

Abstract
G-protein–coupled receptors (GPCRs) constitute the largest family of receptors and major pharmacological targets. Whereas many GPCRs have been shown to form di-/oligomers, the size and stability of such complexes under physiological conditions are largely unknown. Here, we used direct receptor labeling with SNAP-tags and total internal reflection fluorescence microscopy to dynamically monitor single receptors on intact cells and thus compare the spatial arrangement, mobility, and supramolecular organization of three prototypical GPCRs: the β1-adrenergic receptor (β1AR), the β2-adrenergic receptor (β2AR), and the γ-aminobutyric acid (GABAB) receptor. These GPCRs showed very different degrees of di-/oligomerization, lowest for β1ARs (monomers/dimers) and highest for GABAB receptors (prevalently dimers/tetramers of heterodimers). The size of receptor complexes increased with receptor density as a result of transient receptor–receptor interactions. Whereas β1-/β2ARs were apparently freely diffusing on the cell surface, GABAB receptors were prevalently organized into ordered arrays, via interaction with the actin cytoskeleton. Agonist stimulation did not alter receptor di-/oligomerization, but increased the mobility of GABAB receptor complexes. These data provide a spatiotemporal characterization of β1-/β2ARs and GABAB receptors at single-molecule resolution. The results suggest that GPCRs are present on the cell surface in a dynamic equilibrium, with constant formation and dissociation of new receptor complexes that can be targeted, in a ligand-regulated manner, to different cell-surface microdomains.
Keywords: live cell imaging, protein–protein interactions
G-protein–coupled receptors (GPCRs) constitute the largest family of cell-surface receptors and important pharmacological targets (1). Whereas research performed over the past 30 y has revealed in great detail the basic mechanisms and kinetics of GPCR signaling (1, 2), fundamental aspects, such as receptor di-/oligomerization or G-protein coupling and dissociation, remain controversial, mostly due to technical limitations to directly observe these phenomena (3–5).
Although GPCRs were initially thought to be monomeric, evidence accumulated over the past two decades suggests that they can form dimers or oligomers in intact cells (6–10). In a few cases, such as the γ-aminobutyric acid type B (GABAB) receptor and other family-C GPCRs, di-/oligomerization is essential for receptor function (10–12). It is now well established that functional GABAB receptors consist of a GABAB1 subunit (which binds GABA but cannot activate G proteins) and a GABAB2 subunit (which is binding deficient but can signal to G proteins) (10–12). Interestingly, heterodimerization of GABAB1 with GABAB2 masks an endoplasmic reticulum retention signal on the C-terminal tail of GABAB1, and this is required for GABAB1 to reach the cell surface (10–12). For family-A GPCRs, the situation is more controversial, even for well-studied members such as the β1-adrenergic receptor (β1AR) and the β2-adrenergic receptor (β2AR) (6–10). For instance, β2ARs have been suggested to be monomers (13), constitutive dimers (14, 15), or higher-order oligomers (16). Similarly, β1ARs have been suggested to form either stable (15) or transient interactions (16). Moreover, whereas the di-/oligomerization of family-A GPCRs has been proposed to play roles in receptor trafficking (17) and/or signaling (18), it is apparently not required for receptor function (19–21). The current uncertainty on these topics calls for new methods capable of directly monitoring the size and stability of GPCR supramolecular complexes at physiological expression levels in living cells.
Evidence for GPCR di-/oligomerization has been mostly obtained with biochemical methods or with biophysical techniques, such as resonance energy transfer (RET) (6–10). Compared with biochemical methods, fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) have the advantage of analyzing GPCR di-/oligomerization in intact cells. However, even though these methods have provided important insights on GPCR di-/oligomerization, they are based on average proximity measurements and usually require high receptor expression levels. Moreover, an intense debate arose a few years ago on the possible occurrence of RET due to random collisions (13, 22, 23).
Besides BRET and FRET, other optical methods have been proposed to study protein–protein interactions in living cells (4, 16, 24, 25). Of these, single-molecule microscopy has the great potential of directly observing the state and behavior of individual proteins. Interestingly, two recent single-molecule microscopy studies using fluorescent ligands showed dynamic dimerization of M1 muscarinic and N-formyl peptide receptors (26, 27).
Here, we used the SNAP-tag technology (28) to directly label cell-surface GPCRs with small organic fluorophores and visualize individual receptors on the surface of living cells by total internal reflection fluorescence microscopy (TIRF-M). Three prototypical GPCRs, i.e., the β1AR, the β2AR (both family-A GPCRs), and the GABAB receptor (family C), which are implicated in fundamental physiological processes such as heart contraction and neurotransmission and represent major pharmacological targets, were analyzed. This allowed us to precisely monitor over time and thus accurately compare the spatial arrangement, mobility, and supramolecular organization of these GPCRs, both under basal conditions and after agonist stimulation.
Results
Validation of Single-Molecule Analysis.
To directly and efficiently label cell-surface GPCRs with a bright fluorophore, we used the SNAP-tag (28). The SNAP-tag is a 20-kDa protein derived from the enzyme O6-alkylguanine-DNA alkyltransferase (AGT), which can be fused to a protein of interest and covalently labeled with fluorescent benzylguanine (BG) derivatives. We initially calibrated the method, using single molecules of the cell-impermeant Alexafluor 647 BG derivative (Alexa647-BG) spotted on glass coverslips that were imaged by TIRF-M (SI Results and Fig. S1).
Then, we evaluated the possibility of using the SNAP method to visualize single cell-surface proteins in living cells. A construct coding for a monomeric cell-surface receptor, CD86 (16), with an N-terminal SNAP-tag (SNAP-CD86) was used. After transfection of CHO cells with this construct, labeling with saturating concentrations of Alexa647-BG produced a highly specific staining (Fig. S2). When low-expressing cells were visualized by TIRF-M, individual fluorescent particles were visible in SNAP-CD86–transfected (Fig. S2 and Movie S1) but not in mock-transfected cells. Particles were automatically detected and tracked with algorithms developed by Jaqaman et al. (29). This analysis was able to correctly recognize (Fig. 1 A and B, blue circles) and track (Fig. 1 A and B, blue splines) a large majority of particles (typically >90%) present in SNAP-CD86 image sequences (Fig. 1 A and B and Movie S2).
Fig. 1.

Detection and tracking of individual SNAP-tagged proteins on the surface of living cells. (A) Enlarged view of single SNAP-CD86 particles on the surface of a living cell visualized by TIRF-M. Particles were automatically detected and tracked. The current position (blue circle) and trajectory (blue spline) of each particle are indicated. Apparent merging and splitting events are shown as green and red segments, respectively. (Scale bar, 5 μm.) (B) Same tracks on inverted image (white background). Inset, higher magnification.
Next, we exploited the single-molecule data to analyze the size of receptor complexes. In the case of a monomeric protein, particle intensities are expected to be normally distributed with a mean (μ) corresponding to that of single fluorophores. In the case of a mixture of complexes with different size (e.g., monomers, dimers, and oligomers), the distribution of particle intensities is expected to be the sum of n components, each having mean μ⋅n. Thus, a mixed Gaussian fitting was performed on the distribution of particle intensities to retrieve the weight of each underlying component. This analysis was validated on monomeric receptors containing either one (SNAP-CD86) or two (SNAP2×-CD86) SNAP-tags fused to their N termini. The latter represents a valuable control, because virtually all of the receptors are expected to be labeled with two fluorophores and should therefore be detected as “dimers”. In cells transfected with the SNAP-CD86 construct (Fig. S2), a predominant peak with an average intensity of 0.0205 ± 0.0057 (mean ± SD) was observed. These values were almost superimposable to those of Alexa647-BG molecules on glass (compare with Fig. S1) and were correctly detected by the mixed Gaussian fitting analysis as monomers (n = 1). In addition, particles bleached in one step, further confirming that they were single molecules (Fig. S3). An analysis of particle intensities at different expression levels (Fig. S2) revealed that, at all particle densities measured (0.15–0.45 particle/µm2), the monomeric fraction (n = 1) was largely predominant (>85%), with a small component of apparently dimeric particles (n = 2; approximately 5–18%). This component was due to random colocalization of two particles below the resolution limit of our method and not to SNAP-CD86 dimerization, as indicated by the fact that the same fractions of apparent dimers were obtained at 10% labeling efficiency (Fig. S2)—these results were also consistent with those of computer simulations (SI Results, Movie S3 and Fig. S4). Moreover, because these simulations indicated that the method was accurate up to 0.45 particle/μm2 (Fig. S4), only movies with particle densities below this value were analyzed. The analysis of SNAP2×-CD86 image sequences (Fig. S2) showed a largely predominant peak with μ and σ of intensity values approximately corresponding to two times those measured with SNAP-CD86. The mixed Gaussian fitting analysis correctly identified SNAP2×-CD86 particles as dimers. As expected for particles containing two fluorophores, particles were typically bleaching in two steps (Fig. S3). After partial photobleaching, a peak with the intensity of single fluorophores appeared in SNAP2×-CD86 image sequences (Fig. S2, dashed line). Such data obtained after partial photobleaching were used by the mixed Gaussian fitting algorithm to precisely estimate the intensity of single fluorophores in each image sequence (SI Materials and Methods). These results showed that the vast majority SNAP-tags were functional and were labeled with Alexa647-BG, thus excluding the presence of a relevant fraction of unlabeled, “dark” SNAP-tagged receptors. In addition, they indicated that the method was able to efficiently discriminate between populations of “monomeric” and “dimeric” receptors.
β1- and β2ARs Have Different Di-/Oligomerization States.
We then applied our method to two prototypical family-A GPCRs, i.e., β1- and β2ARs. Constructs coding for β1- and β2ARs with the SNAP-tag fused to their N termini were fully functional (Fig. S5). In CHO cells transfected with these constructs (Fig. 2 A and F) and labeled with Alexa647-BG, single fluorescent particles were detected by TIRF-M (Fig. S5). Although similar results were also obtained in HEK293 cells, CHO cells were chosen because they have no detectable β1-/β2ARs and we observed no agonist-dependent clustering of SNAP-tagged β1-/β2ARs. Typical intensity distributions of GPCR particles and mixed Gaussian fits are shown in Fig. 2 B and G. Data obtained at different particle densities were used to generate plots of the distribution of particle sizes over density, where the areas filled with different colors indicate the relative abundance of particles containing the indicated number of receptors (Fig. 2 C and H). Both receptors were present as mixtures of complexes with different size, which was increasing with particle density. The proportion of di-/oligomeric complexes and the dependency on particle density were much higher than in control SNAP-CD86 and simulated image sequences, as expected for true interactions. The effect of density was more prominent for the β1AR, in which case the fraction of monomers was ∼70% at low densities (0.15–0.3 particle/µm2), whereas dimers predominated over monomers and a small fraction of tri-/tetramers at the highest particle densities measured (Fig. 2C). The β2AR had a higher tendency of forming dimers, which constituted ∼60% at low densities (0.15–0.3 particle/µm2), the rest being represented by monomers (Fig. 2H). At higher densities, β2AR contained a mixture of di-, tri-, and tetramers, which at 0.4–0.45 particle/µm2 accounted for ∼50%, 30%, and 15%, respectively (Fig. 2H). The different abundance of monomers between β1- and β2ARs at low densities was highly statistically significant (Fig. 2G, Inset). These results were confirmed by a separate analysis based on the photobleaching steps (Fig. S3). For this purpose, the intensity profile of each particle was fitted with a stepwise model and the number of receptors in each particle was estimated on the basis of the number and size of steps. The results were in very good agreement with those of the mixed Gaussian fitting analyses.
Fig. 2.

Analysis of β1- (A–E) and β2- (F–J) AR oligomerization and lateral mobility by single-molecule TIRF-M. (A and F) Schematic representation of the used SNAP-tagged constructs. (B and G) Representative intensity distributions of Alexa647-labeled particles. Particle densities were 0.24 (B) and 0.25 (G) particle/μm2. Data were fitted with a mixed Gaussian model. A mixed Gaussian fit after partial photobleaching (dashed lines) was used to precisely estimate the intensity of single fluorophores in each image sequence. (G, Inset) Comparison of the fraction of monomeric β1AR and β2AR particles at low density (0.15–0.3 particle/μm2). Each data point represents one cell. *P = 0.0003 by Mann–Whitney test. (C and H) Dependency of the distribution of particle components on particle density. Shown is the cumulative distribution of mono-, di-, tri-, and tetramers of Alexa647-labeled receptors, based on mixed Gaussian fitting analyses like those shown in B and G, as a function of particle density. Data were fitted using third-order polynomial functions to provide an indication of their trend. Each data point represents one cell. [n = 6,181 particles from 27 different cells (C) and 7,419 particles from 30 different cells (H)]. (D and I) Distribution of diffusion coefficients of receptor particles calculated from their mean square displacement (MSD). Insets, MSD plots; shown are the mean (red) as well as the 10% and 90% percentiles (shaded area) of particles that were tracked for at least 3 s; black, data of representative individual particles. (E and J) Effect of the size of GPCR complexes on their lateral diffusion. The size of individual particles was estimated on the basis of the number of bleaching steps. Shown are box plots of diffusion coefficients measured for particles of different size. The boxes encompass the 25% and 75% percentiles and median values are indicated by red lines. Differences in E and J are statistically significant by a Kruskal–Wallis test (P < 0.0001) followed by Dunn’s test (*P < 0.001).
Next we analyzed the movement of receptor particles on the cell surface (Fig. 2 D and I). Plots of their mean square displacement (MSD) against time gave linear relationships, suggesting that the receptors were freely diffusing. MSD data of individual particles indicated no corralling at this temporal resolution. A calculation of their diffusion coefficients from MSD data showed that β1ARs were diffusing quickly on the cell surface (median diffusion coefficient = 0.052 μm2⋅s−1; peak = 0.032–0.043 μm2⋅s−1) (Fig. 2D). β2ARs had a slightly slower diffusion speed (median diffusion coefficient = 0.039 μm2⋅s−1; peak = 0.021–0.032 μm2⋅s−1) (Fig. 2I). Overall, these values are similar to those reported on β2ARs with other methods (30) as well as on other receptors by TIRF-M (26). The speed of receptor diffusion was negatively correlated with the size of receptor complexes (Fig. 2 E and J).
β1- and β2ARs Undergo Transient Interactions.
Because the observed increase of receptor di-/oligomerization with receptor density was suggesting the presence of transient interactions, we attempted to directly observe and characterize such interactions. An example of a transient colocalization between two β1AR particles is shown in Fig. 3 A–C. For each pair of colocalizing particles, the colocalization time (Δt), i.e., the time between merging and splitting, was automatically calculated and Δt values were used to estimate the apparent lifetime of particle colocalizations 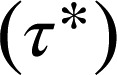 by fitting the data to an exponential decay function. Colocalization of β1ARs should result from both random colocalizations and true receptor–receptor interactions. To distinguish these true interactions from random colocalizations, we simulated particles with the same characteristics (diffusion coefficients, intensity distribution, and bleaching rate) of β1ARs, but showing no interactions. The estimated
by fitting the data to an exponential decay function. Colocalization of β1ARs should result from both random colocalizations and true receptor–receptor interactions. To distinguish these true interactions from random colocalizations, we simulated particles with the same characteristics (diffusion coefficients, intensity distribution, and bleaching rate) of β1ARs, but showing no interactions. The estimated 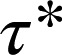 -value for these random colocalizations
-value for these random colocalizations 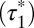 was 1.08 s (95% confidence interval: 1.04–1.12) and served as a background value (Fig. 3D). Almost identical
was 1.08 s (95% confidence interval: 1.04–1.12) and served as a background value (Fig. 3D). Almost identical 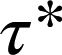 -values (1.01 s; 95% confidence interval, 0.96–1.05) were obtained with control monomeric (SNAP-CD86) receptors. Then, β1AR data were fitted to the sum of two exponential functions, the first one with a lifetime equal to
-values (1.01 s; 95% confidence interval, 0.96–1.05) were obtained with control monomeric (SNAP-CD86) receptors. Then, β1AR data were fitted to the sum of two exponential functions, the first one with a lifetime equal to 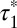 (Fig. 3E). The apparent lifetime of the second component
(Fig. 3E). The apparent lifetime of the second component 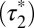 , i.e., the one resulting from receptor–receptor interactions, was estimated to be 5.05 s (95% confidence interval: 4.17–5.93). Similar results (
, i.e., the one resulting from receptor–receptor interactions, was estimated to be 5.05 s (95% confidence interval: 4.17–5.93). Similar results (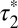 = 4.61 s; 95% confidence interval, 4.02–5.40) were obtained for low-density β2AR movies (Fig. 3F). Knowing
= 4.61 s; 95% confidence interval, 4.02–5.40) were obtained for low-density β2AR movies (Fig. 3F). Knowing 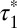 and
and 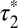 , the true lifetime of receptor–receptor interactions can be roughly estimated as
, the true lifetime of receptor–receptor interactions can be roughly estimated as 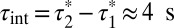 at 20 °C (27).
at 20 °C (27).
Fig. 3.

Dynamic visualization of receptor–receptor interactions. (A) Example of two Alexa647-labeled β1AR particles showing a transient colocalization. A merging event (green) is followed after some frames by a splitting event (red). Images are centered on the particles’ position. (Scale bar, 1 μm.) (B) Same traces as in A on a white background and without centering. (C) Intensity profiles of the traces in A, showing intensity doubling upon merging. (D) Colocalizations between control particles devoid of true interactions. Black, simulated particles with diffusion coefficients, intensity distribution, and bleaching rate analogous to those of β1AR particles. Orange, monomeric SNAP-CD86 receptors. The apparent lifetime of particle colocalizations (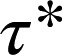 ; 95% confidence intervals in parentheses) was calculated by fitting colocalization time data with an exponential decay function. (E) Lifetime of β1AR colocalizations. Colocalization time data derived from experiments as in A (green) were fitted to the sum (black) of two exponential decays (blue and red, respectively). The obtained apparent lifetimes of particle colocalizations (
; 95% confidence intervals in parentheses) was calculated by fitting colocalization time data with an exponential decay function. (E) Lifetime of β1AR colocalizations. Colocalization time data derived from experiments as in A (green) were fitted to the sum (black) of two exponential decays (blue and red, respectively). The obtained apparent lifetimes of particle colocalizations (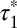 and
and 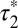 ; 95% confidence intervals in parentheses) were then used to estimate the true lifetime of receptor–receptor interactions. (F) Same as E with low-density (<0.35 particle/μm2) β2AR movies. Data in E and F are from 20 and 8 different cells, respectively. Data in E and F were fitted better with two components than with one, as judged by an F-test (P < 1.0 × 10−8).
; 95% confidence intervals in parentheses) were then used to estimate the true lifetime of receptor–receptor interactions. (F) Same as E with low-density (<0.35 particle/μm2) β2AR movies. Data in E and F are from 20 and 8 different cells, respectively. Data in E and F were fitted better with two components than with one, as judged by an F-test (P < 1.0 × 10−8).
Functional GABAB Heterodimers Form Large Complexes Tethered to the Cortical Actin Cytoskeleton.
We then applied our method to the GABAB receptor, a prototypical family-C GPCR, consisting of heteromers between GABAB1 and GABAB2 subunits (10–12). Initially, we transfected CHO cells with N-terminally SNAP-tagged GABAB1 and GABAB2 subunits (31). As expected, the SNAP-tagged GABAB1 subunit alone did not reach the cell surface, whereas the SNAP-tagged GABAB2 subunit alone was detectable on the cell surface, where it formed a mixture of mono-/di-/oligomers with high lateral mobility (Fig. S6). Thus, cells cotransfected with both SNAP-tagged subunits (Fig. S7) most likely contained a mixture of labeled homomeric (composed of GABAB2) and heteromeric complexes. Therefore, to selectively analyze functional GABAB complexes, we took advantage of the fact that the GABAB1 subunit requires dimerization with the GABAB2 subunit to reach the cell surface and analyzed cells cotransfected with SNAP-tagged GABAB1 and untagged GABAB2 subunits (Fig. 4 A and B). Note that in these experiments the labeling stoichiometry is one fluorophore per heterodimer (h.d.). At low densities (0.15–0.2 particle/µm2), GABAB complexes prevalently consisted of heterodimers (n = 1 h.d.) (Fig. 4 C and D). The proportion of higher-order oligomers increased with particle density. At the highest density analyzed (0.4–0.45 particle/µm2), tetramers (dimers of dimers; n = 2 h.d.) and octamers (tetramers of dimers; n = 4 h.d.) constituted a large fraction (30–40% each) of the detected complexes. Very similar results were obtained after correction for random colocalization (SI Results and Fig. S8). These findings were confirmed by an additional analysis, in which the intensity profiles of each receptor particle were fitted with a stepwise model and the number and size of steps were used to estimate the size of the receptor particles (Fig. S3).
Fig. 4.

Selective analysis of GABAB receptor heterodimers. (A) Schematic representation of the used constructs. Cells were cotransfected with SNAP-GABAB1 and wild-type GABAB2 subunits. (B) Representative image of a cell transfected with both constructs, labeled with Alexa647-BG and visualized by TIRF-M. Particle density = 0.43 particle/μm2. (C) Intensity distribution of the particles in B. Data were fitted with a mixed Gaussian model. (D) Dependency of the distribution of particle components on particle density. Data are based on mixed Gaussian fitting analyses like those shown in C and are represented as in Fig. 2 C and H. Because only the GABAB1 subunit is labeled, one fluorophore (n = 1) corresponds to one heterodimer (h.d.) [n = 4,472 particles from 17 different cells]. (E) Distribution of diffusion coefficients of GABAB heteromeric particles on the cell surface. Inset, MSD plot; shown are the mean (red) as well as the 10% and 90% percentiles (shaded area) of particles that were tracked for at least 3 s; black, representative data of individual mobile and immobile particles. (F) Effect of the size of GABAB heteromeric particles on their lateral diffusion. The size of individual particles was estimated on the basis of the number of bleaching steps. Shown are box plots of diffusion coefficients measured for particles of different size. The boxes encompass the 25% and 75% percentiles and median values are indicated by red lines. Differences are statistically significant by a Kruskal–Wallis test (P < 0.0001) followed by Dunn’s test (*P < 0.001). (G) Image of a cell with ∼15 times higher receptor density than in B, showing GABAB receptors arranged in rows. (H) Interaction of GABAB receptors with the cortical actin cytoskeleton. Cells were cotransfected with SNAP-GABAB1 and wild-type GABAB2 subunits, treated or not (control) with latrunculin A and labeled with Alexa647-BG. After fixation, actin filaments were stained with Alexa488-phalloidin. (Scale bars, 5 μm.) (I) Same as D in cells pretreated with latrunculin A [n = 7,748 particles from 26 different cells].
Next, we evaluated the diffusion speed of functional GABAB complexes. Most particles had limited mobility (median diffusion coefficient = 0.028 μm2⋅s−1; peak < 0.01 μm2⋅s−1; particles with diffusion coefficients <0.01 μm2⋅s−1 = 24.6%; Fig. 4E), which was negatively correlated with particle size (Fig. 4F). Interestingly, GABAB receptors showed a tendency to arrange in rows. This was already visible at receptor densities used for the single-molecule analysis and was even better appreciated at higher receptor densities (Fig. 4G). Moreover, GABAB receptors colocalized with actin fibers stained with fluorescent phalloidin, suggesting an interaction between these receptors and the cortical actin cytoskeleton. Actin depolymerization with latrunculin A abolished the arrangement of GABAB receptors in rows (Fig. 4H), but did not modify the size of GABAB complexes (Fig. 4I).
Agonist Stimulation Increases the Lateral Mobility of GABAB Receptors, but not of β1-/β2ARs.
Labeling the receptors themselves rather than the ligands allowed us to investigate whether the di-/oligomerization or mobility of GPCRs was affected by agonists. CHO cells expressing SNAP-tagged β1-/β2ARs or GABAB receptors were stimulated with 10 μM isoproterenol or 50 μM GABA, respectively. Agonist stimulation had no effect on the di-/oligomerization state of any receptor (Fig. S9) or on the mobility of β1- and β2ARs (Fig. 5 A and B). However, it did increase the mobility of GABAB receptors (median diffusion coefficient from 0.027 to 0.056 μm2⋅s−1; Fig. 5C).
Fig. 5.

Effect of agonists on receptor mobility. (A–C) Cells were transfected with SNAP-β1AR (A), SNAP-β2AR (B), or SNAP-GABAB1 plus wild-type GABAB2 (C) constructs, labeled with Alexa647-BG, and stimulated with the indicated concentrations of agonists 5–10 min before image acquisition. Shown are the distributions of diffusion coefficients of the analyzed receptor particles compared with control nonstimulated cells. Differences in C are statistically significant by a Mann–Whitney test (P < 0.0001).
Discussion
The extent and functional relevance of GPCR di-/oligomerization is highly debated. Whereas monomeric GPCRs, including rhodopsin (19, 20) and the β2AR (21), efficiently activate G proteins, several studies suggest that GPCRs can form di-/oligomers (6–10). The exact size and stability of such complexes are largely unknown. We used single-molecule TIRF-M combined with direct labeling of the receptors with small organic fluorophores via SNAP-tags to compare the spatial arrangement, mobility, and di-/oligomerization state of three GPCRs, i.e., the β1AR, β2AR, and GABAB receptors.
The β1AR was found to be prevalently monomeric at low densities and to form an increasing number of dimers at higher densities, due to the occurrence of transient interactions. In contrast, the β2AR had a higher tendency to form dimers or higher-order oligomers already at lower densities. Because the lifetimes of receptor–receptor interactions appear similar for the two receptors, this difference must result from other factors, such as different efficiencies in converting a “collision” into an interaction, different interactions with other proteins capable of interfering with dimerization, or localizations in different subcellular microdomains, as shown for β1- and β2ARs in cardiomyocytes (31), leading to dissimilar effective densities of the two receptors. Moreover, β1AR data were characterized by a relatively higher cell-to-cell biological variability, which might reflect a more dynamic type of di-/oligomerization. These results are consistent with our recent fluorescence recovery after photobleaching (FRAP) study (16) and provide a direct quantification of the size of these complexes and of their dynamics. They are also consistent with the results of the two previous single-molecule studies, providing further evidence that GPCR dimerization is a dynamic process, in which receptors rapidly associate and dissociate.
GABAB receptors are known to be obligate heterodimers (10–12). Recent FRET measurements suggest that they might organize into larger supramolecular complexes (32, 33). Our study provides a direct observation of such complexes and a precise evaluation of their size. On the one hand, we found GABAB receptors to be present in equilibrium between heterodimers and higher-order oligomers, with a relative preference for tetramers (dimers of dimers) and octamers (tetramers of dimers). Whereas GABAB heterodimers are stable due to strong noncovalent interactions, the dependency of the formation of higher-order oligomers on receptor density suggests that these larger complexes are the result of weaker and likely transient interactions among heterodimers. Moreover, the relative predominance of tetramers and octamers might reflect an ordered and asymmetric spatial organization within the oligomers. On the other hand, the analysis of receptor mobility revealed that GABAB receptor heteromers are largely immobile and arranged into rows on the surface of living cells. The latter phenomenon, which is apparently due to an interaction between the GABAB1 subunit and the actin cytoskeleton, might play a role in the spatial organization of GABAB receptors at synapses. This interaction was seemingly stronger for oligomers than for heterodimers, as suggested by a pronounced difference in their diffusion coefficients. However, the interaction with actin fibers was not responsible for the formation of the observed large GABAB complexes, as indicated by the results of actin depolymerization with latrunculin A. Thus, these large receptor complexes appear to be due to true oligomerization, either via direct receptor–receptor interactions or with intervention of a scaffold. Finally, our results revealed the presence of cell-surface GABAB2 monomers, which is consistent with the lack of an ER-retention signal in the GABAB2 subunit and a different type of interaction involved in GABAB2 homodimerization (34, 35).
The effect of ligands on GPCR di-/oligomerization is controversial (6–8, 10). For instance, previous experiments on the β2AR in intact cells (14) or with purified receptors (36) indicate that agonists have minor effects that could be either due to small changes in steady-state di-/oligomerization or due to conformational changes in individual protomers. No effects of ligands were instead observed in FRAP experiments (16). Our data support the view that agonist binding has no major acute effects on the di-/oligomerization of β1AR, β2AR, and GABAB receptors. In contrast, we observed an increase in the lateral mobility of GABAB receptors after agonist stimulation, which suggests the occurrence of ligand-regulated interactions with the actin cytoskeleton.
The method used in this study is complementary to those based on RET and FRAP. Specifically, it allows to dynamically quantify the size of protein complexes and to identify subpopulations thereof, at receptor densities that do not exceed physiological levels. For instance, considering β2AR complexes, a density of 0.45 particle/μm2 corresponds to ∼1 receptor/μm2, which is in the same range as found in different cell types (37).
Another characteristic of our approach is the use of the SNAP-tag technology (28) to directly label cell-surface GPCRs, which has several advantages compared with the use of fluorescent ligands (26, 27). First, whereas the latter methods are associated with partial labeling and/or possible influences of negative cooperativity in ligand binding (38, 39), virtually complete receptor labeling can be achieved. Second, our approach allows us to analyze ligand-free receptors and to study the effects of agonists or antagonists. Third, the present method can be easily extended to other GPCRs and cell-surface proteins. Although beyond the scope of the present study, a similar approach combined with dual color labeling, e.g., using SNAP- and CLIP-tags (28, 40), might also be used to study interactions between different proteins such as those occurring between receptors and G proteins or among G-protein subunits. A limitation of our method is that because its spatial resolution is dictated by the diffraction of light, it cannot directly distinguish between complexes due to direct receptor–receptor interactions and complexes due to receptor interactions with a scaffold.
Taken together, our study shows that a method based on labeling with SNAP-tags and single-molecule TIRF-M can be used to dynamically quantify and thus compare the spatial arrangement, mobility, and di-/oligomerization of different GPCRs and possibly other cell-surface proteins. Data obtained on β1AR, β2AR, and GABAB receptors indicate that the cell-surface topology and mobility as well as type and degree of receptor di-/oligomerization vary considerably among different receptors. GPCRs appear to be highly dynamic, constantly associating and dissociating with other receptors to form new supramolecular complexes as well as with other proteins to maintain their specific location on the cell surface.
Materials and Methods
Cell Culture and Transfection.
For single-molecule experiments, CHO cells were cultured in DMEM/F12 medium supplemented with 10% (vol/vol) FCS, penicillin, and streptomycin at 37 °C, 5% (vol/vol) CO2. Cells were plated at a density of 3 × 105 cells per well onto 24-mm clean glass coverslips and transfected using the Lipofectamine 2000 reagent (Invitrogen), following the manufacturer’s protocol. For each well, 2 μg DNA and 6 μL Lipofectamine 2000 were used. Cells were analyzed 8–12 h after transfection to achieve low expression levels.
SNAP Labeling.
Cells were labeled with 1 µM Alexa647-BG (Alexafluor 647-SNAP Surface; New England Biolabs) in complete (+FCS) phenol-red–free medium for 30 min at 37 °C. At the end of the incubation, the cells were washed three times with complete phenol-red–free medium and immediately imaged. These conditions resulted in saturating labeling of cell-surface SNAP-tagged receptors (Fig. S2).
Total Internal Reflection Fluorescence Microscopy.
A commercial TIRF microscope (Leica AM TIRF) equipped with an EM-CCD camera (Cascade 512B; Roper Scientific), a 100× oil-immersion objective (HCX PL APO 100×/1.46), and a 635-nm diode laser was used. To avoid photobleaching before image acquisition, cells were searched and focused in bright field and a fine focus adjustment in TIRF mode was performed using only 2% laser power, an intensity insufficient for detecting single molecules. This procedure resulted in negligible photobleaching. Afterward, laser power was set to 83% and image sequences (400–600 frames) were acquired with an exposure time of 50 ms, resulting in the acquisition of an image every 96 ms. The penetration depth of the evanescent field was ∼110 nm. Illumination intensity was homogeneous over the imaged area (maximum difference = 10%). Under these conditions, the photobleaching half-life was 6.82 ± 0.12 s. The microscope was equipped with an incubator and a temperature control unit. Experiments were performed at 20.5 ± 0.3 °C. Cells were imaged in a buffer containing 137 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM Hepes, pH 7.3. Only cells with less than 0.45 receptor particle/μm2 were analyzed.
Additional Methods.
Details about plasmids, coverslip cleaning, determination of Alexa647-BG labeling efficiency, radioligand binding, measurement of cAMP concentrations, Latrunculin A treatment/actin staining, and computational analyses are available in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Bianca Klüpfel, Monika Frank, and Christian Dees for excellent technical support. This work was supported by grants from the European Research Council (Advanced Grant Towards the Quantal Nature of Receptor/cAMP Signals) and the Deutsche Forschungsgemeinschaft (SFB487). T.S. was partially supported by the Royal Golden Jubilee Ph.D. Program.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205798110/-/DCSupplemental.
References
- 1.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 2.Lohse MJ, Bünemann M, Hoffmann C, Vilardaga JP, Nikolaev VO. Monitoring receptor signaling by intramolecular FRET. Curr Opin Pharmacol. 2007;7(5):547–553. doi: 10.1016/j.coph.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Lohse MJ, et al. Optical techniques to analyze real-time activation and signaling of G-protein-coupled receptors. Trends Pharmacol Sci. 2008;29(3):159–165. doi: 10.1016/j.tips.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Qin K, Dong C, Wu G, Lambert NA. Inactive-state preassembly of Gq-coupled receptors and Gq heterotrimers. Nat Chem Biol. 2011;7(10):740–747. doi: 10.1038/nchembio.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Challiss RA, Wess J. Receptors: GPCR-G protein preassembly? Nat Chem Biol. 2011;7(10):657–658. doi: 10.1038/nchembio.665. [DOI] [PubMed] [Google Scholar]
- 6.Angers S, Salahpour A, Bouvier M. Dimerization: An emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 7.Milligan G, Bouvier M. Methods to monitor the quaternary structure of G protein-coupled receptors. FEBS J. 2005;272(12):2914–2925. doi: 10.1111/j.1742-4658.2005.04731.x. [DOI] [PubMed] [Google Scholar]
- 8.Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2(4):274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- 9.Ferré S, et al. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5(3):131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pin JP, et al. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev. 2007;59(1):5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]
- 11.Jones KA, et al. GABAB receptors function as a heteromeric assembly of the subunits GABABR1 and GABABR2. Nature. 1998;396(6712):674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- 12.White JH, et al. Heterodimerization is required for the formation of a functional GABAB receptor. Nature. 1998;396(6712):679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- 13.James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods. 2006;3(12):1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- 14.Angers S, et al. Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET) Proc Natl Acad Sci USA. 2000;97(7):3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. Quantitative assessment of β1- and β2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem. 2002;277(47):44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- 16.Dorsch S, Klotz KN, Engelhardt S, Lohse MJ, Bünemann M. Analysis of receptor oligomerization by FRAP microscopy. Nat Methods. 2009;6(3):225–230. doi: 10.1038/nmeth.1304. [DOI] [PubMed] [Google Scholar]
- 17.Cao TT, Brelot A, von Zastrow M. The composition of the β2- adrenergic receptor oligomer affects its membrane trafficking after ligand-induced endocytosis. Mol Pharmacol. 2005;67(1):288–297. doi: 10.1124/mol.104.003608. [DOI] [PubMed] [Google Scholar]
- 18.Hebert TE, et al. A peptide derived from a β2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem. 1996;271(27):16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- 19.Ernst OP, Gramse V, Kolbe M, Hofmann KP, Heck M. Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. Proc Natl Acad Sci USA. 2007;104(26):10859–10864. doi: 10.1073/pnas.0701967104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whorton MR, et al. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem. 2008;283(7):4387–4394. doi: 10.1074/jbc.M703346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whorton MR, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA. 2007;104(18):7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouvier M, Heveker N, Jockers R, Marullo S, Milligan G. BRET analysis of GPCR oligomerization: Newer does not mean better. Nat Methods. 2007;4(1):3–4, author reply 4. doi: 10.1038/nmeth0107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salahpour A, Masri B. Experimental challenge to a ‘rigorous’ BRET analysis of GPCR oligomerization. Nat Methods. 2007;4(8):599–600, author reply 601. doi: 10.1038/nmeth0807-599. [DOI] [PubMed] [Google Scholar]
- 24.Briddon SJ, Hill SJ. Pharmacology under the microscope: The use of fluorescence correlation spectroscopy to determine the properties of ligand-receptor complexes. Trends Pharmacol Sci. 2007;28(12):637–645. doi: 10.1016/j.tips.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulbrich MH, Isacoff EY. Subunit counting in membrane-bound proteins. Nat Methods. 2007;4(4):319–321. doi: 10.1038/NMETH1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hern JA, et al. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc Natl Acad Sci USA. 2010;107(6):2693–2698. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasai RS, et al. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J Cell Biol. 2011;192(3):463–480. doi: 10.1083/jcb.201009128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keppler A, et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21(1):86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 29.Jaqaman K, et al. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5(8):695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barak LS, et al. Internal trafficking and surface mobility of a functionally intact β2-adrenergic receptor-green fluorescent protein conjugate. Mol Pharmacol. 1997;51(2):177–184. doi: 10.1124/mol.51.2.177. [DOI] [PubMed] [Google Scholar]
- 31.Nikolaev VO, et al. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327(5973):1653–1657. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 32.Maurel D, et al. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: Application to GPCR oligomerization. Nat Methods. 2008;5(6):561–567. doi: 10.1038/nmeth.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Comps-Agrar L, et al. The oligomeric state sets GABAB receptor signalling efficacy. EMBO J. 2011;12:2336–2349. doi: 10.1038/emboj.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pagano A, et al. C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. J Neurosci. 2001;21(4):1189–1202. doi: 10.1523/JNEUROSCI.21-04-01189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kammerer RA, et al. Heterodimerization of a functional GABAB receptor is mediated by parallel coiled-coil alpha-helices. Biochemistry. 1999;38(40):13263–13269. doi: 10.1021/bi991018t. [DOI] [PubMed] [Google Scholar]
- 36.Fung JJ, et al. Ligand-regulated oligomerization of β2-adrenoceptors in a model lipid bilayer. EMBO J. 2009;28(21):3315–3328. doi: 10.1038/emboj.2009.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hegener O, et al. Dynamics of β2-adrenergic receptor-ligand complexes on living cells. Biochemistry. 2004;43(20):6190–6199. doi: 10.1021/bi035928t. [DOI] [PubMed] [Google Scholar]
- 38.Urizar E, et al. Glycoprotein hormone receptors: Link between receptor homodimerization and negative cooperativity. EMBO J. 2005;24(11):1954–1964. doi: 10.1038/sj.emboj.7600686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: Uncharted pharmacological landscapes. Pharmacol Rev. 2010;62(4):701–725. doi: 10.1124/pr.110.002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gautier A, et al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15(2):128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.