

Stimuli-responsive nanoparticles are gaining considerable attention in the field of drug delivery due to their useful physicochemical changes in response to specific triggers, such as pH[1], temperature[2], enzymes[3] or redox conditions[4], present in certain physiological or disease microenvironment of interest. Among these nanoparticles, stimuli-responsive cross-linked micelles (SCMs) represent a versatile nanocarrier system for tumor targeting drug delivery[2c, 4-5]. For instance, SCMs exhibit superior structural stability under physiological condition compared to the non-crosslinked counterpart. As a result, these nanocarriers are able to better retain the encapsulated drug and minimize its premature release while circulating in the blood pool[2c, 4b, 5b]. The introduction of environmentally sensitive crosslinkers makes SCMs responsive to the local environment of the tumor (e.g. tumor extra-cellular pH (6.5-7.2), endosomal/lysosomal pH (4.5–6)[5b, 6], and tumor reductive intra-cellular condition[4-5]). In these instances, the payload drug is released almost exclusively in the cancerous tissue upon accumulation via the well known enhanced permeation and retention (EPR) effect[2c, 4b, 5b].
Remarkable progress in this field has led to the development of SCMs responsive to a single stimulus[4b, 5b]. Various cleavable linkages have been introduced in SCMs, such as reducible disulfide bonds[4b], pH cleavable[6] or hydrolysable ester bonds[2c]. Currently, second generation SCMs able to respond to multiple stimuli are being actively pursued as tools for accomplishing the multistage delivery of drugs to the complex in vivo micro-environment[7]. Boronic acids are well-known to bind diols forming reversible boronate esters that exhibit fast dual responsiveness to external pH and competing diols[8]. Based on this interaction, there has been increasing interest in using boronic acids as building blocks to design carbohydrate sensors[9], nano-reactor[10], drug delivery systems[11] and self-healing materials[12]. Among diols, catechols are an excellent reactant for the formation of complexes with boronic acids, thanks to the favorable syn-peri-planar arrangement of the aromatic hydroxyl groups combined with their electron-donating character[8a, 13]. Herein, we present the first report, to our knowledge, on the synthesis of a novel class of dual-responsive boronate cross-linked micelles (BCM) for drug delivery based on the self-assembly and in situ complexation of boronic-acid containing polymers and catechol containing polymers. We hypothesize that these BCMs will retain the encapsulated drug under physiological conditions, while releasing the payload quickly when triggered by the lower pH of the tumor environment or when exposed to exogenous competing diols.
Additionally, we are also presenting a Förster resonance energy transfer (FRET) reporter system to evaluate the in vivo stability of these micelles. This characterization is usually difficult to accomplish in vivo due to the lack of suitable analytical techniques.
We have previously reported a novel class of micelles for efficient anticancer drug delivery based on linear polyethylene glycol (PEG) and dendritic cholic acids (CA) block copolymers (called telodendrimers)[4b, 14]. In the present work, we have improved the stability of these micelles by crosslinking with boronate esters at the core-shell interface. The crosslinking reactants (i.e. boronic acid and catechol) were introduced on the block copolymers through step-wise peptide chemistry via attachment to a hydrophilic linker and positioned onto adjacent sites of the telodendrimers (Scheme 1, Scheme S1, S2). The peptide chemistry employed provides a facile strategy to synthesize a number of well-defined telodendrimers bearing a defined number and derivative of boronic acids and catechols. Nitro-phenylboronic acid (NBA) and phenylboronic acid (BA) were chosen as reactants to bind the catechol partners. The resulting boronate esters are known to be stable at physiological pH[8a, 15] even in the presence of most competing carbohydrates present in blood[8a, 8b]. The reaction of boronic acid with catechol on distinct telodendrimers in aqueous conditions takes place concomitantly to the self-assembly of the telodendrimers into micelles, resulting in boronate ester crosslinked micelles.
Scheme 1.
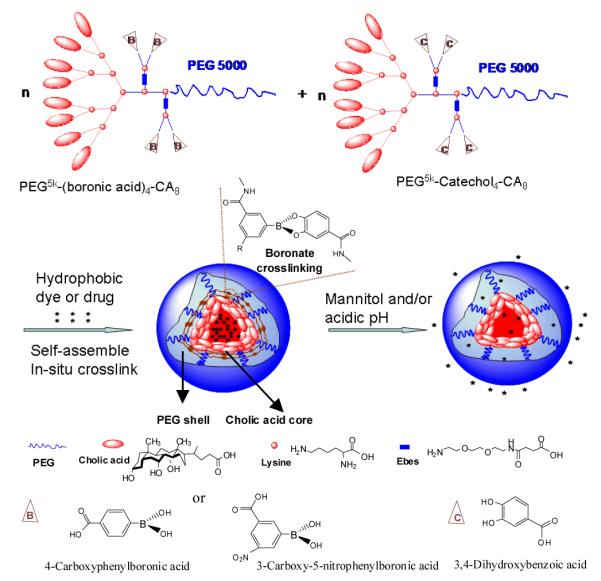
Schematic representation of the telodendrimer pair [PEG5k-(boronic acid/catechol)4-CA8] and the resulting boronate crosslinked micelles (BCM) in response to mannitol and/or acidic pH.
The synthetic schemes of a series of boronic acid and catechol-containing telodendrimers are shown in the Supporting information (Scheme S1-3). A representative crosslinkable telodendrimer pair, PEG5k-NBA4-CA8 and PEG5k-Catechol4-CA8 consist of, respectively, four nitro-phenylboronic acids and four catechols, attached to the α- and ε-amino groups of pendant lysines positioned at adjacent sites between the linear PEG and dendritic octamer of cholic acids (Scheme S1-3). The parent telodendrimer, PEG5k-CA8 was also synthesized[14b] to generate non-crosslinked micelles (NCM) for comparison. The structure of telodendrimers was confirmed by MALDI-TOF MS, 1H-NMR and a colorimetric assay based on the indicator of alizarin red S (ARS)[8a, 16](Table S1, Figure S1-3).
ARS is a catechol dye displaying dramatic changes in color and fluorescence intensity upon binding to boronic acid (Figure S3, S4, Scheme S4)[16]. The introduction of electron withdrawing group onto the phenyl ring of boronic acid stabilizes the boronate form of the acid and lowers the pKa value, which in turn favors boronate ester formation at a higher pH of 7.4[15]. The nitro-phenylboronic acids containing telodendrimer PEG5k-NBA4-CA8 caused more significant color change of ARS (from burgundy to yellow) compared with the equal concentrations of phenylboronic acids containing telodendrimer PEG5k-BA4-CA8, indicating the stronger binding of PEG5k-NBA4-CA8 with ARS (Figure S4). We also observed that the fluorescence intensity of ARS at 580 nm increased significantly upon binding with PEG5k-NBA4-CA8 and PEG5k-BA4-CA8 (Figure S5).
Next we proceeded to verify the formation of boronate crosslinking within BCMs prepared via solvent evaporation method[4b, 14]. The fluorescence spectra of ARS were recorded with micelles comprised of varying ratios of boronic acid- and catechol-containing telodendemers. When the concentrations of ARS and boronic acid containing telodendrimers were fixed at 0.1 mM, the fluorescence of ARS was dramatically suppressed with increasing amounts of PEG5k-Catechol4-CA8 (0 to 0.5 mM) (Figure 1A, S-6). These results are a qualitative indication of the formation of catechol-boronate crosslinking esters as ARS was prevented from complexation with boronic acid containing telodendrimers.
Figure 1.

(A) The fluorescent intensity of ARS (0.1 mM) upon mixing with micelles formed by PEG5k-NBA4-CA8 (0.1 mM) with different ratios of PEG5k-Catechol4-CA8 (0-0.5 mM) in PBS at pH7.4. Excitation: 468 nm. (B) Continuous dynamic light scattering measurements of NCM in SDS and BCM4 in SDS for 120 min, at which time mannitol was added or pH of the solution was adjusted to 5.0 (see arrow). TEM images of BCM4 in PBS(C1), BCM4 in SDS for 120 min (C2), BCM4 in SDS for 120 min and then adjusted the pH of the solution to 5.0 for 20 min (C3), and BCM4 in SDS for 120 min and then treated with mannitol (100 mM) for 20 min (C4), (scale bar: 100 nm).
A series of BCMs were formed using equal molar ratios of the boronic acid-containing telodendrimers and catechol-containing telodendrimers and their physical properties are shown in Table 1. Boronate cross-linking dramatically reduced the critical micelle concentrations (CMC) as compared with NCM (Table 1). The particle sizes for BCMs were all in the range of 22 – 27 nm with narrow distribution (Table 1, Figure 1B, 1C1, S7), which is similar to the parent non-crosslinked PEG5k-CA8 micelles.
Table 1.
Characterization of boronate cross-linked micelles and non-cross-lnked micelles.
| Micelle Formulation |
Telodendrimer pair | Size (nm)[a] |
CMC (μg/mL)[b] |
Stability In SDS[c] |
PTX Content[d] |
|---|---|---|---|---|---|
| BCM1 | PEG5k-BA2-CA8, PEG5k-Catechol2-CA8 |
23 ± 4 | 10.5 | 2 min | 9.9% |
| BCM2 | PEG5k-NBA2-CA8, PEG5k-Catechol2-CA8 |
26 ± 6 | 8.7 | 30 min | 9.8% |
| BCM3 | PEG5k-BA4-CA8, PEG5k-Catechol4-CA8 |
22 ± 3 | 7.4 | 5 min | 9.8% |
| BCM4 | PEG5k-NBA4-CA8, PEG5k-Catechol4-CA8 |
27 ± 5 | 4.2 | Long term | 9.9% |
| NCM | PEG5k-CA8 | 22 ±6 | 50.1 | <10 sec | 10.0% |
Measured by dynamic light scattering (DLS).
Measured via fluorescent method by using pyrene as a probe.
The total period of time that the micelles retained their sizes in SDS, continuously measured by DLS every 10 sec at pH 7.4.
PTX loading content of micelles (drug/polymer, w/w), in the presence of 20 mg/mL of total telodendrimers and 2.0 mg/mL PTX initial loading, measured by HPLC.
We then investigated the interaction of the crosslinked micelles with plasma proteins to simulate potentially destabilizing conditions for in vivo applications. BCM4 still retained size uniformity and narrow distribution peaked at 30 nm when exposed to 50% (v/v) human plasma for 24 h (Figure S-8E). Quite to the opposite, the NCM showed significantly broader size and bimodal distribution with populations at 81 and 237 nm, indicating the formation of large aggregates (Figure S-8B). We further investigated whether boronate cross-linking enhances micellar stability against severe micelle-disrupting conditions. Sodium dodecyl sulfate (SDS), a strong ionic detergent, has been reported to efficiently break down polymeric micelles[17]. Micelle solutions were exposed to an aqueous solution of SDS while continuously monitoring the particle size with dynamic light scattering. The rapid disappearance (< 10 sec) of the particle size signal for the NCM reflects the loss of integrity (Figure 1B, S8A, 8C). The BCM1, BCM2 and BCM3 retained the size in SDS for 2 min, 5 min and 30 min, respectively (Table 1). Despite an initial decrease, the constant particle size was observed over 2 days for BCM4 treated under the same conditions indicating that the cross-linked micelles self-assembled from the telodendrimer pair of PEG5k-NBA4-CA8 and PEG5k-Catechol4-CA8 remained intact (Figure 1B, S8D, 8F and S9). BCM3 and BCM4, containing double the number of boronate esters retained their structural integrity significantly longer in the presence of SDS, when compared to BCM1 and BCM2, respectively. BCM2 and BCM4 crosslinked via nitro phenyl boronate esters were more stable than the corresponding phenyl boronate esters crosslinked micelles BCM1 and BCM3. We further investigated the response to pH- and diol- for BCM4 in the presence of SDS. The particle size signal of BCM4 decreased suddenly (within 2 min) in SDS after 120 min incubation in pH 5.0, indicating that the micelle rapidly dissociated when a critical percentage of boronate bonds were hydrolyzed (Figure 1B, S8G). We found that mannitol (containing three cis-diol pairs) could also efficiently cleave the crosslinking boronate bonds of the BCM4, as evidenced by the rapid reduction in particle size of BCM4 in the presence of SDS and excess of mannitol (100 mM) (Figure 1A, S8H). On the contrary, the size of BCM4 persisted in the presence of both SDS and 100 mM glucose (containing one cis-diol) (Figure S8I). TEM permitted the confirmation that the micellar structure of NCM was disrupted in SDS solution[4b]. The TEM graphs also demonstrated the micellar structure of BCM4 was well retained in SDS at pH 7.4 (Figure 1C2) but was rapidly disrupted in SDS at pH 5.0 or in the presence of 100 mM mannitol (Figure 1C3, 1C4).
We investigated the release profiles from the micelles by using paclitaxel (PTX) as a model drug. The loading content of PTX into NCM and BCMs was above 9.8% (w/w, drug/polymer) while the loading efficiency for all micelles above 98% (Table 1). PTX release from NCM was rapid with almost 30% of PTX released within the first 9 h independently from the pH of the release medium or the presence of diols (Figure S10). PTX release from BCM3 crosslinked via phenyl boronate was significantly slower than NCM but faster than BCM4 with nitro- phenyl boronate crosslinking at pH7.4 (Figure S10A, S10B). PTX release from BCM3 was promoted when decreasing the pH of the medium from 7.4 to 6.5 while that of BCM4 was accelerated at pH 5.5 (Figure S10A). In the presence of glucose at its physiological level (2-10 mM) or even higher concentration (50 mM), PTX release from BCM3 and BCM4 was similar to that in the release media without glucose (Figure S10B). It was noted that PTX release was not sensitive to 10 mM mannitol but could be gradually facilitated as the concentration of mannitol increased up to the range of 50-100 mM (Figure S10B). This could be attributed to the significantly higher affinity of mannitol with boronic acids than that of glucose at the same concentration in physiological conditions. Mannitol is a safe FDA approved drug for diuresis. High blood level of mannitol (> 50 mM) can be achieved clinically based on the recommended dose. In this study, mannitol can be applied in vivo as an on-demand cleavage reagent via systemic intravenous injection to trigger drug release after the drug loaded BCMs have accumulated in tumor sites. In order to simulate the in vivo situations, the PTX release from BCM4 was first incubated under psychological pH for a period of time (e.g. 5hr) and then was triggered with acidic pH and/or mannitol. As shown in Figure 2A, the PTX release from BCM4 was significantly slower than that from NCMs at the initial 5 h. When 100 mM mannitol was added or the pH of the medium was adjusted to 5.0 at the 5 hr time point, there was a burst of drug release from the BCM4. It should be noted that the PTX release can be further accelerated via the combination of 100 mM of mannitol and pH 5.0. This two-stage release strategy can be exploited so that premature drug release can be minimized during circulation in vivo followed by rapid drug release triggered by the acidic tumor microenvironment, or upon micelle exposure to the acidic compartments of cancer cells or by the additional administration of mannitol.
Figure 2.
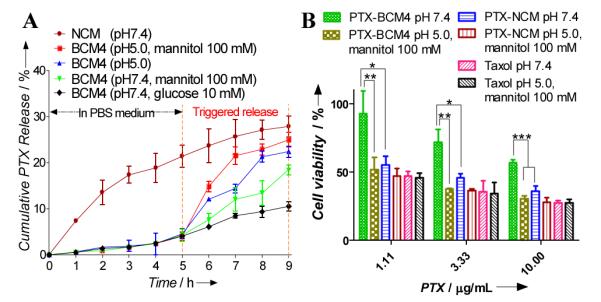
(A) pH- and diol- responsive paclitaxel (PTX) release profiles of BCM4 by treating with diols (mannitiol and glucose) and/or pH 5.0 at 5hr compared with that of NCM. (B) MTT assays showing the viability of SKOV-3 cells after 1hr incubation with Taxol®, PTX-NCM and PTX-BCM4 with or without treatment with 100 mM mannitol at pH5.0, followed by 3 times wash with PBS and additional 23hr incubation. *: p < 0.05, **: p < 0.01, ***: p < 0.001.
PEG5k-NBA4-CA8 and PEG5k-Catechol4-CA8 and empty BCM4 showed no noticeable cytotoxicity to SKOV-3 ovarian cancer cells up to 1.0 mg/mL (Figure S11). Confocal laser scanning microscopic images showed that BCM4 loaded with a hydrophobic near infrared dye (DiD) were taken up by SKOV-3 ovarian cancer cells and mainly localized in the cytoplasmic region after 1 hr incubation (Figure S12). The in vitro anticancer activity of PTX loaded non-cross-linked micelles (PTX-NCM) and PTX loaded BCM4 (PTX-BCM4) was evaluated on SKOV-3 ovarian cancer cells for 1 hr incubation followed by PBS wash and 23 hr further incubation. PTX-NCM showed comparable in vitro anti-tumor effects against SKOV-3 cells as Taxol® (free drug of paclitaxel) (Figure 2B). PTX-BCM4 was found to be considerably less cytotoxic than Taxol® and PTX-NCM at equal dose levels. This reflects the fact that less PTX exposed to cells caused by slower drug release from BCM4 within the cell culture media at pH 7.4 in the presence of 5.5 mM glucose (Figure 2B). There were minimal changes in the toxicity profile of PTX-NCM and free drug triggered with acidic pH and mannitol. In contrast, PTX-BCM4 showed significantly enhanced cancer cell inhibition at pH 5.0 in the presence of mannitol (100 mM) (Figure 2B). As described above, the combination of acidic pH and mannitol facilitated drug release because of cleavage of the crosslinking boronates of BCM4 resulting in enhanced cytotoxicity. Due to the enhanced stability, BCM4 is expected to be able to deliver higher concentration of PTX to tumor site than NCM for in vivo applications. Subsequent release on-demand of should result in better anti-tumor effects.
We then proceeded to develop a FRET system to evaluate the in vivo stability of the crosslinked micelles. FRET is a powerful technique to probe the molecular proximity of a fluorescent donor-acceptor pair [18] and for this reason FRET has been widely applied in the investigation of a variety of biological events[18]. Very recently, a few reports utilized FRET to probe the stability and drug release profile of non-crosslinked micelles[19].
We constructed the FRET reporter system by using a green dye DiO (donor) and a red-orange dye rhodamine B (acceptor) as a FRET pair to investigate the in vivo stability of NCM and BCM4 (Figure 3A, S13). DiO was encapsulated in the core of micelles as the hydrophobic drug surrogate to track the payloads. Rhodamine B was covalently conjugated to the telodendrimers to track the nanocarriers. When the 20 nm non-crosslinked FRET micelle (FRET-NCM) was intact, the proximity between DiO and rhodamine B was within the FRET range allowing efficient energy transfer from DiO to rhodamine B upon excitation of DiO at 480 nm (Figure 3C, S13). The FRET ratio (Irhodamine B/(Irhodamine B + IDiO)) was measured to be 80%, where Irhodamine B and IDiO were fluorescence intensity of rhodamine B at 580 nm and DiO at 530 nm, respectively, in PBS. Upon FRET-NCM dissociation, the FRET ratio was significantly reduced to 21% as result of the separation between DiO and rhodamine B (Figure 3C). A FRET signal was also detected from boronate crosslinked FRET micelles (FRET-BCM4, particle size: 21 nm) with an observed FRET ratio of 89% in PBS (Figure 3D). Significant DiO emission at 530 nm was observed along with dramatic reduction of rhodamine B fluorescence when FRET-BCM4 was diluted 20 times via DMSO, indicating a loss of FRET signal due to the solvation of DiO when the micelle was dissolved. Upon excitation at 480 nm, the rhodamine B signal of NCM and BCM4 labeled with rhodamine B alone was negligible in comparison with the corresponding FRET micelles (Figure S13C, S13D). Therefore, by monitoring the dynamic change of FRET ratio, we were able to monitor the stability of the micelles in real time[4b, 19a]. FRET micelles were injected into nude mice via tail vein and the blood was collected at different time points to investigate their in vivo stability by monitoring FRET efficiency. The FRET ratio of FRET-NCM decreased rapidly to 46% within 1 min post-injection and dropped to 21% after 24 min (Figure 3E, Figure S14). One possible reason is the dissociation of NCM upon extreme dilution in blood stream as the estimated concentration of micelles in blood (100 μg/mL) was close to their CMC (50.1 μg/mL, Table 1)[20]. Moreover, blood flow with a strong shear stress could mix the micelles thoroughly with blood proteins and lipoprotein nanoparticles (e.g. LDL, HDL, VLDL), leading to a fast release of core-loaded DiO dye from the circulating micelles[19a]. In contrast, FRET ratio of FRET-BCM4 decreased much slower than that of FRET-NCM at the same micelle concentration (Figure 3E, Figure S14), indicating the boronate crosslinking greatly enhanced the in vivo stability of the micelles, therefore, will decrease premature payload release.
Figure 3.

Schematic illustration of FRET-NCM in PBS (A) and in DMSO (B) at pH7.4; (C) Fluorescence emission spectra of FRET-NCM in PBS (red line) and DMSO (black line) with 480 nm excitation. (D) Emission spectra of FRET-BCM4 in PBS (red line) and DMSO (black line) with 480 nm excitation. (E)The FRET ratio (Irfhodamine B/(Irhodamine B + IDiO)) in blood of nude mice (n=3) over time after intravenous injection of 100 μL FRET-NCM and FRET-BCM4 (2.0 mg/mL).Excitation: 480 nm. (F) The fluorescence signal changes of rhodamine B conjugated NCM and BCM4 in the blood collected at different time points after intravenous injection in the nude mice (n=3). Excitation: 540 nm. (G) Ex vivo near infrared fluorescence (NIRF) images of SKOV-3 xenograft bearing mouse obtained after intravenous injection of BCM4 co-loaded with PTX and DiD.
Rhodamine B labeled NCM and BCM4 were used to study the in vivo blood elimination kinetics of the micelles (Figure S15). After intravenous injection into mice, rhodamine B signal of NCM was rapidly eliminated from blood circulation and fell into the background level within 10 hr post injection (Figure 3F). Rhodamine B signal of BCM4 in blood was 6 times higher than that of NCM at 10 hr post injection and sustained for more than 24 hr. The above profiles of elimination kinetics indicated that the cross-linked micelles have longer blood circulation time than the non-cross-linked micelles. We further demonstrated DiD and PTX co-loaded BCM4 to be able to preferentially accumulate in SKOV-3 ovarian tumor (Figure S16). Ex vivo imaging at 32 hr post injection confirmed the preferential uptake of BCM4 in tumor compared to normal organs (Figure 3G). This is due to the prolonged in vivo circulation time of the micelles and the size-mediated EPR effect.
In summary, we reported the design and synthesis of a novel class of dual pH- and diol-responsive crosslinked micelles formed by well-defined telodendrimers containing boronic acid and catechol respectively. By tuning the pKa and numbers of boronic acids and catechols in the telodendrimers, we have optimized the stability of the resulting boronate crosslinked micelles as well as their stimuli-response to cis-diols and acidic pH. The release of PTX from the boronate cross-linked micelles was significantly slower than that from non-cross-linked micelles but can be accelerated by the acidic pH and/or mannitol. We developed a highly efficient FRET system and further demonstrated that BCMs exhibited enhanced in vivo stability. This novel nano-carrier platform shows great promise for drug delivery with minimal premature drug release at physiological glucose level (2-10 mM) and physiological pH values (pH 7.4) in blood circulation but can be activated to release drug on demand at acidic tumor microenviroment or in the acidic cellular compartments upon uptake in target tumor cells, and/or by the additional intravenous administration of mannitol as triggering agent.
Supplementary Material
Footnotes
This work was funded by NIH/NCI (R01CA115483 and R01CA140449), NIH/NIBIB (R01EB012569) and DoD BCRP Postdoctoral Award (W81XWH-10-1-0817).
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Dr. Yuanpei Li, Department of Biochemistry and Molecular Medicine, UC Davis Cancer Center, University of California, Davis 2700 Stockton Blvd., Sacramento, California 95817, USA Yuanpei.Li@ucdmc.ucdavis.edu.
Dr. Wenwu Xiao, Department of Biochemistry and Molecular Medicine, UC Davis Cancer Center, University of California, Davis 2700 Stockton Blvd., Sacramento, California 95817, USA.
Dr. Kai Xiao, Department of Biochemistry and Molecular Medicine, UC Davis Cancer Center, University of California, Davis 2700 Stockton Blvd., Sacramento, California 95817, USA
Dr. Lorenzo Berti, Department of Biochemistry and Molecular Medicine, UC Davis Cancer Center, University of California, Davis 2700 Stockton Blvd., Sacramento, California 95817, USA
Prof. Juntao Luo, Department of Pharmacology, SUNY Upstate Cancer Research Institute, SUNY Upstate Medical University, Syracuse, NY 13210, USA.
Harry P. Tseng, Department of Biochemistry and Molecular Medicine, UC Davis Cancer Center, University of California, Davis 2700 Stockton Blvd., Sacramento, California 95817, USA
Gabriel Fung, Department of Biochemistry and Molecular Medicine, UC Davis Cancer Center, University of California, Davis 2700 Stockton Blvd., Sacramento, California 95817, USA.
Prof. Kit S. Lam, Department of Biochemistry and Molecular Medicine, UC Davis Cancer Center, University of California, Davis 2700 Stockton Blvd., Sacramento, California 95817, USA.
References
- [1] a).Zhou K, Wang Y, Huang X, Luby-Phelps K, Sumer BD. J. Gao, Angew Chem. 2011;123:6233–6238. doi: 10.1002/anie.201100884. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:6109–6114. doi: 10.1002/anie.201100884. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee Y, Ishii T, Cabral H, Kim HJ, Seo JH, Nishiyama N, Oshima H, Osada K, Kataoka K. Angew Chem. 2009;121:5413–5416. doi: 10.1002/anie.200900064. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:5309–5312. doi: 10.1002/anie.200900064. [DOI] [PubMed] [Google Scholar]; c) Du JZ, Sun TM, Song WJ, Wu J, Wang J. Angew Chem. 2010;122:3703–3708. [Google Scholar]; Angew Chem Int Ed. 2010;49:3621–3626. doi: 10.1002/anie.200907210. [DOI] [PubMed] [Google Scholar]; d) Xu P, Van Kirk EA, Zhan Y, Murdoch WJ, Radosz M, Shen Y. Angew Chem. 2007;119:5087–5090. doi: 10.1002/anie.200605254. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:4999–5002. doi: 10.1002/anie.200605254. [DOI] [PubMed] [Google Scholar]; e) Lee ES, Na K, Bae YH. Nano Lett. 2005;5:325–329. doi: 10.1021/nl0479987. [DOI] [PubMed] [Google Scholar]
- [2] a).Li Y, Pan S, Zhang W, Du Z. Nanotechnology. 2009;20:065104. doi: 10.1088/0957-4484/20/6/065104. [DOI] [PubMed] [Google Scholar]; b) Chaikof EL, Kim W, Thevenot J, Ibarboure E, Lecommandoux S. Angew Chem. 2010;122:4353–4356. doi: 10.1002/anie.201001356. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:4257–4260. doi: 10.1002/anie.201001356. [DOI] [PubMed] [Google Scholar]; c) Rijcken CJ, Snel CJ, Schiffelers RM, van Nostrum CF, Hennink WE. Biomaterials. 2007;28:5581–5593. doi: 10.1016/j.biomaterials.2007.08.047. [DOI] [PubMed] [Google Scholar]
- [3] a).Ulijn RV, Thornton PD, Mart RJ. Adv Mater. 2007;19:1252–1256. [Google Scholar]; b) Law B, Weissleder R, Tung CH. Bioconjug Chem. 2007;18:1701–1704. doi: 10.1021/bc070054z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4] a).Meng FH, Li YL, Zhu L, Liu ZZ, Cheng R, Cui JH, Ji SJ, Zhong ZY. Angew Chem. 2009;121:10098–10102. doi: 10.1002/anie.200904260. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9914–9918. doi: 10.1002/anie.200904260. [DOI] [PubMed] [Google Scholar]; b) Li Y, Xiao K, Luo J, Xiao W, Lee JS, Gonik AM, Kato J, Dong TA, Lam KS. Biomaterials. 2011;32:6633–6645. doi: 10.1016/j.biomaterials.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5] a).Lee SC, Koo AN, Lee HJ, Kim SE, Chang JH, Park C, Kim C, Park JH. Chem Commun. 2008:6570–6572. doi: 10.1039/b815918a. [DOI] [PubMed] [Google Scholar]; b) Hennink WE, Talelli M, Iman M, Varkouhi AK, Rijcken CJF, Schiffelers RM, Etrych T, Ulbrich K, van Nostrum CF, Lammers T, Storm G. Biomaterials. 2010;31:7797–7804. doi: 10.1016/j.biomaterials.2010.07.005. [DOI] [PubMed] [Google Scholar]
- [6].Xing MMQ, Lu CH, Zhong W. Nanomed-Nanotechnol. 2011;7:80–87. [Google Scholar]
- [7] a).Wei C, Guo J, Wang C. Macromol Rapid Commun. 2011;32:451–455. doi: 10.1002/marc.201000708. [DOI] [PubMed] [Google Scholar]; b) Ma N, Li Y, Xu H, Wang Z, Zhang X. J Am Chem Soc. 2010;132:442–443. doi: 10.1021/ja908124g. [DOI] [PubMed] [Google Scholar]; c) Dai J, Lin S, Cheng D, Zou S, Shuai X. Angew Chem. 2011;123:9576–9580. doi: 10.1002/anie.201103806. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:9404–9408. doi: 10.1002/anie.201103806. [DOI] [PubMed] [Google Scholar]
- [8] a).Springsteen G, Wang BH. Tetrahedron. 2002;58:5291–5300. [Google Scholar]; b) Zhu L, Shabbir SH, Gray M, Lynch VM, Sorey S, Anslyn EV. J Am Chem Soc. 2006;128:1222–1232. doi: 10.1021/ja055817c. [DOI] [PubMed] [Google Scholar]; c) Qin Y, Sukul V, Pagakos D, Cui CZ, Jakle F. Macromolecules. 2005;38:8987–8990. [Google Scholar]; Sumerlin dB. S., Roy D, Cambre JN. Chem Commun. 2008:2477–2479. doi: 10.1039/b802293c. [DOI] [PubMed] [Google Scholar]
- [9] a).Li S, Davis EN, Anderson J, Lin Q, Wang Q. Biomacromolecules. 2009;10:113–118. doi: 10.1021/bm8009768. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cannizzo C, Amigoni-Gerbier S, Larpent C. Polymer. 2005;46:1269–1276. [Google Scholar]; c) van Hest JCM, Kim KT, Cornelissen JJLM, Nolte RJM. J Am Chem Soc. 2009;131:13908–13909. doi: 10.1021/ja905652w. [DOI] [PubMed] [Google Scholar]
- [10].van Hest JCM, Kim KT, Cornelissen JJLM, Nolte RJM. Adv Mater. 2009;21:2787–2791. [Google Scholar]
- [11] a).Wu W, Zhang LZ, Lin Y, Wang JJ, Yao W, Jiang XQ. Macromol Rapid Comm. 2011;32:534–539. doi: 10.1002/marc.201000757. [DOI] [PubMed] [Google Scholar]; b) Shi LQ, Wang BL, Ma RJ, Liu G, Li Y, Liu XJ, An YL. Langmuir. 2009;25:12522–12528. doi: 10.1021/la901776a. [DOI] [PubMed] [Google Scholar]; c) Zhao Y, Trewyn BG, Slowing, Lin VS. J Am Chem Soc. 2009;131:8398–8400. doi: 10.1021/ja901831u. [DOI] [PubMed] [Google Scholar]
- [12].He L, Fullenkamp DE, Rivera JG, Messersmith PB. Chem Commun. 2011;47:7497–7499. doi: 10.1039/c1cc11928a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Marken F, Huang YJ, Jiang YB, Fossey JS, James TD. J Mater Chem. 2010;20:8305–8310. [Google Scholar]
- [14] a).Luo J, Xiao K, Li Y, Lee JS, Shi L, Tan YH, Xing L, Cheng R. Holland, Liu GY, Lam KS. Bioconjug Chem. 2010;21:1216–1224. doi: 10.1021/bc1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao K, Luo J, Fowler WL, Li Y, Lee JS, Xing L, Cheng RH, Wang L, Lam KS. Biomaterials. 2009;30:6006–6016. doi: 10.1016/j.biomaterials.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li Y, Xiao K, Luo J, Lee J, Pan S, Lam KS. J Control Release. 2010;144:314–323. doi: 10.1016/j.jconrel.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS. Biomaterials. 2011;32:3435–3446. doi: 10.1016/j.biomaterials.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mulla HR, Agard NJ, Basu A. Bioorg Med Chem Lett. 2004;14:25–27. doi: 10.1016/j.bmcl.2003.10.017. [DOI] [PubMed] [Google Scholar]
- [16].Springsteen G, Wang B. Chem Commun. 2001:1608–1609. doi: 10.1039/b104895n. [DOI] [PubMed] [Google Scholar]
- [17].Koo AN, Lee HJ, Kim SE, Chang JH, Park C, Kim C, Park JH, Lee SC. Chem Commun. 2008:6570–6572. doi: 10.1039/b815918a. [DOI] [PubMed] [Google Scholar]
- [18] a).Jares-Erijman EA, Jovin TM. Nat Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]; b) Sekar RB, Periasamy A. J Cell Biol. 2003;160:629–633. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sapsford KE, Berti L, Medintz IL. Angew Chem. 2006;118:4676–4704. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:4562–4589. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- [19] a).Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng JX. Langmuir. 2008;24:5213–5217. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]; b) Chen KJ, Chiu YL, Chen YM, Ho YC, Sung HW. Biomaterials. 2011;32:2586–2592. doi: 10.1016/j.biomaterials.2010.11.069. [DOI] [PubMed] [Google Scholar]; c) Park K, Chen HT, Kim SW, Li L, Wang SY, Cheng JX. P Natl Acad Sci USA. 2008;105:6596–6601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cheng JX, Kim S, Shi YZ, Kim JY, Park K. Expert Opin Drug Del. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.