

Abstract
Background
Pulmonary artery hypertension (PAH) is a proliferative disorder associated with enhanced pulmonary artery smooth muscle cell proliferation and suppressed apoptosis. The sustainability of this phenotype required the activation of a prosurvival transcription factor like signal transducers and activators of transcription-3 (STAT3) and nuclear factor of activated T cell (NFAT). Because these factors are implicated in several physiological processes, their inhibition in PAH patients could be associated with detrimental effects. Therefore, a better understanding of the mechanism accounting for their expression/activation in PAH pulmonary artery smooth muscle cells is of great therapeutic interest.
Methods and Results
Using multidisciplinary and translational approaches, we demonstrated that STAT3 activation in both human and experimental models of PAH accounts for the expression of both NFATc2 and the oncoprotein kinase Pim1, which trigger NFATc2 activation. Because Pim1 expression correlates with the severity of PAH in humans and is confined to the PAH pulmonary artery smooth muscle cell, Pim1 was identified as an attractive therapeutic target for PAH. Indeed, specific Pim1 inhibition in vitro decreases pulmonary artery smooth muscle cell proliferation and promotes apoptosis, all of which are sustained by NFATc2 inhibition. In vivo, tissue-specific inhibition of Pim1 by nebulized siRNA reverses monocrotaline-induced PAH in rats, whereas Pim1 knockout mice are resistant to PAH development.
Conclusion
We demonstrated for the first time that inhibition of the inappropriate activation of STAT3/Pim1 axis is a novel, specific, and attractive therapeutic strategy to reverse PAH.
Keywords: apoptosis, hypertension, pulmonary, proliferation
Pulmonary artery (PA) hypertension (PAH) is a proliferative vascular remodeling disease characterized by enhanced inflammation,1 vasoconstriction, and PA smooth muscle cell (PASMC) proliferation and resistance to apoptosis.2 This leads to an increase in pulmonary vascular resistance and eventually to right heart failure.3 The sustainability of this proproliferative and antiapoptotic phenotype is due in part to the activation of a transcription factor, nuclear factor of activated T cells (NFAT).4,5 Nonetheless, the mechanism by which NFAT expression and activation are increased remains elusive.
The signal transducers and activators of transcription (STAT) protein family regulates diverse cellular processes, including growth and survival, and is frequently deregulated in cancer. The family is composed of 7 isoforms (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6), with STAT3 being the most important in cardiovascular diseases.6,7 STATs are activated (ie, phosphorylated [P-STAT] and translocated to the nucleus) in response to cytokines (such as interleukin-6),8 growth factors (such as platelet-derived growth factor),8 or agonists (such as endothelin-1 and angiotensin II),9 all of which are implicated in PAH.10–12 Interestingly, several STAT binding sites have been identified within the promoter regions13 of NFATc1, NFATc2, and NFATc3 (the 3 major NFAT isoforms implicated in PAH5). Although the role of STAT3 in PAH has been suggested,14 its exact function and its effects on NFAT remain to be established.
In cancer, STAT3 promotes the expression of the provirus integration site for Moloney murine leukemia virus (Pim1), a proto-oncogene encoding a serine/threonine protein kinase.15 Overexpression of Pim1 is linked to the development and progression of several cancers by increasing cell proliferation/survival and resistance to apoptosis.16–18 Finally, Pim1 activation enhanced NFATc1 through NFATc4 activity in rat PC12 cells and lymphoid cells,19,20 suggesting a potential role for Pim1 in PAH.
The present study aimed to elucidate the role of STAT3 and Pim1 in the origin of PAH, particularly with regard to the sustainability of the proproliferative and antiapoptotic phenotype mediated by NFAT.
Methods
The online-only Data Supplement gives details on all methods. For human tissue samples, see Table I in the online-only Data Supplement. For supplies and chemicals, see Table II in the online-only Data Supplement.
All human tissues were obtained from the Laval Hospital tissue bank. PAH tissues were from 8 subjects with nonfamilial PAH, all classified within the group 1 of the latest World Health Organization classification with a mean PA pressure >25 mm Hg. Normal lung tissues were obtained from 8 individuals without PAH undergoing lung resection for benign nodules (n=3) and malignant tumor (n=5). Buffy coat samples were obtain from 7 healthy donors, 13 nonfamilial PAH patients (9 idiopathic PAH and 4 scleroderma-PAH), and 7 patients with scleroderma. All experiments were performed in accordance with Laval University and Laval Hospital biosafety and ethics committee (protocol 20142). All patients gave informed consent before the study.
Cell Culture
For all PASMCs (control and PAH), we used cells in the fourth to sixth passage. PAH PASMCs were isolated from <1500-μm-diameter small pulmonary arteries from 3 male patients with PAH defined by a mean PA pressure >25 mm Hg (Table I in the online-only Data Supplement) as previously described.21 Control PASMCs from 5 patients were purchased (Cell Application Group, San Diego, CA). See the Methods section in the online-only Data Supplement.
siRNA and Adenovirus Efficiencies
siRNA and adenovirus efficiencies were evaluated by both quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and immunoblot (see Figure XIC in the online-only Data Supplement).
Statistics
Averaged data are mean±SEM. Normality of our data was assessed by the Shapiro-Wilk normality test. For comparisons between 2 means, we used unpaired Student t test. For comparison between >2 means, we used 1-way ANOVA followed by the Dunn test. For correlation studies, Spearman 2-tailed test was used. In cultured cell– based experiment, n indicates the number of patients; in the in vivo studies, n indicates the number of animals. For survival analysis, a cohort of 20 knockout (KO) and 20 wild-type (WT) mice exposed to chronic hypoxia (CH) or injected with pyrrole-modified monocrotaline (MCTP) were prospectively followed up for 25 days and euthanized after hemodynamic studies on day 25 after MCTP injection or CH exposure. Spontaneous deaths were counted as events, and mice undergoing planed hemodynamic studies and euthanasia were censored. Time-to-event data were plotted with the Kaplan-Meier method, with differences evaluated through the use of the log-rank test. Values of P<0.05 were considered significant.
Results
STAT3 Activation Is Increased in Human PAH PASMCs
STAT3 activation (nuclear translocation of Y705-phosphorylated STAT3) was measured in whole PAs of 8 PAH patients and 8 healthy donors (Table I in the online-only Data Supplement) and in PASMCs isolated from 8 individuals (3 PAH and 5 healthy donors). Compared with control, the percentage of cells presenting a colocalization between P-STAT3 and DAPI was significantly increased in PAs from PAH patients and in PAH PASMCs (Figure 1A). This finding was confirmed in PASMCs by immunoblots measuring the PY705-STAT3/STAT3 ratio normalized to smooth muscle actin (Figure 1A).
Figure 1.

The signal transducers and activators of transcription-3 (STAT3)/Pim1 axis is activated in human pulmonary artery (PA) hypertension (PAH). A, STAT3 activation measured by PY705-STAT3 nuclear localization is significantly increased in both whole PAs and PA smooth muscle cells (SMCs) isolated from PAH patients compared with control. This was confirmed by immunoblots showing a 2.8-fold increase in the PY705-STAT3/STAT3 ratio in PAH PASMCs compared with control PASMCs. B, Pim1 mRNA levels measured by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) are significantly increased in both lungs and PASMCs isolated from PAH patients compared with control patients. This was confirmed by immunoblots in isolated PASMCs. Finally, Pim1 mRNA levels in lungs correlate with PAH severity assessed by pulmonary vascular resistance (PVR). C, PAH PASMCs treated with siSTAT3 had a significant reduction in Pim1 expression (qRT-PCR) compared with the scrambled-treated cells. Endothelin-1– (ET1; 10 nmol/L), angiotensin II– (AngII; 200 nmol/L), platelet-derived growth factor– (PDGF; 30 ng/mL), and tumor necrosis factor– (TNF; 100 ng/mL) treated healthy PASMCs had an increased PY705-STAT3/STAT3 ratio associated with a proportional increase in Pim1 expression measured by immunoblots (P<0.05 vs control group).
Pim1 Expression Is Increased in Human PAH PASMCs and Correlates With PAH Severity
To investigate the role of Pim1 in PAH, Pim1 expression was measured in both lungs (from 8 PAH and 8 control patients) and PASMCs isolated from distal PAs (3 PAH and 5 control patients). Compared with control, 8-fold and 2.5-fold increases in Pim1 mRNA levels (qRT-PCR) were observed in PAH lung tissue and PAH PASMCs, respectively (Figure 1B). This was confirmed at the protein level in distal PAs by immunofluorescence (Figure IA in the online-only Data Supplement) in lung sections with Pim1 in green, smooth muscle actin in red, and nucleus in blue (DAPI). As shown, green fluorescence was significantly increased in PAH. In addition, the rise in Pim1 was localized mainly within the PASMCs as shown by a greater colocalization between smooth muscle actin and Pim1, giving a yellow staining (Figure IA in the online-only Data Supplement). This finding was confirmed in isolated PASMCs with immunoblots (Figure 1B) showing a 2-fold increase in Pim1 protein expression in PAH PASMCs compared with control.
To determine whether Pim1 expression correlates with PAH severity, Pim1 mRNA expression was measured in human lungs with various degrees of PAH, evaluated by pulmonary vascular resistance. As shown, Pim1 mRNA levels correlate positively with pulmonary vascular resistance (Figure 1B). Because PAH is characterized by increased circulating levels of activated T cells5 and endothelial progenitor cells22,23 in which Pim1 is expressed,24,25 Pim1 expression could be used as a PAH marker. Thus, Pim1 mRNA levels were measured in human buffy coat (containing both T cells and endothelial progenitor cells) isolated from healthy donors, patients with idiopathic PAH, patients with scleroderma only, and patients with PAH associated with scleroderma (Table I in the online-only Data Supplement). As in lungs and PAH PASMCs, Pim1 expression was significantly increased in buffy coats of patients with PAH associated or not with scleroderma (Figure IB in the online-only Data Supplement). As in lungs, Pim1 mRNA levels in buffy coats correlate with PAH severity evaluated by both the mean of PA pressure and pulmonary vascular resistance (Figure IB in the online-only Data Supplement). These results further support a role for Pim1 in PAH and suggest that Pim1 may be a good biomarker for the disease.
STAT3 Activation in Human PAH PASMCs Triggers Pim1 Expression
To investigate whether STAT3 plays a role in the regulation of Pim1 in PAH, Pim1 mRNA levels were measured in PAH and control PASMCs in the presence or absence of STAT3 siRNA (siSTAT3). Compared with the scrambled siRNA, siSTAT3 significantly reduced Pim1 expression in PAH PASMCs (Figure 1C), suggesting that STAT3 activation regulates Pim1 expression in PAH PASMCs. To confirm this finding, healthy PASMCs were treated for 48 hours with factors known to activate STAT3 and that are increased in PAH10–12 such as endothelin-1, angiotensin II, platelet-derived growth factor, and tumor necrosis factor. As expected, these factors increase STAT3 activation measured by immunoblots (PY705-STAT3/STAT3 ratio) by 2.8-, 3.2-, 2.5-, and 1.9-fold in endothelin-1–, angiotensin II–, platelet-derived growth factor–, and tumor necrosis factor–treated PASMCs, respectively (Figure 1C), which promoted Pim1 expression (Figure 1C). To determine whether STAT3 binds directly to the Pim1 gene and activates its transcription, we performed an in silico analysis using the ENCODE consortium database.26 This analysis revealed the presence of several STATs binding sites in 3′ of the Pim1 gene, conserved among several species (Figure IIA in the online-only Data Supplement). Chromatin immunoprecipitation coupled with PCR experiments were performed to demonstrate the direct binding of STAT3 in 3′ of the Pim1 gene. The VEGF gene was used as a positive control (with established STAT3 binding sites13), whereas the OR8J1 gene was used as negative control (without STAT3 binding sites13). As predicted in silico, we showed that STAT3 binds directly to Pim1, whereas no bindings were indentified with OR8J1. On the basis of our positive control VEGF, the interaction between STAT3 and Pim1 (which showed greater binding signal) can be considered strong (Figure IIB in the online-only Data Supplement). Among other putative Pim1 activators, STAT1, STAT5,8 and Akt27,28 did not appear to be activated in PAH PASMCs (Figure IIIA in the online-only Data Supplement). Therefore, STAT3 is likely responsible for Pim1 activation in PAH.
Pim1 Promotes Proliferation and Resistance to Mitochondria-Dependent Apoptosis in PAH PASMCs
We treated PAH PASMCs with either Pim1 or STAT3 siRNA and their control-scrambled siRNA and STAT3 inhibitor peptide (50 μmol/L) or its negative control. Compared with healthy PASMCs, PAH PASMCs had a greater proliferation rate (increased percent of proliferating cell nuclear antigen–positive cells) and were resistant to starvation (0.1% FBS)-induced apoptosis (decreased percent of terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling [TUNEL]–positive cells; Figure 2A). Similar to STAT3 inhibition (siRNA and inhibitor peptide), Pim1 inhibition (siRNA) in starved PAH PASMCs increased apoptosis by 8.2-fold, whereas PAH PASMC (10% FBS) proliferation was decreased by 2.4-fold (Figure 2A). These findings were confirmed in healthy PASMCs stimulated for 48 hours with platelet-derived growth factor (30 ng/mL) or endothelin-1 (10 nmol/L; Figure IVA in the online-only Data Supplement), both known proproliferative factors.29–31
Figure 2.

Pim1 is implicated in the regulation of both pulmonary artery (PA) smooth muscle cell (PASMC) proliferation and apoptosis. A, PA hypertension (PAH) PASMCs had increased proliferation (percent proliferating cell nuclear antigen [PCNA]) and decreased apoptosis (percent terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling [TUNEL]) compared with healthy PASMCs. Both signal transducers and activators of transcription-3 inhibition and Pim1 inhibition significantly decrease PAH PASMC proliferation and promote apoptosis. B, Pim1 inhibition in PAH PASMCs reverses ΔΨm hyperpolarization (tetramethylrhodamine methyl-ester [TMRM]), whereas Pim1 activation in healthy PASMCs with adenovirus overexpressing Pim1 promotes it. C, Pim1 inhibition in PAH PASMCs decreases the PS112-Bad/Bad ratio, allowing Bad translocation to the mitochondria (increased colocalization between Bad in green and mitochondria [mitotracker red[ in red giving a yellow pattern).
Pim1 is implicated in mitochondrial membrane potential (ΔΨm) regulation32,33; thus, the increase in apoptosis after Pim1 inhibition might result from the activation of mitochondria-dependent apoptosis. Because the mitochondria transition pore is voltage dependent,34 ΔΨm depolarization is an index of the threshold for mitochondria-dependent apoptosis, and apoptosis is associated with decreased ΔΨm. Using tetramethylrhodamine methyl ester, we measured ΔΨm in PAH PASMCs transfected with Pim1 siRNA and in control PASMCs infected by an adenovirus carrying the Pim1 gene (Ad-green fluorescent protein [GFP]-Pim1-WT). Transfections of PAH PASMCs with Pim1 siRNA caused significant ΔΨm depolarization compared with control scrambled-treated PAH PASMCs. In contrast, infection of control PASMCs with Ad-GFP-Pim1-WT caused a significant ΔΨm hyperpolarization compared with Ad-GFP–treated control PASMCs (Figure 2B). These data confirmed the role of Pim1 in ΔΨm regulation in PAH PASMCs and thus mitochondria-dependent apoptosis. Similar results were obtained with the STAT3 peptide inhibitor (Figure IVB in the online-only Data Supplement). Note that Pim1 inhibition has no effect on control PASMCs (data not shown).
Previous studies revealed that Pim1 inhibits the proapoptotic protein Bad (increased P-Bad), blocking its translocation to the mitochondria and causing ΔΨm hyperpolarization.35 In PAH PASMCs, Pim1 inhibition decreases the P-Bad/total Bad ratio (Figure 2C), promoting Bad (green) mitochondrial (stained with mitotracker red) translocation (greater colocalization in yellow). Control PASMCs infected with Ad-GFP-Pim1-WT showed the opposite effects (Figure 2C). These findings confirmed the implication of Pim1 in the resistance to mitochondria-dependent apoptosis seen in PAH PASMCs.
STAT3 Regulates Pim1 and NFATc2 Expression, and Pim1 Regulates NFATc2 Activation in PAH PASMC
We previously demonstrated that the sustainability of the proproliferative and antiapoptotic phenotype seen in PAH PASMCs is attributed in part to the activation of the transcription factor NFATc2. In PAH, NFATc2 activation leads to the downregulation of important K+ channels like Kv1.5, leading to PASMC depolarization, increasing [Ca2+]I, and promoting PASMC proliferation.4,5,36 NFATc2 activation also triggers the upregulation of Bcl-2, leading to mitochondrial hyperpolarization and apoptosis inhibition.4,5,36 Pim1 is a NFAT activator; thus, the Pim1-dependent NFAT activation could be implicated in PAH. NFATc2 expression (qRT-PCR) and activation (luciferase assay) are increased in PAH PASMCs compared with control PASMCs (Figure 3A). PAH PASMCs treated with siSTAT3 showed a significant decrease in both NFATc2 mRNA and activation levels (Figure 3A), whereas PAH PASMCs treated with siPim1 revealed only a decrease in NFATc2 activation level (similar to that induced by siSTAT3) but not in NFATc2 mRNA level (Figure 3A). This last finding was further confirmed with the NFATc2 nuclear translocation assay. As shown, Pim1 inhibition significantly decreased the amount of NFATc2 (green) colocalizing with DAPI (blue), reducing the yellow staining (Figure V in the online-only Data Supplement). Taken together, these findings suggest that STAT3 activation accounts for NFATc2 expression, whereas Pim1 is required only for the activation of NFAT in PAH PASMCs. These results were further confirmed with the Pim1 inhibitor quercetagetine37 and Pim1 dominant-negative adenovirus (Figure 3A). Quercetagetine mimics Pim1 siRNA (decreases NFAT activation) in PAH PASMCs, whereas overexpression of Pim1 by Ad-GFP-Pim1-WT in control PASMCs enhanced NFAT activation (Figure 3A).
Figure 3.

The signal transducers and activators of transcription-3 (STAT3)/Pim1 axis accounts for nuclear factor of activated T cell-c2 (NFATc2) expression and activation in pulmonary artery hypertension (PAH) pulmonary artery smooth muscle cells (PASMCs). A, NFATc2 expression is increased in PAH PASMCs compared with control PASMCs. STAT3 inhibition decreases NFATc2 expression, whereas Pim1 inhibition has no effects. NFATc2 activation (luciferase assay) is increased in PAH PASMCs compared with control PASMCs. Pim1 inhibition decreases NFAT activation, whereas Pim1 overexpression in control PASMC promotes it. B, Pim1 inhibition in PAH PASMC decreases [Ca2+]i (FLUO3) and (C) Bcl-2 expression (immunofluorescence).
Finally, we provide further evidence that the STAT3/ Pim1-dependent NFAT activation in PAH PASMCs contributes to the proproliferative and antiapoptotic phenotype by increasing [Ca2+]i (Figure 3B), a well-accepted proproliferative factor, and the apoptotic factor Bcl-2 compared with normal PASMCs (Figure 3C). Note that Pim1 inhibition has no effects on [Ca2+]i in control PASMC (data not shown).
STAT3/ Pim1/NFATc2 Axis Is Increased in Rat Experimental PAH
Pim1 expression (qRT-PCR, immunoblots, and immunofluorescence) was quantified in both lungs and PAs isolated from monocrotaline (MCT)-injected rats, an accepted model of PAH.5 Greater Pim1 expression was seen in PAs of rats with severe PAH (>21 days after MCT injection). In contrast, light expression was seen in partially remodeled PAs, and no expression was seen in nonremodeled PAs from rats with mild early PAH (1 to 12 days after MCT; Figure 4A and Figure VIA and VIC in the online-only Data Supplement). As in humans, Pim1 expression is confined within the PASMCs (greater colocalization between Pim1 in green and smooth muscle-actin in red in Figure VIA in the online-only Data Supplement than Pim1 with the endothelial cell marker VE-cadherin in Figure VIB in the online-only Data Supplement) and correlates with PAH severity (Figure 4A). Moreover, Pim1 mRNA levels were measured in rat buffy coats isolated from control rats (mean PA pressure <15 mm Hg), rats with mild PAH (mean PA pressure <30 mm Hg), and rats with severe PAH (mean PA pressure >30 mm Hg). As in distal PAs, Pim1 mRNA levels in buffy coats correlate with PAH severity (Figure VIIA in the online-only Data Supplement). These results further support a role for Pim1 in PAH and suggest that Pim1 may be a good biomarker for the disease.
Figure 4.
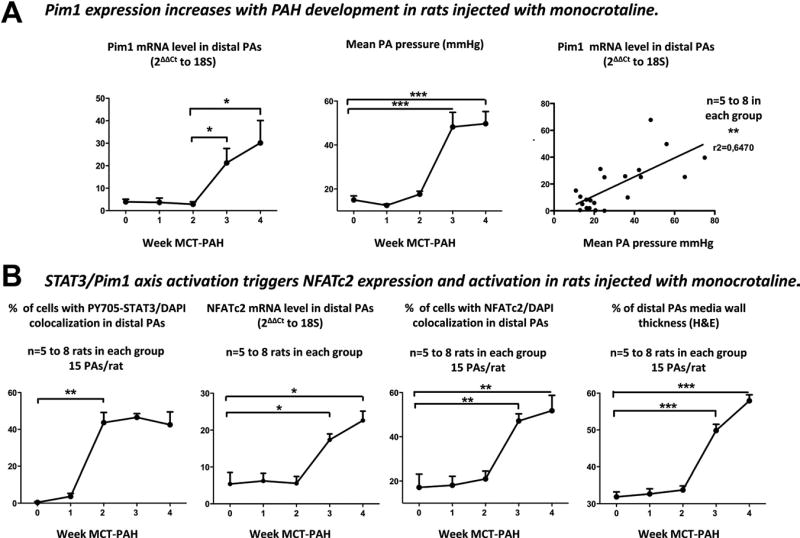
Pim1 expression is increased in the monocrotaline (MCT)-injected rat pulmonary artery (PA) hypertension (PAH) model. A, Heavy Pim1 mRNA expression is seen in PAs of rats with severe PAH (>21 days after MCT injection). In contrast, light expression was seen in mild PAH, and no expression was seen in rats with early PAH. Moreover, Pim1 expression correlates with PAH severity assed by mean PA pressure. B, Signal transducers and activators of transcription-3 (STAT3) activation (nuclear translocation of PY705-STAT3) precedes both Pim1 expression (quantitative reverse-transcription polymerase chain reaction [qRT-PCR]) and nuclear factor of activated T cell-c2 (NFATc2) expression (qRT-PCR) and activation (nuclear translocation), which paralleled both increased PA wall remodeling (hematoxylin and eosin staining) and PA pressure.
Finally, the fact that Pim1 is poorly expressed in other tissues like aorta, kidney, and liver (Figure VIIB in the online-only Data Supplement) makes it an attractive therapeutic target for PAH. To further study the implication of the STAT3/Pim1/NFAT axis activation in the progression of PAH, rats were euthanized at different time points after the subcutaneous injection of MCT. PA pressure and PA wall thickness (hematoxylin and eosin staining) were measured. STAT3 activation (P-STAT3 nuclear translocation in Figure 4B and P-STAT3/STAT3 ratio by immunoblots in Figure VIC in the online-only Data Supplement) increased between weeks 1 and 2 after MCT injection preceding both Pim1 expression and NFAT activation (qRT-PCR and NFATc2 nuclear translocation in Figure 4A and 4B and immunoblots in Figure VIC in the online-only Data Supplement), which increased between weeks 2 and 3 after MCT injection, paralleling the increase in both PA wall remodeling and PA pressure (Figure 4A and 4B). Although these findings do not demonstrate a direct causal link between STAT3 activation and Pim1/NFAT, they confirm the implication of the STAT3/Pim1/NFATc2 in PAH development. These results confirm the implication of a STAT3-dependent activation of Pim1 in the origin of PAH.
Pim1 Inhibition Reverses MCT PAH
Pim1 siRNA (1 nmol) was selectively delivered to the lung of MCT-PAH rats 18 days after MCT injection (when endogenous expression of Pim1 peaks) by intratracheal nebulization. To verify the tissue distribution of our treatment, we nebulized an adenovirus carrying GFP (Figure VIIIA in the online-only Data Supplement) and measured the effect of Pim1 inhibition in several tissues (Figure VIIIB in the online-only Data Supplement). Both techniques revealed that Pim1 inhibition by nebulized siRNA is localized to the PAs, therefore having limited detrimental effect. After confirming that Pim1 siRNA-treated MCT PAH rats had decreased Pim1 expression in both PAs and lungs (qRT-PCR) (Figure VIIIB in the online-only Data Supplement), we showed, using noninvasive measurements (Doppler and echocardiography),5 that Pim1 inhibition increases PA acceleration time and decreases right ventricular wall thickness compared with the scrambled-treated MCT PAH rats (Figure VIIIC in the online-only Data Supplement). These findings were confirmed invasively by PA pressure, PA wall remodeling, right ventricular hypertrophy (right ventricular/left ventricle septum), and general cardiac functions (exercise capacity on treadmill) measurements (Figure 5A).
Figure 5.
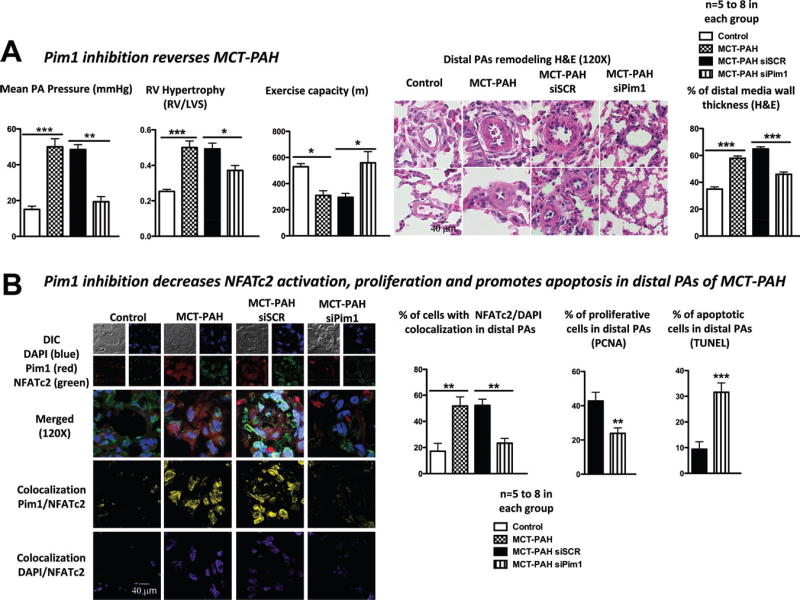
Pim1 inhibition reverses monocrotaline (MCT)–pulmonary artery (PA) hypertension (PAH). A, Pim1 inhibition (1 nebulization of 1 nmol) 18 days after MCT injection decreased mean PA pressure and right ventricular (RV) hypertrophy, improved exercise capacity, and diminished distal PA wall thickness. B, As in vitro, these effects were mediated by the inhibition of Pim1-dependent nuclear factor of activated T cell-c2 (NFATc2) activation showed by a decrease in Pim1/NFATc2 (yellow) and in NFATc2/DAPI (purple) colocalization, decreasing PA smooth muscle cell (PASMC) proliferation (percent proliferating cell nuclear antigen [PCNA]) and increasing apoptosis (percent terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling [TUNEL]).
This finding was associated with a significant decrease in Pim1-dependent NFATc2 activation (decreased Pim1/ NFATc2 [yellow] and NFATc2/DAPI [purple] colocalization), decreasing PASMC proliferation (percent proliferating cell nuclear antigen) and resistance to apoptosis (percent TUNEL; Figure 5B).
Pim1 Knockout Mice Are Resistant to PAH
PAH was induced in mice by intravenous injection (5 mg/kg) of MCTP and by exposure to CH for 15 days (10% O2) as previously described.38 As in rats, the development of PAH was assessed both noninvasively and invasively. Compared with Pim1 KO mice, WT mice injected with MCTP or exposed to CH developed PAH (decreased PA acceleration time and increased right ventricular hypertrophy, PA wall thickness, PA pressure, and mortality) without affecting systemic pressure and cardiac output (Figure 6A and Figure IXA and IXB in the online-only Data Supplement).
Figure 6.

Pim1 knockout (KO) mice are resistant to pyrrole-modified monocrotaline (MCTP) and chronic hypoxia (CH) pulmonary artery (PA) hypertension (PAH). A, Compared with Pim1 KO mice (n=5), MCTP and CH wild-type (WT) mice (n=8) developed PAH (increased PA pressure, right ventricular/left ventricle septum ratio, PA wall thickness) without detrimental effects on both systemic pressure and cardiac output. B, PAH development in MCTP WT and CH WT mice is due to the signal transducers and activators of transcription-3 (STAT3)/Pim1-dependent activation of nuclear factor of activated T cell-c2 (NFATc2). Both STAT3 activation (nuclear translocation assay) and NFATc2 mRNA levels (quantitative reverse-transcription polymerase chain reaction [qRT-PCR]) are increased in WT and KO MCTP and CH mice but, in the absence of Pim1, were not sufficient to trigger PAH.
As in rats, the development of PAH in MCTP-WT and CH-WT mice was associated with the STAT3/Pim1-dependent activation of NFATc2. Interestingly, both STAT3 activation and NFATc2 mRNA levels were also increased in MCTP and CH Pim1 KO mice. However, in the absence of Pim1, they were not sufficient to trigger PAH. This result suggests that the implication of STAT3 in PAH development is mainly Pim1 dependent and that STAT3 activation alone cannot induce PAH. Therefore, Pim1 represents the primary therapeutic target of the STAT3/Pim1 axis.
Discussion
Here, we show that the STAT3/Pim1/NFAT axis is associated with the development of PAH, and we believe we have opened a new avenue of investigation for PAH treatment. Pim1 expression is increased in human and experimental PAH and relies on STAT3 activation (Figure 3). Interestingly, Pim1 expression parallels NFAT activation, PA remodeling, and PA pressure and correlates with PAH severity (Figures 1 and 4).
Unlike STAT3 and NFAT, which are constitutively expressed in several tissues, including normal PAs, Pim1 affects PASMC phenotype only in PAH, thus constituting an interesting therapeutic target for PAH. Notably, inhibition of endogenous Pim1 significantly improves PA pressure, PA medial hypertrophy, and right ventricular hypertrophy without affecting systemic pressure and cardiac output (Figures 5 and 6). We provide direct in vitro and in vivo evidence that the mechanisms by which Pim1 inhibition reverses PAH involve the inhibition of PASMC proliferation within the remodeled PAs (Figure 5) and the depolarization of PASMC mitochondria by promoting Bad activation (Figure 2) and decreasing Bcl-2, both of which increase apoptosis. We believe these effects are sustained by the Pim1-dependent activation of NFAT, a transcription factor that regulates both PASMC proliferation and resistance to apoptosis.5 Because our goal was to identify a new way of reversing established PAH, we focused our research on PASMCs and not endothelial cells, which are affected at the onset of PAH and are downregulated in established PAH. Nonetheless, because previous studies reported that Pim1 is implicated in embryonic stem cell differentiation into endothelial cells,25 Pim1 might be also implicated in endothelium-related vascular lesions like plexiform lesions, which are seen in patients with PAH.39
Our study suggests that circulating vasoactive factors like angiotensin, endothelin-1, platelet-derived growth factor, or cytokines, which are elevated in both human and experimental PAH, activate STAT3. Once activated, STAT3 increases NFATc2 and Pim1 expression (Figure 7). Cross-talk between STAT3 and NFAT is currently a hot topic because both signaling pathways are involved in the pathogenesis of cancer and cardiovascular diseases.40,41 In the present study, we demonstrated for the first time in the pulmonary vasculature that STAT3 regulates NFATc2 expression and indirectly (via Pim1) NFATc2 activity. Indeed, we showed that STAT3 inhibition decreases NFATc2 expression in human PAH, whereas in experimental PAH, STAT3 activation precedes NFAT expression. Moreover, we showed that both STAT3 and Pim1 inhibition similarly inhibit NFAT activation and that in experimental PAH, Pim1 expression parallels NFAT expression and activation. Although further experiments are required to determine whether STAT3 directly regulates Pim1 and NFAT, these findings demonstrate the implication of the STAT3/Pim1 axis in the regulation of both NFAT expression and activation in PAH. These findings might not be limited to pulmonary vasculature because chromatin immunoprecipitation sequence studies in several cell lines demonstrated the presence of several STAT response elements within the NFATc1, NFATc2, and NFATc3 promoter regions.13
Figure 7.
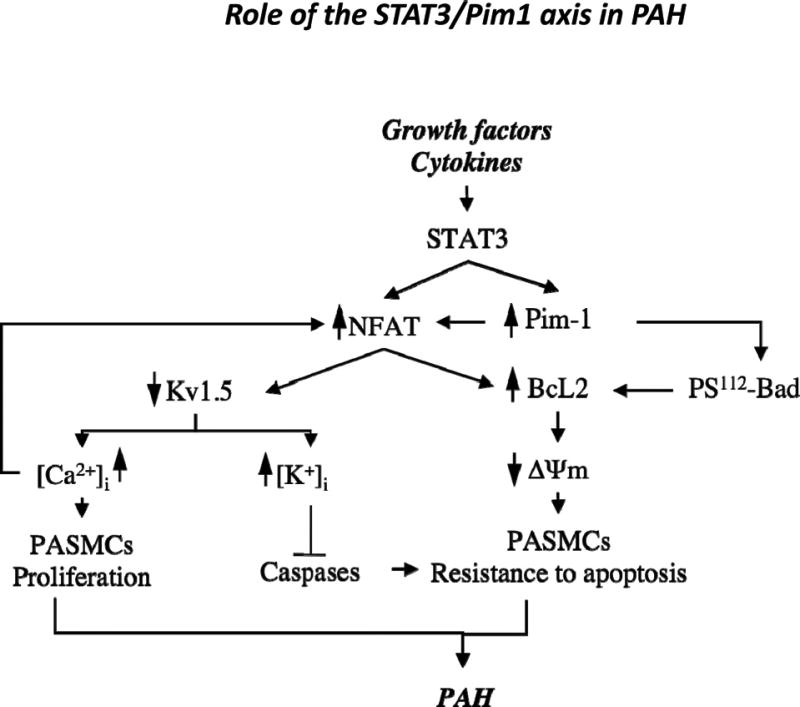
Proposed mechanism of the role of the signal transducers and activators of transcription-3 (STAT3)/Pim1 axis in pulmonary artery (PA) hypertension (PAH). Schematic representation of STAT3/Pim1 implication in PAH pathogenesis. Increased circulating growth factors, agonists, and cytokines trigger STAT3 activation, resulting in increased nuclear factor of activated T cell-c2 (NFATc2) expression and increased Pim1 activation. Once activated, Pim1 triggers NFATc2 activation (nuclear translocation), promoting [Ca2+]i-dependent PA smooth muscle cell (PASMC) proliferation and inhibiting mitochondria-dependent apoptosis through a Bcl2-dependent mechanism. The NFATc2-dependent effect on Bcl2 will be reinforced by Pim1-dependent inhibition of Bad.
Although STAT3 is an accepted Pim1 activator,42 the Akt pathway has been demonstrated as the main Pim1 activator in cardiomyocytes.32 However, in our study, the Akt pathway was not activated in PAH PASMCs (Figure IIIA in the online-only Data Supplement), suggesting that in our model Pim1 activation is Akt independent and relies on STAT3. Once activated, Pim1 is able to interact with NFAT, triggering its activation independently of the calcium/calcineurin axis and without affecting its nuclear localization.19,20 Once activated, NFAT regulates multiple genes that might positively reinforce its activation. For example, the downregulation of Kv1.5 will lead to PASMC depolarization and opening of L-type Ca++ channels and will sustain the increase in [Ca++]i, and thus calcineurin-dependent NFAT activation. Therefore, once activated, the system will remain functional, explaining why only PAH PASMCs have activated NFAT and why this activation is sustained in cultured conditions.
We demonstrated that STAT3 activation in PAH accounts for a Pim1-dependent activation of NFAT sustaining PAH PASMC proliferation and resistance to apoptosis (Figure 7). Interestingly, the fact that Pim1 activation remains high despite the activation of NFAT suggests that its role in PAH might not be limited to NFAT activation. In fact, we demonstrated that by inhibiting Bad, it decreases mitochondria-dependent apoptosis. In addition, by phosphorylating Cdc25 and its associated kinase C-TAK1 and by downregulating p27kip1, Pim1 enhances cell cycle progression at the G2/M phase.43– 45 Thus, Pim1 is implicated in several mechanisms promoting cell proliferation and survival that might be implicated in PAH pathogenesis.
Among the 3 components of the STAT3/Pim1/NFAT axis, we believe that Pim1 inhibition might represent a unique, specific, and novel way to reverse PAH without affecting physiological processes in which both STAT3 and NFAT are involved, such as the immune response. This is reinforced by our findings in Pim1 KO mice that showed that the implication of STAT3 in the origin of PAH relies mainly on Pim1 activation because STAT3 activation in Pim1 KO mice is not sufficient to induce PAH (Figure 6). This interesting finding needs to be confirmed in several other models, including human PASMCs, which will be the subject of a future study.
Finally, the fact that the pulmonary circulation is selectively diseased in human PAH is a major therapeutic challenge. The majority of drugs targeting the vasculature will, if given systemically, affect the healthy normal circulation as well, thereby limiting efficacy. Discovery of factors selectively expressed in the PAs, in addition to methods of delivering treatment selectively to the pulmonary circulation (such as inhaled delivery of drugs or genes), is critical. The airway administration of a Pim1 siRNA satisfies both requirements to ensure selective targeting of the diseased circulation. The concept that inducing apoptosis to remove the excess cells obstructing the PAs is beneficial in PAH is supported by other studies showing that serine-elastase inhibitors46 and Rho-kinase inhibitors47 cause regression of established PAH by inducing PASMC apoptosis. The novelty of our findings is that our therapeutic intervention is localized within the remodeled PAs and affects only the proliferative PASMCs expressing Pim1; quiescent PASMCs and surrounding cells are not affected. Because Pim1 is poorly expressed in healthy blood vessels and has limited and nonlethal implication in physiological processes (Pim1 KO mice are healthy and fertile; only erythrocyte microcytosis was reported48), it represents an ideal therapeutic target. Therefore (and as suggested by our findings), Pim1 inhibition will have limited toxicity, unlike STAT3 and NFAT inhibitors, for example. Finally, in addition to being an interesting therapeutic target, we demonstrated that Pim1 could indeed be an interesting and specific PAH biomarker in humans. Elevated Pim1 expression in the buffy coat seems to be specific to PAH because Pim1 is not elevated in other inflammatory diseases alone like scleroderma (which is a common cause of PAH). Because of their close relationships with Pim1, both STAT3 and NFAT could be considered putative biomarkers as well. However, because of their expression in several other tissues and under multiple conditions, their activations are not specific to PAH.49
Conclusions
PAH is a rapidly lethal disease for which treatments are limited. We believe that our findings will open the door to new avenues of investigation and potential future therapies for PAH. Our results will also lead to a better understanding of the regulation of apoptosis and proliferation by STAT3 and Pim1, which will benefit many other human diseases like cancer. Therefore, we describe Pim1 as a novel and specific therapeutic target and potential bio-marker for PAH.
Supplementary Material
CLINICAL PERSPECTIVE.
The contribution of vasoconstriction to the pathophysiology of pulmonary arterial hypertension (PAH) has been overemphasized, resulting in an excessive focus on vasodilator therapy. Only 20% of patients respond to vasodilator therapy. Recently, we have learned that PAH is due to remodeling of distal pulmonary arteries, leading to increased vascular resistance and right ventricular failure. The sustainability of this phenotype is attributed to the increased activation of transcription factor like nuclear factor of activated T cells. Its inhibition as a therapeutic approach to treat PAH is difficult because it regulates many physiological processes like immune response. In this article, using a multidisciplinary approach and large number of human tissues, we demonstrated that nuclear factor of activated T cell expression and activation are regulated by the proto-oncogene Pim1. Because Pim1 expression is restricted to PAH patients and is not present in normal conditions, it represents a safe and efficient way of inhibiting nuclear factor of activated T cells in PAH patients. We showed that suppression of Pim1 in human PAH pulmonary artery smooth muscle cells blocks nuclear factor of activated T cells and reverses the PAH phenotype, whereas Pim1 upregulation in normal pulmonary artery smooth muscle cells recapitulates it. In vivo, we showed that Pim1 knockout mice are resistant to multiple experimental models of pulmonary hypertension and that Pim1 inhibition reverses PAH in rodents. Finally, because Pim1 expression is restricted to PAH patients, we provide strong evidence that Pim1 expression correlates with PAH severity in humans. In conclusion, Pim1 is an attractive therapeutic target and a new biomarker of PAH.
Acknowledgments
Pim1 KO mice were provided by A. Berns from The Netherlands Cancer Institute, Amsterdam. We appreciate the contribution of Jacques Huot, Josée Lavoie, and Eric Paquet. We wish to thank Christine Racine and Sabrina Biardel from the Respiratory Health Network tissue bank for providing the patients’ lung tissue, serum, and buffy coats.
Sources of Funding
This work was supported by Heart and Stroke Foundation of Canada, Canadian Institute for Health Research, and Canadian Research Chair to Dr Bonnet.
Footnotes
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.110.963314/DC1.
Disclosures
None.
References
- 1.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “work in progress”. Circulation. 2000;102:2781–2791. doi: 10.1161/01.cir.102.22.2781. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad S. Pulmonary hypertension and right heart failure. Chest. 1995;108:1773. doi: 10.1378/chest.108.6.1773. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–185. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Grote K, Luchtefeld M, Schieffer B. JANUS under stress: role of JAK/STAT signaling pathway in vascular diseases. Vascul Pharmacol. 2005;43:357–363. doi: 10.1016/j.vph.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 9.Banes-Berceli AK, Ketsawatsomkron P, Ogbi S, Patel B, Pollock DM, Marrero MB. Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation. Am J Physiol Heart Circ Physiol. 2007;293:H1291–H1299. doi: 10.1152/ajpheart.00181.2007. [DOI] [PubMed] [Google Scholar]
- 10.Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM, Hu F, Ballabh P, Podlutsky A, Losonczy G, de Cabo R, Mathew R, Wolin MS, Ungvari Z. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension. 2009;54:668– 675. doi: 10.1161/HYPERTENSIONAHA.109.133397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frasch HF, Marshall C, Marshall BE. Endothelin-1 is elevated in monocrotaline pulmonary hypertension. Am J Physiol. 1999;276:L304–L310. doi: 10.1152/ajplung.1999.276.2.L304. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 14.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104:1184–1191. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- 15.Padma R, Nagarajan L. The human PIM1 gene product is a protein serine kinase. Cancer Res. 1991;51:2486–2489. [PubMed] [Google Scholar]
- 16.Beier UH, Weise JB, Laudien M, Sauerwein H, Gorogh T. Overexpression of Pim1 in head and neck squamous cell carcinomas. Int J Oncol. 2007;30:1381–1387. [PubMed] [Google Scholar]
- 17.Chiang WF, Yen CY, Lin CN, Liaw GA, Chiu CT, Hsia YJ, Liu SY. Up-regulation of a serine-threonine kinase proto-oncogene Pim1 in oral squamous cell carcinoma. Int J Oral Maxillofac Surg. 2006;35:740–745. doi: 10.1016/j.ijom.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 18.Cibull TL, Jones TD, Li L, Eble JN, Ann Baldridge L, Malott SR, Luo Y, Cheng L. Overexpression of Pim1 during progression of prostatic adenocarcinoma. J Clin Pathol. 2006;59:285–288. doi: 10.1136/jcp.2005.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glazova M, Aho TL, Palmetshofer A, Murashov A, Scheinin M, Koskinen PJ. Pim1 kinase enhances NFATc activity and neuroendocrine functions in PC12 cells. Brain Res Mol Brain Res. 2005;138:116–123. doi: 10.1016/j.molbrainres.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Rainio EM, Sandholm J, Koskinen PJ. Cutting edge: transcriptional activity of NFATc1 is enhanced by the Pim1 kinase. J Immunol. 2002;168:1524–1527. doi: 10.4049/jimmunol.168.4.1524. [DOI] [PubMed] [Google Scholar]
- 21.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115:1479–1491. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diller GP, Thum T, Wilkins MR, Wharton J. Endothelial progenitor cells in pulmonary arterial hypertension. Trends Cardiovasc Med. 2010;20:22–29. doi: 10.1016/j.tcm.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Diller GP, van Eijl S, Okonko DO, Howard LS, Ali O, Thum T, Wort SJ, Bedard E, Gibbs JS, Bauersachs J, Hobbs AJ, Wilkins MR, Gatzoulis MA, Wharton J. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117:3020–3030. doi: 10.1161/CIRCULATIONAHA.108.769646. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt T, Karsunky H, Rodel B, Zevnik B, Elsasser HP, Moroy T. Evidence implicating Gfi-1 and Pim1 in pre-T-cell differentiation steps associated with beta-selection. EMBO J. 1998;17:5349–5359. doi: 10.1093/emboj/17.18.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zippo A, De Robertis A, Bardelli M, Galvagni F, Oliviero S. Identification of Flk-1 target genes in vasculogenesis: Pim1 is required for endothelial and mural cell differentiation in vitro. Blood. 2004;103:4536– 4544. doi: 10.1182/blood-2003-11-3827. [DOI] [PubMed] [Google Scholar]
- 26.Rosenbloom KR, Dreszer TR, Pheasant M, Barber GP, Meyer LR, Pohl A, Raney BJ, Wang T, Hinrichs AS, Zweig AS, Fujita PA, Learned K, Rhead B, Smith KE, Kuhn RM, Karolchik D, Haussler D, Kent WJ. ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010;38:D620–D625. doi: 10.1093/nar/gkp961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morcinek JC, Weisser C, Geissinger E, Schartl M, Wellbrock C. Activation of STAT5 triggers proliferation and contributes to anti-apoptotic signalling mediated by the oncogenic Xmrk kinase. Oncogene. 2002;21:1668–1678. doi: 10.1038/sj.onc.1205148. [DOI] [PubMed] [Google Scholar]
- 28.Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and Pim1 genes in human T cells. Blood. 1999;93:1980–1991. [PubMed] [Google Scholar]
- 29.Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest. 2005;115:2691–2694. doi: 10.1172/JCI26593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka H, Sukhova G, Schwartz D, Libby P. Proliferating arterial smooth muscle cells after balloon injury express TNF-alpha but not interleukin-1 or basic fibroblast growth factor. Arterioscler Thromb Vasc Biol. 1996;16:12–18. doi: 10.1161/01.atv.16.1.12. [DOI] [PubMed] [Google Scholar]
- 31.Zhang F, Hu Y, Xu Q, Ye S. Different effects of angiotensin II and angiotensin-(1-7) on vascular smooth muscle cell proliferation and migration. PLoS One. 2010;5:e12323. doi: 10.1371/journal.pone.0012323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, Siddiqi S, Emmanuel GN, Wu W, Fischer K, Martindale JJ, Glembotski CC, Leri A, Kajstura J, Magnuson N, Berns A, Beretta RM, Houser SR, Schaefer EM, Anversa P, Sussman MA. Pim1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007;13:1467–1475. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- 33.Sussman MA. Mitochondrial integrity: preservation through Akt/Pim1 kinase signaling in the cardiomyocyte. Expert Rev Cardiovasc Ther. 2009;7:929–938. doi: 10.1586/erc.09.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 35.Hu XF, Li J, Vandervalk S, Wang Z, Magnuson NS, Xing PX. PIM1-specific mAb suppresses human and mouse tumor growth by decreasing PIM1 levels, reducing Akt phosphorylation, and activating apoptosis. J Clin Invest. 2009;119:362–375. doi: 10.1172/JCI33216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonnet S, Paulin R, Sutendra G, Dromparis P, Roy M, Watson KO, Nagendran J, Haromy A, Dyck JR, Michelakis ED. Dehydroepian-drosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-{beta}/NFAT axis. Circulation. 2009;120:1231–1240. doi: 10.1161/CIRCULATIONAHA.109.848911. [DOI] [PubMed] [Google Scholar]
- 37.Holder S, Zemskova M, Zhang C, Tabrizizad M, Bremer R, Neidigh JW, Lilly MB. Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase. Mol Cancer Ther. 2007;6:163–172. doi: 10.1158/1535-7163.MCT-06-0397. [DOI] [PubMed] [Google Scholar]
- 38.Raoul W, Wagner-Ballon O, Saber G, Hulin A, Marcos E, Giraudier S, Vainchenker W, Adnot S, Eddahibi S, Maitre B. Effects of bone marrow-derived cells on monocrotaline- and hypoxia-induced pulmonary hypertension in mice. Respir Res. 2007;8:8. doi: 10.1186/1465-9921-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, Yang J, Suntharalingam J, Soon E, Exley A, Stewart S, Hecker M, Zhu Z, Gehling U, Seeger W, Pepke-Zaba J, Morrell NW. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180:780–787. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lagunas L, Clipstone NA. Deregulated NFATc1 activity transforms murine fibroblasts via an autocrine growth factor-mediated Stat3-dependent pathway. J Cell Biochem. 2009;108:237–248. doi: 10.1002/jcb.22245. [DOI] [PubMed] [Google Scholar]
- 41.Manukyan I, Galatioto J, Mascareno E, Bhaduri S, Siddiqui MA. Cross-talk between calcineurin/NFAT and Jak/STAT signaling induces cardioprotective alphaB-crystallin gene expression in response to hypertrophic stimuli. J Cell Mol Med. 2010;14:1707–1716. doi: 10.1111/j.1582-4934.2009.00804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zemskova M, Sahakian E, Bashkirova S, Lilly M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J Biol Chem. 2008;283:20635–20644. doi: 10.1074/jbc.M709479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bachmann M, Hennemann H, Xing PX, Hoffmann I, Moroy T. The oncogenic serine/threonine kinase Pim1 phosphorylates and inhibits the activity of Cdc25C-associated kinase 1 (C-TAK1): a novel role for Pim1 at the G2/M cell cycle checkpoint. J Biol Chem. 2004;279:48319– 48328. doi: 10.1074/jbc.M404440200. [DOI] [PubMed] [Google Scholar]
- 44.Bachmann M, Kosan C, Xing PX, Montenarh M, Hoffmann I, Moroy T. The oncogenic serine/threonine kinase Pim1 directly phosphorylates and activates the G2/M specific phosphatase Cdc25C. Int J Biochem Cell Biol. 2006;38:430– 443. doi: 10.1016/j.biocel.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 45.Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008;68:5076–5085. doi: 10.1158/0008-5472.CAN-08-0634. [DOI] [PubMed] [Google Scholar]
- 46.Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105:516–521. doi: 10.1161/hc0402.102866. [DOI] [PubMed] [Google Scholar]
- 47.Barman SA, Zhu S, White RE. RhoA/Rho-kinase signaling: a therapeutic target in pulmonary hypertension. Vasc Health Risk Manag. 2009;5:663–671. doi: 10.2147/vhrm.s4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laird PW, van der Lugt NM, Clarke A, Domen J, Linders K, McWhir J, Berns A, Hooper M. In vivo analysis of Pim1 deficiency. Nucleic Acids Res. 1993;21:4750– 4755. doi: 10.1093/nar/21.20.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.