

Abstract
During wound repair, epidermal cells at the edge of an injury establish front-rear polarity through orchestrated changes in their cytoskeleton and adhesion structures. The polarity and directed migration of such cells is determined by the assembly, extension, and stabilization of a lamellipodium. Actinin-4 associates with lamellipodia and has been implicated in regulating lamellipodial structure, function and assembly. To study the functions of actinin-4 in human keratinocytes, we used shRNA to generate knockdown cells and compared their motility behavior and matrix adhesion assembly to scrambled shRNA treated control keratinocytes. Actinin-4 knockdown keratinocytes lack polarity, assemble multiple lamellipodia with a 2× increased area over controls, display reduced activity of the actin remodeling protein cofilin, and fail to migrate in a directional manner. This motility defect is rescued by plating knockdown cells on preformed laminin-332 matrix. In actinin-4-knockdown keratinocytes, focal contact area is increased by 25%, and hemidesmosome proteins are mislocalized. Specifically, α6β4 integrin localizes to large lamellipodial extensions, displays reduced dynamics, and fails to recruit its bullous pemphigoid antigen binding partners. Together, our data indicate a role for actinin-4 in regulating the steering mechanism of keratinocytes via profound effects on their matrix adhesion sites.—Hamill, K. J., Hopkinson, S. B., Skalli, O., Jones, J. C. R. Actinin-4 in keratinocytes regulates motility via an effect on lamellipodia stability and matrix adhesions.
Keywords: focal contact, hemidesmosome, laminin, integrin
Members of the actinin family of proteins cross-link actin filaments (1, 2). Four major isoforms have been identified: two muscle-specific isoforms, actinin-2 and 3 (ACTN2 and ACTN3), which are located in Z disks in striated muscle cells and are found in dense plaques in smooth muscle, and two nonmuscle isoforms, actinin-1 and -4 (ACTN1 and ACTN4) (2–5). Each share a common domain structure consisting of an actin-binding domain consisting of two calponin homology domains, followed by a pleckstrin homology domain and two EF-hand calcium regulation domains (3).
The nonmuscle actinin isoforms, ACTN1 and ACTN4, exhibit 80% nucleotide and 87% amino acid similarity (3). Both are found in sheet-like lamellipodial extensions of migrating cells (3, 6, 7). However, the cellular location and function of these isoforms is also tissue, cell type, and indeed context specific. Specifically, ACTN1 decorates microfilaments and is found associated with adherens cell–cell junctions and focal contact cell-matrix attachment sites via interaction with β1 integrin (8, 9). Although ACTN4 also localizes along actin stress fibers and at sites of cell-cell contact, it is found in the nucleus in certain cells and interacts with collagen type XVII [Col XVII; bullous pemphigoid antigen 2 (BPAG2)], a transmembrane component of epithelial cell matrix adhesion devices termed hemidesmosomes (3, 9, 10). ACTN4 has also been described as occasionally recruited to focal contacts in fibroblasts and, in contrast to ACTN1, is highly concentrated at the leading edge of motile cells (3, 11, 12).
Localization to lamellipodia suggests that ACTN4 plays a role in migration (13, 14). Indeed, both nonmuscle actinins have been reported to support or inhibit migration, depending on cell or tissue type. For example, down-regulation of ACTN1 expression leads to increased motility and tumorigenicity of 3T3 fibroblasts but reduced motility of glioblastoma multiform cells. while not influencing the migration of astrocytoma cell lines (6, 15–17). Genetic ablation of ACTN4 results in an increase in lymphocyte chemotaxis (18). In contrast, ACTN4 knockdown results in decreased motility or decreased invasion potential of ovarian carcinoma cells, oral squamous cell carcinoma cell lines, glioblastoma multiforme cells, and astrocytoma lines (6, 17, 19, 20). Consistent with these findings, increased ACTN4 expression is correlated with poor prognosis in ovarian clear-cell adenocarcinomas, bladder cancer invasion, and ductal carcinoma of the pancreas (21–23). In addition, an increase in ACTN4 expression is associated with infiltrative histological phenotype and poor prognosis in ovarian cancer (24).
The above analyses emphasize that the function of actinins in motility is contingent on cellular context. This finding led us to evaluate the effect of down-regulation of ACTN4 on the motility behavior of keratinocytes. Keratinocytes, like other epithelial cells, express ACTN1 and ACTN4. However, unlike fibroblasts and a number of epithelial cell types, they also assemble two distinct matrix adhesion structures, namely focal contacts and hemidesmosome-rich protein complexes, both of which are known to influence cell motility phenotype (25–33). We utilized shRNA technology to knockdown expression of ACTN4 in human epidermal keratinocytes and then assessed the consequences on keratinocyte motile behavior, assembly of focal contacts and hemidesmosome protein complexes.
MATERIALS AND METHODS
Cell culture
Immortalized human epidermal keratinocytes (iHEKs) and β4 integrin-deficient keratinocytes derived from a patient with junctional epidermolysis bullosa (JEB) with pyloric atresia (JEB cells) stably expressing full-length, green fluorescent protein (GFP)-tagged β4 integrin (JEBβ4FL) were described previously (25). The cells were maintained in defined keratinocyte serum-free medium supplemented with a 1% penicillin/streptomycin mixture (Invitrogen Corp., Carlsbad, CA, USA) and grown at 37°C. ACTN4-knockdown keratinocytes were generated using previously described lentiviral shRNAs (17). iHEKs or JEBβ4 cells (5×105) were seeded overnight in 6-well dishes, then infected with lentivirus encoding ACTN4 shRNA or a scrambled shRNA at a multiplicity of infection (MOI) of 0.5 in culture medium supplemented with polybrene (8 μg/ml; Invitrogen). The following day, the medium of the infected cells was aspirated and replaced with fresh medium containing puromycin (0.5 μg/ml) for selection of stable transfectants. In the case of the iHEKs, multiple individual clones were isolated. ACTN4 knockdown was confirmed by SDS-PAGE immunoblotting.
ACTN4 and GFP construct generation
Cloning of full-length ACTN4 was described previously (10). Through site directed mutagenesis, four silent mutations were introduced at the shRNA target site using the QuickChange XL Site-Directed Mutagenesis Kit from Stratagene (La Jolla, CA, USA) following the procedure of the manufacturer. The refractory ACTN4 construct or the mRNA for enhanced green fluorescent protein (EGFP) encoded by pEGFP-N1 (Clontech, Palo Alto, CA, USA), was subcloned into pENTR4 (Invitrogen) and then used in an LR recombination reaction to transfer the cassette into the pAd/CMV/V5-DESTadenoviral vector. Following amplification, the adenoviral expression clones were introduced into 293A cells by lipofectamine-mediated transfection. After 10–12 d, the crude viral lysates were harvested and used to amplify the adenovirus as described previously (25). The amplified viral stocks were titered, and epithelial cells were infected at an MOI of 1:50 in cell medium.
Antibodies, chemicals, and other reagents
Mouse monoclonal antibodies against β4 integrin (3E1) and α3 integrin (P1B5) were purchased from Millipore (Billerica, MA, USA). Mouse monoclonal antibodies against Talin and against laminin-332 (GB3) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Harlan Sera Lab Ltd. (Bicester, UK), respectively. Rabbit monoclonal antibodies against ACTN4, ACTN1, paxillin, plectin, and actin were obtained from Epitomics, Inc. (Burlingame, CA, USA). Rabbit polyclonal antibodies against phospho-cofilin (Ser3), total cofilin, and Lamin A/C were obtained from Cell Signaling (Beverly, MA, USA). Antibodies against β4 integrin (CD104) were purchased from BD Biosciences (San Jose, CA, USA). 5E, a human mAb against bullous pemphigoid antigen 1e (BPAG1e), was a gift from Dr. Takashi Hashimoto (Keio University, Tokyo, Japan; ref. 34). A mouse monoclonal antibody against BPAG1e (10C5) was characterized previously (35). The mouse IgM mAb preparation (1804b) and rabbit polyclonal antibody (J17) against the very amino-terminal domain of Col XVII were described previously (36–38). Secondary antibodies conjugated with various fluorochromes or horseradish peroxidase were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA). Rhodamine-conjugated phalloidin was obtained from Invitrogen. Adenovirus expressing GFP-tagged Xenopus cofilin was generously provided by Dr. James Bamburg (University of Colorado, Boulder, CO, USA).
SDS-PAGE and Western immunoblotting
Cell extracts were prepared as described previously (39, 40). Protein samples were processed for SDS-PAGE and immunoblotting as detailed elsewhere (39, 41). Western immunoblots were scanned and quantified using a MetaMorph imaging system (Universal Imaging Corp.; Molecular Devices, Downingtown, PA, USA) as detailed elsewhere (25).
Immunofluorescence microscopy
Cells on glass coverslips were processed as detailed elsewhere (25). All preparations were viewed with a confocal laser scanning microscope (UV LSM 510; Zeiss Inc., Thornwood, NY, USA). Images were exported as TIF files, and figures were prepared using Adobe Photoshop and Illustrator software (Adobe Systems, San Jose, CA, USA).
Observations of live cells, motility assays, and kymography
Cell surface area, lamellipodial area, and lamellipodial protrusion number were measured from images of individual cells captured at ×60 objective and measured using MetaMorph software (Universal Imaging). Single-cell motility was measured as detailed previously (25). Briefly, cells were plated onto uncoated 35-mm glass-bottomed culture dishes (MatTek Corp., Ashland, MA, USA) 18–24 h prior to cell motility assays. Cells were viewed on a Nikon TE2000 inverted microscope (Nikon Inc., Melville, NY, USA). Images were taken at 2-min intervals over 2 h, and cell motility behavior was analyzed by a MetaMorph imaging system. For each cell line, means of total distance migrated (speed), and processivity (max distance from origin/total distance) were calculated. For kymography, cells were plated as above and images taken using a ×60 objective every 5 s for 15 min. From the resultant stack of images, a composite from each of the frames beneath a 1-pixel-wide line in the direction of migration was constructed using MetaMorph software. Measurements of lamellipodia extension distance and persistence were measured from the resultant composite (42).
Fluorescence-activated cell sorting (FACS)
For flow cytometry, freshly trypsinized cells were resuspended in PBS containing a 50% dilution of normal goat serum, incubated with monoclonal antibodies against α3 integrin (P1B5) or β4 integrin (3E1) for 45 min at room temperature, washed with PBS, incubated with FITC-conjugated goat anti-mouse IgG for 45 min, washed, resuspended in PBS, and analyzed using a Beckman Coulter Elite PCS sorter (Beckman Coulter, Fullerton, CA, USA). For negative controls, primary antibody was omitted from the above procedure.
Focal contact area
Keratinocytes were plated overnight on glass coverslips, then processed for indirect immunofluorescence microscopy with antibodies against paxillin. Images of individual cells were captured at ×90 view, 2× binning. ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA) was used to threshold staining above background levels, and, from the resultant image, particles of size 2–100 pixels2 were analyzed in regions of lamellipodial extension. At least 3 coverslips, >30 cells/coverslip, were analyzed per line.
Rac activity assay
Quantitative analysis of Rac 1, 2, and 3 activation was performed using a G-LISA Rac activation assay (Cytoskeleton, Denver, CO, USA). Briefly, cell lysates were prepared from the cell lines 24 h following plating at 50% confluence, Samples at a normalized total protein concentration of 0.5 mg/ml were used for determination of Rac activity in a 96-well plate format according to the manufacturer's instructions using an ELx808 ultramicroplate spectrophotometer (Bio-Tek Instruments, Winooski, VT, USA).
Fluorescence recovery after photobleaching (FRAP)
FRAP studies were performed as described elsewhere (43, 44). Time-lapse observations were made using a LSM 510 confocal microscope (Zeiss) under the following conditions: ×100, 1.4 numerical aperture oil-immersion objective, maximum power 25 mW, tube current 5.1 A (31% laser power), pinhole 1.33 Airy units (optical slice 1.0 μm). GFP images were acquired by excitation at 488 nm and emission at 515–545 nm. Cell regions were bleached at the plane of the membrane at 488 nm, 100% laser power, using the minimum number of iterations to cause complete bleaching (20–40). Recovery was monitored at 31% laser power at 1-min intervals. For quantitative analyses, the fluorescence intensity of the photobleached region, the extracellular background intensity, and the intracellular brightest intensity were determined using MetaMorph 4.0 software (Universal Imaging). All data were analyzed using Microsoft Excel (Microsoft, Redmond, WA, USA). Data were adjusted for sample fading as detailed elsewhere (43, 44).
Statistical analyses
Experiments were performed at minimum in triplicate, and results represent means ± sd or means ± se as indicated. ANOVA and Student t tests were performed on data sets using GraphPad Prism (GraphPad, La Jolla, CA, USA), and differences between samples were deemed significant at values of P < 0.05.
RESULTS
Lentiviral-mediated shRNA knockdown of ACTN4 in human epidermal keratinocytes
In order to investigate the functions of actinins in human keratinocytes, we utilized lentiviral-mediated delivery of shRNAs targeting the message encoding ACTN4 to generate stable knockdown cells. The design, validation, specificity, and characterization of these lentiviral/shRNA constructs were described previously (17). Multiple individual clones exhibiting a range of knockdown of ACTN4 in iHEKs were isolated for further analyses. Specifically, we derived clones ACTN4shRNA-1, -2, and -3 exhibiting, respectively, 10, 15, and 5% ACTN4 expression relative to parental cell levels (Fig. 1A). ACTN1 expression level is unaffected by ACTN4 knockdown (Fig. 1A). As a control, we also expressed a scrambled shRNA in keratinocytes. Expression of this shRNA does not affect ACTN1 or ACTN4 expression levels (Supplemental Fig. S1A). As an additional control, we generated an ACTN4 expression construct that is refractory to the shRNA and utilized an adenoviral delivery mechanism to restore ACTN4 expression in ACTN4-knockdown cells (Supplemental Fig. S1B).
Figure 1.
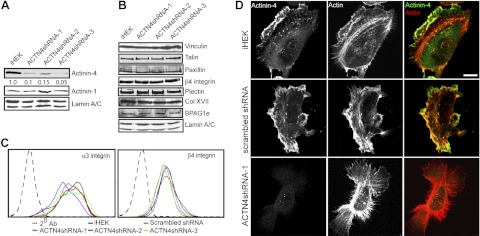
Knockdown of ACTN4 in human keratinocytes. iHEKs were infected with a lentivirus encoding shRNA targeting ACTN4, and clonal lines were established through antibiotic-resistance selection. Blots were quantified by densitometry; relative ACTN4 levels are indicated for each lane. A, B) Total protein lysates from control iHEKs and 3 ACTN4-knockdown clones (ACTN4shRNA-1, -2, and -3) were prepared for immunoblotting and probed with antibodies against ACTN4, ACTN1, vinculin, talin, paxillin, β4 integrin, plectin, Col XVI, BPAG1e, and lamin A/C, as indicated. C) iHEKs (black traces), iHEKs expressing a scrambled shRNA (maroon traces), and ACTN4-knockdown clones (red, blue, and green traces) were processed for FACS using antibodies against β4 integrin and α3 integrin. Black dotted tracing indicates cells prepared using secondary antibody alone. D) Control iHEKs, iHEKs expressing a scrambled shRNA, and ACTN4shRNA-1 clones were seeded overnight on glass coverslips, then processed for indirect immunofluorescence microscopy with antibodies against ACTN4 (green in merge), together with rhodamine-conjugated phalloidin (actin, red). Scale bar = 10 μm.
Total protein levels of the major focal contact proteins, paxillin, talin, and vinculin, and the hemidesmosome complex proteins β4 integrin, plectin, Col XII, and BPAG1e are unchanged relative to control iHEKs in ACTN4-knockdown cells (Fig. 1B). However, cell surface expression of α3 integrin, a focal contact associated integrin expressed by iHEKs, is slightly reduced compared with controls, as determined by FACS analyses (Fig. 1C). Cell surface expression levels of β4 integrin were unchanged relative to iHEKs or iHEKs expressing a scrambled shRNA (Fig. 1C).
Indirect immunofluorescence microscopical analyses reveal that the distribution of ACTN4 in wild-type iHEKs or iHEKs expressing a scrambled shRNA is largely restricted to ruffles at the leading front of lamellipodial extensions, while staining is greatly diminished in ACTN4-knockdown cells (Fig. 1D). This staining pattern is similar to that described in the colon carcinoma SW480 line but more restricted than that reported in uterine fibroblasts, where ACTN4 predominantly decorates actin stress fibers (3). Moreover, no evidence of focal contact staining was observed (12).
Interestingly, phalloidin labeling of filamentous actin in ACTN4-knockdown clones appears markedly different from that in wild-type cells (Fig. 1D). Stress fiber/cortical actin bundling is enhanced in the former cells, which also possess an increased number of large lamellipodial extensions (Fig. 1D). To determine whether the observed differences in cell shape and surface area were indeed reflective of the entire cell population, images of live individual cells plated overnight on glass-bottomed dishes were captured, and cell surface area, lamellipodial area, and number of lamellipodial protrusions were determined (representative images in Fig. 2A, quantification in Fig. 2B, C). Results indicate that the ACTN4-knockdown lines exhibit a significantly increased cell and lamellipodial area and have significantly reduced intrinsic polarity than parental iHEK or iHEK induced to express a scrambled shRNA control (Fig. 2B, C).
Figure 2.
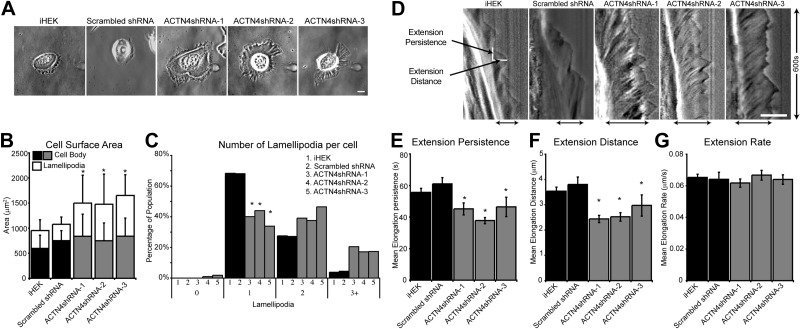
ACTN4-knockdown keratinocytes display polarity and lamellipodial dynamics defects. iHEKs and iHEKs stably infected with lentivirus encoding shRNA targeting ACTN4 or a scrambled shRNA were plated overnight on glass-bottomed dishes and then imaged by phase-contrast microscopy. A) Representative images of each clone. B) Mean ± se cell surface area was determined from images from 3 independent experiments, 50–100 cells/group. Solid bars represent cell body area; open bars represent lamellipodial extension area. C) Each cell was scored for the number of lamellipodial protrusions. Percentages of cells projecting 0, 1, 2, or 3+ protrusions are shown. D–G) Phase-contrast images of individual keratinocytes were acquired every 5 s over 10 min. Kymographs were generated as a montage of the images beneath a 1-pixel-wide line drawn in the direction of migration, with time on the vertical axes. D) Representative kymographs for each line. Black arrows at bottom indicate lamellipodia. White lines on the iHEK kymograph indicate measurements taken for each protrusion event. E–G) Plots of time extending (extension persistence; E), distance elongated (extension distance; F), and extension rate (G). Values are means ± se from 25–50 cells/line in three separate studies. Scale bars = 10 μm. *P < 0.05 vs. iHEK and scrambled shRNA groups.
Actinin-deficient keratinocytes display impaired lamellipodial dynamics
The lamellipodial localization of ACTN4 and the observed changes to lamellipodial area and number in ACTN4-knockdown clones suggested the possibility that ACTN4 is involved in the regulation of lamellipodial extension/protrusion. Thus, we next analyzed lamellipodial dynamics in iHEKs, iHEKs expressing a scrambled shRNA control and ACTN4-knockdown clones. To do so, we imaged individual keratinocytes every 5 s over 10 min and generated a kymograph of a 1-pixel-wide line drawn in the direction of the major lamellipodial protrusion (representative kymographs in Fig. 2D; ref. 42). From these kymographs, we measured the time spent elongating (extension persistence, Fig. 2E) and the length of the extension event (extension distance, Fig. 2F) for each individual protrusion event. From the ratio of distance/persistence we derived the rate of extension (Fig. 2G). Compared to controls, ACTN4-knockdown keratinocytes demonstrate significantly reduced mean persistence and distance measurements, with no significant difference in extension rate (Fig. 2E–G). These defects are rescued through expression of the refractory ACTN4 mRNA but not through expression of a GFP-encoding construct (Supplemental Figs. S1B and S2C).
ACTN4-deficient keratinocytes fail to migrate processively
Reduced lamellipodial extension persistence also correlates with reduced intrinsic directionality of gross migration (42). We therefore tracked the motile behavior of individual keratinocytes imaged every 2 min over 1 h (see representative vector diagrams, Fig. 3A). Results indicate that while total distance migrated is unchanged (migration speed, Fig. 3B), the ratio of distance migrated from origin to total distance migrated (processivity; Fig. 3C) is significantly reduced in the ACTN4-knockdown clones relative to controls. This condition indicates a loss of ability to establish and/or maintain polarity. This motility defect is ameliorated through expression of the refractory ACTN4 mRNA but not through expression of GFP (Supplemental Fig. S1D).
Figure 3.
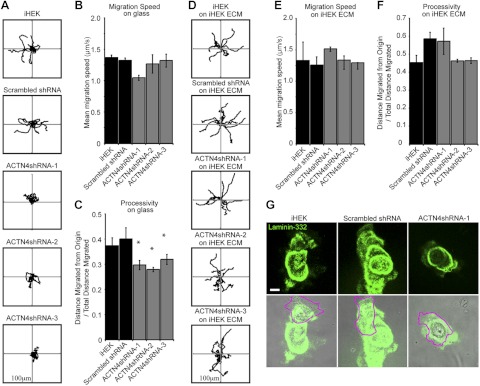
ACTN4-knockdown keratinocytes display motility defects. A) Indicated clones and lines were plated at low density overnight onto glass-bottomed dishes and then imaged every 2 min over 1 h. Cells were tracked, and representative migration paths of 10 individual cells were plotted, with each line representing a discrete cell. B) Mean migration speed was calculated from ≥3 independent assays, 15–50 cells/assay. C) Mean processivity of migration was calculated as the ratio of maximum distance from origin/total distance migrated, where 1 = linear migration path. D–F) Indicated clones and lines were plated for 2 h onto matrix deposited by iHEKs and then imaged every 2 min over 1 h. D) Migration paths of individual cells were plotted as in panel A. E, F) Speed (E) and processivity (F) were calculated as in panels B and C, respectively; 3 independent assays with 15–50 cells/assay. G) Indicated cells were plated overnight at low density onto glass coverslips, then fixed and stained with antibodies against laminin-332. Bottom panels show overlays on phase-contrast images, with cell boundaries outlined in magenta. Scale bar = 10 μm. Axes in panels A, D = 100 μm. *P < 0.05 vs. iHEK and scrambled shRNA groups.
Previously, we demonstrated that keratinocytes exhibiting normal migration speed but defective processivity can be rescued by plating them onto a preformed laminin-322-rich extracellular matrix (25). We therefore assessed the motile behavior of ACTN4-knockdown clones plated onto laminin-332-rich matrix preparations derived from wild-type iHEKs. In sharp contrast to their motile behavior, when plated overnight on glass, ACTN4-knockdown cells plated onto iHEK matrix show the same level of processivity as wild-type iHEKs or scrambled shRNA-expressing iHEKs plated on iHEK matrix (Fig. 3D–F). This finding suggests that the matrix deposited by ACTN4-deficient cells differs from that of wild-type iHEKs. To investigate this, we stained keratinocytes plated overnight on uncoated glass coverslips with antibodies against laminin-332 (Fig. 3G). ACTN4-knockdown keratinocytes deposit laminin-332 in roughly circular patterns, which contrasts sharply to the more linear paths of laminin-332 deposited by control iHEKs (Fig. 3E).
Focal contacts and adhesions are stabilized in ACTN4-knockdown cells
Knockdown of ACTN4 in murine lung fibroblasts previously has been demonstrated to lead to an increase in cell spreading accompanied by an increase in focal contact size (45). Thus, we next analyzed focal contact size by staining ACTN4-knockdown keratinocytes with a paxillin antibody probe (Fig. 4A). Consistent with published findings, the focal contacts of ACTN4-deficient cells are significantly larger than those of wild-type keratinocytes or iHEKs induced to express a scrambled shRNA (Fig. 4B).
Figure 4.
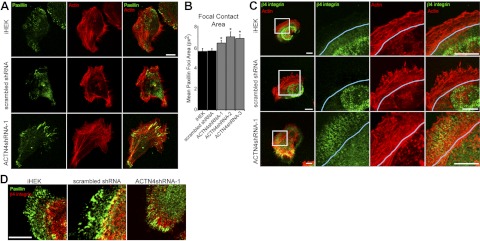
ACTN4-knockdown keratinocytes display increased focal contact area. Control iHEKs, iHEKs expressing scrambled shRNA, and ACTN4shRNA-1 cells were plated overnight onto glass coverslips and then processed for indirect immunofluorescence microscopy with antibodies against paxillin (green) together with rhodamine-conjugated phalloidin (actin, red). B) Mean ± se focal contact area was determined from images captured from the indicated cell clones and lines stained for paxillin; 3 separate experiments, 15–40 cells/experiment. *P < 0.05 vs. iHEK. C) Control iHEKs, iHEKs expressing scrambled shRNA, and ACTN4shRNA-1 cells were plated overnight onto glass coverslips and then processed for indirect immunofluorescence microscopy with antibodies against β4 integrin (green) together with rhodamine-conjugated phalloidin (actin, red). Blue line indicates base of lamellipodium. D) Same keratinocyte clones and lines were processed for indirect immunofluorescence microscopy using antibodies against paxillin (green) in combination with β4 integrin antibodies (red). Scale bars = 10 μm.
Hemidesmosomal proteins are mislocalized in ACTN4-knockdown keratinocytes
In addition to focal contacts, keratinocytes utilize a second cell-matrix attachment complex, termed the hemidesmosome. In culture, human keratinocytes assemble what appear to be immature hemidesmosomes at the ultrastructural level (38). However, these immature hemidesmosomes contain all major hemidesmosome components, including the bullous pemphigoid (BP) antigens (BPAG1e and Col XVII), plectin, and α6β4 integrin (46, 47). We therefore next investigated whether hemidesmosome protein localization is modified in cells with reduced ACTN4 expression. In control cells plated at low density, β4 integrin is found in arcs within the constraints of the cortical actin ring at the base of the lamellipodium and are not present in lamellipodial extensions (Fig. 4C). In contrast, in ACTN4-knockdown keratinocytes, the spatial organization of α6β4 integrin is dramatically altered relative to wild-type keratinocytes, with β4 integrin staining extending into each lamellipodium (Fig. 4C). Costaining for focal contacts (paxillin; Fig. 4D) with antibodies against β4 integrin reveals that α6β4 integrin is interspersed between sites of focal contacts within each lamellipodium in ACTN4-deficient keratinocytes.
Recruitment of plectin to β4 integrin is unaffected by ACTN4 knockdown (Fig. 5A). However, interestingly, a reduced association is found between α6β4 integrin and both BP antigens (BPAG1e and Col XVII) in ACTN4-deficient keratinocytes, which indicates that hemidesmosome maturation is reduced in ACTN4-deficient cells (Fig. 5B). Defective BP antigen recruitment to α6β4 integrin is rescued through reexpression of ACTN4 in the knockdown cells (Supplemental Fig. S1E; only Col XVII is shown).
Figure 5.
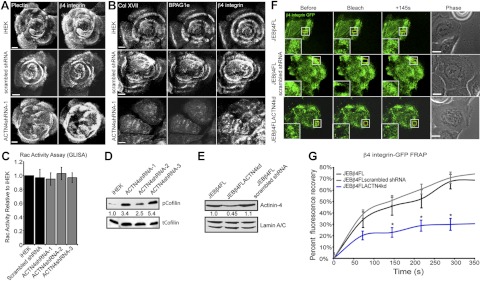
Hemidesmosome-protein rich complex assembly is impaired in ACTN4-deficient keratinocytes. A, B) Control iHEKs, iHEKs expressing scrambled shRNA, or ACTN4shRNA-1 cells were plated onto glass coverslips and 48 h later were processed for double- or triple-label indirect immunofluorescence microscopy using antibodies against either plectin and β4 integrin (A) or against Col XVII, BPAG1e, and β4 integrin (B) as indicated. C) Rac activity levels of the indicated lines were determined by G-LISA. Results are plotted as means ± se from 5 independent assays. D) Total protein extracts from iHEKs and ACTN4shRNA-1, -2, and -3 cells were probed with antibodies against Ser3-phosphorylated cofilin (pcofilin) or total cofilin (tcofilin) as indicated. Blots were scanned, and relative expression levels of p-cofilin, normalized to total cofilin, were determined by densitometry. E–G) JEB keratinocytes expressing full-length β4 integrin tagged with GFP (JEBβ4FL) were infected with lentivirus encoding shRNA targeting ACTN4 (JEBβ4FLACN4kd) or a scrambled shRNA (JEBβ4FL scrambled shRNA). E) Total protein extracts were immunoblotted with antibodies against ACTN4 and lamin A/C. Blots were scanned, and relative expression levels of ACTN4 were determined by densitometry. F, G) JEBβ4FL JEBβ4FLACN4kd and JEBβ4FL scrambled shRNA keratinocytes were plated onto glass-bottomed dishes, grown to confluence then scratch wounded. Regions within clusters of β4 integrin-GFP were bleached 6 h following wounding in cells at the wound margins. F) Representative images before bleaching (left panels), immediately after bleaching (second panels), and 145 s after bleaching (third panels), along with phase-contrast images (right panels). Insets: boxed regions at higher magnification. G) Mean ± se percentage of pre-bleach-level fluorescence recovery for each cell line was determined from >15 assays/cell type. Scale bars = 10 μm. *P < 0.05 vs. JEBβ4FL.
Cofilin activity is reduced in ACTN4-knockdown keratinocytes
The increased lamellipodial area and reduced lamellipodial dynamics of ACTN4 cells indicates that actin remodeling is misregulated in these cells. Previously, we have identified that cells deficient for β4 integrin or exhibiting knockdown in BPAG1e expression display reduced activity of the RhoGTPase Rac1 and its downstream effector protein cofilin (25, 48). Since we observe aberrant α6β4 integrin/BP complex formation in the ACTN4-knockdown skin cells, we predicted that Rac and cofilin activation would be reduced in such cells. To test this possibility, we utilized a Rac G-LISA to measure Rac activity levels (Fig. 5C) and probed whole-cell extracts of iHEKs and the ACTN4-knockdown keratinocytes with antibodies against Ser3-phosphorylated cofilin (inactive) and total cofilin to determine cofilin activity levels (Fig. 5D and ref. 49). These analyses reveal that ACTN4-knockdown cells display Rac activity levels that are not significantly different from control iHEKs or iHEKs expressing scrambled shRNA (Fig. 5C). However, cofilin activity is reduced, with all knockdown clones displaying phosphorylation levels that are significantly higher than control iHEKs. Next, we asked whether increasing the level of cofilin in ACTN4-knockdown cells is sufficient to restore their lamellipodial dynamics and cell motility defects. ACTN4shRNA-1 cells infected with adenovirus encoding cofilin display significantly increased lamellipodial persistence and extension distance values compared with uninfected ACTN4shRNA-1 cells and ACTN4shRNA-1 cells expressing a GFP construct (Supplemental Fig. S1B, C). Moreover, cofilin expression ameliorates the motility defect of ACTN4shRNA-1 cells (Supplemental Fig. S1D).
β4 Integrin dynamics are reduced in ACTN4-knockdown cells
The mislocalization of α6β4 integrin and hemidesmosomal components in ACTN4-deficient keratinocytes and reduced actin remodeling, indicated by reduced cofilin activity, raise the possibility that β4 dynamics are affected by ACTN4 knockdown. To assess whether this is the case, we utilized a previously described patient-derived keratinocyte line that lacks endogenous β4 integrin due to nonsense mutations in ITGB4 (JEB) but has been induced to stably express a GFP-tagged form of β4 integrin through retroviral mediated integration (JEBβ4FL) (25). Through lentiviral delivery of shRNA into this line, we first generated cells exhibiting stable knockdown of ACTN4 (JEBβ4FLACTN4kd) or expressing a scrambled shRNA (JEBβ4FL scrambled shRNA; Fig. 5E). FRAP rates were then determined for the GFP-tagged β4 integrin in these lines at regions displaying distinct β4 integrin organization in cells moving into a scratch wound (Fig. 5F, G). Knockdown of ACTN4 leads to a reduction in fluorescence recovery rates relative to controls, indicating that β4 integrin dynamics are impaired in such cells (Fig. 5G).
DISCUSSION
In the current study, we assayed the consequences of ACTN4 knockdown on skin cell motility and the organization of keratinocyte matrix adhesion structures. Knockdown of ACTN4 perturbs the directionality of keratinocyte motility, results in an increase in focal contact surface area, and leads to multifaceted changes in hemidesmosome-like protein complexes. Specifically, in ACTN4-knockdown cells, we demonstrate mislocalization of α6β4 integrin from the base to the main body of the lamellipodium, reduced β4 integrin dynamics, and reduced recruitment of the BP antigens to α6β4 integrin clusters.
Focal contacts play an important role in migration by providing traction forces for moving cells and acting as the hubs for signals that are required for motility (50–52). Since the focal contacts in ACTN4-deficient cells are larger than those in wild-type cells, one might assume that these cells would exhibit retarded migration rates. However, surprisingly, the overall speed of ACTN4-knockdown cells is not reduced relative to wild-type cells, although the cells fail to maintain polarity, and therefore their net migration rate is reduced. Intriguingly, α6β4 integrin is mislocalized in ACTN4-knockdown cells and is found in lamellipodial extensions, a cellular location that is normally exclusively restricted to focal contacts. We speculate that the mislocalized α6β4 integrin stabilizes interaction of the entire lamellipodium with the cell substrate, facilitates lamellipodial enlargement, and contributes to the aberrant dynamics of these important cell surface extensions. Moreover, it is possible that the observed increased focal contact area is an indirect consequence of this α6β4 integrin mediated stabilization of the lamellipodia. Indeed, the consequence of ACTN4 knockdown on focal contacts and hemidesmosome protein–rich complexes supports the notion that these distinct matrix adhesion structures crosstalk (53). Ozawa and Tsuruta (53) demonstrated that focal contact dynamics and size are increased when keratinocytes are treated with antibodies, which perturb α6 or β4 integrin. Likewise, here we show that α6β4 integrin mislocalization and failure to associate with the BP antigens in ACTN4-knockdown skin cells is accompanied by an increase in focal contact surface area.
Why is α6β4 integrin mislocalized in ACTN4-deficient cells, and why does it show reduced interaction with the BP antigens? One possible explanation is that α6β4 integrin associates either directly or indirectly with actin, such that in wild-type cells, α6β4 integrin is restricted to the base of the lamellipodium by actin bundles. In ACTN4 depleted cells, this association is presumably lost or modified. In this scenario, actin no longer restrains α6β4 integrin, which then moves into the lamellipodium. This possibility would be consistent with studies in which α6β4 integrin-actin interactions have been demonstrated in certain cancer cells and with evidence that α6β4 integrin dynamics are retarded by the actin cytoskeleton (26, 28, 44). In this regard, ACTN4 has been reported to bind Col XVII, which mediates the association of BPAG1e with α6β4 integrin at the site of hemidesmosomes in intact skin and in hemidesmosome protein–rich complexes in cultured keratinocytes (10). Thus, ACTN4 may mediate the link between actin and α6β4 integrin via Col XVII and, hence, restrict hemidesmosome protein complexes to the base of the lamellipodium. Consistent with this notion, there is diminished BP antigen localization with α6β4 integrin in ACTN4-knockdown cells, even though plectin recruitment is unaffected. Moreover, this result also suggests that ACTN4 is involved in targeting BP antigens to the cell surface. One assumes that this is actin cytoskeleton and motor molecule dependent, although this will require confirmation.
The large lamellipodial protrusions with reduced dynamics projected by ACTN4-knockdown keratinocytes are also likely to be a result of changes in signaling to the actin cytoskeleton. Lamellipodial formation and dynamics are dependent on the activity of the RhoGTPase Rac and its downstream effector proteins. In ACTN4-knockdown cells, Rac activity is unaffected. Nonetheless, we observe a reduction in the activity of cofilin. Cofilin activity is usually restricted to the base of a lamellipodia, where it severs actin filaments and leads to their depolymerization, thereby effectively replenishing the G-actin pool and allowing further extension of the lamellipodia (54–56). Therefore, we hypothesize that in the ACTN4-knockdown cells, lamellipodia extend and are stabilized, potentially by mislocalized α6β4 integrin. Thereafter, further extension is rate limited by deficient cofilin activity and G-actin availability, leading to oversized, stable lamellipodia. Consistent with these data, restoration of cofilin activity via adenoviral means is sufficient to rescue lamellipodial dynamics to control levels in the ACTN4-knockdown cells. However, our findings raise an additional question: why cofilin is inactive in cells with active Rac, since our prior studies indicate that Rac signals to activate cofilin in motile skin cells (25, 48, 57). A number of potential explanations address this, including a decreased activity of the cofilin phosphatases (slingshot 1, 2, and 3), increased LIM-kinase 1 activity, or mislocalizaton of either cofilin, Rac, or their potential scaffolding proteins, such as α6β4 integrin (57–59). Further investigation will be required to resolve this question.
Stabilization of the lamellipodium in the ACTN4-knockdown cells is one possible explanation for their lack of processivity. An alternative explanation relates to their matrix deposition. Previously, we demonstrated that in keratinocytes where cofilin is inactive, through either absence of α6β4 integrin, knockdown of BPAG1e, or expression of dominant negative forms of cofilin or slingshots, laminin 332 is deposited aberrantly in circular rather than more linear tracks (25, 48, 57). Based on these results, we speculate that the defect in processivity of the ACTN4-deficient cells is, at least in part, a consequence of a failure of α6β4 integrin and its associated proteins to regulate the deposition of a matrix that can support directed migration. Our finding that the motility defect of ACTN4-knockdown cells is corrected by plating them onto a laminin-332 matrix deposited by iHEK cells and the rescue of ACTN4-knockdown cells by overexpression of cofilin is consistent with this hypothesis.
In summary, our data indicate that ACTN4 not only mediates the assembly and maturation of cell-matrix adhesive devices but, in so doing, regulates signal pathways involved in directing the deposition of a matrix that favors directionality. Indeed, our data demonstrate that ACTN4 is required for efficient migration by being an important component of the steering mechanism of a keratinocyte.
Supplementary Material
Acknowledgments
A Dermatology Foundation Career Development Award (to K.J.H.), as well as grants K99AR060242 (K.J.H.), and R01AR054184 (J.C.R.J.) from the U.S. National Institute of Arthritis and Musculoskeletal and Skin Diseases supported this project.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Arthritis and Musculoskeletal and Skin Diseases or the U.S. National Institutes of Health. Imaging work was performed at the Northwestern University Cell Imaging Facility, generously supported by grant NCI CCSG P30 CA060553, awarded to the Robert H. Lurie Comprehensive Cancer Center. FACS analyses were performed at the Northwestern University Flow Cytometry Facility, supported by a Cancer Center Support grant (NCI CA060553).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ACTN1–4
- actinin-1–4
- BP
- bullous pemphigoid
- BPAG1e
- bullous pemphigoid antigen 1e
- Col XVII
- collagen type XVII
- FACS
- fluorescence-activated cell sorting
- FRAP
- fluorescence recovery after photobleaching
- GFP
- green fluorescent protein
- iHEK
- immortalized human epidermal keratinocyte
- JEB
- junctional epidermolysis bullosa
- MOI
- multiplicity of infection
REFERENCES
- 1. Baron M. D., Davison M. D., Jones P., Critchley D. R. (1987) The sequence of chick alpha-actinin reveals homologies to spectrin and calmodulin. J. Biol. Chem. 262, 17623–17629 [PubMed] [Google Scholar]
- 2. Youssoufian H., McAfee M., Kwiatkowski D. J. (1990) Cloning and chromosomal localization of the human cytoskeletal alpha-actinin gene reveals linkage to the beta-spectrin gene. Am. J. Hum. Genet. 47, 62–72 [PMC free article] [PubMed] [Google Scholar]
- 3. Honda K., Yamada T., Endo R., Ino Y., Gotoh M., Tsuda H., Yamada Y., Chiba H., Hirohashi S. (1998) Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J. Cell Biol. 140, 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beggs A. H., Byers T. J., Knoll J. H., Boyce F. M., Bruns G. A., Kunkel L. M. (1992) Cloning and characterization of two human skeletal muscle alpha-actinin genes located on chromosomes 1 and 11. J. Biol. Chem. 267, 9281–9288 [PubMed] [Google Scholar]
- 5. Millake D. B., Blanchard A. D., Patel B., Critchley D. R. (1989) The cDNA sequence of a human placental alpha-actinin. Nucleic Acids Res. 17, 6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sen S., Dong M., Kumar S. (2009) Isoform-specific contributions of alpha-actinin to glioma cell mechanobiology. PLoS One 4, e8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamaji S., Suzuki A., Kanamori H., Mishima W., Yoshimi R., Takasaki H., Takabayashi M., Fujimaki K., Fujisawa S., Ohno S., Ishigatsubo Y. (2004) Affixin interacts with alpha-actinin and mediates integrin signaling for reorganization of F-actin induced by initial cell-substrate interaction. J. Cell Biol. 165, 539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Otey C. A., Pavalko F. M., Burridge K. (1990) An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J. Cell Biol. 111, 721–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vallenius T., Luukko K., Makela T. P. (2000) CLP-36 PDZ-LIM protein associates with nonmuscle alpha-actinin-1 and alpha-actinin-4. J. Biol. Chem. 275, 11100–11105 [DOI] [PubMed] [Google Scholar]
- 10. Gonzalez A. M., Otey C., Edlund M., Jones J. C. (2001) Interactions of a hemidesmosome component and actinin family members. J. Cell Sci. 114, 4197–4206 [DOI] [PubMed] [Google Scholar]
- 11. Honda K., Yamada T., Hayashida Y., Idogawa M., Sato S., Hasegawa F., Ino Y., Ono M., Hirohashi S. (2005) Actinin-4 increases cell motility and promotes lymph node metastasis of colorectal cancer. Gastroenterology 128, 51–62 [DOI] [PubMed] [Google Scholar]
- 12. Galler A. B., Garcia Arguinzonis M. I., Baumgartner W., Kuhn M., Smolenski A., Simm A., Reinhard M. (2006) VASP-dependent regulation of actin cytoskeleton rigidity, cell adhesion, and detachment. Histochem. Cell Biol. 125, 457–474 [DOI] [PubMed] [Google Scholar]
- 13. Ridley A. J. (2011) Life at the leading edge. Cell 145, 1012–1022 [DOI] [PubMed] [Google Scholar]
- 14. Small J. V., Stradal T., Vignal E., Rottner K. (2002) The lamellipodium: where motility begins. Trends Cell Biol. 12, 112–120 [DOI] [PubMed] [Google Scholar]
- 15. Gluck U., Ben-Ze'ev A. (1994) Modulation of alpha-actinin levels affects cell motility and confers tumorigenicity on 3T3 cells. J. Cell Sci. 107(Pt. 7), 1773–1782 [DOI] [PubMed] [Google Scholar]
- 16. Gluck U., Kwiatkowski D. J., Ben-Ze'ev A. (1993) Suppression of tumorigenicity in simian virus 40-transformed 3T3 cells transfected with alpha-actinin cDNA. Proc. Natl. Acad. Sci. U. S. A. 90, 383–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quick Q., Skalli O. (2010) Alpha-actinin 1 and alpha-actinin 4: contrasting roles in the survival, motility, and RhoA signaling of astrocytoma cells. Exp. Cell. Res. 316, 1137–1147 [DOI] [PubMed] [Google Scholar]
- 18. Kos C. H., Le T. C., Sinha S., Henderson J. M., Kim S. H., Sugimoto H., Kalluri R., Gerszten R. E., Pollak M. R. (2003) Mice deficient in alpha-actinin-4 have severe glomerular disease. J. Clin. Invest. 111, 1683–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barbolina M. V., Adley B. P., Kelly D. L., Fought A. J., Scholtens D. M., Shea L. D., Stack M. S. (2008) Motility-related actinin alpha-4 is associated with advanced and metastatic ovarian carcinoma. Lab. Invest. 88, 602–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamada S., Yanamoto S., Yoshida H., Yoshitomi I., Kawasaki G., Mizuno A., Nemoto T. K. (2010) RNAi-mediated down-regulation of alpha-actinin-4 decreases invasion potential in oral squamous cell carcinoma. Int. J. Oral Maxillofac. Surg. 39, 61–67 [DOI] [PubMed] [Google Scholar]
- 21. Yamamoto S., Tsuda H., Honda K., Takano M., Tamai S., Imoto I., Inazawa J., Yamada T., Matsubara O. (2012) ACTN4 gene amplification and actinin-4 protein overexpression drive tumour development and histological progression in a high-grade subset of ovarian clear-cell adenocarcinomas. Histopathology 60, 1073–1083 [DOI] [PubMed] [Google Scholar]
- 22. Koizumi T., Nakatsuji H., Fukawa T., Avirmed S., Fukumori T., Takahashi M., Kanayama H. (2010) The role of actinin-4 in bladder cancer invasion. Urology 75, 357–364 [DOI] [PubMed] [Google Scholar]
- 23. Kikuchi S., Honda K., Tsuda H., Hiraoka N., Imoto I., Kosuge T., Umaki T., Onozato K., Shitashige M., Yamaguchi U., Ono M., Tsuchida A., Aoki T., Inazawa J., Hirohashi S., Yamada T. (2008) Expression and gene amplification of actinin-4 in invasive ductal carcinoma of the pancreas. Clin. Cancer Res. 14, 5348–5356 [DOI] [PubMed] [Google Scholar]
- 24. Yamamoto S., Tsuda H., Honda K., Kita T., Takano M., Tamai S., Inazawa J., Yamada T., Matsubara O. (2007) Actinin-4 expression in ovarian cancer: a novel prognostic indicator independent of clinical stage and histological type. Mod. Pathol. 20, 1278–1285 [DOI] [PubMed] [Google Scholar]
- 25. Sehgal B. U., Debiase P., Matzno S., Chew T.-L., Claiborne J. N., Hopkinson S. B., Russell A., Marinkovich P. M., Jones J. C. R. (2006) Integrin β4 regulates migratory behavior of keratinocytes by determining laminin-332 (laminin-5) matrix organization. J. Biol. Chem. 281, 35487–35498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rabinovitz I., Mercurio A. M. (1997) The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin-containing motility structures. J. Cell Biol. 139, 1873–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pullar C. E., Baier B. S., Kariya Y., Russell A. J., Horst B. A., Marinkovich M. P., Isseroff R. R. (2006) Beta4 integrin and epidermal growth factor coordinately regulate electric field-mediated directional migration via Rac1. Mol. Biol. Cell 17, 4925–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rabinovitz I., Toker A., Mercurio A. M. (1999) Protein kinase C-dependent mobilization of the alpha6beta4 integrin from hemidesmosomes and its association with actin-rich cell protrusions drive the chemotactic migration of carcinoma cells. J. Cell Biol. 146, 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsuruta D., Hashimoto T., Hamill K. J., Jones J. C. (2011) Hemidesmosomes and focal contact proteins: functions and cross-talk in keratinocytes, bullous diseases and wound healing. J. Dermatol. Sci. 62, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pelham R. J., Wang Y. L. (1997) Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. U. S. A. 94, 13661–13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schafer C., Born S., Mohl C., Houben S., Kirchgessner N., Merkel R., Hoffmann B. (2010) The key feature for early migratory processes: Dependence of adhesion, actin bundles, force generation and transmission on filopodia. Cell Adh. Migr. 4, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sakamoto S., McCann R. O., Dhir R., Kyprianou N. (2010) Talin1 promotes tumor invasion and metastasis via focal adhesion signaling and anoikis resistance. Cancer Res. 70, 1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xia N., Thodeti C. K., Hunt T. P., Xu Q., Ho M., Whitesides G. M., Westervelt R., Ingber D. E. (2008) Directional control of cell motility through focal adhesion positioning and spatial control of Rac activation. FASEB J. 22, 1649–1659 [DOI] [PubMed] [Google Scholar]
- 34. Hashimoto T., Amagai M., Ebihara T., Gamou S., Shimizu N., Tsubata T., Hasegawa A., Miki K., Nishikawa T. (1993) Further analyses of epitopes for human monoclonal anti-basement membrane zone antibodies produced by stable human hybridoma cell lines constructed with Epstein-Barr virus transformants. J. Invest. Dermatol. 100, 310–315 [DOI] [PubMed] [Google Scholar]
- 35. Klatte D. H., Jones J. C. (1994) Purification of the 230-kD bullous pemphigoid antigen (BP230) from bovine tongue mucosa: structural analyses and assessment of BP230 tissue distribution using a new monoclonal antibody. J. Invest. Dermatol. 102, 39–44 [DOI] [PubMed] [Google Scholar]
- 36. Hopkinson S. B., Baker S. E., Jones J. C. R. (1995) Molecular genetic studies of a human epidermal autoantigen (the 180-kD bullous pemphigoid antigen/BP180): identification of functionally important sequences within the BP180 molecule and evidence for an interaction between BP180 and alpha 6 integrin. J. Cell Biol. 130, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzales M., Haan K., Baker S. E., Fitchmun M. I., Todorov I., Weitzman S., Jones J. C. R. (1999) A cell signal pathway involving laminin-5, α3β1 integrin, and mitogen-activated protein kinase can regulate epithelial cell proliferation. Mol. Biol. Cell 10, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goldfinger L. E., Stack M. S., Jones J. C. R. (1998) Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J. Cell Biol. 141, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riddelle K. S., Green K. J., Jones J. C. (1991) Formation of hemidesmosomes in vitro by a transformed rat bladder cell line. J. Cell Biol. 112, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langhofer M., Hopkinson S. B., Jones J. C. (1993) The matrix secreted by 804G cells contains laminin-related components that participate in hemidesmosome assembly in vitro. J. Cell Sci. 105(Pt. 3), 753–764 [DOI] [PubMed] [Google Scholar]
- 41. Harlow E., Lane D. (1988) Antibodies: A Laboratory Manual, CSHL Press, Woodbury, NY, USA [Google Scholar]
- 42. Hinz B., Alt W., Johnen C., Herzog V., Kaiser H. W. (1999) Quantifying lamella dynamics of cultured cells by SACED, a new computer-assisted motion analysis. Exp. Cell Res. 251, 234–243 [DOI] [PubMed] [Google Scholar]
- 43. Tsuruta D., Gonzales M., Hopkinson S. B., Otey C., Khuon S., Goldman R. D., Jones J. C. (2002) Microfilament-dependent movement of the beta3 integrin subunit within focal contacts of endothelial cells. FASEB J. 16, 866–868 [DOI] [PubMed] [Google Scholar]
- 44. Tsuruta D., Hopkinson S. B., Jones J. C. (2003) Hemidesmosome protein dynamics in live epithelial cells. Cell Motil. Cytoskeleton 54, 122–134 [DOI] [PubMed] [Google Scholar]
- 45. Shao H., Wang J. H., Pollak M. R., Wells A. (2010) Alpha-actinin-4 is essential for maintaining the spreading, motility and contractility of fibroblasts. PLoS One 5, e13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carter W. G., Kaur P., Gil S. G., Gahr P. J., Wayner E. A. (1990) Distinct functions for integrins alpha 3 beta 1 in focal adhesions and alpha 6 beta 4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J. Cell Biol. 111, 3141–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ozawa T., Tsuruta D., Jones J. C., Ishii M., Ikeda K., Harada T., Aoyama Y., Kawada A., Kobayashi H. (2010) Dynamic relationship of focal contacts and hemidesmosome protein complexes in live cells. J. Invest. Dermatol. 130, 1624–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hamill K. J., Hopkinson S. B., DeBiase P., Jones J. C. (2009) BPAG1e maintains keratinocyte polarity through beta4 integrin-mediated modulation of Rac1 and cofilin activities. Mol. Biol. Cell 20, 2954–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Agnew B. J., Minamide L. S., Bamburg J. R. (1995) Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J. Biol. Chem. 270, 17582–17587 [DOI] [PubMed] [Google Scholar]
- 50. Gardel M. L., Schneider I. C., Aratyn-Schaus Y., Waterman C. M. (2010) Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Beningo K. A., Dembo M., Kaverina I., Small J. V., Wang Y. L. (2001) Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153, 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dembo M., Wang Y. L. (1999) Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys. J. 76, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ozawa T., Tsuruta D. (2011) Comparative study of the dynamics of focal contacts in live epithelial and mesenchymal cells. Med. Mol. Morphol. 44, 27–33 [DOI] [PubMed] [Google Scholar]
- 54. Carlier M. F., Laurent V., Santolini J., Melki R., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D. (1997) Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J. Cell Biol. 136, 1307–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carlier M. F., Ressad F., Pantaloni D. (1999) Control of actin dynamics in cell motility. Role of ADF/cofilin. J. Biol. Chem. 274, 33827–33830 [DOI] [PubMed] [Google Scholar]
- 56. Pollard T. D., Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 57. Kligys K., Claiborne J. N., DeBiase P., Hopkinson S. B., Mizuno K., Jones J. C. R. (2007) The Slingshot family of phosphatases mediates Rac1 regulation of cofilin phosphorylation, laminin-332 organization and motility behavior of keratinocytes. J. Biol. Chem. 282, 32520–32528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang N., Higuchi O., Ohashi K., Nagata K., Wada A., Kangawa K., Nishida E., Mizuno K. (1998) Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812 [DOI] [PubMed] [Google Scholar]
- 59. Jovceva E., Larsen M. R., Waterfield M. D., Baum B., Timms J. F. (2007) Dynamic cofilin phosphorylation in the control of lamellipodial actin homeostasis. J. Cell Sci. 120, 1888–1897 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.