(2-aminoethyl)amino]-naphthalene-1-sulfonic acid, from the quenching group, DABCYL [4-(4-dimethylaminophenylazo)benzoyl], to give an increase in fluorescence. The fluorescence was measured at a setting of 480-nm excitation and 510-nm emission.
Affinity maturation of E3 scFv
Affinity maturation of E3 scFv was done according to the literature described by Colby et al. (22). Briefly, the Mut E3 yeast display library was generated by random mutagenesis of WT-E3 scFv, followed by gap repair homologous recombination after electroporation of Mut PCR product and NcoI- and NotI-digested pYD vector (Invitrogen, Carlsbad, CA, USA) into competent yeast EB 100. Primers used for error-prone PCR were forwand, 5′-AGTAACGTTTGTCAGTAATTGC-3′, and reverse, 5′-GTCGATTTTGTTACATCTACAC-3′. The scFvs from the Mut E3 yeast display library were expressed and the potential high-affinity yeast-scFv FAP binders were selected by sorting after staining with biotin-FAP, followed by antibiotin 488 or streptavidin-PE. The sorting was performed for 4 rounds with decreasing concentrations of biotin-FAP. Single clones were picked randomly after the third and fourth round of sorting, and the FAP binding ability of individual clones was compared with WT E3 by flow cytometry. The clones with higher binding to FAP were sequenced and cloned into pCYN vector for further analysis.
Site-directed mutagenesis of Mut 1, 2, and 3 E3 scFv
Site-directed mutagenesis was performed using a QuickChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA), according to the manufacturer's instruction. The primers used for mutations were AA#107 Y→H, 5′-GACCAATACCGCGTTGATCACTATGATGGTAGTGGTTC-3′; AA#33 D→G, 5′-CTGGATACCGCTTCAACGGCTACTACATACACTGGCTG-3′; and AA#102 Q→R, 5′-CTGTGCGAAAGACCGATACCGCGTTGATCAC-3′. The mutation-containing pCYN plasmids were checked by sequencing before transformation into TG1 cells for scFv expression.
Production of fibroblast 3D matrix, fibronectin staining, and analysis of fiber orientation
The production of in vivo-like fibroblast 3D matrix and indirect immunofluorescent staining of fibronectin were done as described previously (17, 23). Briefly, mouse embryo fibroblast NIH3T3 was stably transfected with mouse FAP vector (NIH3T3-FAP) or empty vector (NIH3T3-Mock). The expression of mFAP was induced by 2 μg/ml doxycycline 2 d prior to matrix production. Then, NIH3T3-Mock or NIH3T3-FAP cells were seeded on glass coverslips, and confluent fibroblastic cultures were treated every other day with fresh medium supplemented with doxycycline, 50 μg/ml ascorbic acid, and WT or Mut 2 E3 scFvs for 8 d. The same molar concentration of mouse IgG was used as a negative control. Unextracted cell matrices on coverslips were fixed and permeabilized before blocking with M.O.M mouse IgG blocking solution (Vector Laboratories, Burlingame, CA, USA) containing 20% donkey serum for 1 h at room temperature. Rabbit anti-mouse fibronectin antibody (Abcam, Cambridge, MA, USA) was incubated at 25 μg/ml at 4°C overnight, followed by donkey anti-rabbit, Cy5-conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA, USA) at 15 μg/ml for 1 h at room temperature.
The analysis of fibronectin fiber orientation was performed according to the method published previously by Amatangelo et al. (23). Five images/experiment (z stack of 0.5-μm-thick z slices) were captured using a Perkin-Elmer spinning-disc microscope (PerkinElmer Life Sciences, Waltham, MA, USA) mounted on a Nikon TE-2000S microscope (Optical Apparatus, Ardmore, PA, USA). The z slices were reconstituted as 3D-overlay maximum-projection images using MetaMorph offline imaging analysis software (Molecular Devices, Sunnyvale, CA, USA). Flattened binary images were subjected to autothreshold, and fiber orientation was measured using the integrated morphometry analysis function. Fiber orientation angles were rounded to the nearest 0.1°, and the mode angle was determined as the angle to which the maximum number of fibers was oriented and set to 0°. The fiber distribution was achieved by calculating the percentage of fibers arranged in parallel ±10° of the mode angle for each region analyzed. The results shown are representative of two independent experiments.
Statistical analysis
The data from 3D matrix fiber distribution were analyzed using multinomial regression. The angles were categorized as <−20, =−10, =0, =10, and ≥20. The number of fibers in each angle category was regressed to the treatment, adjusted by the date of the experiment, duplications and images. All tests were 2-sided, with a value of P < 0.05 considered significant.
RESULTS
Gelatin competitively inhibits FAP substrate cleavage
Gelatin has previously been established as a FAP substrate in multiple reports (11, 24–26). We assessed the ability of gelatin to competitively attenuate the cleavage of the fluorescent Ala-Pro-AFC substrate by recombinant FAP. As shown in Fig. 1, gelatin itself had no intrinsic fluorescent activity, nor did it cleave the Ala-Pro-AFC substrate. However when 2% gelatin was incubated with FAP and Ala-Pro-AFC, nearly 75% inhibition of the fluorescent substrate cleavage by FAP was seen. Serial dilutions of gelatin demonstrated that inhibition of FAP by gelatin was concentration dependent (Fig. 1). This is consistent with gelatin functioning as a competitive inhibitor of FAP activity and allows the potential of gelatin to block the access of the catalytic site of FAP. Thus, we aimed to exploit this competitive inhibition of FAP substrate to identify inhibitory antibodies using phage display techniques.
Figure 1.

Competitive inhibition of the fluorescent substrate by gelatin. Gelatin inhibition of 3 nM FAP enzymatic activity was performed by incubating serial dilutions of gelatin in reaction buffer prior to the addition of the fluorescent substrate Ala-Pro-AFC. Nearly 75% inhibition of FAP DPP activity can be seen. Thus, gelatin competitively attenuates cleavage of the fluorescent Ala-Pro-AFC substrate by recombinant FAP.
Panning strategy to identify scFvs that inhibit FAP enzymatic activity
The strategy to identify inhibitory antibodies is shown in Fig. 2. The first round of panning of the phage display library aimed to identify antibodies that bound to the range of FAP epitopes. For the second round, a functional screen was utilized, whereby gelatin was added to the FAP-coated immunotubes prior to incubation with the amplified first-round eluted phages, which contained a mixture of anti-FAP antibodies. Gelatin was utilized to block access of the desired anti-FAP antibodies to the serine catalytic triad. Antibodies that did not bind to FAP in the presence of gelatin, i.e., the depleted supernatant from the panning step, were repanned against FAP in the absence of gelatin to select those binders that targeted the epitopes previously occupied by gelatin. Eluted phages from the repanning (round 2B) were amplified for a third round of panning.
Figure 2.

Panning strategy for identification of scFv antibodies that block substrates. Human scFv phage display library was incubated with FAP-coated immunotubes. The first round of panning selected for all scFvs that bind to FAP. This includes scFvs that bind to the catalytic site of FAP (bright green) and all other noncatalytic epitopes of FAP (other colors). In the second round of panning, gelatin was incubated to competitively block access of FAP catalytic sites to the catalytic scFvs (bright green). This strategy led to the separation of these scFvs as the non-catalytic-site scFvs bound to the FAP-coated immunotubes, while the desired catalytic site FAP binders (bright green) remained in the supernatant. This supernatant containing the desired catalytic site FAP binders was then collected and repanned with FAP in round 2B in the absence of gelatin to enrich for scFvs binding to the FAP binding site. In round 3, the FAP catalytic site binders were amplified. Starting from round 2B, FAP scFv binders were expressed and evaluated for their ability to inhibit the DPP enzymatic activity.
Identification of phage display antibodies that bind to FAP
Selections were monitored by determining the number of phages bound to FAP following each of three rounds of panning. As shown in Table 1, the titer of eluted phage bound to FAP increased by 105-fold following 3 rounds of selection, suggesting effective amplification of FAP-targeting phage antibodies. Antibodies binding to FAP were identified by ELISA from randomly selected single colonies from the second and third rounds of panning. As shown in Table 2, 25% (48/192) of selected clones from round 2 were positive, while 35% (67/192) of clones from round 3 were positive, with greater OD values of scFvs from the latter rounds. This allowed us to identify >50 distinct scFvs by PCR fingerprinting, of which 40 produced unique open reading frame sequences. The scFvs selected in round 2 tended to have greater diversity as determined by deduced amino acid sequences subjected to cluster analysis (data not shown). All 40 scFvs were expressed for further characterization.
Table 1.
Eluted and amplified phages in rounds 1–3
| Round | Eluted phages (cfu/ml) | Amplified phages (cfu/ml) |
|---|---|---|
| 1 | 3 × 103 | 4.5 × 1012 |
| 2 | 6 × 105 | 2.5 × 1012 |
| 3 | 1.5 × 108 | 1 × 1012 |
Table 2.
Characteristics of scFvs that target FAP
| Round | ScFv clones picked | ScFv-positive binders by ELISA, >5× baseline | Mean ELISA [OD (range)] | ScFvs cloned | ScFvs expressed | ScFvs inhibiting FAP enzymatic activity |
|---|---|---|---|---|---|---|
| 2 | 192 | 48 | 0.853 (0.45–2.55) | 29 | 25 | 1 |
| 3 | 192 | 67 | 0.976 (0.44–4.86) | 24 | 15 | 0 |
Inhibition of FAP enzymatic activity by moderate-affinity scFv
FAP enzymatic DPP activity assay was performed with all 40 scFvs identified from phage display. Only one scFv, named E3, was found to inhibit the FAP cleavage of the fluorescent substrate, as shown in Fig. 3. E3 was identified from the 25 scFvs in round 2, where the functional screen of the depleted gelatin library was employed. E3 at 17.85 μM (100 μg) resulted in ∼35% inhibition of FAP enzymatic activity. The inhibitory capability of E3 was confirmed in two separate experiments. E8 is representative of the 39 scFvs that did not inhibit enzymatic activity at all concentrations tested (Fig. 3).
Figure 3.

E3 scFv inhibits FAP enzymatic activity in vitro. The anti-FAP scFv antibodies were incubated with 60 ng FAP-ECD prior to the addition of the Ala-Pro-AFC substrate. The inhibition of DPP activity by the scFv E3 is evidenced by the lower fluorescence activity compared to the antibody E8 that binds to FAP but does not inhibit its enzymatic activity. Approximately 35% inhibition of FAP enzymatic activity is seen at 17.85 μM (100 μg) of E3.
The E3 and E8 anti-FAP scFvs were also evaluated for their ability to bind recombinant FAP by surface plasmon resonance and FAP in the context of being expressed on the cell surface via flow cytometry. Both E3 and E8 scFvs (1 μM) bound specifically to immobilized FAP-ECD, as depicted in the sensorgrams in Fig. 4A, B. Preliminary characterization of the binding affinities of these scFvs suggested that the KD of E3 is ∼2 × 10−7 M, whereas the KD of E8 is ∼6 × 10−9 M (data not shown). As measured by flow cytometry (Fig. 4C), E8 is capable of binding to HEK-FAP-transfected cells, as demonstrated by the right shift of the open curve compared to the closed curve (HEK-vector control). Consistent with its low affinity and monovalent binding, E3 failed to bind HEK-FAP cells when measured by flow cytometry. However, its ability to inhibit FAP enzymatic activity suggests that E3 can bind the native conformation of FAP. Therefore, we sought to improve E3 affinity in an effort to enhance its functional characteristics.
Figure 4.
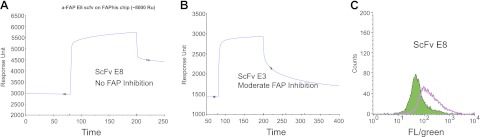
Representative binding characteristics of scFvs, including the FAP inhibitor E3. Surface plasmon resonance was used to determine the binding of scFvs to FAP. ScFv association/dissociation rates and affinities of the antibodies for FAP were determined by passing serial diluted samples of the scFv over a FAP-ECD immobilized CM5 chip. A, B) Sensorgrams demonstrate significant FAP binding by the scFvs E8 (A) and E3 (B). C) FACS analyses also confirmed the binding of the scFv E8 to HEK-FAP cells (open curve) in contrast to negative control (shaded curve).
Identification of Mut 2 E3 scFv through yeast affinity maturation
A Mut E3 yeast display library consisting of 7.8 × 106 clones was constructed as described in Materials and Methods. After 4 rounds of selection, 50 colonies that demonstrated greater FAP-binding than WT-E3 scFvs on flow cytometry were randomly picked. Thirty-seven were identified as unique clones by DNA sequence analysis (data not shown). Among them, the yeast display clone named Mut E3 4-12′ consistently showed high affinity FAP binding. Figure 5 demonstrates that the FAP binding of Mut E3 4-12 was increased by ∼100-fold (bottom right quadrant) compared to that of WT-E3 when expressed on the yeast surface fused to the agglutinin receptors. However, these 37 high-affinity FAP binders were unable to produce enough soluble scFvs for further analysis when expressed either in TG1 bacteria cells (cloned in pCYN vector) or in X-33 yeast cells (pPICZα vector). Using sequence analysis of all the Mut clones, we found that the difficulty of expression and purification might be due to frequent framework mutations that could cause failure of correct protein folding and secretion (data not shown). Because the complementarity-determining regions (CDRs) of the scFv are primarily responsible for antigen recognition (27), we introduced different combinations of CDR mutations based on the Mut E3 4-12 sequence into pCYN-WT E3 by site-directed mutagenesis. Only 3 Mut E3 4-12-derived variants, named E3 Mut1, Mut2, and Mut3, could be expressed in sufficient quantities by TG1 cells (cloned in pCYN vector). The comparison of amino acid changes of Mut E3 4-12, its derived variants, with WT-E3 scFv are shown in Fig. 6A. The ability of Mut E3 scFvs to inhibit FAP enzymatic activity was compared to WT-E3 scFv. Different from other postprolyl peptidases, FAP not only has dipeptidyl peptidase exopeptidase activity, but also endopeptidase activity. FAP-specific endopeptidase activity was tested using a FRET peptide substrate, while Alo-pro-AFC was used to test the dipeptidyl peptidase activity. As shown in Fig. 6B, the ability of E3 Mut 2 scFvs in inhibiting FAP endopeptidase activity was found to be ∼3-fold higher than that of WT-E3 scFvs at 17.85 μM (100 μg). Similar inhibitory effects of the Mut 2 scFvs were seen for FAP dipeptidyl peptidase activity with Ala-Pro-AFC substrate (data not shown). However, neither E3 Mut 1 nor Mut 3 scFvs showed greater inhibitory effects than the parental WT-E3 scFvs (data not shown). The affinity of WT and Mut 2-E3 scFvs was determined by equilibrium-binding analysis on a BIAcore (Fig. 7). Plotting response units at equilibrium vs. scFv concentration demonstrates that the binding affinity of WT-E3 measured in this manner (5×10−7 M) correlated well with previous data (see above). When measured in this manner, the KD of E3 Mut 2 scFvs was 1.4 × 10−7 M, 4-fold higher than that of the WT-E3, consistent with its improved ability to inhibit FAP activity.
Figure 5.

Comparison of the binding ability of FAP between WT E3 and Mut E3 in yeast display system. After 4 rounds of sorting, individual yeast clones were randomly picked from the Mut E3 library. Protein expression was induced, and yeast cells with different scFvs displayed on the cell surface were incubated with biotin-FAP, followed by anti-biotin 488. Binding ability of FAP was compared between yeast cells displayed with WT E3 scFv (A) and Mut E3 4-12 scFv (B), one of the best clones selected.
Figure 6.

Mut 2-E3 scFv enhanced the inhibitory effect of WT-E3 scFv on FAP enzyme activity. A) Amino acid mutations on CDR regions of Mut E3 4-12 scFv were compared with those of WT-E3 scFv. E3 Mut 1, 2, and 3 were created by introducing the CDR mutations into WT-E3 scFv based on the sequence of Mut E3 4-12. B) WT and Mut 2 E3 anti-FAP scFv antibodies were incubated with 120 ng FAP-ECD prior to the addition of endopeptidase substrate FRET. Inhibition of FAP endopeptidase activity by the scFv E3 is evidenced by the dose-dependent decrease in fluorescence intensity. At 17.85 μM E3 scFvs (100 μg), Mut 2 scFvs inhibited the activity of FAP to 30% of what WT-E3 achieved.
Figure 7.

Comparison of the affinities of Mut 2 and WT-E3 scFvs. The affinities of Mut 2 and WT-E3 scFvs for FAP were compared by BIAcore technology. Response unit was recorded with various concentrations of Mut 2 and WT-E3 scFvs passing over a FAP-ECD-immobilized CM5 chip.
Effect of FAP inhibitory scFvs on the formation of fibroblast 3D matrices
The in vivo-like 3D matrix system was used as a platform to assess whether enzymatic inhibitory anti-mFAP scFvs can affect the formation of the fibroblast-derived 3D matrix, more specifically, the orientation of ECM fibers (17, 23). The percentage of fibronectin fibers oriented within 10° of the mode angle (−10° to 10°) in NIH3T3-Mock or NIH3T3-FAP matrices is shown in Fig. 8. In Fig. 8A, NIH3T3-Mock was used to derive matrices, while in Fig. 8B, the matrices were made from NIH3T3-FAP cells, all without any antibodies applied. Mouse IgG was used as a negative antibody control to NIH3T3-FAP matrices in Fig. 8C, while WT or Mut 2 E3 scFvs were applied to NIH3T3-FAP matrices in Fig. 8D, E, respectively. Consistent with our previous reports (17), 3D fibroblast matrices enriched with FAP showed a more directional and parallel fiber pattern compared to FAP-negative matrices. The percentage of parallel fibers within 10° of the mode angle in NIH3T3-FAP matrices (Fig. 8B) was 40.44%, in contrast to 29.57% in NIH3T3-Mock matrices (Fig. 8A). Compared to no antibody control (Fig. 8B), the application of both WT (Fig. 8D) and Mut 2 E3 scFvs (Fig. 8E) significantly decreased the percentage of parallel fibronectin fibers in FAP-positive NIH3T3 matrices to 34.06 and 36.15%, respectively (P<0.001). The FAP matrices formation was not significantly different between two negative controls, mouse IgG (Fig. 8C) or no antibody control (Fig. 8B), indicating the disorganized FAP matrices in Fig. 8D, E are likely caused by the addition of inhibitory anti-FAP scFvs.
Figure 8.
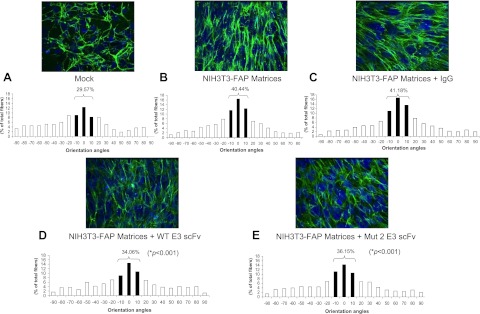
Effects of anti-FAP scFvs on fibronectin organization in fibroblast-derived 3D matrices. Fibronectin fibers were analyzed by immunofluorescence in unextracted 3D matrices formed by stably transfected NIH3T3 FAP cells (B–E) or vector control (mock; A). During the formation of FAP matrices, 3 μM of mouse IgG (NIH3T3-FAP matrices + IgG; C), WT E3 scFv (NIH3T3-FAP matrices + WT E3 scFv; D) or Mut 2 E3 scFv (NIH3T3-FAP matrices + Mut 2 scFv; E) was added to the medium for 8 d. Orientation of fibronectin fibers was measured (see Materials and Methods for details). Numbers indicate the percentage of fibers positioned within 10 ° of the mode fiber orientation angle (P value between anti-FAP scFv-treated 3D matrix and nontreated control). *P < 0.05.
DISCUSSION
We have identified the first inhibitory scFv antibodies of FAP enzymatic activity. A competitive inhibitor of FAP activity, gelatin, was utilized in a deletion panning technique to identify inhibitory antibodies. Using subtraction techniques, we exploited gelatin as a competitive inhibitor of the FAP substrate to identify inhibitory antibodies of the “spent” library that binds to FAP, but are blocked by gelatin. Functional screens have been previously utilized, for example, to select antibodies that internalize via endocytosis (28, 29). However, this is the first report that we are aware of that used a functional screen to identify inhibitory antibodies of a protease.
Although we were able to identify a significant number of antibodies that bind to FAP, the identification of functional antibodies which inhibit FAP activity proved to be challenging, as we were only able to identify a single inhibitory scFv antibody with appropriate binding characteristics. A number of potential obstacles may exist that limit the robust ability to easily identify inhibitory antibodies. First, given the structure of FAP, inhibitory antibodies most likely do not bind directly to the catalytic site, but rather physically block substrate passage through the small openings (which are ∼8-10 Å in diameter) leading to a deep cavern of the catalytic domain (30). Thus, the “entrance” to the catalytic domain cavern is an epitope that largely consists of open space, which may lead to significant binding difficulties. Second, the presence of two access points for the catalytic domain, which may function as an entrance and outlet for large natural substrates (31), may artificially abolish the effects of inhibitory antibodies, as the blockage of one entrance site may be negated by access of the small fluorescent substrate through the alternative port. Finally, the relatively small size and low affinity of the scFvs may provide suboptimal blockage of the entrance to the catalytic site. Therefore, affinity maturation of the initially identified FAP inhibitory E3 scFvs was performed using a yeast display system aiming to enhance the inhibitory effects of E3 by increasing its affinity. Thirty-seven potential FAP scFv binders were identified from the Mut E3 yeast display library. Despite the problem of expression and purification of E3 4-12, one of the best of 37 identified, we were able to express some clones that had a WT-E3 backbone but contained CDR mutations of E3 4-12. This led to the finding of Mut 2 E3 scFv that has higher affinity for FAP and enhanced inhibitory effect of FAP endopeptidase activity than those of WT-E3. Reengineering of Mut 2 E3 scFv into an intact IgG may further improve its ability to inhibit enzymatic activity. Although the affinity of Mut 2 E3 scFv is relatively modest (KD of 1.4×10−7 M), the scFv itself or its derived IgG may still be a useful clinical reagent for investigating in vivo targeting of FAP-positive tumor stroma as the threshold level for biological effect is unknown (32).
It is well known that tumor-associated stromal ECMs influence tumorigenesis (33). Interactions between cancer cells and stroma are critical for growth and invasiveness of epithelial tumors. Various cell invasion strategies have been characterized, such as “mesenchymal invasion” featuring spindled cells that invade following the direction of ECM fibers (34). A novel 3D system derived from tumor-associated fibroblasts was established that mimics the in vivo stromagenic features of fibroblasts and their matrices (23). Adapting the same 3D matrix system using FAP-positive or -negative NIH3T3 cells, we demonstrated FAP-enriched fibroblast matrices had the same characteristic parallel fiber orientation as the ones made from tumor-associated fibroblasts (17). The velocity and directionality of tumor cells were enhanced on FAP matrices. In addition, small molecular inhibitors of FAP enzymatic activity resulted in disorganization of ECM fibers in FAP matrices and interfered with cancer cell movement (17). In the present study, the application of inhibitory anti-FAP scFvs during matrix formation significantly disrupted the organization of ECM fiber distribution in FAP matrices. In contrast, the negative control antibody did not show any effect on the matrix formation. These suggest our inhibitory anti-FAP scFvs have the potential of disrupting cancer invasion by normalizing tumor-associated matrices.
There are many theoretical advantages for targeting the tumor stroma, which comprises ∼20-50% of the tumor mass, rather than tumor cells (7). The close physical proximity of the tumor stromal cells to the tumor capillaries allows for easier accessibility of circulating proteins in the blood to reach the stromal target (35). In addition, stromal cells have fewer genetic alterations compared to transformed malignant cells (36), and thus the reactive stromal cells may express target antigens conserved across different histological cancer types and in both primary and metastatic tumors (8, 37). Therefore, treatment strategies aimed at the tumor stroma could potentially be effective in both localized and advanced disease settings, as well as in multiple cancer subtypes (38). Our previous report of FAP inhibitory rabbit sera showed moderate tumor growth attention in mouse models (13). This discovery of functional inhibitory anti-FAP scFvs from a human phage display library provides more insight for future development of therapeutic antibodies that target the tumor stroma to interfere with cancer cell motility and invasion.
Acknowledgments
This study was supported by U.S. National Institutes of Health grants CA090468, CA103991-01, and CA09035-28; the Frank Strick Foundation; the Bernard A. and Rebecca S. Bernard Foundation; and an appropriation from the Commonwealth of Pennsylvania and the American Cancer Society.
Footnotes
- 3D
- 3-dimensional
- Ala-Pro-AFC
- alanine-proline-7-amido-4-trifluoromethylcoumarin
- CDR
- complementarity determining region
- CM
- carboxymethylated
- DPP
- dipeptidyl peptidase
- ECD
- extracellular domain
- ECM
- extracellular matrix
- FAP
- fibroblast activation protein
- FRET
- fluorescence resonance energy transfer
- HEK293
- human embryonic kidney 293
- Mut
- mutant
- OD
- optical density
- RU
- resonance unit
- scFv
- single-chain variable fragment
- WT
- wild type
REFERENCES
- 1. Iozzo R. V. (1995) Tumor stroma as a regulator of neoplastic behavior. Agonistic and antagonistic elements embedded in the same connective tissue. Lab. Invest. 73, 157–160 [PubMed] [Google Scholar]
- 2. Liotta L. A. (1986) Tumor invasion and metastases—role of the extracellular matrix: Rhoads Memorial Award lecture. Cancer Res. 46, 1–7 [PubMed] [Google Scholar]
- 3. Roskelley C. D., Bissell M. J. (2002) The dominance of the microenvironment in breast and ovarian cancer. Semin. Cancer Biol. 12, 97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruiter D., Bogenrieder T., Elder D., Herlyn M. (2002) Melanoma-stroma interactions: structural and functional aspects. Lancet Oncol. 3, 35–43 [DOI] [PubMed] [Google Scholar]
- 5. Rettig W. J., Chesa P. G., Beresford H. R., Feickert H. J., Jennings M. T., Cohen J., Oettgen H. F., Old L. J. (1986) Differential expression of cell surface antigens and glial fibrillary acidic protein in human astrocytoma subsets. Cancer Res. 46, 6406–6412 [PubMed] [Google Scholar]
- 6. Rettig W. J., Garin-Chesa P., Beresford H. R., Oettgen H. F., Melamed M. R., Old L. J. (1988) Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. U. S. A. 85, 3110–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garin-Chesa P., Old L. J., Rettig W. J. (1990) Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. U. S. A. 87, 7235–7239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scanlan M. J., Raj B. K., Calvo B., Garin-Chesa P., Sanz-Moncasi M. P., Healey J. H., Old L. J., Rettig W. J. (1994) Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc. Natl. Acad. Sci. U. S. A. 91, 5657–5661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niedermeyer J., Scanlan M. J., Garin-Chesa P., Daiber C., Fiebig H. H., Old L. J., Rettig W. J., Schnapp A. (1997) Mouse fibroblast activation protein: molecular cloning, alternative splicing and expression in the reactive stroma of epithelial cancers. Int. J. Cancer 71, 383–389 [DOI] [PubMed] [Google Scholar]
- 10. Niedermeyer J., Enenkel B., Park J. E., Lenter M., Rettig W. J., Damm K., Schnapp A. (1998) Mouse fibroblast-activation protein–conserved Fap gene organization and biochemical function as a serine protease. Eur. J. Biochem. 254, 650–654 [DOI] [PubMed] [Google Scholar]
- 11. Park J. E., Lenter M. C., Zimmermann R. N., Garin-Chesa P., Old L. J., Rettig W. J. (1999) Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 274, 36505–36512 [DOI] [PubMed] [Google Scholar]
- 12. Henry L. R., Lee H. O., Lee J. S., Klein-Szanto A., Watts P., Ross E. A., Chen W. T., Cheng J. D. (2007) Clinical implications of fibroblast activation protein in patients with colon cancer. Clin. Cancer Res. 13, 1736–1741 [DOI] [PubMed] [Google Scholar]
- 13. Cheng J. D., Dunbrack R. L., Jr., Valianou M., Rogatko A., Alpaugh R. K., Weiner L. M. (2002) Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. 62, 4767–4772 [PubMed] [Google Scholar]
- 14. Huang Y., Wang S., Kelly T. (2004) Seprase promotes rapid tumor growth and increased microvessel density in a mouse model of human breast cancer. Cancer Res. 64, 2712–2716 [DOI] [PubMed] [Google Scholar]
- 15. Cheng J. D., Valianou M., Canutescu A. A., Jaffe E. K., Lee H. O., Wang H., Lai J. H., Bachovchin W. W., Weiner L. M. (2005) Abrogation of fibroblast activation protein enzymatic activity attenuates tumor growth. Mol. Cancer Ther. 4, 351–360 [DOI] [PubMed] [Google Scholar]
- 16. Kraman M., Bambrough P. J., Arnold J. N., Roberts E. W., Magiera L., Jones J. O., Gopinathan A., Tuveson D. A., Fearon D. T. (2010) Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830 [DOI] [PubMed] [Google Scholar]
- 17. Lee H. O., Mullins S. R., Franco-Barraza J., Valianou M., Cukierman E., Cheng J. D. (2011) FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 11, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sui J., Li W., Murakami A., Tamin A., Matthews L. J., Wong S. K., Moore M. J., Tallarico A. S., Olurinde M., Choe H., Anderson L. J., Bellini W. J., Farzan M., Marasco W. A. (2004) Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. U. S. A. 101, 2536–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harrison J. L., Williams S. C., Winter G., Nissim A. (1996) Screening of phage antibody libraries. Methods Enzymol. 267, 83–109 [DOI] [PubMed] [Google Scholar]
- 20. Schier R., Marks J. D., Wolf E. J., Apell G., Wong C., McCartney J. E., Bookman M. A., Huston J. S., Houston L. L., Weiner L. M., Adams G. P. (1995) In vitro and in vivo characterization of a human anti-c-erbB-2 single-chain Fv isolated from a filamentous phage antibody library. Immunotechnology 1, 73–81 [DOI] [PubMed] [Google Scholar]
- 21. Yuan Q. A., Simmons H. H., Robinson M. K., Russeva M., Marasco W. A., Adams G. P. (2006) Development of engineered antibodies specific for the Mullerian inhibiting substance type II receptor: a promising candidate for targeted therapy of ovarian cancer. Mol. Cancer Ther. 5, 2096–2105 [DOI] [PubMed] [Google Scholar]
- 22. Colby D. W., Kellogg B. A., Graff C. P., Yeung Y. A., Swers J. S., Wittrup K. D. (2004) Engineering antibody affinity by yeast surface display. Methods Enzymol. 388, 348–358 [DOI] [PubMed] [Google Scholar]
- 23. Amatangelo M. D., Bassi D. E., Klein-Szanto A. J., Cukierman E. (2005) Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am. J. Pathol. 167, 475–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun S., Albright C. F., Fish B. H., George H. J., Selling B. H., Hollis G. F., Wynn R. (2002) Expression, purification, and kinetic characterization of full-length human fibroblast activation protein. Protein Expr. Purif. 24, 274–281 [DOI] [PubMed] [Google Scholar]
- 25. Levy M. T., McCaughan G. W., Abbott C. A., Park J. E., Cunningham A. M., Muller E., Rettig W. J., Gorrell M. D. (1999) Fibroblast activation protein: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology 29, 1768–1778 [DOI] [PubMed] [Google Scholar]
- 26. Goldstein L. A., Ghersi G., Pineiro-Sanchez M. L., Salamone M., Yeh Y., Flessate D., Chen W. T. (1997) Molecular cloning of seprase: a serine integral membrane protease from human melanoma. Biochim. Biophys. Acta 1361, 11–19 [DOI] [PubMed] [Google Scholar]
- 27. Carter P. J. (2006) Potent antibody therapeutics by design. Nat. Rev. Immunol. 6, 343–357 [DOI] [PubMed] [Google Scholar]
- 28. Crnogorac-Jurcevic T., Efthimiou E., Nielsen T., Loader J., Terris B., Stamp G., Baron A., Scarpa A., Lemoine N. R. (2002) Expression profiling of microdissected pancreatic adenocarcinomas. Oncogene 21, 4587–4594 [DOI] [PubMed] [Google Scholar]
- 29. Poul M. A., Becerril B., Nielsen U. B., Morisson P., Marks J. D. (2000) Selection of tumor-specific internalizing human antibodies from phage libraries. J. Mol. Biol. 301, 1149–1161 [DOI] [PubMed] [Google Scholar]
- 30. Aertgeerts K., Levin I., Shi L., Snell G. P., Jennings A., Prasad G. S., Zhang Y., Kraus M. L., Salakian S., Sridhar V., Wijnands R., Tennant M. G. (2005) Structural and kinetic analysis of the substrate specificity of human fibroblast activation protein alpha. J. Biol. Chem. 280, 19441–19444 [DOI] [PubMed] [Google Scholar]
- 31. Wolf B. B., Quan C., Tran T., Wiesmann C., Sutherlin D. (2008) On the edge of validation–cancer protease fibroblast activation protein. Mini Rev. Med. Chem. 8, 719–727 [DOI] [PubMed] [Google Scholar]
- 32. Adams G. P., Schier R., McCall A. M., Simmons H. H., Horak E. M., Alpaugh R. K., Marks J. D., Weiner L. M. (2001) High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 61, 4750–4755 [PubMed] [Google Scholar]
- 33. Radisky D., Muschler J., Bissell M. J. (2002) Order and disorder: the role of extracellular matrix in epithelial cancer. Cancer Invest. 20, 139–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Condeelis J., Segall J. E. (2003) Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 3, 921–930 [DOI] [PubMed] [Google Scholar]
- 35. Rettig W. J., Garin-Chesa P., Healey J. H., Su S. L., Ozer H. L., Schwab M., Albino A. P., Old L. J. (1993) Regulation and heteromeric structure of the fibroblast activation protein in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer Res. 53, 3327–3335 [PubMed] [Google Scholar]
- 36. Kurose K., Hoshaw-Woodard S., Adeyinka A., Lemeshow S., Watson P. H., Eng C. (2001) Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: clues to tumour-microenvironment interactions. Hum. Mol. Genet. 10, 1907–1913 [DOI] [PubMed] [Google Scholar]
- 37. Welt S., Divgi C. R., Scott A. M., Garin-Chesa P., Finn R. D., Graham M., Carswell E. A., Cohen A., Larson S. M., Old L. J., Rettig W. J. (1994) Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J. Clin. Oncol. 12, 1193–1203 [DOI] [PubMed] [Google Scholar]
- 38. Scott A. M., Wiseman G., Welt S., Adjei A., Lee F. T., Hopkins W., Divgi C. R., Hanson L. H., Mitchell P., Gansen D. N., Larson S. M., Ingle J. N., Hoffman E. W., Tanswell P., Ritter G., Cohen L. S., Bette P., Arvay L., Amelsberg A., Vlock D., Rettig W. J., Old L. J. (2003) A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 9, 1639–1647 [PubMed] [Google Scholar]