

Abstract
Sickle cell disease (SCD) is a globally distributed hereditary red blood cell (RBC) disorder. One of the hallmarks of SCD is the presence of circulating dense RBCs, which are important in SCD-related clinical manifestations. In human dense sickle cells, we found reduced calpastatin activity and protein expression compared to either healthy RBCs or unfractionated sickle cells, suggesting an imbalance between activator and inhibitor of calpain-1 in favor of activator in dense sickle cells. Calpain-1 is a nonlysosomal cysteine proteinase that modulates multiple cell functions through the selective cleavage of proteins. To investigate the relevance of this observation in vivo, we evaluated the effects of the orally active inhibitor of calpain-1, BDA-410 (30 mg/kg/d), on RBCs from SAD mice, a mouse model for SCD. In SAD mice, BDA-410 improved RBC morphology, reduced RBC density (D20; from 1106±0.001 to 1100±0.001 g/ml; P<0.05) and increased RBC-K+ content (from 364±10 to 429±12.3 mmol/kg Hb; P<0.05), markedly reduced the activity of the Ca2+-activated K+channel (Gardos channel), and decreased membrane association of peroxiredoxin-2. The inhibitory effect of calphostin C, a specific inhibitor of protein kinase C (PKC), on the Gardos channel was eliminated after BDA-410 treatment, which suggests that calpain-1 inhibition affects the PKC-dependent fraction of the Gardos channel. BDA-410 prevented hypoxia-induced RBC dehydration and K+ loss in SAD mice. These data suggest a potential role of BDA-410 as a novel therapeutic agent for treatment of SCD.—De Franceschi, L., Franco, R. S., Bertoldi, M., Brugnara, C., Matté, A., Siciliano, A., Wieschhaus, A. J., Chishti, A. H., Joiner, C. H. Pharmacological inhibition of calpain-1 prevents red cell dehydration and reduces Gardos channel activity in a mouse model of sickle cell disease.
Keywords: peroxiredoxin-2 · BDA-410 · protease
Sickle cell disease (SCD) is a globally distributed hereditary red blood cell (RBC) disorder, which results from a point mutation in codon 6 of the β-globin gene that causes the insertion of valine in place of glutamic acid, which produces a defective form of hemoglobin, hemoglobin S (HbS). Studies of the kinetics of HbS polymerization following deoxygenation show a high-order exponential dependence on hemoglobin (Hb) concentration, thus highlighting a crucial role for cellular HbS concentration in sickling (1). Pathophysiological studies have shown that the dense, dehydrated RBCs typically observed in SCD have an extremely short life span (2) and play a central role in the acute and chronic clinical manifestations of SCD, in which intravascular sickling in capillaries and small vessels leads to vasoocclusion and impaired blood flow, with ischemic cell damage in a variety of organs and tissues (3–8). Two main cation transport systems have been identified in RBC dehydration: the K-Cl cotransport and the Ca2+activated K+ channel (5, 9–13). However, the relative contributions of the two transport systems to RBC dehydration in SCD are still not completely understood. Recent progress on the pathogenesis of SCD has revealed a complexity that presents multiple opportunities for developing new experimental therapies, potentially targeting several pathological pathways (14).
Calpains are ubiquitous calcium-dependent cysteine proteases that selectively cleave proteins in response to calcium (15, 16). Human RBCs contain only calpain-1, and its activity is regulated by activator protein and the endogenous inhibitor, calpastatin (17–19). In sickle cells, calpain-1 expression is similar to that observed in normal erythrocytes (20); however, less is known about its functional regulation. In RBCs, calpain-1 targets membrane and cytoskeleton proteins, which causes limited degradation that modulates the activities or function of these proteins or membrane associated enzymes (21–25). In particular, calpain-1 is involved in activation of the Ca2+ pump ATPase through the removal of a 14-kDa peptide, thereby regulating RBC Ca2+ content (17, 21, 26). Oxidative stress by peroxynitrite has also been shown to activate calpain-1 in RBCs (27). Recently, a specific calpain-1 inhibitor, BDA-410, has been developed (28). BDA-410 is an orally active molecule, which showed beneficial effects in a mouse model for Alzheimer's disease (29) and delayed the progression of malaria in a model of Plasmodium chabaudi infection (30).
Here, we have characterized calpastatin activity in human sickle cells, showing that calpain-1 inhibition is decreased in dehydrated sickle cells, which suggests an imbalance between activator/inhibitor of calpain-1 in favor of activator in dense sickle cells. We then asked whether the exogenous inhibition of calpain-1 by the calpain-1 inhibitor BDA-410 might be beneficial in sickle cell hematological phenotype. We show amelioration of sickle cell features, which suggests that BDA-410 has potential as a novel therapeutic tool to be explored in future studies for treatment of SCD.
MATERIALS AND METHODS
In vitro studies on human RBCs
Blood was obtained with informed consent from patients with homozygous SCD who were not being treated with hydroxyurea and had not been transfused for ≥3 mo. Dense, dehydrated sickle cells (fraction VI) were isolated using a discontinuous Optiprep gradient (Sigma-Aldrich, St. Louis, MO, USA)and had a density >1.10 g/cm3 (31). RBC suspensions containing 100 μl of cells were washed 3 times with 150 mM NaCl and, after complete removal of the supernatant, were lysed by adding 900 μl of lysis buffer (10 mM Tris-HCl, 0.5 mM dithothreitol, 1 mM EDTA, 1 mM EGTA; Sigma-Aldrich) followed by two fast-freeze/thaw cycles using dry ice and alcohol. The freeze/thaw cycles help ensure complete lysis of the dehydrated population of sickle cells. The lysate was spun at high speed to pellet membranes, and the supernatant was isolated for further assay. Lysate supernatant (3 μl) was added to lysis buffer (697 μl), and the optical density (OD) at 415 nm (OD415) was determined. The remainder of the lysate supernatant was heated at 100°C for 5 min. Calpastatin has been shown to be stable in such heated extracts. After centrifugation to pellet the precipitated proteins, the clear supernatant was stored at −20°C until use. Inhibition of exogenous calpain-1 activity was determined by adding up to 30 μl of extract to an assay system containing 0.6 μg purified human calpain-1 (μ-calpain, C-6108; Sigma-Aldrich), 1 μg fluorescent casein substrate (casein-bodipy FL, E6638; Molecular Probes/Invitrogen, Carlsbad, CA, USA), and 0.7 mM Ca2+ (final free concentration) in a total volume of 200 μl. Small variations in lysate OD415 were compensated by adjusting the volume of extract added to the reaction mixture. Ca2+-dependent protease activity was determined in a fluorescent plate reader, which measured the amount of fluorescence as the casein was digested by calpain-1, resulting in dequenching of the bodipy dye. The amount of calpastatin protein in the extracts was examined by immunoblot analysis, using 4–20% agarose gels (58511; Cambrex, Karlskoga, Sweden), anti-human calpastatin (Serotec, Kidlington, UK), and ECL (Amersham, Little Chalfont, UK) detection.
Effects of BDA-410 on SCD model Hbbsingle/single SAD1 (SAD) mice
Transgenic SAD mice aged between 3 and 6 mo (body weight 25–30 g) were used as a mouse model for SCD, and age-matched C57B6/2J mice were used as wild-type (WT) controls (5, 12, 32, 33). Mice from both strains were divided into 4 groups of 6 mice each: 2 groups from each strain were treated with BDA-410 at the dosage of 30 mg/kg/d by gavage 1×/d for 14 d, while the others received vehicle only (cremophore/PEG-400/water 10:10:80 mixture, homogenized; PEG and cremophore from Sigma-Aldrich) (5, 6, 12, 32, 33). The dosage of BDA-410 was chosen based on previous in vivo studies with this molecule (28–30). Hematological parameters, RBC density profile, cation content, and Ca2+-activated K+ channel activity were evaluated at baseline and after 7 and 14 d of therapy. Blood sampling and vehicle administration have been previously shown not to affect these blood parameters (5, 7, 12, 32, 33).
Hypoxia study
In a separate study, SAD mice and control mice (n=6) were treated with BDA-410 at the dosage of 30 mg/kg/d by gavage 1×/d for 14 d and exposed to hypoxia 8% oxygen for 48 h (5, 7, 12, 32, 33). Oxygen pressure inside the cage was constantly monitored by an oxygen electrode, and mice had free access to water and food. Hematological parameters, RBC density profile, and cation content were evaluated before and after hypoxia, as previously reported (5, 7, 12, 32, 33). Control groups of untreated SAD and control mice also were exposed to hypoxia under the same conditions.
Measurement of hematological parameters, RBC cation content, Ca2+-activated Rb+ influx, and K-Cl cotransport in RBCs
These studies were performed as previously reported on a total of 150 μl blood drawn at indicated time intervals from each ether anesthetized mouse by retroorbital venipuncture into heparinized microhematocrit tubes (5, 12). Hb levels were determined by spectroscopic measurement of the cyanmet derivative. The hematocrit (Hct) was determined in a microhematocrit centrifuge. Density distribution curves were obtained using phthalate esters in microhematocrit tubes (5, 7, 12, 32, 33). RBC cation content was determined on washed RBCs by atomic absorption spectrometer ANALYST 2000 (Perkin-Elmer, Branchburg, NJ, USA) using standards in double-distilled water (5, 7, 12, 32, 33). The Ca2+-activated K+ channel (Gardos channel) activity was evaluated on whole blood as Rb+ influx, as described previously (5, 7, 12, 32, 33). Whenever indicated, the activity of the Gardos channel was evaluated in RBCs from BDA-410-treated and untreated mice in the presence of the protein kinase inhibitor calphostin C (1 μM; refs. 34–36). The K-Cl cotransport activity was evaluated in fresh RBCs as chloride- and volume-dependent K+ efflux (37).
RBC ghost preparation and peroxiredoxin-2 (Prx2) immunoblot analysis
RBC ghosts were prepared by lysing 1 vol of packed RBCs in 10 vol of ice-cold phosphate lysis buffer [PLB; 5 mM Na2HPO4, pH 8.0, containing a protease inhibitor cocktail tablet (Roche, Mannheim, Germany), 3 mM benzamidine (Sigma-Aldrich), and 1 mM Na3VO4 (Sigma-Aldrich); ref. 36] and in the presence of n-ethylmaleimide (NEM; 100 mM; Sigma-Aldrich; refs. 36, 38–40). Samples were incubated for 10 min on ice and centrifuged for 10 min at 12,000 g, 4°C. Ghosts were then washed 4 times (centrifugation at 12,000 g, 4°C) with PLB until they appeared almost white. Ghosts were solubilized in sample buffer (50 mM Tris, pH 6.8; 100 mM β-mercaptoethanol; 2% v/v SDS; 10% v/v glycerol; and a few grains of bromphenol blue; all from Sigma-Aldrich) in the presence of NEM (100 mM) and loaded on 10% gels (36, 38, 39). The following specific antibodies were used for immunoblot analysis: anti-Prx2 (Lab Frontier, Seoul, Korea) and anti-actin (Sigma-Aldrich). Secondary antibodies were from GE Healthcare (Little Chalfont, UK). ECL-Plus (Amersham) was used as detecting system. PRDX2 was also studied by cytofluorimetric analysis on RBCs fixed, permeabilized, and probed with anti-PRDX2 (41), kindly provided by Ho Zoo Chae (School of Biological Science and Technology, Chonnam National University, Gwangju, Korea) as described previously (42). The RBCs were then subjected to cytofluorimetric analysis using FACS-Cato cytometer (BD Biosciences, Milan, Italy). Data were analyzed with FACS DIVA software (BD Bioscience).
Bioinformatics analysis of calpain-1 prediction sites on Prx2 or Ca2+-activated K+ channel
A search for calpain-1 cleavage sites was carried out with the tool provided by the Calpain for Modulatory Proteolysis Data Bank (CaMPDB) project developed by duVerle et al. (43). In analyzing the amino acid sequence of Prx2 or Ca2+-activated K+ channel, we set a high score (>0.1) in order to avoid false-positive results as much as possible.
Statistical analysis
Data from in vivo studies with BDA-410 were analyzed by t test, comparing the treatment results against baseline values obtained from the same mice. Differences were considered significant at P < 0.05.
RESULTS
Human dense sickle cells show reduced calpain-1 inhibition and reduced calpastatin protein compared to controls
Extracts from unfractionated normal or sickle cells inhibited exogenous calpain-1 activity in a dose-dependent manner, as shown for a typical experiment in Fig. 1A, reflecting the activity of endogenous inhibitor calpastatin. However, extracts from dense (>1.10 g/cm3) sickle cells were not inhibitory and, in contrast, caused an increase in exogenous calpain-1 activity that exhibited a saturation behavior with increasing extract volume. This finding suggests the presence in the extract of an activating ligand that forms a complex with calpain-1. Addition of 15 μl of extract (volumes were corrected to adjust for small variations in lysate Hb concentration) resulted in 65 ± 16% calpain-1 activity for unfractionated sickle cells and 147 ± 36% activity for dense cells (P<0.001, n=8; Fig. 1B). In addition, we found a reduction in the amount of calpastatin, the endogenous calpain-1 inhibitor, in dense sickle cells compared to normal erythrocytes (Fig. 1C). In dense sickle cells, the increased calpain-1 activity after addition of extract from dense cells may be due to the imbalance between calpain-1 activator protein and calpastatin, in favor of the calpain-1 activator (18, 19). Since calpastatin is itself a substrate of calpain-1, these results are consistent with a cellular history of in vivo calpain-1 activation in dense sickle cells.
Figure 1.

Human dense sickle cells show reduced calpastatin activity. A) Inhibition of exogenous calpain-1 by heat-stable extracts. Up to 30 μl of extract was added to the reaction mixture described in Materials and Methods. Extracts were prepared from unfractioned normal RBCs (unfx AA; solid diamonds), unfractionated sickle cells (unfx SS; open squares), and dense sickle cells (>1.10 g/cm3, corresponding to fraction VI; fx VI SS; open triangles; refs. 11, 31). B) Comparison of inhibitory ability of 15 μl of heat-stable extract prepared from unfractionated (unfx) and dense (>1.10 g/cm3; corresponding to fraction VI; fx VI SS) sickle cells (n=8; P<0.001) and added to the reaction mixture described in Materials and Methods. The value of >100% for the extracts prepared from dense sickle cells indicates the presence of a calpain-1 stimulating activity in these cells. C) Immunoblot analysis of heat-stable extract showing decreased calpastatin protein in the dehydrated (fx VI) sickle cells compared to unfractionated sickle cells (n=3).
In vivo calpain-1 inhibition by BDA-410 ameliorates RBC pathology in SCD model SAD mice
Based on these findings, we investigated whether oral administration of the calpain-1 inhibitor BDA-410 affects the hematological phenotype of SCD. To address this question, we administered BDA-410 to SAD mice for SCD that exhibits RBC dehydration. The main advantages of SAD mice for this purpose are the absence of thalassemic features, previous successful use to study the mechanisms involved in generation of dense dehydrated RBCs, and lack of the intense hemolysis that obscures RBC dehydration in more severe SCD mouse models (5, 6, 12, 32, 33). SAD and WT mice underwent 14 d treatment with the calpain-1 specific inhibitor BDA-410 at the dosage of 30 mg/kg/d, administered by gavage. Hematological parameters were evaluated at baseline and at 7 and 14 d of treatment. No changes in body weight were observed during the treatment period. As shown in Table 1, BDA-410 induced a significant increase in Hct at 7 and 14 d of treatment in SAD mice but not in WT mice. Also, no changes were found in vehicle-treated mice (data not shown). No changes in Hb levels were present in BDA-410 treated WT and SAD mice compared to either baseline values. Mean cell volume (MCV) values significantly increased in both mouse strains at 14 d of treatment (Table 1). Mean cellular Hb content (MCHC) values significantly decreased at 7 and 14 d of treatment in SAD mice (Table 1). In both mouse strains, reticulocyte counts were unchanged during BDA-410 treatment. No changes in any of the hematological parameters compared to baseline values were noted in WT and SAD mice treated with vehicle only (Supplemental Table S1).
Table 1.
Hematological data of WT and SCD model SAD mice treated with the calpain inhibitor BDA-410
| Parameter | WT mice |
SAD mice |
||||
|---|---|---|---|---|---|---|
| Baseline | BDA-410, 7 d | BDA-410, 14 d | Baseline | BDA-410, 7 d | BDA-410, 14 d | |
| n | 6 | 6 | 6 | 6 | 6 | 6 |
| Hct (%) | 45.1 ± 1.0 | 46.3 ± 1.2 | 47.1 ± 1.4 | 44.7 ± 0.2 | 46.1 ± 1* | 46.6 ± 0.5* |
| Hb (g/dl) | 14.3 ± 0.8 | 14.9 ± 0.5 | 14.7 ± 0.6 | 13.8 ± 0.5 | 14.2 ± 0.8 | 14.8 ± 0.7 |
| MCV (fl) | 46.8 ± 0.3 | 50.8 ± 0.9* | 51.1 ± 0.4* | 45.7 ± 0.3 | 46.5 ± 0.2 | 48.9 ± 0.6* |
| MCH (pg) | 15.1 ± 0.4 | 15.2 ± 1 | 14.4 ± 0.4 | 14.7 ± 0.2 | 15.2 ± 0.3 | 15.2 ± 0.8 |
| MCHC (g/dl) | 30.2 ± 1 | 30.1 ± 0.3 | 28.1 ± 0.7 | 34.0 ± 0.9 | 31.7 ± 0.3* | 30.8 ± 0.2* |
| Retics (%) | 3.5 ± 0.2 | 3.6 ± 0.8 | 3.97 ± 0.5 | 4.5 ± 0.2 | 3.9 ± 1 | 5.2 ± 0.5 |
| D20 | 1095 ± 0.001 | 1092 ± 0.0005* | 1091 ± 0.001* | 1106 ± 0.001 | 1102 ± 0.0005* | 1100 ± 0.001* |
Data are presented as means ± sd (n=6). Hct, hematocrit; Hb, hemoglobin; MCV, mean cell volume; MCHC, mean cellular hemoglobin content; retics, reticulocytes; D20, erythrocyte density (densest 20% fraction of RBCs with higher density than phthalate phase).
P < 0.05 vs. untreated mice.
As shown in Fig. 2A, BDA-410 administration had a time-dependent beneficial effect on RBC morphology in SAD mice compared to before treatment. The changes in RBC indices and morphology observed in SAD mice were associated with a leftward shift in the RBC density profile in SAD mice treated with BDA-410 (Table 1 and Fig. 2B, right panel), reflecting improved RBC hydration. We also observed a leftward shift of the densest 20% fraction of RBCs (D20) in WT mice (Table 1 and Fig. 2B, left panel). RBC K+ content was significantly reduced in untreated SAD mice compared to WT mice, with no major difference in RBC Na+ content (Fig. 2C). During BDA-410 treatment, the RBC K+ content in SAD mouse RBCs increased significantly compared to baseline values, while no changes were evident in WT mice (Fig. 2C). No significant changes in RBC Na+ content were evident (Fig. 2C).
Figure 2.

BDA-410 treatment ameliorates sickle cell morphology, reduces the dense RBC fraction in sickle cell SAD mice, and increases sickle cell K+ content. A) Representative pictures of RBCs drawn from a WT mouse and an SCD model SAD mouse at baseline and after 7 and 14 d of treatment with BDA-410 at the dosage of 30 mg/kg/d. RBC pictures were captured with a Leica DM6000 (Leica Microsystems, Wetzlar, Germany), ×63/0.8 dry objective. B) Density profiles were obtained at baseline and after 7 and 14 d of treatment with BDA-410 at the dosage of 30 mg/kg/d. Plots show average ± se density profiles (n=6). Left panel: effects of BDA410 on RBC density profiles from WT mice. Right panel: effects of BDA410 on RBC density profiles from SAD mice. C) Effects of pharmacological inhibition of calpain-1 on RBC cation content from WT and SAD mice. RBC Na+ and K+ content are shown at baseline and after 14 dof treatment with BDA-410 at the dosage of 30 mg/kg/d. Data are presented as means ± sd (n=6); *P < 0.05 vs. WT mice; °P < 0.05 vs. untreated mice.
BDA-410 treatment reduces the activity of the Ca2+-activated K+ channel
Since human dense sickle cells showed evidence of increased calpain-1 activity, and we observed amelioration of RBC indices and dehydration in SAD mice treated with the calpain-1 inhibitor BDA-410, we used a bioinformatics approach (43) to search for possible calpain-1 targets on membrane ion transport systems involved in regulation of RBC hydration. It is difficult to predict specificity of protease sites because calpain-1 appears to recognize the overall 3-dimensional structure of its substrates more than the primary structure. In addition, calpain-1 generally cleaves substrates by cutting their interdomain regions, indicating its modulatory functions for substrate proteins (44, 45). A search for calpain-1 cleavage sites via the CaMPDB tool (43) on the Ca2+-activated K+ channel (Gardos channel) identified two putative cleavage sites at high score (>0.15; Fig. 3A). These are localized at Glu288, at the end of the α-helix that mediates membrane insertion, and a number of residues (Arg333, Arg336, Leu343, AL344, His346, Phe348 and Arg360) in the 330–360 region in the cytoplasmic domain, which overlaps with a domain that is critical for phosphorylation-dependent activation (residues 355–413; ref. 46) and with the region that mediates both Ca2+-dependent and -independent binding of calmodulin (residues 286–384; refs. 47, 48). Deletions in this region led to impaired calmodulin binding and loss-of-function effects (47, 49), arguing that bioinformatics prediction could be functionally important. We evaluated the activity of the Gardos channel in mice treated with BDA-410. As shown in Fig. 3B, the activity of the Gardos channel is significantly increased in untreated SAD mouse RBCs compared to WT. The BDA-410 treatment in vivo markedly reduced Gardos channel activity in SAD mice compared to baseline values (Fig. 3B). Previous studies by us and others have shown that the activity of the Gardos channel might be regulated by phosphorylation/dephosphorylation events, involving protein kinase C (PKC) and phosphatases, whose identity is still unknown (34–36). We evaluated the effects of the PKC inhibitor, calphostin C, on Gardos channel activity in BDA-410 treated and untreated mice. As shown in Fig. 3B, in untreated SAD mice calphostin C significantly reduced the activity of the Gardos channel, as previously reported (34–36). However, in RBCs from BDA-410 treated SAD mice, calphostin C did not further reduce the activity of the Gardos channel, suggesting that the fraction of the Gardos channel inhibited by calpain-1 inhibitor is most likely related to PKC channel phosphorylation. We also evaluated the activity of K-Cl cotransport in SAD mice with and without BDA-410, but did not observe significant changes in K-Cl cotransport activity on BDA-410 treatment (data not shown).
Figure 3.

BDA-410 treatment reduces the activity of the Gardos channel and the amount of Prx2 associated with the membrane. A) Putative calpain-specific cleavage sites on Ca2+-dependent K+ channel. Putative domains are highlighted in cyan, the residues involved in blue. B) Ca2+-activated K+ channel (Gardos channel) activity in RBCs from WT and SAD mice at baseline and after 14 d treatment with BDA-410 at the dosage of 30 mg/kg/d. RBCs from a subgroup of treated and untreated BDA-410 (14 d) mice were treated in vitro with the protein kinase inhibitor calphostin C (1 μM). Data are presented as means ± sd (n=12; calphostin C experiments n=4); *P < 0.05 vs. WT mice; °P < 0.05 vs. untreated mice. C) Top panel: putative calpain-specific cleavage sites on Prx2. Putative domains are highlighted in cyan, the residues involved in blue. In Prx2, the positions of the catalytic cysteine residues are reported (yellow). Bottom panel: effects of BDA-410 treatment (14 d treatment) on RBC membrane recruitment of Prx2 in WT and transgenic SAD mouse RBCs. Immunoblot analysis of RBC membranes with specific antibody anti-Prx2 (Prx2). NT, nontreated groups. Vertical lines have been inserted to indicate a repositioned gel line. Data are representative for 6 experiments (on 6-cm gels). Actin was used as a loading control.
Previous studies have proposed a functional connection between activation of the Gardos channel and the membrane associated protein Prx2, formerly termed calpromotin, although mechanism remains unknown (50, 51). Recently, we have reported increased membrane association of Prx2 in SAD mouse RBCs compared to WT RBCs (39). Analysis by the CaMPDB (43) of Prx2 sequence identified two putative calpain cleavage sites (Fig. 3C, top panel) with high score (>0.1; Ala85 and the triplex Asp181/Thr182/Ile183) in the C-terminal domain near the catalytic Cys residue (Cys172). Deletion of the C-terminal arm of PRDX2 led to an enzyme unable to respond to oxidative stress (52), lacking hyperoxidation regulation. Thus, functional connection seems to emerge among calpain activity, Prx2 cytosol to membrane partition, and Gardos activation. We then evaluated Prx2 membrane association in WT and SAD mice before and after BDA-410 treatment. As shown in the bottom panel of Fig. 3C, Prx2 associated with the membrane is higher in SAD RBC membranes compared to WT. BDA-410 treatment significantly reduced the amount of Prx2 associated with the membrane in SAD mice, while no differences were observed in treated WT mice (Fig. 3C, bottom panel, lanes 2, 4) as also shown by flow-cytometric analysis (Supplemental Fig. S1).
BDA-410 treatment prevents hypoxia-induced RBC dehydration in SAD mice
SAD mice that had been subjected to BDA-410 treatment for 14 d, and untreated SAD mice, were exposed to hypoxia (8% oxygen) for 48 h, followed by 2 h of reoxygenation (12, 33) to mimic sickle cell-related vasoocclusive events. Untreated SAD mice exhibited hypoxia-induced RBC dehydration (Fig. 4A and Table 1) and RBC K+ loss (Fig. 4B and Table 1), which were ameliorated by BDA-410 treatment (D20 1110±0.0005 in untreated SAD mice vs. 1104±0.001 in BDA-410 treated SAD mice; P<0.05, n=6). As previously reported, no changes in reticulocyte count were observed in either mouse group after exposure to hypoxia (5, 12, 32, 33).
Figure 4.
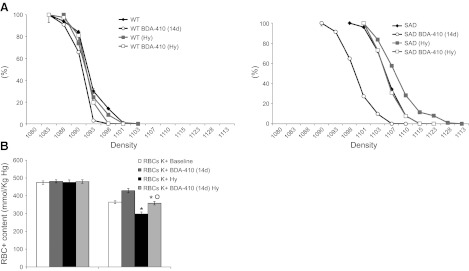
BDA-410 treatment prevents the hypoxia-induced RBC dehydration and K+ loss in SCD model SAD mice. A) Left panel: density profiles of RBCs from WT mice were obtained at baseline, after 14 d of treatment with BDA-410 at the dosage of 30 mg/kg/d, and after 48 h hypoxia (8% oxygen; Hy). Right panel: density profiles of RBCs from SAD mice were obtained at baseline and after 14 d of treatment with BDA-410 at the dosage of 30 mg/kg/d, and after 48 h hypoxia (8% oxygen; Hy). Plots show average ± se density profiles (n=6). B) Effects of pharmacological inhibition of calpain-1 on RBC K+ content from WT and SAD mice exposed to hypoxia (8% oxygen; Hy). RBC K+ contents are shown at baseline, after 14 d of treatment with BDA-410 at the dosage of 30 mg/kg/d, and after 48 h hypoxia. Data are presented as means ± sd (n=6). *P < 0.05 vs. WT mice; °P < 0.05 vs. untreated mice.
DISCUSSION
Calpain-1 is a nonlysosomal calcium-dependent protease that modulates multiple cell functions in various cell models through the selective cleavage of proteins. In human sickle cells, we found increased calpain-1 activation in dense sickle cells and reduced activity of its main inhibitor, calpastatin. In the light of these data, we examined the action of BDA-410, a selective inhibitor of calpain-1, with structural features similar to those of the natural endogenous calpain-1 inhibitor, calpastatin, in sickle cells. We hypothesized that the inhibition of calpain-1 by BDA-410 might prevent the loss of membrane substrate protein function in sickle cells. In SAD mice, treatment with BDA-410 in vivo reduced the percentage of dense sickle cells, increased the RBC K+ content and ameliorated RBC indices and morphology (Fig. 2), suggesting that calpain-1 modulates the activity of membrane transport systems involved in regulation of RBC hydration. Studies on sickle cells have shown that two transport systems contribute to the generation of dense sickle cell: the K-Cl cotrasport and the Ca2+-activated K+ channel (Gardos) (7, 9, 10, 53). Since we did not find significant differences in the activity of the K-Cl cotransport in SAD mouse RBCs as a result of BDA-410 treatment, we focused on the Gardos channel: using a bioinformatics tool, we indeed found potential calpain-1 cleavage sites on the Gardos channel protein sequence, which are placed in the C-terminal region of the channel, overlapping a sequence known to be subjected to modulation by phosphorylation and involved in calmodulin binding (46–48). Based on our finding that the activity of the Gardos channel was significantly decreased in treated SAD mice compared to untreated SAD mice, we suggest that calpain-1 is involved in enhancing Gardos channel activity. The inhibitory effect of calphostin C on the RBC Gardos channel that is seen in untreated SAD mice is no longer present after BDA-410 treatment, suggesting that calpain-1 inhibition affects the PKC-dependent fraction of the Gardos channel activity. In other cell models, it has been shown that PKC family members can be activated by calpain-1 through a limited proteolysis (54, 55). In human erythrocytes, Al and Cohen (56) have reported that a calpain-1 mediated proteolyis of PKC is required for substrate phosphorylation on the RBC membrane. In sickle cells under deoxygenation, increased PKC activity in RBC membrane has been reported (57). In our model, we speculate that BDA-410 might inhibit the Gardos channel indirectly by preventing the PKC calpain-mediated activation. This could explain the lack of calphostin C inhibition of the Gardos channel activity in SAD mice treated with the BDA-410 inhibitor. However, we cannot exclude a possible effect of BDA-410 on the phosphatases involved in modulation of the Gardos channel, and this activity might be enhanced by calpain-1 inhibition (24, 58).
In sickle cells, the activation of the Gardos channel has also been linked to membrane association of Prx2, but the effect exerted by Prx2 on Gardos channel remains to be elucidated (50, 51). Our bioinformatics analysis revealed the presence of calpain-1 cleavage sites on Prx2. These are located in the C-terminal segment of Prx2, downstream from the catalytic cysteine residue (Cys172) that is important for antioxidant action. In SAD mice, Prx2 membrane association was increased compared to WT and markedly reduced in BDA-410-treated SAD mouse RBCs. While the increase in Prx2 membrane association could be related to the higher oxidative stress experienced by SAD mice, the decrease following calpain-1 inhibitor implies an influence of calpain-1 activity on Prx2 cytosol/membrane transitions. Since Prx2 and Gardos channel have putative calpain-1 binding sites, and their functions are affected by the calpain-1 inhibitor, one could hypothesize that calpain-1 is involved in the Prx2-Gardos channel network. A possible functional connection between Prx2 and Gardos channel could be linked to the role of Prx2 in protecting crucial cysteine residues from oxidation. The Gardos channel, immediately after the pore region, presents a motif (C265XC267XXXXXXC274C275) adjacent to the pore region that could behave as a functional mimic of protein disulfide isomerase (59). In this view, Prx2 could switch on/off the thiol-redox center of the Gardos channel, similarly to that described for Prx1 in controlling neuronal differentiation by thiol-redox dependent activation of glycerophosphodiester phosphodiesterase-2 (GDE2; ref. 60).
BDA-410 treatment results in multiple beneficial effects on sickle erythrocytes: in particular, the reduction of dense RBCs may have significant implications for the development and pathological features of SCD. In vitro and in vivo studies in animal models for SCD have suggested a crucial role of dehydrated RBCs in the pathogenesis of vasoocclusive events: the dense, dehydrated RBCs might be easily trapped in postcapillary venules, promoting microvascular obstruction (9–11, 53). Thus, the possibility to interfere with the generation of dense RBCs might be a useful additional therapeutic approach in SCD. Our studies indicate that an imbalance exists between calpain-1 activator and inhibitor, which favors calpain-1 activation in SCD. The beneficial effects of BDA-410 on hematological phenotype of a mouse model for SCD suggest a potential role of BDA-410 as a new therapeutic agent to be further characterized in SCD.
Supplementary Material
Acknowledgments
This work was supported by ex-40% Ministro dell'Istruzione, dell'Università e della Ricerca (MIUR), University of Verona, to L.D.F. and by U.S. National Institutes of Health grants HL089517 and HL095050 and a Tufts Collaborates grant to A.H.C.
The authors are grateful to Drs. Narihiko Yoshii and Hiroshi Kinoshita. A.H.C. proposed the evaluation of BDA-410 in SCD mice. A.H.C. and C.H.J. served as equal senior authors of the manuscript.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- CaMPDB
- Calpain for Modulatory Proteolysis Data Bank
- D20
- densest 20% fraction of red blood cells
- Hb
- hemoglobin
- Hct
- hematocrit
- MCV
- mean cell volume
- MCHC
- mean cellular hemoglobin content
- NEM
- n-ethylmaleimide
- OD
- optical density
- PKC
- protein kinase C
- PLB
- phosphate lysis buffer
- Prx2
- peroxiredoxin-2
- RBC
- red blood cell
- SAD
- Hbbsingle/single SAD1
- SCD
- sickle cell disease
- WT
- wild-type
REFERENCES
- 1. Eaton W. A., Hofrichter J. (1990) Sickle cell hemoglobin polymerization. Adv. Protein Chem. 40, 63–279 [DOI] [PubMed] [Google Scholar]
- 2. Franco R. S., Yasin Z., Lohmann J. M., Palascak M. B., Nemeth T. A., Weiner M., Joiner C. H., Rucknagel D. L. (2000) The survival characteristics of dense sickle cells. Blood 96, 3610–3617 [PubMed] [Google Scholar]
- 3. Solovey A. A., Solovey A. N., Harkness J., Hebbel R. P. (2001) Modulation of endothelial cell activation in sickle cell disease: a pilot study. Blood 97, 1937–1941 [DOI] [PubMed] [Google Scholar]
- 4. Hebbel R. P. (2008) Adhesion of sickle red cells to endothelium: myths and future directions. Transfus. Clin. Biol. 15, 14–18 [DOI] [PubMed] [Google Scholar]
- 5. De Franceschi L., Brugnara C., Rouyer-Fessard P., Jouault H., Beuzard Y. (1999) Formation of dense erythrocytes in SAD mice exposed to chronic hypoxia: evaluation of different therapeutic regimens and of a combination of oral clotrimazole and magnesium therapies. Blood 94, 4307–4313 [PubMed] [Google Scholar]
- 6. Siciliano A., Malpeli G., Platt O. S., Lebouef C., Janin A., Scarpa A., Olivieri O., Amato E., Corrocher R., Beuzard Y., De Franceschi L. (2011) Abnormal modulation of cell protective systems in response to ischemic/reperfusion injury is important in the development of mouse sickle cell hepatopathy. Haematologica 96, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sabaa N., de Franceschi L., Bonnin P., Castier Y., Malpeli G., Debbabi H., Galaup A., Maier-Redelsperger M., Vandermeersch S., Scarpa A., Janin A., Levy B., Girot R., Beuzard Y., Leboeuf C., Henri A., Germain S., Dussaule J. C., Tharaux P. L. (2008) Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J. Clin. Invest. 118, 1924–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Franceschi L., Cappellini M. D., Olivieri O. (2011) Thrombosis and sickle cell disease. Semin. Thromb. Hemostasis 37, 226–236 [DOI] [PubMed] [Google Scholar]
- 9. Joiner C. H., Rettig R. K., Jiang M., Franco R. S. (2004) KCl cotransport mediates abnormal sulfhydryl-dependent volume regulation in sickle reticulocytes. Blood 104, 2954–2960 [DOI] [PubMed] [Google Scholar]
- 10. Joiner C. H., Rettig R. K., Jiang M., Risinger M., Franco R. S. (2007) Urea stimulation of KCl cotransport induces abnormal volume reduction in sickle reticulocytes. Blood 109, 1728–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McGoron A. J., Joiner C. H., Palascak M. B., Claussen W. J., Franco R. S. (2000) Dehydration of mature and immature sickle red blood cells during fast oxygenation/deoxygenation cycles: role of KCl cotransport and extracellular calcium. Blood 95, 2164–2168 [PubMed] [Google Scholar]
- 12. Stocker J. W., De Franceschi L., McNaughton-Smith G. A., Corrocher R., Beuzard Y., Brugnara C. (2003) ICA-17043, a novel Gardos channel blocker, prevents sickled red blood cell dehydration in vitro and in vivo in SAD mice. Blood 101, 2412–2418 [DOI] [PubMed] [Google Scholar]
- 13. Brugnara C., Van Ha T., Tosteson D. C. (1989) Acid pH induces formation of dense cells in sickle erythrocytes. Blood 74, 487–495 [PubMed] [Google Scholar]
- 14. Hebbel R. P., Vercellotti G. M., Pace B. S., Solovey A. N., Kollander R., Abanonu C. F., Nguyen J., Vineyard J. V., Belcher J. D., Abdulla F., Osifuye S., Eaton J. W., Kelm R. J., Jr., Slungaard A. (2010) The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood 115, 2483–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuchay S. M., Kim N., Grunz E. A., Fay W. P., Chishti A. H. (2007) Double knockouts reveal that protein tyrosine phosphatase 1B is a physiological target of calpain-1 in platelets. Mol. Cell. Biol. 27, 6038–6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanna R. A., Campbell R. L., Davies P. L. (2008) Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456, 409–412 [DOI] [PubMed] [Google Scholar]
- 17. Salamino F., De Tullio R., Mengotti P., Viotti P. L., Melloni E., Pontremoli S. (1993) Site-directed activation of calpain is promoted by a membrane-associated natural activator protein. Biochem. J. 290(Pt. 1), 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Melloni E., Michetti M., Salamino F., Pontremoli S. (1998) Molecular and functional properties of a calpain activator protein specific for mu-isoforms. J. Biol. Chem. 273, 12827–12831 [DOI] [PubMed] [Google Scholar]
- 19. Melloni E., Minafra R., Salamino F., Pontremoli S. (2000) Properties and intracellular localization of calpain activator protein. Biochem. Biophys. Res. Commun. 272, 472–476 [DOI] [PubMed] [Google Scholar]
- 20. Olorunsogo O. O., Agbolade F. O., Owojuyigbe S. O., Adebisi J. A., Adebayo A. O., Okunade W. G. (1990) Comparative action of calpain on erythrocyte Ca2(+)-pumping ATPase in sickle cell anaemia, essential hypertension and kwashiorkor. Biosci. Rep. 10, 281–291 [DOI] [PubMed] [Google Scholar]
- 21. Falchetto R., Vorherr T., Carafoli E. (1992) The calmodulin-binding site of the plasma membrane Ca2+ pump interacts with the transduction domain of the enzyme. Protein Sci. 1, 1613–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boivin P., Galand C., Dhermy D. (1990) In vitro digestion of spectrin, protein 4.1 and ankyrin by erythrocyte calcium dependent neutral protease (calpain I). Int. J. Biochem. 22, 1479–1489 [DOI] [PubMed] [Google Scholar]
- 23. Mehdi S., Angelastro M. R., Wiseman J. S., Bey P. (1988) Inhibition of the proteolysis of rat erythrocyte membrane proteins by a synthetic inhibitor of calpain. Biochem. Biophys. Res. Commun. 157, 1117–1123 [DOI] [PubMed] [Google Scholar]
- 24. Pontremoli S., Sparatore B., Salamino F., De Tullio R., Pontremoli R., Melloni E. (1988) The role of calpain in the selective increased phosphorylation of the anion-transport protein in red cell of hypertensive subjects. Biochem. Biophys. Res. Commun. 151, 590–597 [DOI] [PubMed] [Google Scholar]
- 25. Wieschhaus A., Khan A., Zaidi A., Rogalin H., Hanada T., Liu F., De Franceschi L., Brugnara C., Rivera A., Chishti A. H. (2012) Calpain-1 knockout reveals broad effects on erythrocyte deformability and physiology. [E-pub ahead of print] Biochem. J. doi: 10.1042/BJ20121008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang K. K., Roufogalis B. D., Villalobo A. (1989) Calpain I activates Ca2+ transport by the reconstituted erythrocyte Ca2+ pump. J. Membr. Biol. 112, 233–245 [DOI] [PubMed] [Google Scholar]
- 27. Matarrese P., Straface E., Pietraforte D., Gambardella L., Vona R., Maccaglia A., Minetti M., Malorni W. (2005) Peroxynitrite induces senescence and apoptosis of red blood cells through the activation of aspartyl and cysteinyl proteases. FASEB J. 19, 416–418 [DOI] [PubMed] [Google Scholar]
- 28. Battaglia F., Trinchese F., Liu S., Walter S., Nixon R. A., Arancio O. (2003) Calpain inhibitors, a treatment for Alzheimer's disease: position paper. J. Mol. Neurosci. 20, 357–362 [DOI] [PubMed] [Google Scholar]
- 29. Trinchese F., Fa M., Liu S., Zhang H., Hidalgo A., Schmidt S. D., Yamaguchi H., Yoshii N., Mathews P. M., Nixon R. A., Arancio O. (2008) Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Invest. 118, 2796–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li X., Chen H., Jeong J. J., Chishti A. H. (2007) BDA-410: a novel synthetic calpain inhibitor active against blood stage malaria. Mol. Biochem. Parasitol. 155, 26–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yasin Z., Witting S., Palascak M. B., Joiner C. H., Rucknagel D. L., Franco R. S. (2003) Phosphatidylserine externalization in sickle red blood cells: associations with cell age, density, and hemoglobin F. Blood 102, 365–370 [DOI] [PubMed] [Google Scholar]
- 32. Bennekou P., de Franceschi L., Pedersen O., Lian L., Asakura T., Evans G., Brugnara C., Christophersen P. (2001) Treatment with NS3623, a novel Cl-conductance blocker, ameliorates erythrocyte dehydration in transgenic SAD mice: a possible new therapeutic approach for sickle cell disease. Blood 97, 1451–1457 [DOI] [PubMed] [Google Scholar]
- 33. de Franceschi L., Baron A., Scarpa A., Adrie C., Janin A., Barbi S., Kister J., Rouyer-Fessard P., Corrocher R., Leboulch P., Beuzard Y. (2003) Inhaled nitric oxide protects transgenic SAD mice from sickle cell disease-specific lung injury induced by hypoxia/reoxygenation. Blood 102, 1087–1096 [DOI] [PubMed] [Google Scholar]
- 34. Rivera A., Jarolim P., Brugnara C. (2002) Modulation of Gardos channel activity by cytokines in sickle erythrocytes. Blood 99, 357–603 [DOI] [PubMed] [Google Scholar]
- 35. Rivera A., Rotter M. A., Brugnara C. (1999) Endothelins activate Ca(2+)-gated K(+) channels via endothelin B receptors in CD-1 mouse erythrocytes. Am. J. Physiol. 277, C746–C754 [DOI] [PubMed] [Google Scholar]
- 36. De Franceschi L., Biondani A., Carta F., Turrini F., Laudanna C., Deana R., Brunati A. M., Turretta L., Iolascon A., Perrotta S., Elson A., Bulato C., Brugnara C. (2008) PTPepsilon has a critical role in signaling transduction pathways and phosphoprotein network topology in red cells. Proteomics 8, 4695–4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Franceschi L., Beuzard Y., Brugnara C. (1995) Sulfhydryl oxidation and activation of red cell K(+)-Cl- cotransport in the transgenic SAD mouse. Am. J. Physiol. 269, C899–906 [DOI] [PubMed] [Google Scholar]
- 38. Matte A., Low P. S., Turrini F., Bertoldi M., Campanella M. E., Spano D., Pantaleo A., Siciliano A., De Franceschi L. (2010) Peroxiredoxin-2 expression is increased in beta-thalassemic mouse red cells but is displaced from the membrane as a marker of oxidative stress. Free Radic. Biol. Med. 49, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Biondani A., Turrini F., Carta F., Matte A., Filippini A., Siciliano A., Beuzard Y., De Franceschi L. (2008) Heat-shock protein-27, -70 and peroxiredoxin-II show molecular chaperone function in sickle red cells: Evidence from transgenic sickle cell mouse model. Proteomics Clin. Appl. 2, 706–719 [DOI] [PubMed] [Google Scholar]
- 40. De Franceschi L., Bertoldi M., De Falco L., Santos Franco S., Ronzoni L., Turrini F., Colancecco A., Camaschella C., Cappellini M. D., Iolascon A. (2011) Oxidative stress modulates heme synthesis and induces peroxiredoxin-2 as a novel cytoprotective response in beta-thalassemic erythropoiesis. Haematologica 96, 1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koo K. H., Lee S., Jeong S. Y., Kim E. T., Kim H. J., Kim K., Song K., Chae H. Z. (2002) Regulation of thioredoxin peroxidase activity by C-terminal truncation. Arch. Biochem. Biophys. 397, 312–318 [DOI] [PubMed] [Google Scholar]
- 42. Matte A., Low P. S., Turrini F., Bertoldi M., Campanella M. E., Spano D., Pantaleo A., Siciliano A., De Franceschi L. (2010) Peroxiredoxin-2 expression is increased in beta-thalassemic mouse red cells but is displaced from the membrane as a marker of oxidative stress. Free Radic. Biol. Med. 49, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DuVerle D. A., Ono Y., Sorimachi H., Mamitsuka H. (2011) Calpain cleavage prediction using multiple kernel learning. PLoS One 6, e19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saido T. C., Sorimachi H., Suzuki K. (1994) Calpain: new perspectives in molecular diversity and physiological-pathological involvement. FASEB J. 8, 814–822 [PubMed] [Google Scholar]
- 45. Sorimachi H., Ishiura S., Suzuki K. (1997) Structure and physiological function of calpains. Biochem. J. 328 (Pt 3), 721–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gerlach A. C., Syme C. A., Giltinan L., Adelman J. P., Devor D. C. (2001) ATP-dependent activation of the intermediate conductance, Ca2+-activated K+ channel, hIK1, is conferred by a C-terminal domain. J. Biol. Chem. 276, 10963–10970 [DOI] [PubMed] [Google Scholar]
- 47. Fanger C. M., Ghanshani S., Logsdon N. J., Rauer H., Kalman K., Zhou J., Beckingham K., Chandy K. G., Cahalan M. D., Aiyar J. (1999) Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J. Biol. Chem. 274, 5746–5754 [DOI] [PubMed] [Google Scholar]
- 48. Joiner W. J., Khanna R., Schlichter L. C., Kaczmarek L. K. (2001) Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J. Biol. Chem. 276, 37980–37985 [DOI] [PubMed] [Google Scholar]
- 49. Syme C. A., Hamilton K. L., Jones H. M., Gerlach A. C., Giltinan L., Papworth G. D., Watkins S. C., Bradbury N. A., Devor D. C. (2003) Trafficking of the Ca2+-activated K+ channel, hIK1, is dependent upon a C-terminal leucine zipper. J. Biol. Chem. 278, 8476–8486 [DOI] [PubMed] [Google Scholar]
- 50. Moore R. B., Mankad M. V., Shriver S. K., Mankad V. N., Plishker G. A. (1991) Reconstitution of Ca(2+)-dependent K+ transport in erythrocyte membrane vesicles requires a cytoplasmic protein. J. Biol. Chem. 266, 18964–18968 [PubMed] [Google Scholar]
- 51. Moore R. B., Shriver S. K., Jenkins L. D., Mankad V. N., Shah A. K., Plishker G. A. (1997) Calpromotin, a cytoplasmic protein, is associated with the formation of dense cells in sickle cell anemia. Am. J. Hematol. 56, 100–106 [DOI] [PubMed] [Google Scholar]
- 52. Moon J. C., Hah Y. S., Kim W. Y., Jung B. G., Jang H. H., Lee J. R., Kim S. Y., Lee Y. M., Jeon M. G., Kim C. W., Cho M. J., Lee S. Y. (2005) Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J. Biol. Chem. 280, 28775–28784 [DOI] [PubMed] [Google Scholar]
- 53. De Franceschi L., Corrocher R. (2004) Established and experimental treatments for sickle cell disease. Haematologica 89, 348–356 [PubMed] [Google Scholar]
- 54. Kishimoto A., Kajikawa N., Shiota M., Nishizuka Y. (1983) Proteolytic activation of calcium-activated, phospholipid-dependent protein kinase by calcium-dependent neutral protease. J. Biol. Chem. 258, 1156–1164 [PubMed] [Google Scholar]
- 55. Kishimoto A., Mikawa K., Hashimoto K., Yasuda I., Tanaka S., Tominaga M., Kuroda T., Nishizuka Y. (1989) Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain). J. Biol. Chem. 264, 4088–4092 [PubMed] [Google Scholar]
- 56. Al Z., Cohen C. M. (1993) Phorbol 12-myristate 13-acetate-stimulated phosphorylation of erythrocyte membrane skeletal proteins is blocked by calpain inhibitors: possible role of protein kinase M. Biochem. J. 296(Pt. 3), 675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fathallah H., Sauvage M., Romero J. R., Canessa M., Giraud F. (1997) Effects of PKC alpha activation on Ca2+ pump and K(Ca) channel in deoxygenated sickle cells. Am. J. Physiol. 273, C1206–C1214 [DOI] [PubMed] [Google Scholar]
- 58. Ciana A., Minetti G., Balduini C. (2004) Phosphotyrosine phosphatases acting on band 3 in human erythrocytes of different age: PTP1B processing during cell ageing. Bioelectrochemistry 62, 169–173 [DOI] [PubMed] [Google Scholar]
- 59. Woycechowsky K. J., Raines R. T. (2003) The CXC motif: a functional mimic of protein disulfide isomerase. Biochemistry 42, 5387–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yan Y., Sabharwal P., Rao M., Sockanathan S. (2009) The antioxidant enzyme Prdx1 controls neuronal differentiation by thiol-redox-dependent activation of GDE2. Cell 138, 1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.