

Abstract
Many cancer cells have increased rates of aerobic glycolysis, a phenomenon termed the Warburg effect. In addition, in tumors there is a predominance of expression of the M2 isoform of pyruvate kinase (PKM2). M2 expression was previously shown to be necessary for aerobic glycolysis and to provide a growth advantage to tumors. We report that knockdown of pyruvate kinase in tumor cells leads to a decrease in the levels of pyruvate kinase activity and an increase in the pyruvate kinase substrate phosphoenolpyruvate. However, lactate production from glucose, although reduced, was not fully inhibited. Furthermore, we are unique in reporting increased serine and glycine biosynthesis from both glucose and glutamine following pyruvate kinase knockdown. Although pyruvate kinase knockdown results in modest impairment of proliferation in vitro, in vivo growth of established xenograft tumors is unaffected by PKM2 absence. Our findings indicate that PKM2 is dispensable for tumor maintenance and growth in vivo, suggesting that other metabolic pathways bypass its function.
Many cancer cells have increased rates of glucose uptake with a concomitant decrease in oxidative phosphorylation, even in the presence of oxygen. This phenomenon of aerobic glycolysis with increased lactate production has been termed the Warburg effect (1). Previous work suggested that expression of pyruvate kinase M2 (PKM2) is a key event in determining this metabolic phenotype, and tumor expression of M2 provides a proliferative advantage in vitro and in vivo (2). In addition, some tyrosine kinases involved in cancer might also be responsible for regulation of the Warburg effect, as it has been shown that they can phosphorylate glycolytic enzymes, including PKM2, and that this phosphorylation may regulate PKM2 activity and promote the Warburg effect and tumor growth (3, 4).
Glucose taken up by cells is phosphorylated by hexokinase and subsequently catabolized via glycolysis to phosphoenolpyruvate (PEP). PK catalyzes the dephosphorylation of PEP to pyruvate, generating a molecule of ATP independent of oxygen supply. PK is a tetrameric enzyme encoded by four isozymes (L, R, M1, and M2) that differ in their kinetic properties and tissue expression distribution. The R and the L isoforms are encoded by the same gene but expressed under the control of different tissue promoters, leading to type L expression in tissues with gluconeogenesis, such as the liver, kidney, and small intestine, and type R expression in erythrocytes. The M1 isoform (PKM1) is expressed in muscle and brain, and the M2 isoform, differing only by the differential splicing of one exon, is expressed during embryogenesis, in adipose tissue and pancreatic islets, and is the predominant form found in cancer cells. PKM1 has high affinity for PEP, is not allosterically regulated, and is constitutively active in a tetrameric state. In contrast, PKM2 is allosterically regulated by metabolic intermediates such as fructose-1,6-bisphosphate (FBP) and exists either as a dimer with low affinity for PEP, or as a FBP-bound tetramer with high affinity for PEP (5).
The enzymatic activity of the PKM2 isoform may be inhibited by phosphotyrosine binding, and mutation of the residue involved in this phosphotyrosine binding led to reduced rates of glycolysis, increased oxygen consumption, and impaired tumor cell proliferation in vitro (6). In addition, the oncogenic form of fibroblast growth factor receptor type 1 has been recently shown to directly phosphorylate PKM2 on tyrosine residue 105. This process results in inhibition of the formation of active, tetrameric PKM2 by disruption of binding of coactivator FBP (3). Constitutive expression of a PKM2 mutant, where Y105 was substituted with phenylalanine, led to decreased tumor initiation and growth in an in vivo xenograft model, accompanied by a switch to higher oxidative phosphorylation and reduced glycolysis. Thus, M2 expression seems to be necessary for aerobic glycolysis and to provide an advantage for tumor growth. A recent study has shed light into the mechanism by which PKM2 affects the balance between glycolysis and oxidative phosphorylation. Luo et al. have demonstrated that prolyl hydroxylation of PKM2 by PHD3 stimulates the function of PKM2 as a coactivator of HIF-1 transactivation, leading to HIF-dependent metabolic reprogramming (7).
However, the exact role of PKM2 in tumor growth and maintenance is not yet fully understood. Data available only address the effects of lack of fully functional PKM2 during tumor initiation, but not the impact of PKM2 inhibition in established tumors, which would be of interest therapeutically. Here we address the role of PKM2 in modulation of tumor growth and maintenance. We further characterize the metabolic adaptation that cells undergo in the absence of PKM2.
Results
Accumulation of Phosphoenolpyruvate in Cells Lacking PK.
To gain insight as to whether the predominant expression of PKM2 observed in tumors is necessary for cancer cell proliferation, we investigated the role of PKM2 in tumor cell growth and maintenance. We used short hairpin RNAs (shRNAs) to deplete M2, M1, or both PK isoforms in HCT116 cancer cells. Specific shRNAs to M2 were designed to its unique exon 10 (sh1408 and sh1411), whereas shRNAs targeting both M1 and M2 isoforms (sh566 and sh1021), or only M1 (sh1493) were obtained from The Research Consortium collection (Fig. 1A). We could verify specificity of the knockdown by quantitative RT-PCR (qRT-PCR), as sh1493 only targeted PKM1 mRNA levels (Fig. 1B, Lower), and sh1408 and sh1411 only PKM2 (Fig. 1B, Upper). All other shRNAs decreased the levels of PKM1 and PKM2 to the same extent. Protein knockdown of PKM2 was equally efficient in cells expressing PKM2-targeting shRNAs, except sh1411 (specific to PKM2), which was less efficient (Fig. 1C). PK activity in the cells was drastically decreased by the four shRNAs that targeted PKM2, either alone (sh1408 and sh1411) or in combination with PKM1 (sh566 and sh10221) (Fig. 1D), although the effect was less pronounced with sh1411 (specific to PKM2), in keeping with it being a less effective PKM2 shRNA (Fig. 1C). Additionally, specific knockdown of PKM1 (sh1493) did not affect PK activity in HCT116 cells (Fig. 1D) and was only slightly decreased in RKO cells (Fig. S1), although the knockdown of PKM1 in these lines was similar to that achieved with the dual specificity shRNAs targeting both PKM1 and PKM2. This finding is in line with the predominant expression of M2 over M1 and indicates that most PK activity in these lines is driven by PKM2. The decrease in PK activity correlated with an increase in the levels of cellular PEP (Fig. 1E and Fig. S1), with a higher accumulation of PEP in those cells where PK activity was most drastically reduced (dual M1 and M2 sh566 and sh1021). It should be noted that there are minimally detectable levels of PKM1 in HCT116 and RKO cells at the mRNA level (Fig. 1B). This level of PKM1 contributes to a small but significant portion of the PK activity in these cells, as can be observed in the difference in PEP levels after knockdown of both PKM1 and PKM2 vs. only PKM2 (sh566 and sh1021 vs. sh1408 and sh1411) (Fig. 1E).
Fig. 1.

Knockdown of PKM2 leads to decreased PK activity, accumulation of phosphoenolpyruvate, decreased glycolytic rates, and increased oxygen consumption in HCT116 cancer cells. (A) Schematic representation of PKM1 and PKM2 with the alternatively spliced exons 9 and 10. The location of the shRNAs is specified by a star. sh566 and sh1021 knockdown PKM1 and PKM2. sh1493 is specific to PKM1 and sh1408 and sh1411 are specific for PKM2. (B) qRT-PCR quantification of the levels of PKM2 (Upper) and PKM1 (Lower) mRNA 7 d after shRNA knockdown. Specificity of the shRNAs is shown at the top of the panel. sh566 and sh1021 knock down both PKM1 and PKM2, sh1493 only PKM1, and sh1408 and sh1411 PKM2. (C) Immunoblotting shows shRNA-mediated stable knockdown of endogenous PKM2 protein 4 and 7 d after infection. (D) PK catalytic activity from cell lysates generated from HCT116 cells expressing the different shRNAs targeting PK. Asterisks indicate P = 0.0022 (sh1411 vs. shAlk), P < 0.0001 (sh1408 vs. shNSQ), and P = 0.0023 (sh566 vs. sh1408). (E) PEP levels from cellular lysates. PEP levels are obtained by IC-MS/MS and values depicted in an arbitrary unit that reflects the integrated peak area of a MS signal. Data were normalized by total protein content in the cells. Error bars represent SD of the mean from triplicate. Asterisks indicate P = 0.0392 (sh566 vs. sh1411), P = 0.0029 (sh1021 vs. sh1408), P = 0.0021 (sh1021 vs. sh1411), and P = 0.0192 (sh1408 vs. shAlk).
Accumulation of Metabolic Intermediates and Utilization of Alternative Energy Pathways to Generate Glycolytic Metabolites in HCT116 Colon Carcinoma Cells upon PKM2 Knockdown.
Metabolic labeling studies in which cells were induced with doxycycline for 7 d to knock down PKM, then cultured with either 13C6-glucose or 13C5-glutamine for 8 h, were performed in HCT116 cells. In addition to causing greatly decreased cellular PK activity (Fig. 2A) and an increased accumulation of PEP (Fig. 2C), PKM knockdown significantly decreased the production of 13C3-lactate and inhibited 13C6-glucose uptake (Fig. 2B). Although the release of 13C3-lactate into the media is diminished in PKM knockdown cells, PKM knockdown did not completely prevent the conversion of glucose into pyruvate, as evidenced by the presence, albeit decreased, of 13C3-alanine (Fig. 2C), and of 13C2-glutamate and 13C2-aspartate (Fig. S2B) in addition to 13C3-lactate. Interestingly, in PKM knockdown cells the reduction in 13C2-glutamate and 13C2-aspartate, both derived via the tricarboxylic acid (TCA) cycle, is significantly smaller than that observed for the 13C3-lactate (5–15% 13C2-glutamate vs. ∼50% 13C3-lactate), suggesting that despite a reduction in available pyruvate, carbon flux through pyruvate dehydrogenase is only moderately effected.
Fig. 2.
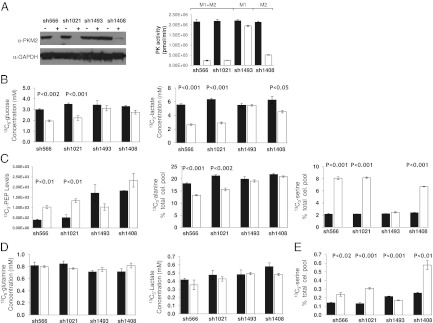
Decreased glucose uptake and lactate production and accumulation of metabolic intermediates in HCT116 colon carcinoma cells upon PKM2 knockdown. (A, Left) Immunoblotting shows doxycycline- (Dox) inducible, shRNA-mediated knockdown of endogenous PKM2 protein in HCT116 cells 7 d after infection with lentivirus containing shRNAs as indicated. (Right) PK catalytic activity from cell lysates from same experiment. Closed bars, no doxycycline, open bars, with doxycycline. (B) Consumed 13C6-glucose (mM, Left) and released 13C3-lactate (mM, Right) in media from PKM1/2 knockdown HCT116 cultures incubated with 13C6-glucose for 8 h, as determined by NMR. Approximately 6% of glucose in the naive media was unlabeled. This value remained constant for all samples at the 8-h time point. (C) Intracellular metabolites derived from 13C6-glucose; (Left) 13C3-PEP levels obtained by IC-MS/MS and values depicted in an arbitrary units. 13C3-alanine (Center), and 13C3-serine (Right); values were determined by LC-MS/MS and are expressed as a percent of the total cellular pool for each metabolite. (D) Consumed 13C5-glutamine (mM, Left) and released 13C3-lactate (mM, Right) in media from PKM1/2 knockdown HCT116 cultures incubated with 13C5-glutamine for 8 h as determined by NMR. (E) 13C3-serine as percent of the total cellular serine pool from cellular lysates of cells cultured for 8 h with 13C5-glutamine. Approximetly 8% of glutamine in the naive media was unlabeled. Measurements were made by NMR (media) or LC-MS/MS (cell lysates). Error bars represent SE of the mean from triplicate wells. P values depicted. Closed bars indicate cells without doxycycline, and open bars cells treated with doxycycline in all graphs.
Because 13C3-PEP accumulates in these cells (Fig. 2C), we analyzed other upstream metabolites to investigate the fate of the 13C6-glucose in the PKM knockdown cells. We observed a three- to fourfold increase in the levels of 13C3-serine and 13C3-glycine derived from 13C6-glucose in the PKM knockdown cells (Fig. 2C and Fig. S2C). Furthermore, the PKM knockdown resulted in a significant release of 13C3-serine and 13C2-glycine into the media (Fig. S2A). In cells incubated with 13C5-glutamine, neither glutamine consumption nor the production of 13C5-glutamine–derived 13C3-lactate was significantly affected by PKM knockdown (Fig. 2D). However, there was an increase in13C5-glutamine–derived 13C3-serine following PKM knockdown (Fig. 2E).
Knockdown of PKM2 Leads to Impaired in Vitro Cellular Proliferation in HCT116 and RKO Colon Carcinoma Cells but No Effect on in Vivo Tumor Xenograft Growth.
To elucidate the role of PKM1 and PKM2 in tumor cell proliferation and viability, we studied the effects of PKM knockdown on the proliferation of HCT116 and RKO colon carcinoma cells (Figs. 3 and 4), as well as A549 lung adenocarcinoma cells (Fig. S3). In HCT116 cells, only knockdown with dual (PKM1/M2) specificity shRNAs had an effect on the proliferation of tumor cells in clonogenic survival assays (Fig. 3A) or as a monolayer (sh566 and sh1021) (Fig. 3B). The same was also observed in the case of RKO cells (Fig. 4 B and C). In contrast, knockdown of only PKM2 using sh1408 and sh1411 or only of PKM1 using sh1493 had little or no effect on cellular proliferation in vitro. These results suggest that robust inhibition of PKM2 activity alone in tumor cells is not sufficient to impact cell proliferation in vitro, and that the residual PKM1 activity in cells where only PKM2 is knocked down allows for tumor cell survival. Inhibition of both M1 and M2 isoforms of PK is thus necessary to impact tumor cell proliferation and is associated with an in vitro growth dependence. These data correlate with the observations presented above, where knockdown of both isoforms led to the most drastic reduction in PK catalytic activity as well as PEP accumulation in the cells (Fig. 1 D and E). These data further suggest that any residual PK activity in cells can mediate their survival, either via use of oxidative phosphorylation with reduced glycolysis or by the use of an alternative energy source.
Fig. 3.

Knockdown of PKM2 leads to impaired in vitro cellular proliferation in HCT116 colon carcinoma cells but no effect on in vivo tumor xenograft growth. (A) Clonogenic survival assays of HCT116 and (B) monolayer proliferation of HCT116 cells upon knockdown of PKM1 and PKM2. Proliferation was measured at 4 and 7 d after lentiviral infection and selection. Error bars represent SD of the mean from octoplicate wells. (C) PEP levels from tumor extracts from D. Each point represents a separate animal. Asterisks indicate P = 0.0057 (WT veh vs. sh1021 Dox), P = 0.0325 (WT Dox vs. sh1021 Dox), and P = 0.0317 (WT Dox vs. sh1408 Dox). (D) Immunoblotting revealing PKM2 levels in HCT116 tumor xenografts harvested from mice at 28–32 d postimplantation. (E) PK catalytic activity from HCT116 tumor xenografts expressing the different shRNAs targeting PK. (F) Mean tumor volume (TVol) from HCT116 xenografts expressing shRNAs to PKM1/2 as indicated. Xenografts were allowed to grow for 14 d, then dosed with doxycycline or vehicle daily for 14 d. Each point represents the mean tumor volume of eight mice per group ± SEM.
Fig. 4.

Knockdown of PKM1/2 leads to impaired in vitro cellular proliferation in RKO colon carcinoma cells but no effect on in vivo tumor xenograft growth in nude mice. (A) Immunoblotting shows doxycycline-inducible, shRNA-mediated knockdown of endogenous PKM2 protein in RKO cells at 4 and 7 d after infection with lentivirus containing shRNAs. (B) Cellular proliferation and (C) colony formation capacity of the RKO cells upon doxycycline induction. Proliferation was measured at 4 and 7 d after lentiviral infection as described above. Error bars represent SD of the mean from octoplicate wells. (D) Immunoblotting revealing PKM2 levels in RKO tumor xenografts harvested from mice dosed with doxycycline or vehicle daily for 14 d. PKM2 is the band indicated by the arrow. Each lane is a separate animal. (E) PEP levels from tumor extracts from D. Each point represents a separate animal. *P = 0.0008 (sh1021 Veh vs. sh1021 Dox). (F) Mean tumor volume (TVol) from RKO xenografts expressing shRNAs to PKM1/2 as indicated. Each point represents the mean tumor volume of eight mice per group ± SEM.
To understand the role of PKM1/M2 in in vivo tumor growth and maintenance and in an effort to validate the potential role of PKM2 as a therapeutic target, nude mice were injected subcutaneously with either HCT116 (Fig. 3), RKO (Fig. 4), or A549 (Fig. S3) cells expressing doxycycline-inducible shRNAs to PKM1/M2 or specific for PKM1, respectively. Xenografts were allowed to grow in vivo for 14, 7, and 16 d, respectively, at which point doxycycline was given to the mice daily until the end of the study, when mice were killed. PKM2 knockdown was very efficient in HCT116 (Fig. 3D) and RKO (Fig. 4D) tumor xenografts expressing the shRNAs targeting PKM2, whereas PKM2 protein levels were unaffected in the control, uninduced tumors. Moreover, PEP levels were highly increased in these tumors (Figs. 3C and 4E), reflecting the impairment of PK activity in these tumors and thus accumulation of its substrate.
Despite robust knockdown and impairment of PK activity in the tumor xenografts, tumor volume in the absence of PKM1/M2 or PKM2 was not significantly affected. This finding was observed in tumor xenografts from HCT116 (Fig. 3F), RKO (Fig. 4F), and A549 cells (Fig. S3E). These results demonstrate that, although there is a dependence on PK activity for cancer cell growth in vitro, this dependence is not maintained in vivo, and PK is not necessary for tumor maintenance and growth in vivo.
Discussion
We show that PKM2 is dispensable for tumor maintenance and growth in vivo. Knockdown of both M1 and M2 isoforms of PK in vitro in HCT116, RKO, and A549 cells led to a significant decrease of PK activity, accompanied by a concomitant and proportional increase in PEP levels and an impairment of tumor cell growth. However, in vivo, despite a depletion of total PK activity comparable to that observed in vitro (as measured by enzymatic PK activity as well as PEP levels), knockdown of PKM2/M1 in already established tumors had no significant effect on in vivo xenograft growth from HCT116, RKO or A549 cancer cells. Christofk et al. (2) suggested that expression of PKM2 confers a selective growth advantage to tumor cells in vivo. When they compared the growth of tumor xenografts of cells expressing wild-type M1 to that of wild-type M2, they indeed observed that the M2-expressing cells formed larger tumors faster than those expressing M1. In a similar manner, Hitosugi et al. (3) showed that cells expressing wild-type M2 formed larger tumors than cells expressing the Y105F mutant, which cannot be tyrosine-phosphorylated at this site and thereby maintains an activity profile similar to PKM1. In neither case did these experiments address the more therapeutically relevant question of tumor maintenance; instead, the role of PK in the initial growth and establishment of tumor xenografts was addressed. In our experiments, we asked whether PKM2 was required for maintenance and growth of established tumors by allowing the tumor xenografts to grow to a certain size before knocking down PKM2 or PKM2 in combination with PKM1. Our results are consistent with recently published data from Lv et al., where they describe that PKM2 is acetylated on lysine 305, leading to decreased PKM2 activity and its lysosomal-dependent degradation (8). Expression of mutant PKM2 K305Q, which mimics acetyl modification of the protein at this residue and leads to reduced PKM2 activity by as much as 95%, promotes H1299 cellular proliferation and in vivo tumor xenograft growth. In agreement with our results, greatly decreased PKM2 activity not only does not abolish tumor growth in vitro and in vivo, but may even promote it. The authors also observe a decrease in lactate and an increase in other glycolytic metabolites in these cells (9).
What is not yet understood is how xenograft tumor growth is largely unaffected by the absence of PKM2 and whether the tumors that survive are less glycolytic or what their metabolic phenotype is like. It is indeed possible that the tumor viability is rescued by alternative metabolic pathways that come into play upon reduction or inhibition of glycolysis by PKM2 knockdown and have the ability to bypass/complement its requirement. One possibility is the use of glutamine as an alternative energy source. Glutamine is the most abundant nutrient in plasma together with glucose, and glutaminolysis, the metabolism of glutamine to lactate, is considered a hallmark of tumor cell metabolism (10–12). Glutaminase overexpression has been reported in a variety of tumors, and glucose deprivation results in increased glutamine consumption in cancer cells (10, 13). Glutamine is used by the cells to generate fatty acids, for nucleotide biosynthesis, to replenish TCA cycle intermediates (such as oxaloacetate), and up to 60% of glutamine has been reported to be used for the production of lactate and alanine (7). Under the conditions of the current study glutamine utilization did not appear to be affected by PKM knockdown. Furthermore, PKM2 knockdown only had a moderate effect on carbon flux into the TCA cycle, as indicated by 13C-label incorporation into glutamate and aspartate. This finding suggests that PKM knockdown does not significantly impact cellular energy production and could explain why we did not see an increased dependence on substrates other than glucose in our in vitro experiments. It is possible, however, that under longer incubation times an increased dependence on glutamine or other substrates may become apparent. Recent studies have also suggested that PKM2 promotes tumorigenesis via effects it has in the nucleus as a transactivator for HIF-1 and β-catenin–dependent transcription, respectively (7, 14). Although we do not see effect of PK knockdown in tumorigenesis in vivo, we did test for the levels of HIF-1 targets hexokinase 2 (HK2) and pyruvate dehydrogenase kinase 1, as well as β-catenin target cyclin D1 in the xenografts from mice upon PK knockdown, and we did not observe any differences (Fig. S4).
In our experiments, 13C3-lactate derived from 13C6-glucose was reduced by 50%, although PKM2 protein was reduced by over 95% after the knockdown, indicating the use of an alternative metabolic pathway for the conversion of PEP into pyruvate. Recently, Vander Heiden et al. (15, 16) have proposed that under conditions of reduced PK activity, phosphoglycerate mutase1 (PGAM1) can act to convert PEP to pyruvate in a process that is independent of enolase and pyruvate kinase. The mechanism for PGAM1-mediated conversion of PEP to pyruvate is not fully characterized, but is thought to involve the transfer of the PEP phosphate to the catalytic histidine of PGAM (15, 16).Vander Heiden et al. (16) predicted that, as a consequence of increased PGAM phosphorylation, there would be an increased redirection of glycolytic carbon into the serine and glycine biosynthesis. Consistent with this finding, a significant increase in glucose-derived serine and glycine, both intracellular and released into the media, was observed following PKM knockdown. The conversion of glutamine into pyruvate occurs independently of glycolysis through the malate pathway (11). The utilization of glutamine was not affected by PKM knockdown nor its conversion into pyruvate, as indicated by 13C3-lactate. PKM knockdown, however, increased the conversion glutamine-derived pyruvate into serine, as would be predicted by Vander Heiden et al. (16). Although making only a small contribution to the total serine pool, the increase in glutamine-derived serine following PK knockdown provides further support for the hypothesis that PGAM can provide an alternative glycolytic pathway. To our knowledge this finding is unique.
Our results show that PK is not necessary for in vivo tumor maintenance and demonstrate the metabolic plasticity of tumor cells, which can produce lactate from glucose even in the almost complete absence of PK activity. PKM2 has been reported to be less active than PKM1 and decreased overall PK activity is suggested to increase the availability of metabolic intermediates for anabolism and cell growth (9, 10). One alternative therapeutic notion that has arisen recently is based on the fact that there seems to be an advantage for the tumors to keep PK activity low or even degrade PK under high glucose conditions. Keeping PKM2 constantly active in the tumors [e.g., by an agonist (17)] might block biosynthesis and continuously drive glycolysis (16). This approach may, however, be problematic because the consequences of continuous ATP generation in tumors are unknown. Nevertheless, our experiments suggest that inhibition of PKM2 in tumors might not be a desirable or effective therapeutic strategy.
Materials and Methods
Cell Culture.
A549 cells were cultured in RPMI 1640, RKO cells in DMEM low glucose, HCT116 cells in Mac Coy’s media. The lentiviral constructs for PKM2 and PKM1 sh566, sh1021 and sh1493 were purchased from Sigma (clones TRCL0000074882, TRCL0000074878, TRCL0000074884). Specific shRNA sequences targeting only PKM2 were designed in house and are as follows: sh1408 5′-CCGGCTACCACTTGCAATTATTTGACTCGAGTCAAATAATTGCAAGTGGTAGTTTTTG-3′, and sh1411 5′-CCGGCCACTTGCAATTATTTGAGGACTCGAGTCCTCAAATAATTGCAAGTGGTTTTTG-3′. Lentiviruses were generated by transfecting HEK293 FT cells with lentiviral vector encoding shRNA, pVSVG and Δ8.7. Hct116, RKO, and A549 cells were infected and selected with puromycin. For cells infected with the inducible vector, doxycycline was added at the time of plating. In the cell proliferation assay cells were seeded 4 d after infection and grown for 4–7 d, followed by fixation and methylene blue staining and quantified at 650 nm. For the clonogenic survival assays, cells were plated and after 15 d, plates were fixed and stained with 0.2% Crystal violet (Sigma).
Antibodies.
Antibody for PKM2 was from Cell Signaling (# 3198); antibodies for tubulin and GAPDH were from Sigma (# T4026 and WHOOO2591M1); and secondary antibodies goat anti-rabbit were from Jackson Immunoresearch (# 111–035-046) and goat anti-mouse from DAKO (# P0447).
Immunoblotting.
Cells were lysed in cold RIPA buffer (Tris•HCl pH7.4 50 mM, NaCl 150 mM, EDTA pH 8 5 mM, EGTA 1 mM) supplemented with 2 mM sodium vanadate (Sigma) and 1× protease inhibitors (Calbiochem). Pulverized tumor tissue was resuspended in RIPA. Five to 20 μg of total protein from each lysate were separated by SDS/PAGE and transferred onto a PVDF membrane, probed with the indicated antibodies.
PK Enzymatic Activity Assay.
PK activity was measured in an LDH-coupled enzymatic assay as described previously (16), either from cell extracts or from tumor xenografts.
qRT-PCR.
RNA extractions were done following the RNeasy kit from Qiagen. One microgram of RNA was reverse-transcribed and cDNA used in the qPCR (SsoFast Evagreen; BioRad). PKM1 primer sequences: 5′ CTA TCC TCT GGA GGC TGT GC 3′ and 5′ CCA TGA GGT CTG TGG AGT GA 3′ and PKM2 primer sequences: 5′ CCA CTT GCA ATT ATT TGA GGA A 3′ and 5′ GTG AGC AGA CCT GCC AGA CT 3′. qPCR was performed in an iQ5 device (BioRad) and analysis using iQ5 software (BioRad).
Ion Chromatography-Tandem Mass Spectrometry Analysis of PEP.
Cells were extracted three times with 500 µL cold methanol. Extracts were dried under N2 stream and resuspended in 200 µL water. The pellets left were used to normalize the IC-MS/MS data. Pulverized tumor samples were resuspended in 100% methanol. Ion chromatography-tandem mass spectrometry (IC-MS/MS) analysis of PEP levels was done using a method as described in refs. 18 and 19 with some modifications.
Analysis of 13C-Labeled Metabolites.
HCT-116 cells were cultured in glucose or glutamine-free DMEM media supplemented with 13C6-glucose or 13C5-glutamine. Cell pellets were lysed in methanol:water (1:1 vol/vol) via three cycles of freeze-thawing. All intracellular 13C-labeled intermediates were quantified using LC-MS/MS with an API4000 triple quadruple MS system (AB Sciex). Analytes of interests were individually tuned and the most sensitive MRM was selected for each analyte. Conditioned media samples were prepared for analysis by NMR spectroscopy by the addition of 5 μL of D2O (CIL) containing 10 mM 3-(trimethylsilyl) propionic-2,2,3,3-d4,acid (TMSP) to 15 μL of media. NMR spectra were acquired using Bruker-600 Avance spectrometer. TMSP was used as a chemical shift and quantitation reference. 13C6-glucose and 13C5-glutamine consumption, as well as 13C3-lactate production, was determined from the integrated NMR signals of these metabolites in both naive and conditioned media. Values obtained by NMR spectroscopy were normalized to cell number.
Tumor Xenografts and in Vivo Efficacy.
Female athymic nude mice (Harlan) were kept in a pathogen-controlled environment and handled in accordance with the Swiss law for animal protection. Nude mice were inoculated subcutaneously with either 3 × 106 HCT116, 2 × 106 A549, or 5 × 105 RKO cells. Tumors were allowed to grow for 7, 16, and 14 d, respectively, after which doxycycline was administered daily, and tumor formation was assessed every 2–3 d. Tumors were dissected, weighed, and lyophilized 2–3 wk after start of doxycycline treatment.
Statistical Analyses.
Statistical analyses were performed in Excel or GraphPad Prism. The PK activity, PEP, and other metabolite levels from IC-MS/MS and NMR were analyzed by two-tailed Student t test. For PK activity and PEP levels, significance was determined by P < 0.05, and the actual P values are indicated in the figure legends. For the rest of metabolites analyzed by NMR, significance is calculated comparing samples with and without doxycycline, and P value indicated on the graph pairs.
Supplementary Material
Footnotes
Conflict of interest statement: All of the authors are employees and shareholders of Novartis Pharma AG (Switzerland) or Novartis Institutes for BioMedical Research, Inc.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212780110/-/DCSupplemental.
References
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 3.Hitosugi T, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2(97):ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper JA, Reiss NA, Schwartz RJ, Hunter T. Three glycolytic enzymes are phosphorylated at tyrosine in cells transformed by Rous sarcoma virus. Nature. 1983;302(5905):218–223. doi: 10.1038/302218a0. [DOI] [PubMed] [Google Scholar]
- 5.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15(4):300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452(7184):181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 7.Luo W, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145(5):732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lv L, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42(6):719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macintyre AN, Rathmell JC. PKM2 and the tricky balance of growth and energy in cancer. Mol Cell. 2011;42(6):713–714. doi: 10.1016/j.molcel.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazurek S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43(7):969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 11.DeBerardinis RJ, et al. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang CV. PKM2 tyrosine phosphorylation and glutamine metabolism signal a different view of the Warburg effect. Sci Signal. 2009;2(97):pe75. doi: 10.1126/scisignal.297pe75. [DOI] [PubMed] [Google Scholar]
- 13.Yang C, et al. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69(20):7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480(7375):118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vander Heiden MG, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329(5998):1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vander Heiden MG, et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol. 2011;76:325–334. doi: 10.1101/sqb.2012.76.010900. [DOI] [PubMed] [Google Scholar]
- 17.Jiang JK, et al. Evaluation of thieno[3,2-b]pyrrole[3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg Med Chem Lett. 2010;20(11):3387–3393. doi: 10.1016/j.bmcl.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimada N, Shinagawa T, Ishii S. Modulation of M2-type pyruvate kinase activity by the cytoplasmic PML tumor suppressor protein. Genes Cells. 2008;13(3):245–254. doi: 10.1111/j.1365-2443.2008.01165.x. [DOI] [PubMed] [Google Scholar]
- 19.Sekiguchi Y, Mitsuhashi N, Kokaji T, Miyakoda H, Mimura T. Development of a comprehensive analytical method for phosphate metabolites in plants by ion chromatography coupled with tandem mass spectrometry. J Chromatogr A. 2005;1085(1):131–136. doi: 10.1016/j.chroma.2005.01.098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.