

Abstract
BACKGROUND
Acute myeloid leukemia (AML) is a heterogeneous disease with respect to presentation and clinical outcome. The prognostic value of recently identified somatic mutations has not been systematically evaluated in a phase 3 trial of treatment for AML.
METHODS
We performed a mutational analysis of 18 genes in 398 patients younger than 60 years of age who had AML and who were randomly assigned to receive induction therapy with high-dose or standard-dose daunorubicin. We validated our prognostic findings in an independent set of 104 patients.
RESULTS
We identified at least one somatic alteration in 97.3% of the patients. We found that internal tandem duplication in FLT3 (FLT3-ITD), partial tandem duplication in MLL (MLL-PTD), and mutations in ASXL1 and PHF6 were associated with reduced overall survival (P = 0.001 for FLT3-ITD, P = 0.009 for MLL-PTD, P = 0.05 for ASXL1, and P = 0.006 for PHF6); CEBPA and IDH2 mutations were associated with improved overall survival (P = 0.05 for CEBPA and P = 0.01 for IDH2). The favorable effect of NPM1 mutations was restricted to patients with co-occurring NPM1 and IDH1 or IDH2 mutations. We identified genetic predictors of outcome that improved risk stratification among patients with AML, independently of age, white-cell count, induction dose, and post-remission therapy, and validated the significance of these predictors in an independent cohort. High-dose daunorubicin, as compared with standard-dose daunorubicin, improved the rate of survival among patients with DNMT3A or NPM1 mutations or MLL translocations (P = 0.001) but not among patients with wild-type DNMT3A, NPM1, and MLL (P = 0.67).
CONCLUSIONS
We found that DNMT3A and NPM1 mutations and MLL translocations predicted an improved outcome with high-dose induction chemotherapy in patients with AML. These findings suggest that mutational profiling could potentially be used for risk stratification and to inform prognostic and therapeutic decisions regarding patients with AML. (Funded by the National Cancer Institute and others.)
Previous studies have highlighted the clinical and biologic heterogeneity of acute myeloid leukemia (AML).1-4 However, a relatively small number of cytogenetic and molecular lesions have sufficient relevance to influence clinical practice.5 The prognostic relevance of cytogenetic abnormalities has led to the widespread adoption of risk stratification, with patients divided into three cytogenetically defined risk groups with significant differences in overall survival.6 More recently, FLT3, NPM1, and CEBPA mutational analysis was shown to improve risk stratification for patients who do not have karyotypic abnormalities.7 Although progress has been made in defining prognostic markers for AML, a substantial percentage of patients lack a specific abnormality of prognostic significance. In addition, there is considerable heterogeneity in the outcome for individual patients in each risk group.
Recent studies have identified novel recurrent somatic mutations in patients with AML. These include mutations in TET2,8,9 ASXL1,10 IDH1 or IDH2,11-13 DNMT3A,4,14 and PHF6.15 Retrospective analyses suggest that a subset of these mutations may have prognostic significance in AML,4,14,16 although these findings have not been validated with detailed clinical and mutational annotation in large, homogeneously treated cohorts of patients with AML. In addition, the question of whether mutational profiling of a larger set of genes, including these novel disease alleles, improves prognostication in AML has not been investigated in a clinical trial cohort.
A recent phase 3 clinical trial (E1900; Clinical Trials.gov number, NCT00049517) from the Eastern Cooperative Oncology Group (ECOG) showed that induction therapy with cytarabine plus 90 mg of daunorubicin per square meter of body-surface area, as compared with cytarabine plus 45 mg of daunorubicin per square meter, improved the outcomes in patients with newly diagnosed AML who were 17 to 60 years of age17; a similar study in patients who were older than 60 years of age showed that dose-intensified daunorubicin improved overall survival in patients 60 to 65 years of age.18 We hypothesized that integrated mutational analysis of all known molecular alterations occurring in more than 5% of patients with AML would allow us to identify novel molecular markers of outcome in AML and to identify molecularly defined subgroups of patients who would benefit from dose-intensified induction chemotherapy.
METHODS
PATIENTS
We performed mutational analysis on diagnostic samples obtained from patients in the ECOG E1900 trial. All patients provided written informed consent. The test cohort (398 patients) comprised all patients in the E1900 trial for whom viably frozen cells were available for DNA extraction and mutational profiling. The validation cohort (104 patients) comprised a second set of patients for whom samples were banked in Trizol reagent (Invitrogen), which was used to extract DNA for mutational studies. The clinical characteristics of the patients we studied, as compared with the complete E1900 trial cohort, are provided in Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org. The median follow-up time for the patients included in the analysis, calculated from the time of randomization for induction therapy, was 47.4 months. Cytogenetic analysis, fluorescence in situ hybridization, and reverse-transcriptase–polymerase-chain-reaction (RT-PCR) assays for recurrent cytogenetic lesions were performed as described initially by Slovak et al.6 and as used previously,17 with central review by the ECOG Cytogenetic Subcommittee.
MUTATIONAL ANALYSIS
The source of the DNA was bone marrow in the case of 55.2% of the samples (277 of 502) and peripheral blood in the case of 44.8% (225 of 502). We sequenced the entire coding regions of TET2, ASXL1, DNMT3A, CEBPA, PHF6, WT1, TP53, EZH2, RUNX1, and PTEN and the regions of previously described mutations for FLT3, NPM1, HRAS, KRAS, NRAS, KIT, IDH1, and IDH2. The genomic coordinates and sequences of all the primers used in this study are provided in Table S2 in the Supplementary Appendix. Paired remission DNA (i.e., DNA from patients who had a complete remission after induction chemotherapy) was available from 241 of the 398 participants in the test cohort and from 65 of the 104 in the validation cohort. Data on variants that could not be validated as bona fide somatic mutations owing to unavailable remission DNA and the absence of reports of the mutations in the published literature of somatic mutations were censored with respect to mutational status for that specific gene. Further details of the sequencing methods are available in the Supplementary Appendix.
STATISTICAL ANALYSIS
The mutual exclusivity of pairs of mutations was evaluated with the use of two-by-two contingency tables and Fisher’s exact test. The association between mutations and cytogenetic risk classification was tested with the use of the chi-square test. Hierarchical clustering was performed with the use of the Lance–Williams dissimilarity formula and the complete-linkage algorithm. Survival time was measured from the date of randomization to the date of death for patients who died and to the date of the last follow-up for those who were alive at the time of the analysis. Survival probabilities were estimated with the use of the Kaplan–Meier method and were compared between patients with a mutation and those without mutant alleles by means of the log-rank test. Multivariate analyses were conducted with the use of the Cox model with forward selection. We checked the proportional-hazards assumption by testing for a nonzero slope in a regression of the scaled Schoenfeld residuals on functions of time (Table S3 in the Supplementary Appendix). When necessary, such as in the analyses performed in various subsets, the results of the univariate analyses were used to select the variables to be included in the forward variable search. Final multivariate models informed the development of novel risk-classification rules. When so indicated, P values were adjusted to control the family-wise error rate with the use of the complete null distribution approximated by resam-pling obtained through the PROC MULTTEST program in SAS or the multtest library in R.19 The only exception was the adjustment in tests of the effect of mutations on the response to the induction dose, for which a step-down Holm procedure was used to correct for multiple testing. All analyses were performed with the use of SAS software, version 9.2 (www.sas.com), and the R statistical package, version 2.12 (www.r-project.org).
RESULTS
FREQUENCY OF GENETIC ALTERATIONS
Somatic alterations were identified in 97.3% of the patients. Figure 1 shows the frequency of somatic mutations in the entire cohort and the interrelationships among the various mutations, as represented visually with the use of a Circos plot. Data for all molecular subsets are provided in Figures S1 and S2 and Tables S4 and S5 in the Supplementary Appendix. In particular, mutational heterogeneity was greater in patients with intermediate-risk AML than in patients with favorable-risk or unfavorable-risk risk AML (P = 0.01) (Fig. S2D in the Supplementary Appendix).
Figure 1. Mutational Complexity of Acute Myeloid Leukemia (AML).
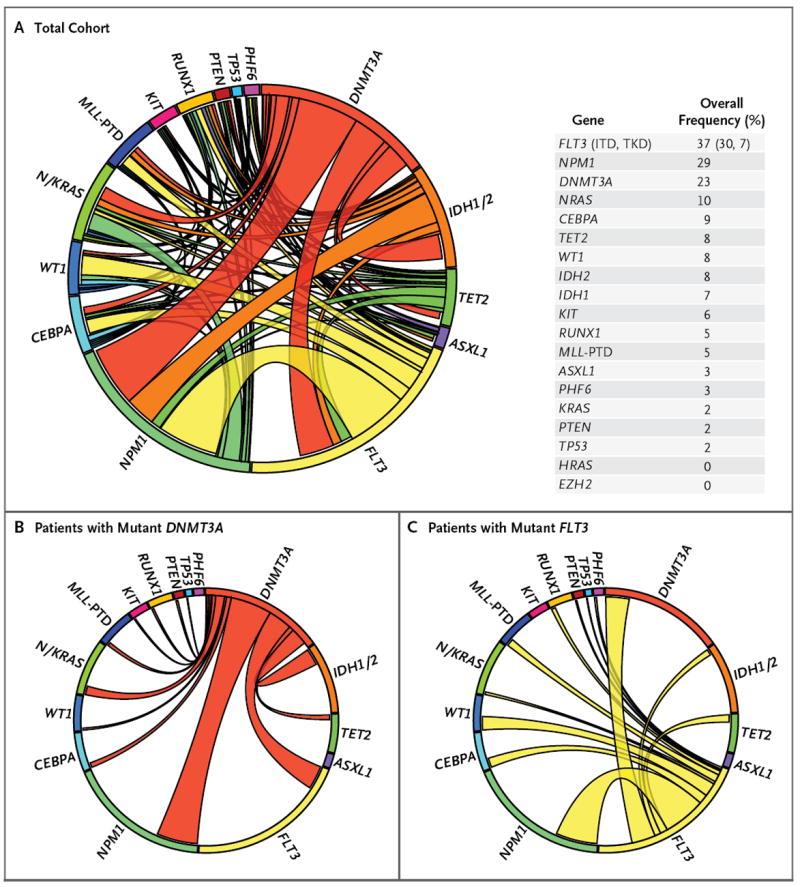
A Circos diagram (Panel A) depicts the relative frequency and pairwise co-occurrence of mutations in patients with newly diagnosed AML who were enrolled in the Eastern Cooperative Oncology Group E1900 clinical trial. The length of the arc corresponds to the frequency of mutations in the first gene, and the width of the ribbon corresponds to the percentage of patients who also had a mutation in the second gene. Pairwise co-occurrence of mutations is denoted only once, beginning with the first gene in the clockwise direction. Panel A also shows the frequency of mutations in the test cohort. Panels B and C show the mutational events in patients with mutant DNMT3A and mutant FLT3, respectively. Since, for clarity, only pairwise mutations are encoded, the arc length was adjusted to maintain the relative size of the arc, and the correct proportion of patients with only a single mutant allele is represented by the not-otherwise-occupied space within each mutational subset (all panels). ITD denotes internal tandem duplication, PTD partial tandem duplication, and TKD tyrosine kinase domain.
MUTATIONAL COMPLEMENTATION GROUPS
Integrated mutational analysis allowed us to identify frequently co-occurring mutations and mutations that were mutually exclusive in the E1900 patient cohort (Table S6 in the Supplementary Appendix). In addition to noting frequent co-occurrence of KIT mutations with core-binding–factor alterations t(8;21) and inv(16)/t(16;16), we found significant co-occurrence of IDH1 and IDH2 mutations with NPM1 mutations and of DNMT3A mutations with NPM1, FLT3, and IDH1 alleles (P<0.001 for all comparisons) (Table S7 in the Supplementary Appendix). We recently reported that IDH1 and IDH2 mutations were mutually exclusive with TET2 mutations20; detailed mutational analysis revealed that IDH1 and IDH2 mutations were also mutually exclusive with WT1 mutations (P<0.001) (Fig. S3 and Table S8 in the Supplementary Appendix). We also observed that DNMT3A mutations and MLL translocations were mutually exclusive (P<0.01).
MOLECULAR DETERMINANTS OF OVERALL SURVIVAL
Univariate analysis revealed, as previously described,21,22 that FLT3 internal tandem duplication (FLT3-ITD) mutations and MLL partial tandem duplication (MLL-PTD) mutations were associated with reduced overall survival (P = 0.001 for FLT3-ITD and P = 0.009 for MLL-PTD) (Table S9 in the Supplementary Appendix), whereas CEBPA mutations and core-binding–factor alterations t(8;21) and inv(16)/t(16;16) were associated with improved overall survival (P = 0.05 for CEBPA and P<0.001 for the core-binding–factor alterations).2,23 In addition, PHF6 and ASXL1 mutations were associated with reduced overall survival (P = 0.006 for PHF6 and P = 0.05 for ASXL1) (Fig. S4 in the Supplementary Appendix). IDH2 mutations were associated with an improved rate of overall survival in the entire test cohort (3-year rate, 66%; P = 0.01) (Fig. S5 in the Supplementary Appendix). The favorable effect of IDH2 mutations was found exclusively in patients with IDH2 R140Q mutations (P = 0.009) (Fig. S5 in the Supplementary Appendix). All the findings in the univariate analysis were also significant in the multivariate analysis (P<0.05, with adjustment for age, white-cell count, transplantation status [did vs. did not undergo stem-cell transplantation], and cytogenetic characteristics) (Table S9 in the Supplementary Appendix), with the exception of the findings for MLL-PTD, PHF6, and ASXL1 mutations. KIT mutations were associated with reduced overall survival among patients who were positive for the t(8;21) core-binding–factor alteration (P = 0.006) but not among patients with the inv(16)/t(16;16) alteration (P = 0.19) (Fig. S6 in the Supplementary Appendix).
PROGNOSTIC VALUE OF MOLECULAR ALTERATIONS IN INTERMEDIATE-RISK AML
Among patients with intermediate-risk AML as defined by cytogenetic analysis (Table S10 in the Supplementary Appendix), FLT3-ITD mutations were associated with reduced overall survival (P = 0.008), a finding that is consistent with the results of previous studies.21ASXL1 and PHF6 mutations were associated with reduced survival, and IDH2 R140Q mutations with improved survival, among patients with intermediate-risk AML (Table S10 in the Supplementary Appendix), an effect similar to that in the entire cohort. In addition, we found that TET2 mutations were associated with reduced overall survival among patients with intermediate-risk AML (P = 0.007) (Fig. S7 in the Supplementary Appendix).
Multivariate analysis revealed that FLT3-ITD mutations constituted the primary predictor of outcome in patients with intermediate-risk AML (adjusted P<0.001). A subsequent multivariate analysis according to FLT3-ITD status showed that in patients with wild-type FLT3-ITD, mutations in TET2, ASXL1, PHF6, and MLL-PTD were independently associated with an adverse outcome. Patients with intermediate-risk AML who had both NPM1 and IDH1 or IDH2 mutations had an improved 3-year rate of overall survival, as compared with patients who had mutant NPM1 and both wild-type IDH1 and wild-type IDH2 (89% vs. 31%, P<0.001) (Fig. S8 in the Supplementary Appendix). We then classified patients with intermediate-risk AML who had wild-type FLT3-ITD into three categories, with marked differences in the 3-year rate of overall survival (adjusted P<0.001): patients with IDH1 or IDH2 mutations and NPM1 mutations (overall survival, 89%); patients with TET2, ASXL1, PHF6, or MLL-PTD mutations (overall survival, 6.3%); and patients with wild-type TET2, ASXL1, PHF6, and MLL-PTD, without co-occurring IDH or NPM1 mutations (overall survival, 46.2%) (Fig. 2A). Similar results were obtained when the analysis was restricted to patients with a normal karyotype (Fig. S9A in the Supplementary Appendix).
Figure 2. Multivariate Risk Classification of Patients with Intermediate-Risk AML.
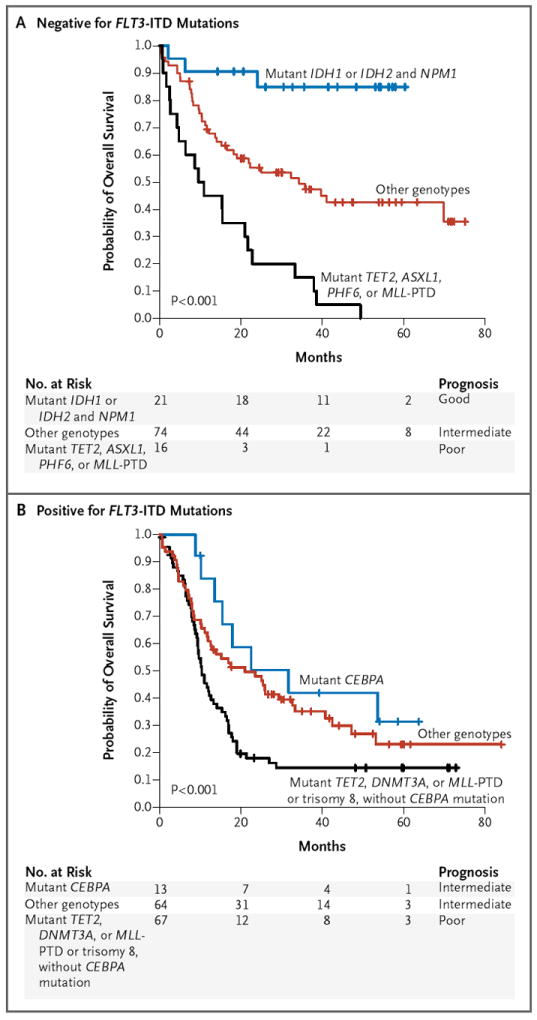
Kaplan–Meier estimates of overall survival are shown for the risk stratification of patients with intermediate-risk AML (with P values for the comparison of all curves). Among patients with wild-type FLT3-ITD (Panel A), there were three categories: patients with mutant TET2, ASXL1, PHF6, or MLL-PTD, who had a poor prognosis for overall survival; those with mutant IDH1 or IDH2 and mutant NPM1, who had a good prognosis for overall survival; and those with any other genotypes, who had an intermediate prognosis for survival. Among patients who were positive for FLT3-ITD mutations (Panel B), there were also three categories of patients: those with mutant TET2, DNMT3A, or MLL-PTD or trisomy 8 without mutant CEBPA, who had a poor prognosis for overall survival; those with mutant CEBPA, who had an intermediate prognosis for survival; and those with any other genotypes, who also had an intermediate prognosis for survival.
In patients with intermediate-risk AML who had mutant FLT3-ITD, we found that CEBPA mutations were associated with an improved outcome and that trisomy 8 and TET2, DNMT3A, and MLL-PTD mutations were associated with an adverse outcome. We used these data to classify patients with intermediate-risk AML who had mutant FLT3-ITD into three categories. The first category included patients with trisomy 8 or TET2, DNMT3A, or MLL-PTD mutations, which were associated with an adverse outcome (3-year rate of overall survival, 14.5%); this rate of survival was significantly lower than the rates among patients in the second category, those with wild-type CEBPA, TET2, DNMT3A, and MLL-PTD (overall survival, 35.2%; P<0.001), and patients in the third category, those with CEBPA mutations (overall survival, 42%; P<0.001) (Fig. 2B). The rate of survival among patients with intermediate-risk AML who had mutant FLT3-ITD and wild-type CEBPA, TET2, DNMT3A, and MLL-PTD did not differ significantly from the rate among patients with mutant FLT3-ITD and mutant CEBPA (P = 0.34), suggesting that the presence of mutations associated with an unfavorable-risk profile more precisely identifies patients with mutant FLT3-ITD who will have adverse outcomes of AML than does the absence of CEBPA mutations alone. These same three risk categories also had significant prognostic value in patients with AML who had mutant FLT3-ITD and a normal karyotype (Fig. S9B in the Supplementary Appendix).
PROGNOSTIC SCHEMA WITH INTEGRATED MUTATIONAL AND CYTOGENETIC PROFILING
These results allowed us to develop a prognostic schema that integrated our findings from the comprehensive mutational analysis with cytogenetic data to identify three risk groups: a group with a favorable-risk profile (median survival, not reached; 3-year rate of overall survival, 64%), a group with an intermediate-risk profile (median survival, 25.4 months; 3-year rate of overall survival, 42%), and a group with an adverse-risk profile (median survival, 10.1 months; 3-year rate of overall survival, 12%) (Fig. 3A and 3B, and Table S11 in the Supplementary Appendix). In multivariate analysis, the mutational prognostic schema predicted the outcome independently of age, white-cell count, induction dose, and transplantation status (adjusted P<0.001). Our classification held true regardless of the type of post-remission therapy (autologous or allogeneic transplantation or consolidation chemotherapy alone) (Fig. S10 in the Supplementary Appendix).
Figure 3. Revised Risk Stratification of Patients with AML on the Basis of Integrated Genetic Analysis.
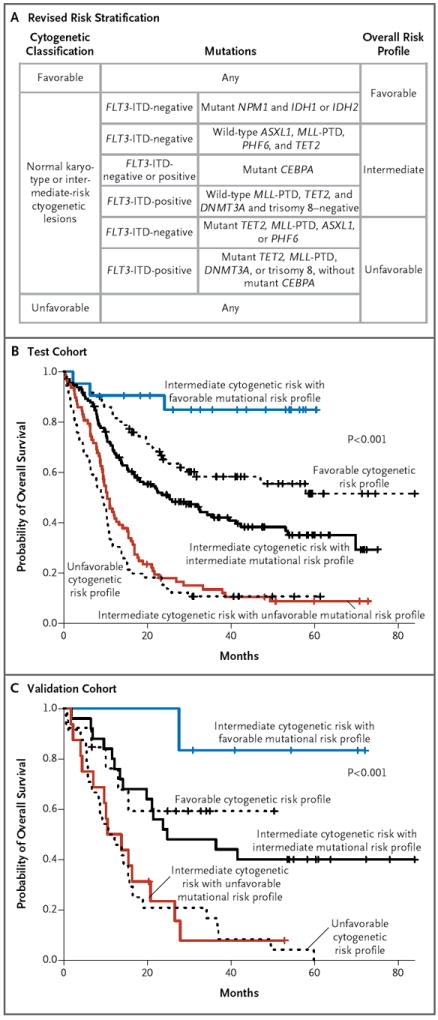
Panel A shows a revised risk stratification on the basis of integrated cytogenetic and mutational analysis. The final overall risk groups are shown on the right. Panel B shows the effect of integrated mutational analysis on risk stratification in the test cohort of patients with AML (with P values for the comparison of all curves), and Panel C shows the reproducibility of the genetic prognostic schema in an independent cohort of 104 patients from the E1900 trial (with P values for the comparison of all curves). In Panels B and C, the black curves show the patients whose risk classification remained unchanged, the blue curve shows patients who were reclassified from intermediate risk to favorable risk, and the red curve shows patients who were reclassified from intermediate risk to unfavorable risk.
Given the number of variables in our prognostic classification, we tested the reproducibility of this predictor in an independent cohort of 104 patients from the ECOG E1900 trial. Mutational analysis of the validation cohort confirmed the reproducibility of our prognostic schema for predicting the outcome in patients with AML (adjusted P<0.001) (Fig. 3C). The predictive value of the mutational prognostic schema was independent of risk with respect to treatment-related death (defined as death within 30 days after initiation of treatment) or lack of response to induction chemotherapy (i.e., lack of a complete remission) in the test cohort and in the combined test and validation cohorts (Table S12 in the Supplementary Appendix).
GENETIC PREDICTORS OF RESPONSE TO INDUCTION CHEMOTHERAPY
Recent studies have shown that mutant DNMT3A was associated with adverse outcomes in patients with AML.4,14 However, we found that DNMT3A mutations were not associated with adverse outcomes in the ECOG E1900 cohort (Fig. 4A) (P = 0.15). In the ECOG E1900 trial, patients were randomly assigned to induction therapy with cytarabine plus either 45 mg of daunorubicin per square meter or 90 mg of daunorubicin per square meter.17 We therefore hypothesized that high-dose daunorubicin improved the outcomes in patients with daunorubicin who had DNMT3A mutations. Indeed, we found that DNMT3A mutational status had a significant effect on the outcome with dose-intensive chemotherapy (Fig. 4B) (P = 0.02). We then assessed the effects of DNMT3A mutational status on the outcome according to treatment group and found that high-dose daunorubicin was associated with an improved rate of survival among patients with mutant DNMT3A (P = 0.04) (Fig. S11A in the Supplementary Appendix) but not among patients with wild-type DNMT3A (P = 0.15) (Fig. S11B in the Supplementary Appendix). In addition, univariate analysis revealed that dose-intensified induction therapy was associated with an improved outcome in patients with AML who had MLL translocations (P = 0.01; P = 0.06 with adjustment for multiple testing) (Fig. S11C and S11D in the Supplementary Appendix) and in those who had NPM1 mutations (P = 0.01; P = 0.10 with adjustment for multiple testing) (Fig. S11E and S11F and Table S13 in the Supplementary Appendix). Because the adjusted P values for NPM1 mutations and MLL translocations (P≤0.10) are close to statistical significance, they should be studied further in prospective trials.
Figure 4. Molecular Determinants of Response to High-Dose Daunorubicin Induction Chemotherapy.
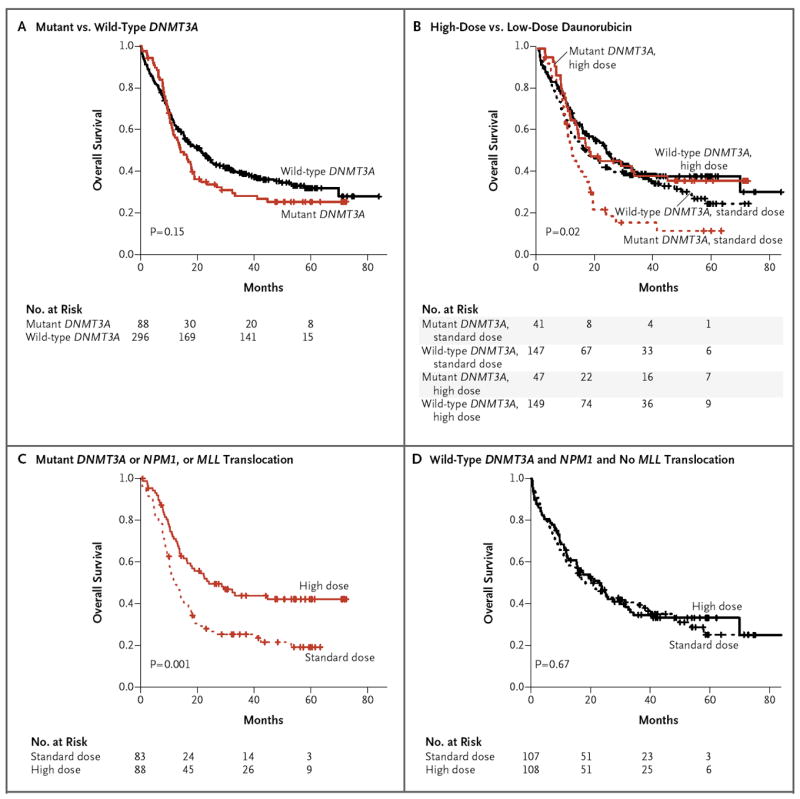
Kaplan–Meier estimates of the probability of overall survival are shown in the entire cohort according to DNMT3A mutational status (Panel A) and according to DNMT3A status and receipt of high-dose or standard-dose daunorubicin (Panel B). The P value in Panel B is for the comparison across all groups. The rates of overall survival according to receipt of high-dose or standard-dose daunorubicin are shown among patients with DNMT3A or NPM1 mutations or MLL translocations (Panel C) and among patients without DNMT3A and NPM1 mutations and with no MLL translocations (Panel D).
We then separated the patients in our cohort into two groups: patients with mutations in DNMT3A or NPM1 or with MLL translocations and patients with wild-type DNMT3A and NPM1 and no MLL translocations. Dose-intensive induction therapy was associated with a marked improvement in the rate of survival among patients who were positive for DNMT3A or NPM1 mutations or MLL translocations (P = 0.001) (Fig. 4C) but not among patients with wild-type DNMT3A and NPM1 and no MLL translocations (P = 0.67) (Fig. 4D). This finding was independent of the clinical covariates of age, white-cell count, and status with respect to transplantation, treatment-related death, and response to chemotherapy (adjusted P = 0.008 and P = 0.34 for patients with mutant and wild-type genes, respectively), suggesting that high-dose anthracycline chemotherapy provides a benefit in genetically defined subgroups of patients with AML.
COMPREHENSIVE MUTATIONAL PROFILING FOR RISK STRATIFICATION AND CLINICAL MANAGEMENT OF AML
On the basis of cytogenetic classification alone, 63% of the patients in the ECOG E1900 cohort were categorized as having an intermediate-risk AML profile (3-year rate of overall survival, 36%), whereas 19% of the patients were classified as having a favorable-risk profile (3-year rate of overall survival, 58%) and 18% as having an unfavorable-risk profile (3-year rate of overall survival, 11%) (Fig. 5A). Mutational analysis allowed for the separation of patients with cytogenetically defined intermediate-risk AML into three subgroups with markedly different outcomes: a subgroup of patients with a favorable mutational risk profile (3-year rate of overall survival, 85%), a subgroup of patients with an unfavorable mutational risk profile (3-year rate of overall survival, 13%), and a subgroup of patients who remained at intermediate risk (3-year rate of overall survival, 42%). Integrating mutational and cytogenetic analyses reduced the proportion of patients with intermediate risk from 63%, as assessed by means of cytogenetic analysis alone, to 35% with inclusion of mutational data. Likewise, the proportion of patients ultimately classified as having a favorable-risk AML profile increased from 19% to 26% (3-year rate of overall survival, 64%), and the proportion of patients classified as having an unfavorable-risk profile increased from 18% to 39% (3-year rate of overall survival, 12%). Mutational analysis also revealed that patients with a mutation in DNMT3A or NPM1 or a MLL translocation had improved overall survival with high-dose chemotherapy, as compared with standard-dose chemotherapy (3-year rate of overall survival, 44% vs. 25%) (Fig. 5B), showing that mutational analysis can identify specific genetically defined subgroups of patients who benefit from high-dose induction chemotherapy.
Figure 5. Comprehensive Mutational Profiling for Risk Stratification and Clinical Management of AML.
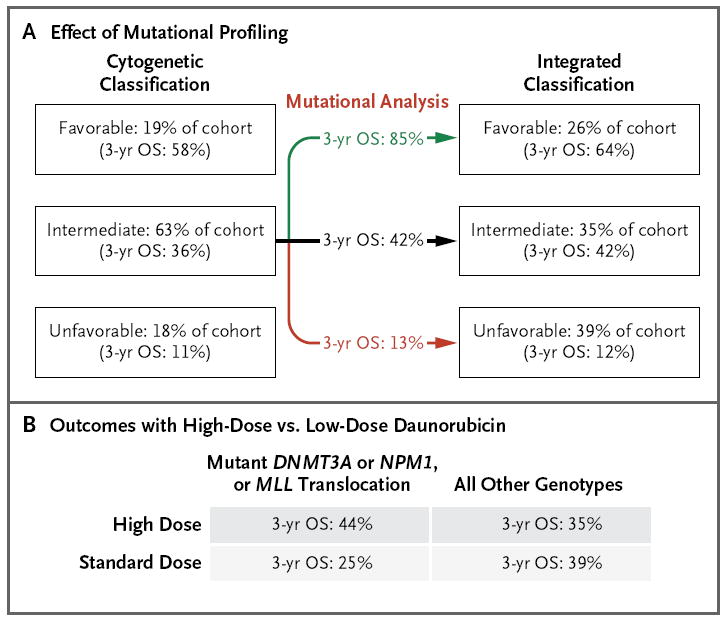
The use of mutational profiling delineates subgroups of patients with intermediate-risk AML, as defined according to cytogenetic analysis, who have markedly divergent prognoses and reassigns a substantial proportion of them to favorable-risk or unfavorable-risk categories (Panel A). In addition, mutational profiling identifies genetically defined subgroups of patients with AML who have improved outcomes with high-dose anthracycline induction chemotherapy (Panel B). OS denotes overall survival.
DISCUSSION
We describe the frequency of various mutations and the prognostic usefulness and therapeutic relevance of integrated mutational profiling in 398 patients from the ECOG E1900 phase 3 clinical trial and the validation of these data in an independent cohort of 104 patients from the same trial.17 Previous studies have suggested that mutational analysis of CEBPA, NPM1, and FLT3-ITD can be used to stratify risk among patients with intermediate-risk AML.7 Using data from a large cohort of patients treated in a single clinical trial, we found that more extensive mutational analysis can better discriminate patients with AML into various prognostic groups (Fig. 3). For example, patients without FLT3-ITD mutations and with both mutant NPM1 and IDH represent a favorable-risk subset defined by a specific mutational genotype, whereas patients who were negative for FLT3-ITD mutations and had mutant NPM1 without concurrent IDH mutations had a much less favorable outcome — particularly if those patients had concurrent mutations associated with an unfavorable-risk profile. We also found that TET2, ASXL1, MLL-PTD, PHF6, and DNMT3A mutations can identify patients with intermediate-risk AML who will have an adverse outcome. Taken together, these data show that mutational analysis of a larger set of genetic alterations than that currently used in the clinic setting could be used to retrospectively classify patients with AML into more precise subgroups with favorable-risk, intermediate-risk, or unfavorable-risk profiles, with marked differences in the overall outcome. This approach could be used to identify an additional subgroup of patients who would have a mutationally defined favorable outcome with induction and consolidation therapy alone and a subgroup of patients with mutationally defined unfavorable risk who would potentially be candidates for allogeneic stem-cell transplantation or participation in a clinical trial, given the prediction of a poor outcome with standard AML therapy (Fig. 5A).
The two recent randomized trials examining the benefits of intensification of the dose of anthracycline in patients with AML showed that more intensive induction chemotherapy improved the outcomes.17,18 An evaluation of the data from the overall cohort in the E1900 trial showed that the distribution of patients among genetic risk categories in both treatment groups was similar to the distribution in our cohort of 502 patients (P = 0.41 by Pearson’s chi-square test). However, the initial reports of the two recent trials did not identify whether dose-intensified induction therapy improved the outcomes in various AML subgroups. We found that intensification of the dose of anthracycline markedly improved the outcomes in patients with mutations in DNMT3A or NPM1 or with MLL translocations, suggesting that mutational profiling can be used to determine which patients will benefit from dose-intensive induction therapy (Fig. 5B). Future studies will be needed to determine whether there are additional subgroups of patients with AML who will benefit from dose-intensified induction therapy and whether dose-intensified induction therapy improves the outcome in patients with AML who are older than 60 years of age and have mutations in DNMT3A or NPM1 or MLL translocations.
We also identified mutational combinations that occur commonly in patients with AML and those that rarely, if ever, co-occur; these findings are consistent with the existence of additional mutational complementation groups with gene mutations that are mutually exclusive with one another in large patient cohorts. For example, the observation that TET2 and IDH mutations were mutually exclusive in this AML cohort led to functional studies linking IDH mutations and loss-of-function TET2 mutations in a shared mechanism of hematopoietic transformation.20
The data in this study show a way in which integrated mutational profiling of a clinical trial cohort can advance our understanding of the biologic characteristics of AML, improve current prognostic models, and inform therapeutic decisions. Most important, these data indicate that more detailed genetic analysis may lead to improved risk stratification and identification of patients who can benefit from more intensive induction chemotherapy. The challenge is to provide genetic information in a timely and affordable way and show that this information could prospectively influence treatment decisions.
Supplementary Material
Acknowledgments
Supported by grants from the National Cancer Institute (NCI) Physical Sciences Oncology Center (U54CA143798-01, to Dr. Levine), the Gabrielle’s Angel Fund (to Dr. Levine), and the Starr Cancer Consortium (to Drs. Levine and Melnick) and funding from the Peter Solomon Fund at Memorial Sloan-Kettering Cancer Center. Mr. Patel is supported by an American Society of Hematology Trainee Research Award; Dr. Figueroa is funded by a Leukemia and Lymphoma Society Special Fellow Award; Drs. Van Vlierberghe and Abdel-Wahab are American Society of Hematology Basic Research Fellows; Dr. Van Vlierberghe is supported by the Fund for Scientific Research (FWO) Flanders; Dr. Melnick is funded by a Leukemia and Lymphoma Society Specialized Center of Research Award and Translational Research Program Award and is a Burroughs Wellcome Clinical Translational Scholar and a Scholar of the Leukemia and Lymphoma Society; Drs. Figueroa and Melnick are also supported by the Sackler Center for Biomedical and Physical Sciences; Dr. Levine is the recipient of an Early Career Award from the Howard Hughes Medical Institute; Dr. Paietta is supported by NCI grants CA21115 and CA114737; Dr. Gallagher is supported by NCI grant CA56771; and Dr. Ferrando is funded by a Leukemia and Lymphoma Society Scholar Award.
We thank Martin Carroll, Charles Sawyers, Craig Thompson, and Stephen Nimer for their insight and suggestions.
APPENDIX
the Human Oncology and Pathogenesis Program (J.P.P., I.D., S.T., O.A., K.H., A.H., O.A.-W., R.L.L.), the Departments of Epidemiology and Biostatistics (M.G.) and Medicine (M.R.M.B.), the Genomics Core Laboratory (J.C., A.V.), the Bioinformatics Core (N.D.S.), and the Leukemia Service, Department of Medicine (M.S.T., O.A.-W., R.L.L.), Memorial Sloan-Kettering Cancer Center, the Department of Medicine, Hematology/Oncology Division, Weill Cornell Medical College (M.E.F., A.M.), Montefiore Medical Center (J.R., R.E.G., E.P.), the Institute for Cancer Genetics and the Departments of Pediatrics and Pathology, Columbia University (P.V.V., A.F.), and the Immunology Program, Sloan-Kettering Institute (M.R.M.B.) — all in New York; the Department of Blood and Bone Marrow Transplantation, H. Lee Moffitt Cancer Center, Tampa, FL (H.F.); the Department of Biostatistics, Dana–Farber Cancer Institute, Boston (Z.S.); the Department of Pathology, Stanford Hospital and Clinics, Palo Alto, CA (A.C.); the Department of Medical and Molecular Genetics, Indiana University School of Medicine, Bloomington (G.V.); Allina Cytogenetics Laboratory, Abbott Northwestern Hospital, Minneapolis (R.R.H.); the Departments of Laboratory Medicine and Pathology (R.P.K.) and Hematology and Oncology (M.L.), Mayo Clinic College of Medicine, Rochester, MN; the Division of Hematology–Oncology, Case Western Reserve University School of Medicine, Cleveland (H.M.L.); Shaare Zedek Medical Center and Technion, Israel Institute of Technology, Jerusalem (J.M.R.); and the Department of Medicine, University of Pennsylvania School of Medicine, Philadelphia (S.L.)
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–28. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- 2.Byrd JC, Mrózek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–36. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 3.Bullinger L, Döhner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–16. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- 4.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–86. doi: 10.1200/JCO.2010.30.2554. Erratum, J Clin Oncol 2011;29:1798. [DOI] [PubMed] [Google Scholar]
- 6.Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–83. [PubMed] [Google Scholar]
- 7.Schlenk RF, Döhner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 8.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 9.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–7. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metzeler KH, Becker H, Maharry K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118:6920–9. doi: 10.1182/blood-2011-08-368225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–55. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan XJ, Xu J, Gu ZH, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 15.Van Vlierberghe P, Patel J, Abdel-Wahab O, et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011;25:130–4. doi: 10.1038/leu.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metzeler KH, Maharry K, Radmacher MD, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–81. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361:1249–59. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Löwenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med. 2009;361:1235–48. doi: 10.1056/NEJMoa0901409. [DOI] [PubMed] [Google Scholar]
- 19.Westfall PH, Young SS. Resampling-based multiple testing. New York: Wiley; 1993. [Google Scholar]
- 20.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fröhling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–80. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 22.Caligiuri MA, Strout MP, Lawrence D, et al. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res. 1998;58:55–9. [PubMed] [Google Scholar]
- 23.Fröhling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol. 2004;22:624–33. doi: 10.1200/JCO.2004.06.060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.