

Abstract
12/15-Lipoxygenase (LOX) activity is elevated in vascular diseases associated with impaired nitric oxide (⋅NO) bioactivity, such as hypertension and atherosclerosis. In this study, primary porcine monocytes expressing 12/15-LOX, rat A10 smooth muscle cells transfected with murine 12/15-LOX, and purified porcine 12/15-LOX all consumed ⋅NO in the presence of lipid substrate. Suppression of LOX diene conjugation by ⋅NO was also found, although the lipid product profile was unchanged. ⋅NO consumption by porcine monocytes was inhibited by the LOX inhibitor, eicosatetraynoic acid. Rates of arachidonate (AA)- or linoleate (LA)-dependent ⋅NO depletion by porcine monocytes (2.68 ± 0.03 nmol ⋅ min−1 ⋅ 106 cells−1 and 1.5 ± 0.25 nmol ⋅ min−1 ⋅ 106 cells−1, respectively) were several-fold greater than rates of ⋅NO generation by cytokine-activated macrophages (0.1–0.2 nmol ⋅ min−1 ⋅ 106 cells−1) and LA-dependent ⋅NO consumption by primary porcine monocytes inhibited ⋅NO activation of soluble guanylate cyclase. These data indicate that catalytic ⋅NO consumption by 12/15-LOX modulates monocyte ⋅NO signaling and suggest that LOXs may contribute to vascular dysfunction not only by the bioactivity of their lipid products, but also by serving as catalytic sinks for ⋅NO in the vasculature.
Keywords: arachidonate, linoleate, hypertension, atherosclerosis
Nitric oxide (⋅NO) is a central mediator of vascular tone, with strict control of ⋅NO essential for blood pressure regulation (1, 2). Reaction with oxyhemoglobin (oxyHb) has generally been viewed the major fate of vascular ⋅NO; however, recent work has shown that hemoglobin sequestration, combined with flow, decreases the rate of ⋅NO reaction with oxyHb, rendering it less effective as an ⋅NO scavenger (3–5). In vascular disease, accelerated ⋅NO loss is often seen (6–8). In hypertension, a role for superoxide (O2⨪) has been found; however, this is only responsible for a proportion of ⋅NO removal, because O2⨪ scavengers incompletely normalize blood pressure (8). Finally, the biological half-life of ⋅NO under oxyHb-free conditions (0.1–3 sec) is shorter than expected rates of ⋅NO oxidation (9, 10). These observations suggest that cell-dependent ⋅NO scavenging occurs under both physiological and pathophysiological conditions.
Vascular ⋅NO consumption may occur via reaction with free radicals formed during turnover of enzymes that are up-regulated during disease. For example, we have shown that 15-lipoxygenases (LOX) and prostaglandin H synthase (PGHS) catalytically consume ⋅NO (11, 12). A central role for oxymyoglobin in regulating coronary blood flow and cardiac contractility has been recently shown (13). Finally, the heme peroxidases, myeloperoxidase (MPO), lactoperoxidase (LPO), and eosinophil peroxidase (EPO) scavenge ⋅NO as a reducing peroxidase substrate (14).
LOXs are non-heme iron-containing enzymes that catalyze the oxidation of unsaturated fatty acids (15, 16). Leukocyte-type 12-LOX, also known as 12/15-LOX, inserts oxygen primarily at C12 of arachidonate (AA) forming 12S-hydroperoxy-5Z,8Z,10E,14Z-eicosatetraenoic acid [12(S)HPETE] (15, 17), whereas 15-LOX forms 15S-hydroperoxy-5Z,8Z,11Z,13E-eicosatetraenoic acid [15(S)HPETE]. Unique among LOXs, 15-LOX and 12/15-LOX can oxidize membrane-bound fatty acids, and also linoleate (LA) (18). 15- and 12/15-LOXs play a central role in vascular disease, because (i) 15-LOX mRNA, protein, and lipid products are found in atheroma, (ii) inhibition of 15-LOX prevents atherosclerosis in rabbits, and (iii) functional inactivation of the 12/15-LOX gene slows down aortic lipid deposition in apo E-deficient mice, and inhibits streptozotocin-induced diabetes (19–25). Furthermore, in vivo, 12/15-LOX expression is required for neointimal thickening in balloon-injured rat aortae (26, 27). 12/15-LOX is elevated in smooth muscle of angiotensin (ang) II-dependent hypertensive rats (28–30). Also, ang II up-regulates 12/15-LOX in mouse macrophages and porcine smooth muscle in vitro via the AT1 receptor (31, 32). Inhibition of 12/15-LOX by nonspecific inhibitors reduces blood pressure in hypertensive rats, and ang II-induced smooth muscle hypertrophy (33–36). The mechanism(s) by which 12/15-LOX contributes to vascular tone and smooth muscle proliferation is unclear.
Previously, purified rabbit and human 15-LOX was found to catalytically consume ⋅NO and inhibit soluble guanylate cyclase (sGC) activation in vitro (11). In this study, the concept of tissue regulation of ⋅NO by LOX is extended to 12/15-LOX, and we show that activity of this isoform in primary monocytes is sufficient to prevent sGC activation by 2 μM ⋅NO. These studies demonstrate a novel mechanism by which cellular LOXs can contribute to vascular disease, independently of their lipid product bioactivity, namely by attenuating the vasodilatory and antiinflammatory actions of ⋅NO.
Experimental Procedures
Materials.
Puromycin was from CLONTECH. 12(S)HPETE, 15(S)HPETE, 12S-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid [12(S)HETE], 5,8,11,14-eicosatetraynoic acid (ETYA), AA, and porcine 12/15-LOX were from Alexis (Nottingham, U.K.). Biotrak cGMP radioimmunoassay kits were from Amersham Pharmacia. LA was from Nu Chek Prep (Elysian, MN). Lymphoprep was from Nycomed (Oslo). Cell culture media was from GIBCO Life Technologies. Unless otherwise stated, other reagents were from Sigma.
Generation of 12/15-LOX-Transfected Rat Smooth Muscle Cells.
Rat smooth muscle A10 cells from American Type Culture Collection, cultured in DMEM, 15% (vol/vol) FCS, 100 μg/ml penicillin, 100 units/ml streptomycin, and 2 mM glutamine, were cotransfected with plasmids encoding mouse 12/15-LOX cDNA (pcDNAm12LO, a gift from Dr. C. Funk, University of Pennsylvania, Philadelphia) and pPUR [selection vector that confers puromycin resistance (25:1 ratio); Promega] using lipotaxi (Boehringer Mannheim). The empty vector pcDNA1 (Invitrogen) without the 12/15-LOX cDNA insert was separately cotransfected with pPUR. About 6 h posttransfection, cells were placed in recovery medium containing 1% (vol/vol) FCS and fed the next day with complete medium. Cells were split 1:12, 48 h post transfection, and then 24 h later selection initiated by using 2.0 μg/ml puromycin. After resistant colonies developed (3–4 weeks), clones were picked by using cloning rings and expanded. Clones were checked for 12/15-LOX overexpression by Western blotting and 12(S)HETE generation. Stable transfectants were maintained in complete medium with 1 μg/ml puromycin.
Isolation of Porcine Monocytes.
Porcine monocytes were isolated as described (37). Briefly, whole blood was diluted into PBS (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4) at 10:1, blood:PBS (vol/vol) containing 10 units/ml heparin and 0.5 mM EDTA then mixed 1:1 (vol/vol) with PBS, 0.8% (wt/vol) trisodium citrate, and 2% (wt/vol) dextran T-70 (pH 7.4) and left to sediment for 60 min. The upper plasma layer was underlaid with ice-cold Lymphoprep [2:1, plasma:Lymphoprep (vol/vol)]. This was centrifuged (800 × g, 20 min, 4°C) before harvesting the interface and diluting 1:1 (vol/vol) with PBS, 0.4% (wt/vol) trisodium citrate (pH 7.4; PBS/citrate). Cells were pelletted (400 × g, 10 min, 4°C) and then washed (recentrifuged at 400 × g, 10 min, 4°C, followed by resuspension in PBS/citrate) four times. Cells were seeded at 108 per 75 cm2 flask in Medium 199 with 15% (vol/vol) newborn calf serum, 2 mM l-glutamine, 100 μg/ml penicillin, 100 units/ml streptomycin, and incubated at 37°C, 5% CO2 for 2 h. Cells were washed twice with sterile PBS (wt/vol). After a further 1-h incubation in medium, cells were washed four times, then incubated 12–16 h in medium.
Western Blotting of LOX.
Immunoblotting was performed as described (38). Briefly, porcine monocytes or A10 cells in PBS containing protease inhibitors were assayed for protein (39), then diluted into SDS-sample buffer [250 mM Tris, 40% (vol/vol) glycerol, pH 6.75, 8% (wt/vol) SDS, 20% (vol/vol) 2-mercaptoethanol, 0.005% (wt/vol) bromophenol blue] and boiled for 10 min. Samples (15–30 μg protein) were separated on a 10% SDS-polyacrylamide gel, and transferred onto a nitrocellulose membrane. Membranes were probed with polyclonal anti-rabbit reticulocyte 15-LOX antibody (1:1,500) and visualized by using enhanced chemiluminescence (Amersham Pharmacia) after incubation with a horseradish peroxidase-conjugated anti-guinea pig secondary antibody.
RNA Extraction and Reverse Transcriptase (RT)-PCR of Porcine 12/15-LOX.
Total RNA was extracted from porcine monocytes by using RNA Isolator (Genosys, The Woodlands, TX). Total RNA (1.2 μg) was reverse transcribed by using random hexamers (Amersham Pharmacia) and 240 units of Superscript MMLV reverse transcriptase (Life Technologies, Paisley, Scotland) for 1 h at 42°C in a total volume of 20 μl. Control samples lacking RNA (− RNA) or reverse transcriptase (− RT) were set up in parallel.
RT-PCR was performed as follows: Each 25 μl reaction consisted of 0.1 μg cDNA, 20 μM forward primer, 20 μM reverse primer, 0.1 mM dNTP mixture, 1.5 mM MgCl2, and 5 units of Taq polymerase (BIOTAQ, Bioline, U.K.) in PCR buffer [16 mM (NH4)2SO4, 67 mM Tris⋅HCl, 0.01% (vol/vol) Tween-20, pH 8.8]. Porcine leukocyte 12/15-LOX primers (MWG Biotech, Milton Keynes, U.K.) corresponded to previously published sequences: forward primer, 5′-TTCAGTGTAGACGTGTCGGAG-3′ (position 145–165); reverse primer, 5′-ATGTATGCCGGTGCTGGCTATATTTAG-3′ (position 451–477; ref. 32). Porcine monocyte cDNA was amplified by touchdown PCR, with an initial denaturation step at 94°C for 5 min, followed by 40 cycles of 1 min at 94°C, 2 min starting at 69°C (with a 0.5°C drop in temperature between cycles), and 72°C for 2 min, with a final extension of 72°C for 5 min. PCR products were then amplified with a second round of PCR of 94°C for 5 min followed by 30 cycles of 94°C for 1 min, 53°C for 2 min, 72°C for 2 min, and a final step of 72°C for 5 min. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control: forward primer, 5′-CCCATCACCATCTTCCAGGAG-3′ (position 211–231); reverse primer, 5′-GTTGTCATGGATGACCTTGGC-3′ (position 475–495); with conditions as for second round PCR outlined above. PCR products were visualized on a 1.5% (wt/vol) agarose gel treated with ethidium bromide (2.0 μg/g agarose), using a Bio-Rad GelDoc UV illuminator.
RIA of LOX Products in Transfected A10 Cells.
Cells were incubated in serum-free medium overnight, then in fresh serum-free medium for 30 min at 37°C. Cells and supernatants were extracted as described (33, 40). First, cell pellets were deacylated by treatment with 1.5 ml 0.2 M NaOH in methanol, 50 μM n-propyl gallate under N2 for 45 min. The solution was then diluted to 15% methanol and extracted, and LOX products in the cell pellets and supernatants were quantified by using a specific RIA (41).
Measurement of ⋅NO Consumption.
Anaerobic solutions of ⋅NO (1.9 mM) were prepared by bubbling ⋅NO gas through N2-saturated, deionized water. Any ⋅NO2 was removed by bubbling ⋅NO through 1 M NaOH. ⋅NO was measured by using a selective ⋅NO sensor (IsoNo, WPI, Sarasota, FL). Electrode calibration was performed by measuring ⋅NO release from S-nitrosoacetylpenacillamine (SNAP) as in the electrode manual. When ⋅NO loss was not linear, rates were determined as first order rate constants (kobs) calculated by determining the slope of ln[⋅NO] versus time. For all kobs determined, the square of the Pearson product moment correlation coefficient (r) for the slope of the replotted data were always greater than 0.9, confirming that the reaction followed first order kinetics. For all ⋅NO consumption rates, data are the means ± SD (n = 3).
For measurement of ⋅NO consumption by porcine 12/15-LOX, 3.8 μM ⋅NO was added to 100 mM potassium phosphate buffer (pH 7.4), 100 μM diethylenetriaminepentaacetic acid (DTPA), 0.2% (wt/vol) sodium cholate containing 500 μM LA or AA at 37°C with stirring. LOX (2.8 milliunits) was added 1 min before ⋅NO (1 unit = 1 μmol/min substrate oxidized). For measurement of ⋅NO uptake by A10 cells, confluent monolayers were trypsinized, washed three times in ice-cold PBS (pH 7.4), counted, and kept on ice in PBS. For assay, 106 cells were added to 0.5 ml Krebs buffer (50 mM Hepes, 100 mM NaCl, 5 mM KCl, 1 mM NaH2PO4 ⋅ 2H2O, 1 mM CaCl2, 2 mM glucose, 1 mM MgCl2, pH 7.4) containing 100 μM DTPA in the ⋅NO electrode chamber at 37°C with stirring. ⋅NO (1.9 μM) was then added and rates of decay monitored with/without additions of 500 μM LA. Porcine monocytes were harvested by trypsinization and gentle scraping before washing in PBS and finally resuspending in Krebs buffer (pH 7.4). Cells (0.5 × 106) in 0.5 ml Krebs buffer containing 100 μM DTPA and 10 μM diphenylene iodonium (DPI) were placed in the chamber of the ⋅NO electrode at 37°C with stirring. In some experiments, cells were preincubated for 10 min with 10 μM indomethacin or 200 μM eicosatetraynoic acid (ETYA). ⋅NO (1.9 μM) was added, and rates of ⋅NO decay monitored with/without additions of 150 μM AA or LA.
Conjugated Diene Generation by 12/15-LOX.
Conjugated diene was measured spectrophotometrically at 235 nm (ɛ235 = 28 mM−1⋅cm−1). Seven milliunits of 12/15-LOX was added to 1 ml of 100 mM phosphate buffer (pH 7.4, 37°C) containing 100 μM DTPA, 500 μM LA, and 0.2% (wt/vol) sodium cholate. Where LOX was added, 3.8 μM ⋅NO was added at the same time.
Reverse-Phase HPLC of HPETEs.
Sequential additions (12 total) of 3.8 μM ⋅NO were made to 1 ml phosphate buffer (100 mM, pH 7.4) containing 100 μM AA, 100 μM DTPA, and 0.2% (wt/vol) sodium cholate at 37°C with stirring. Following each ⋅NO addition, 2.8 milliunits of 12/15-LOX was added and ⋅NO consumption monitored. As ⋅NO decay slowed because of self-inactivation of 12/15-LOX, more ⋅NO and 12/15-LOX was added (total 12/15-LOX per sample was 28 milliunits). Control samples were generated by sequential additions of 12/15-LOX to 1 ml of phosphate buffer (100 mM, pH 7.4) containing 100 μM AA, 100 μM DTPA, and 0.2% (wt/vol) sodium cholate in the absence of ⋅NO. Contaminating nitrite (NO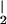 ) was removed by adding an equal volume of 1% (wt/vol) sulfanilamide, 3 M HCl, and 0.02% (wt/vol) N-(1-napthyl)-ethylenediamine to each sample. Lipids were twice extracted with two volumes of diethyl ether. Extracts were pooled and dried over sodium sulfate (30 min, 4°C), then under a stream of nitrogen. Samples were reconstituted in 200 μl of methanol and stored under nitrogen at −80°C. Lipids were separated on a 150 mm × 4.6 mm, 5 μm C18 column (Waters Spherisorb), using a gradient of 50% B to 90% B over 20 min (A: water:acetonitrile:acetic acid, 75:25:0.1; B: methanol:acetonitrile:acetic acid, 60:40:0.1) at 1 ml/min−1. Absorbance was monitored at 235 nm (conjugated dienes) and 205 nm (fatty acids). HPETE products were identified by use of 12(S)HPETE and 15(S)HPETE standards run in parallel under the same conditions.
) was removed by adding an equal volume of 1% (wt/vol) sulfanilamide, 3 M HCl, and 0.02% (wt/vol) N-(1-napthyl)-ethylenediamine to each sample. Lipids were twice extracted with two volumes of diethyl ether. Extracts were pooled and dried over sodium sulfate (30 min, 4°C), then under a stream of nitrogen. Samples were reconstituted in 200 μl of methanol and stored under nitrogen at −80°C. Lipids were separated on a 150 mm × 4.6 mm, 5 μm C18 column (Waters Spherisorb), using a gradient of 50% B to 90% B over 20 min (A: water:acetonitrile:acetic acid, 75:25:0.1; B: methanol:acetonitrile:acetic acid, 60:40:0.1) at 1 ml/min−1. Absorbance was monitored at 235 nm (conjugated dienes) and 205 nm (fatty acids). HPETE products were identified by use of 12(S)HPETE and 15(S)HPETE standards run in parallel under the same conditions.
cGMP Determination.
Porcine monocytes harvested by trypsinization and scraping were washed in PBS then resuspended in Krebs buffer (pH 7.4). Cells (2 × 106) in 0.5 ml Krebs buffer containing 100 μM DTPA, 10 μM DPI, and 1 mM isobutylmethylxanthine (IBMX) were placed in the chamber of the ⋅NO electrode at 37°C with stirring. ⋅NO (1.9 μM) was added, followed by 150 μM LA or 3 μM oxyHb. Samples were incubated for 5 min, then 100 μl was removed and added to 195 μl of ice-cold ethanol for (cGMP) analysis, using a commercial RIA according to the manufacturer's instructions (Amersham Pharmacia).
Results
Expression of 12/15-LOX in Porcine Monocytes.
To examine for ⋅NO uptake by 12/15-LOX in primary vascular cells, porcine monocytes were chosen because they constitutively express this isoform (42, 43). To confirm LOX expression, Western blotting and RT-PCR were carried out on the cells. As shown, immunoreactivity toward an anti-rabbit 15-LOX antibody was found for both isolated porcine monocytes and the commercially available porcine 12/15-LOX (Fig. 1A). Further evidence for expression of 12/15-LOX in porcine monocytes is shown by detection of a PCR product of the expected size (333 bp) for porcine 12/15-LOX in cDNA derived from total porcine monocyte RNA (ref. 32; Fig. 1B). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers used in parallel to ensure cDNA integrity also yielded a product of the expected size (284 bp; Fig. 1C).
Figure 1.

Porcine monocytes express 12/15-LOX. (A) 20 μg monocyte protein, and porcine 12/15-LOX as positive control, were analyzed by Western blot as described in Experimental Procedures. (B) RT-PCR of cDNA derived from total porcine monocyte RNA probed with specific porcine leukocyte 12/15-LOX primers as in Experimental Procedures. (C) RT-PCR of porcine leukocyte cDNA probed with human GAPDH primers as in Experimental Procedures. Each RT-PCR includes minus RNA and minus RT control reactions.
Loss of ⋅NO in the Electrode.
⋅NO (1–4 μM) decay in Krebs buffer or PBS at 37°C followed first order kinetics with a kobs of 6.65 ± 0.25 × 10−3 sec−1. Whereas ⋅NO decay through reaction with oxygen is second order, ⋅NO loss herein was primarily through diffusion into the gas phase and oxidation by the electrode. kobs was used to calculate background ⋅NO losses, which were subtracted from enzyme- or cell-dependent ⋅NO uptake rates.
Consumption of ⋅NO by Porcine Monocytes.
Porcine monocytes consumed ⋅NO at low rates in the absence of stimuli (data not shown). This was fully inhibited by either 3,000 units/ml superoxide dismutase (SOD) or 10 μM DPI, indicating that NADPH oxidase-derived O2⨪ was responsible. To selectively evaluate 12/15-LOX-dependent ⋅NO uptake, DPI was included in all subsequent experiments. Also, 10 μM indomethacin was routinely included because monocytes may constitutively express PGHS-1. Addition of 150 μM LA or AA to porcine monocytes induced an accelerated rate of ⋅NO consumption (1.50 ± 0.25 nmol ⋅ min−1 ⋅ 106 cells−1 or 2.68 ± 0.03 nmol ⋅ min−1 ⋅ 106 cells−1, respectively; Fig. 2A). Inclusion of the LOX inhibitor ETYA (200 μM) led to a slight acceleration of the background rate of ⋅NO consumption, most likely due to the hydrophobicity effects described (ref. 3; Fig. 2B). However, preincubation of monocytes with ETYA for 10 min fully blocked both LA- and AA-stimulated ⋅NO consumption (Fig. 2B). Omission of indomethacin resulted in ≈20% faster rates of ⋅NO consumption when AA was used as substrate, but had no effect on LA-stimulated rates (data not shown). These data suggest that porcine monocytes consume biologically significant quantities of ⋅NO via 12/15-LOX catalysis.
Figure 2.

LOX substrates stimulate ⋅NO consumption and impair activation of sGC in porcine monocytes. (A) Porcine monocytes (0.5 × 106) in 0.5 ml Krebs buffer containing 100 μM DTPA, 10 μM DPI, and 10 μM indomethacin were placed in the chamber of the ⋅NO electrode at 37°C with stirring and incubated for 10 min. ⋅NO (1.9 μM) was then added and ⋅NO decay monitored with/without addition of 150 μM AA or LA as shown by arrows. (B) Samples were identical to A, but cells were preincubated with 200 μM ETYA for 10 min before ⋅NO addition. (C) Porcine monocytes (2 × 106) in 0.5 ml Krebs buffer containing 100 μM DTPA, 10 μM DPI, and 1 mM isobutylmethylxanthine (IBMX) were incubated with 1.9 μM ⋅NO with/without 150 μM LA or 3 μM oxyHb. After 5 min, reactions were stopped and cGMP determined. Results of a representative experiment repeated at least three times are shown (n = 3, mean ± SD). *, P < 0.05 compared with ⋅NO alone, using ANOVA and Bonferroni Dunn post hoc test analysis.
Consumption of ⋅NO Prevents Activation of Porcine Monocyte sGC.
To determine whether unsaturated fatty acid-stimulated consumption of ⋅NO can modulate ⋅NO bioactivity, cGMP generation was determined following a 5 min incubation of monocytes with ⋅NO with/without LA. The nonselective phosphodiesterase inhibitor, isobutylmethylxanthine (IBMX), was included to minimize cGMP hydrolysis. Following incubation with 1.9 μM ⋅NO, significant elevations in monocyte cGMP were found. When 150 μM LA was added immediately following ⋅NO, cGMP generation was significantly suppressed, whereas LA alone did not alter basal cGMP synthesis (Fig. 2C). Addition of 3 μM oxyhemoglobin to scavenge ⋅NO fully blocked ⋅NO activation of sGC.
Consumption of ⋅NO by 12/15-LOX-Transfected Smooth Muscle Cells.
Because ETYA used to inhibit porcine monocyte LOX activity is not a highly specific LOX inhibitor, rat smooth muscle cells (A10) overexpressing murine 12/15-LOX were generated. To confirm the presence of active 12/15-LOX, 12-HETE synthesis was measured and found to be ≈6-fold greater in 12/15-LOX-containing cells than null-transfected controls (Fig. 3B). Also, Western blotting of A10 lysates demonstrated LOX protein only in 12/15-LOX-transfected cells (Fig. 3C). The presence of A10 cells (2 × 106 ml−1) in the sample chamber of the ⋅NO electrode resulted in slightly increased ⋅NO decay rates (kobs = 8.03 ± 1.79 × 10−3 sec−1; Fig. 3A) over background, which were similar in both cell types. However, on addition of LA to 12/15-LOX-containing cells, the rate of ⋅NO consumption increased significantly (0.22 ± 0.08 nmol·min−1·106 cells−1 after background correction). In contrast, null-transfected A10 cells did not display LA-dependent ⋅NO consumption (Fig. 3A). These data confirm that cellular 12/15-LOX consumes ⋅NO in the presence of fatty-acid substrates.
Figure 3.

LA stimulates ⋅NO consumption by 12/15-LOX-transfected A10 cells. (A) ⋅NO (1.9 μM) was added to cells (106) in 0.5 ml Krebs buffer containing 100 μM DTPA in the ⋅NO electrode chamber at 37°C with stirring. Rates of decay monitored with/without additions of 500 μM LA as shown by arrows (LA). (i) No cells, (ii) 12/15-LOX-tranfected cells, (iii) 12/15-LOX-tranfected cells with LA, (iv) null-transfected cells with LA. Results of a representative experiment, repeated at least three times, are shown. (B) 12-HETE generation was measured by using an RIA as in Experimental Procedures. (C) Western blot of 30 μg 12/15-LOX- or null-transfected A10 cell lysates, and porcine 12/15-LOX (as positive control) analyzed as in Experimental Procedures.
Consumption of ⋅NO by Porcine 12/15-LOX.
To examine the effect of ⋅NO consumption on the catalytic activity of 12/15-LOX, a purified porcine 12/15-LOX was used (44). In the absence of fatty acids, 12/15-LOX did not increase rates of ⋅NO decay (Fig. 4 A and B). However, accelerated rates of ⋅NO consumption were observed in the presence of 500 μM LA or AA (2.50 ± 0.83 nmoles·min−1⋅mg−1 or 3.72 ± 0.70 nmoles·min−1⋅mg−1, respectively; mean ± SD, n = 3; Fig. 4A). ⋅NO consumption required the presence of both 12/15-LOX and unsaturated fatty acids (Fig. 4B), indicating that 12/15-LOX consumes ⋅NO during dioxygenase turnover. To examine the effect of ⋅NO on 12/15-LOX activity, rates of LOX-catalyzed diene conjugation were determined in the presence and absence of ⋅NO. As shown in Fig. 4D, ⋅NO inhibited LOX activity by ≈20%. Analysis of LOX products by reverse phase HPLC confirmed ⋅NO suppression of LOX (Fig. 4C) and also indicated that 12-HPETE remained the major product of LOX turnover in the presence of ⋅NO.
Figure 4.

⋅NO suppresses 12/15-LOX activity without changing lipid product profile. (A) ⋅NO (3.8 μM) was added as shown by asterisks to 100 mM phosphate buffer (pH 7.4), 100 μM DTPA, and 0.2% (wt/vol) cholate at 37°C with stirring, containing 500 μM LA or AA. Porcine 12/15-LOX (2.8 mU) was added 1 min before ⋅NO. (i) Control with no fatty acids, (ii) 500 μM LA, (iii) 500 μM AA. (B) Conditions were as in A, but without the addition of 12/15-LOX. (C) LOX products were separated by reverse-phase HPLC as in Experimental Procedures. (i) 12-HPETE generated by 12/15-LOX in the absence of ⋅NO, (ii) 12-HPETE generated by 12/15-LOX in the presence of ⋅NO, (iii) 500 ng 15(S)HPETE standard, (iv) 500 ng 12(S)HPETE standard. (D) Conjugated diene generation by porcine 12/15-LOX was measured spectrophotometrically at 235 nm as in Experimental Procedures. Where LOX was added, 3.8 μM ⋅NO was added at the same time. Results of a representative experiment repeated at least three times are shown. *, P < 0.05 compared with control, using an independent t test.
Discussion
Porcine and murine 12/15-LOX catalytically consumed ⋅NO in the presence of unsaturated fatty acid substrates, with LA-stimulated ⋅NO removal preventing activation of sGC in primary porcine monocytes. Elevation of cGMP in monocytes and macrophages prevents chemotaxis, cytokine release, and IgG complex uptake (45–47). Therefore, catalytic consumption of ⋅NO by 12/15-LOX will attenuate the multifaceted antiinflammatory properties of ⋅NO. These findings establish 12/15-LOX as a potential catalytic sink for ⋅NO in vivo, similar to myoglobin, 15-LOX, PGHS-1, and myeloperoxidase (MPO) (11–14). They also reveal an additional mechanism for the prohypertensive and proatherogenic activity of 12/15-LOX that is distinct from generation of bioactive lipid products.
12/15-LOX expression is induced in macrophages and smooth muscle cells by ang II through the AT1 receptor with several lines of evidence suggesting a role for this isoform in the pathogenesis of ang II-induced hypertension and neointima formation after balloon injury (26–35). Mechanisms by which 12/15-LOX contributes to vascular dysfunction have not been characterized, but could include (i) vasoconstrictive effects of HPETE/HETEs, which occur at relatively high concentrations of 0.5–1 μM (48, 49), and (ii) prevention of ⋅NO signaling by catalytic ⋅NO consumption as shown in this study.
15-LOXs, including rabbit, human, and soybean isoforms, catalytically consume ⋅NO in the presence of unsaturated lipids through radical termination between ⋅NO and an enzyme-bound lipid peroxyl radical (EredLOO⋅; ref. 11). The product of this reaction, an alkyl peroxynitrite (LOONO), then undergoes hydrolysis, forming the expected lipid hydroperoxide (LOOH; ref. 11). Consumption of ⋅NO by 15-LOXs coincides with reversible suppression of turnover through formation of the inactive reduced enzyme (Ered), which requires reactivation through a rate-limiting step (11, 50). The porcine 12/15-LOX undergoes irreversible self-inactivation after 2–3 min of dioxygenase turnover; therefore, detailed mechanistic studies on this isoform were not possible. However, unsaturated fatty acid-stimulated ⋅NO consumption, suppression of dioxygenase activity, and a lack of changes in lipid product profile are identical to the interactions of ⋅NO with 15-LOXs observed (Fig. 4; ref. 11).
Monocytes have several enzymes that can potentially consume ⋅NO via different mechanisms, including PGHS, NADPH oxidase, LOX, and MPO. Preliminary studies showed that isolated porcine monocytes consume ⋅NO under basal conditions through reaction with O2⨪ (data not shown). Published rates of maximal O2⨪ generation by phorbol-stimulated monocytes are 0.5–2 nmol ⋅ min−1 ⋅ 106 cells−1 (51–53). Comparison with LA- or AA-stimulated ⋅NO uptake in this study (2.68 ± 0.03 nmol ⋅ min−1 ⋅ 106 cells−1 and 1.5 ± 0.25 nmol ⋅ min−1 ⋅ 106 cells−1, respectively) indicates that these pathways would cause similar rates of ⋅NO consumption by monocytes when turning over at maximal rates. Their relative importance in controlling ⋅NO bioactivity in vivo will therefore depend on their activities under inflammatory-activated conditions, and local concentrations of superoxide dismutase (SOD). Although the PGHS inhibitor indomethacin had no effect on LA-catalyzed ⋅NO consumption by porcine monocytes, AA-stimulated rates were inhibited by 20%. This observation suggests that PGHS may also consume ⋅NO in monocytes, although at lower rates.
A role for 12/15-LOX in catalyzing ⋅NO consumption by monocytes was indicated by substrate dependency, inhibition with ETYA, and the observations of LOX protein and mRNA expression (Figs. 1 and 2). Further support was obtained by using 12/15-LOX-transfected A10 smooth muscle cells, which also consumed ⋅NO on addition of LA (Fig. 3). Comparisons of 12/15-LOX protein levels on Western blots indicated that porcine monocytes express higher levels of this enzyme than transfected A10 cells (data not shown). This difference in expression is reflected in the 75% lower rates of LA-stimulated ⋅NO consumption by these cells.
Rates of LA- or AA-stimulated ⋅NO consumption by porcine monocytes were considerably greater than documented rates of ⋅NO generation by iNOS in cytokine-activated rat peritoneal macrophages or murine RAW264.7 macrophages (e.g., 0.1 and 0.2 nmol ⋅ min−1 ⋅ 106 cells−1, respectively; refs. 54 and 55), indicating that LOX-dependent ⋅NO consumption will have a significant impact on ⋅NO bioactivity. In this study, LOX turnover was initiated by exogenous addition of substrates, because acutely activating agonists for cellular 12/15- or 15-LOX have not been identified (Fig. 2). The presence of specific LOX products in early atherosclerotic lesions indicates that LOX catalysis occurs in vivo, although precise control mechanisms remain to be elucidated (19–21). Herein, inclusion of the metal chelator DTPA prevents secondary nonenzymatic generation of additional lipid radicals. In vivo, where LOX-derived LOOH decomposes in advanced atheroma, the consumption of ⋅NO by nonenzymatic lipid oxidation will further amplify rates of LOX-dependent ⋅NO removal.
In summary, we reveal that 12/15-LOX catalytically consumes ⋅NO and that this process impairs ⋅NO signaling in primary porcine monocytes. Under basal conditions, ⋅NO plays a central role in maintaining vascular homeostasis (56–59). Thus, stimulation of ⋅NO consumption by monocytes will contribute to proatherogenic conditions by increasing leukocyte adhesion and margination. These findings have implications for our understanding of the role of LOX enzymes in the development of atherosclerosis and hypertension and reveal 12/15-LOX as a potential catalytic sink for ⋅NO in vivo.
Acknowledgments
The authors thank Drs. C. George, S. Hiscox, and S. A. Jones. This work was supported by the British Heart Foundation (V.B.O., M.J.L., and M.J.C.), Wellcome Trust (V.B.O.), National Institutes of Health (Grants RO1-HL64937, RO1-HL58115, and P6-HL58418 to B.A.F. and Grant PO1 HL55798 to R.N.), Juvenile Diabetes Foundation (R.N.), Deutsche Forschungsgemeinschaft (KU161/7-1 to H.K.), and European Union (BHM44-CT98-3191 to H.K.).
Abbreviations
- LOX
lipoxygenase
- ⋅NO
nitric oxide
- O2⨪
superoxide
- HPETE
hydroperoxyeicosatetraenoic acid
- HETE
hydroxyeicosatetraenoic acid
- sGC
soluble guanylate cyclase
- ETYA
eicosatetraynoic acid
- AA
arachidonate
- LA
linoleate
- ang II
angiotensin II
- RT
reverse transcriptase
- DPI
diphenylene iodonium
- oxyHb
oxyhemoglobin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Moncada S, Higgs E A. Eur J Clin Invest. 1991;21:361–374. doi: 10.1111/j.1365-2362.1991.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 2.Arnold W P, Mittal C K, Katsuki S, Murad F. Proc Natl Acad Sci USA. 1997;74:3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu X, Miller M J S, Joshi M S, Sadowska-Krowicka H, Clark D A, Lancaster J R. J Biol Chem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 4.Liao J C, Hein T W, Vaughn M W, Huang K T, Kuo L. Proc Natl Acad Sci USA. 1999;96:8757–8761. doi: 10.1073/pnas.96.15.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaughn M W, Huang K T, Kuo L, Liao J C. J Biol Chem. 2000;275:2342–2348. doi: 10.1074/jbc.275.4.2342. [DOI] [PubMed] [Google Scholar]
- 6.Verbeuren T J, Jordaens F H, Van Hove C E, Van Hoydonck A E, Herman A G. Eur J Pharm. 1990;191:173–184. doi: 10.1016/0014-2999(90)94145-n. [DOI] [PubMed] [Google Scholar]
- 7.Minor R L, Myers P R, Guerra R, Bates J N, Harrison D G. J Clin Invest. 1990;86:2109–2116. doi: 10.1172/JCI114949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White C R, Brock T A, Chang L Y, Crapo J, Briscoe P, Ku D, Bradley W A, Gianturco S H, Gore J, Freeman B A, et al. Proc Natl Acad Sci USA. 1994;91:1044–1048. doi: 10.1073/pnas.91.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ignarro L J, Buga G M, Byrns R E, Wood K S, Chaudhri G. J Pharmacol Exp Ther. 1988;246:218–226. [PubMed] [Google Scholar]
- 10.Kelm M, Schrader J. Circ Res. 1990;66:1561–1575. doi: 10.1161/01.res.66.6.1561. [DOI] [PubMed] [Google Scholar]
- 11.O'Donnell V B, Taylor K B, Parthasarthy S, Kühn H, Koesling D, Freibe K B, Bloodsworth A, Darley-Usmar V M, Freeman B A. J Biol Chem. 1999;274:20083–20091. doi: 10.1074/jbc.274.29.20083. [DOI] [PubMed] [Google Scholar]
- 12.O'Donnell V B, Coles B, Lewis M J, Crews B C, Marnett L J, Freeman B A. J Biol Chem. 2000;275:38239–38244. doi: 10.1074/jbc.M001802200. [DOI] [PubMed] [Google Scholar]
- 13.Flögel U, Merz M W, Gödecke A, Decking U K M, Schrader J. Proc Natl Acad Sci USA. 2001;98:735–740. doi: 10.1073/pnas.011460298. . (First Published January 2, 2001; 10.1073/pnas.011460298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abu-Soud H, Hazen S L. J Biol Chem. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 15.Kühn H, Thiele B J. FEBS Lett. 1999;449:7–11. doi: 10.1016/s0014-5793(99)00396-8. [DOI] [PubMed] [Google Scholar]
- 16.Hamberg M, Samuellsson B. Proc Natl Acad Sci USA. 1974;71:3400–3404. doi: 10.1073/pnas.71.9.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brash A R. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 18.Schewe T, Rapoport S M, Kühn H. Adv Enzymol Rel Areas Mol Biol. 1986;58:273–311. doi: 10.1002/9780470123041.ch7. [DOI] [PubMed] [Google Scholar]
- 19.Kühn H, Belkner J, Zaiss S, Fährenklemper T, Wohfeil S. J Exp Med. 1994;179:1903–1911. doi: 10.1084/jem.179.6.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yla-Herttuala S, Rosenfeld M E, Parthasarathy S, Glass C K, Sigal E, Sarkioia T, Witztum J T, Steinberg D. J Clin Invest. 1991;87:1146–1152. doi: 10.1172/JCI115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Folcik V A, Nivar-Aristy R A, Krajewski L P, Cathcart M K. J Clin Invest. 1995;96:504–510. doi: 10.1172/JCI118062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belkner J, Stender H, Kühn H. J Biol Chem. 1998;273:23225–23232. doi: 10.1074/jbc.273.36.23225. [DOI] [PubMed] [Google Scholar]
- 23.Sendobry S M, Cornicelli J A, Welch K, Tait B, Trivedi B K, Colbry N, Dyer R D, Feinmark S J, Daughtery A. Br J Pharmacol. 1997;120:1199–1206. doi: 10.1038/sj.bjp.0701007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cyrus T, Witzum J L, Rader D J, Tangirala R, Fazio S, Linton M F, Funk C D. J Clin Invest. 1999;103:1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bleich D, Chen S, Zipser B, Sun D, Funk C D, Nadler J L. J Clin Invest. 1999;103:1431–1436. doi: 10.1172/JCI5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Natarajan R, Pei H, Gu J L, Sarma J S, Nadler J. Cardiovasc Res. 1999;41:481–499. doi: 10.1016/s0008-6363(98)00312-5. [DOI] [PubMed] [Google Scholar]
- 27.Gu J L, Pei H, Nadler J L, Rossi J J, Natarajan R. Circulation. 2001;103:1446–1452. doi: 10.1161/01.cir.103.10.1446. [DOI] [PubMed] [Google Scholar]
- 28.Stern N, Kisch E S, Knoll E. Hypertension. 1996;27:1149–1152. doi: 10.1161/01.hyp.27.5.1149. [DOI] [PubMed] [Google Scholar]
- 29.Chang W C, Su G W. Biochem Biophys Res Commun. 1985;127:642–648. doi: 10.1016/s0006-291x(85)80209-6. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki M, Hori M T, Hino T, Golub M S, Tuck M L. Am J Hypertens. 1997;10:371–378. [PubMed] [Google Scholar]
- 31.Scheidegger K J, Butler S, Witzum J L. J Biol Chem. 1997;272:21609–21615. doi: 10.1074/jbc.272.34.21609. [DOI] [PubMed] [Google Scholar]
- 32.Natarajan R, Gu J L, Rossi J, Gonzales N, Lanting L, Xu L, Nadler J. Proc Natl Acad Sci USA. 1993;90:4947–4951. doi: 10.1073/pnas.90.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nozawa K, Tuck M L, Golub M, Eggena P, Nadler J L, Stern N. Am J Physiol. 1990;259:H1774–H1780. doi: 10.1152/ajpheart.1990.259.6.H1774. [DOI] [PubMed] [Google Scholar]
- 34.Stern N, Nozawa K, Golub M, Eggena P, Knoll E, Tuck M L. Am J Hypertens. 1993;6:52–58. doi: 10.1093/ajh/6.1.52. [DOI] [PubMed] [Google Scholar]
- 35.Lin L, Balazy M, Pagani P J, Nasjletti A. Circ Res. 1994;74:197–205. doi: 10.1161/01.res.74.2.197. [DOI] [PubMed] [Google Scholar]
- 36.Natarajan R, Gonzales N, Lanting L, Nadler J. Hypertension. 1994;23:I142–I147. doi: 10.1161/01.hyp.23.1_suppl.i142. [DOI] [PubMed] [Google Scholar]
- 37.Ponec M, Kempenaar J A, Havekes L, van der Schroeff J G, Emeis J J, Vermeer B J. Biochim Biophys Acta. 1981;666:405–410. doi: 10.1016/0005-2760(81)90299-x. [DOI] [PubMed] [Google Scholar]
- 38.Towbin H, Staehelin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Natarajan R, Dunn W D, Stern N, Nadler J. Mol Cell Endocrinol. 1990;72:73–80. doi: 10.1016/0303-7207(90)90096-q. [DOI] [PubMed] [Google Scholar]
- 41.Nadler J L, Natarajan R, Stern N. J Clin Invest. 1987;80:1763–1769. doi: 10.1172/JCI113269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshimoto T, Suzuki H, Yamamoto S, Takai T, Yokoyama C, Tanabe T. Proc Natl Acad Sci USA. 1990;87:2142–2146. doi: 10.1073/pnas.87.6.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maruyama T, Ueda N, Yoshimoto T, Yamamoto S, Komatsu N, Watanabe K. J Histochem Cytochem. 1989;37:1125–1131. doi: 10.1177/37.7.2499621. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi Y, Glasgow W C, Suzuki H, Taketani Y, Yamamoto S, Anton M, Kuhn H, Brash A R. Eur J Biochem. 1993;218:165–171. doi: 10.1111/j.1432-1033.1993.tb18362.x. [DOI] [PubMed] [Google Scholar]
- 45.Bath P M. Eur J Clin Pharmacol. 2000;45:53–58. doi: 10.1007/BF00315350. [DOI] [PubMed] [Google Scholar]
- 46.Mattana J, Singhal P C. Am J Physiol. 1993;265:C92–C98. doi: 10.1152/ajpcell.1993.265.1.C92. [DOI] [PubMed] [Google Scholar]
- 47.Kiemer A K, Hartung T, Vollmar A M. J Immunol. 2000;165:175–181. doi: 10.4049/jimmunol.165.1.175. [DOI] [PubMed] [Google Scholar]
- 48.Uski T K, Hogestatt E D. Gen Pharm. 1992;23:109–113. doi: 10.1016/0306-3623(92)90056-p. [DOI] [PubMed] [Google Scholar]
- 49.Nishiyama M, Okamoto H, Watanabe T, Hori T, Sasaki T, Kirino T, Shimizu T. Eur J Pharmacol. 1998;341:57–63. doi: 10.1016/s0014-2999(97)01353-8. [DOI] [PubMed] [Google Scholar]
- 50.Wiesner R, Rathmann J, Holshutter H G, Stosser R, Mader K, Nolting H, Kühn H. FEBS Lett. 1996;389:229–232. doi: 10.1016/0014-5793(96)00591-1. [DOI] [PubMed] [Google Scholar]
- 51.Pabst M J, Hedegaard H B, Johnston R B. J Immunol. 1982;128:123–128. [PubMed] [Google Scholar]
- 52.Reiss M, Roos D. J Clin Invest. 1978;61:480–488. doi: 10.1172/JCI108959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kitagawa S, Takaku F, Sakamoth S. J Immunol. 1980;125:359–364. [PubMed] [Google Scholar]
- 54.Ischiropoulous H, Zhu L, Beckman J S. Arch Biochem Biophys. 1992;289:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 55.Lewis R S, Tamir S, Tannenbaum S R, Deen W M. J Biol Chem. 1995;270:29350–29355. doi: 10.1074/jbc.270.49.29350. [DOI] [PubMed] [Google Scholar]
- 56.Neviere R, Guery B, Mordon S, Zerimech F, Charre S, Wattel F, Chopin C. Am J Physiol. 2000;278:H1783–H1790. doi: 10.1152/ajpheart.2000.278.6.H1783. [DOI] [PubMed] [Google Scholar]
- 57.Katsuyama K, Shichiri M, Marumo F, Hirata Y. Arterioscler Thromb Vasc Biol. 1998;18:1796–1802. doi: 10.1161/01.atv.18.11.1796. [DOI] [PubMed] [Google Scholar]
- 58.de Graaf J C, Banga J D, Moncada S, Palmer R M, de Groot P G, Sixma J J. Circulation. 1992;85:2284–2290. doi: 10.1161/01.cir.85.6.2284. [DOI] [PubMed] [Google Scholar]
- 59.Banick P D, Chen Q, Xu Y A, Thom S R. J Cell Physiol. 1997;172:12–24. doi: 10.1002/(SICI)1097-4652(199707)172:1<12::AID-JCP2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]