

Abstract
Activation of type 1 cannabinoid receptors (CB1R) decreases GABA and glutamate release in cortical and subcortical regions, with complex outcomes on cortical network activity. To date there have been few attempts to disentangle the region- and cell-specific mechanisms underlying the effects of cannabinoids on cortical network activity in vivo. Here we addressed this issue by combining in vivo electrophysiological recordings with local and systemic pharmacological manipulations in conditional mutant mice lacking CB1R expression in different neuronal populations. First we report that cannabinoids induce hypersynchronous thalamocortical oscillations while decreasing the amplitude of faster cortical oscillations. Then we demonstrate that CB1R at striatonigral synapses (basal ganglia direct pathway) mediate the thalamocortical hypersynchrony, whereas activation of CB1R expressed in cortical glutamatergic neurons decreases cortical synchrony. Finally we show that activation of CB1 expressed in cortical glutamatergic neurons limits the cannabinoid-induced thalamocortical hypersynchrony. By reporting that CB1R activations in cortical and subcortical regions have contrasting effects on cortical synchrony, our study bridges the gap between cellular and in vivo network effects of cannabinoids. Incidentally, the thalamocortical hypersynchrony we report suggests a potential mechanism to explain the sensory “high” experienced during recreational consumption of marijuana.
Keywords: cannabis, high-voltage spindles, striatum, substantia nigra, electrocorticograms
Cannabinoids are a family of compounds that activate cannabinoid receptors, which are well known for their psychotropic effects and therapeutic potentials. The type 1 cannabinoid receptor (CB1R) is massively expressed in the brain at the synaptic terminals of excitatory and inhibitory neurons, and its activation by exogenous or endogenous cannabinoids decreases neurotransmitter release (1–3). One largely unanswered question is how the elementary decreases in excitatory and inhibitory synaptic transmissions induced by systemic intake of cannabinoids interact to produce alterations in neuronal network activity in vivo. A number of recent studies combining systemic injections of CB1R agonists and antagonists with electrophysiological recordings in the hippocampus and neocortex of awake or anesthetized rodents have shown that a hallmark of cannabinoids effects on neuronal network activity is a decrease in synchrony (4–9). Specifically, systemic CB1R activation has been shown to decrease (i) the amplitude of the hippocampal θ rhythm (4, 7, 8), (ii) the amplitude of γ oscillations in the hippocampus (4, 7, 10), enthorinal cortex (8), and prefrontal cortex (7), (iii) the incidence of hippocampal ripples (4, 6, 11), and (iv) spiking correlation in the hippocampus and prefrontal cortex (4, 5, 7, 9).
In contrast to the consistent reports that cannabinoids dampen cortical network oscillations, other findings suggest that they could also increase synchrony. First, CB1R are predominantly expressed by GABAergic forebrain neurons (12), and their activation leads to decreased GABA release (1). Therefore, cannabinoids might be expected to increase network synchrony or generate excessive firing activity, in a similar manner to GABA receptors antagonists, which favor convulsive seizures (13, 14). Second, several studies have reported that cannabinoids are proconvulsant in experimental models of epilepsy (15–17). The concurrent but unbalanced activation of the CB1R at the synaptic terminals of inhibitory and excitatory neurons could be one explanation for these seemingly contradictory observations. Additionally, the high level of CB1R expression in subcortical regions (18–20) leaves open the possibility that at least a part of the cannabinoid-induced alterations in cortical network activity has extracortical origins. To date, the potential cell type- and region-specific impact of CB1R activation on in vivo cortical network activity has received little attention. Here we specifically addressed this issue by comparing the impact of systemic and local injections of the CB1R agonist CP55940 on cortical network oscillations recorded from freely moving mice lacking CB1R expression in distinct neuronal populations. The results reveal the cell- and region-specific mechanisms underlying a dual modulation of cortical synchrony by exogenous cannabinoids and pave the way to a refined understanding of the cognitive alterations associated with marijuana consumption (21, 22).
Results
Systemic CB1R Activation Induces Thalamocortical High-Voltage Spindles While Decreasing Fast Electrocorticogram Oscillations.
To investigate in vivo the cell type-specific impact of systemic CB1R activation on cortical network activity, we recorded neocortical electrocorticograms (ECoG) before and after i.p. injections of the high-affinity CB1R agonist CP55940 (0.3 mg/kg of body weight). ECoG were recorded bilaterally above the somatosensory cortex, in the home cage of two groups of mice: C57BL/6N mice (n = 9) and conditional mutant mice that lack CB1R expression in specific neuronal populations [n = 33, including WT littermates (19, 23, 24)]. All ECoG shown and compared in this study were taken from periods of immobility, to avoid biased quantification of network activity due to decreased locomotor activity induced by the systemic injection of cannabinoids (19) (Fig. S1). We first examined and quantified the impact of CP55940 on the oscillatory content of the ECoG from C57BL/6N mice (Fig. 1). After CP55940 injections the ECoG displayed numerous oscillatory bursts, with a shape, frequency (∼5 Hz), length (∼1–2 s), and amplitude (> 0.5 mV) characteristic of thalamocortical high-voltage spindles (HVS, also referred to as spike-and-wave discharges/seizures) (Fig. 1 A–C; arrowheads in Fig. 1C) (25–27). As expected for this type of oscillation, HVS were sparse during control recordings (27) and, after CP55940 injections, were strictly restricted to periods of immobility (28). A quantitative routine developed to compare HVS before and after CP55940 injections (Experimental Procedures, Fig. 1 B and C, and Fig. S2) showed that, across animals and experiments, HVS incidence and power reliably and strongly increased after CP55940 injections (Fig. 1D). This effect was dependent on CB1R activation because it was completely reversed (Fig. 1D and Fig. S3) and prevented (Fig. 1E) by i.p. injections of the CB1R antagonist AM251 (3 mg/kg). Cannabinoids have been previously shown to decrease the power of neocortical local field potential oscillations over a wide range of frequencies (7, 8). Therefore, we next quantified the effect of CP55940 on ECoG oscillations in a frequency band distinct from the one of the cannabinoid-induced HVS. As expected, CP55940 injections reliably decreased the amplitude of fast (>12 Hz) ECoG oscillations (Fig. 1F) (7, 8), an effect that was also reversed (Fig. 1F and Fig. S3) and blocked (Fig. 1G) by AM251 injections. Taken together these data show that systemic CB1R activation, although decreasing synchrony of fast ECoG oscillations, generated hypersynchronous oscillatory bursts characteristic of thalamocortical HVS.
Fig. 1.

Systemic CB1R activation induces HVS while decreasing the amplitude of faster neocortical oscillations. (A–C) Single experiment showing the effects of an i.p. injection of the CB1R agonist CP55940 (Right), or its vehicle alone (Left), on a mouse ECoG recorded during immobility epochs. (A) Thirty minutes ECoG (black traces) and HVS incidence (colored rasters). (Scale bars, 1 mV and 5 min.) (B) Thirty seconds ECoG taken from rectangles in A. (Middle) Time–frequency power spectrograms (1-s sliding windows, steps of 0.2 s). (Bottom) ECoG power values in the HVS frequency band (4–6 Hz). Dashed line is HVS detection threshold. Circles identify windows detected as part of an HVS. (C) Similar to B, but 10 s ECoG are from dashed rectangles in B. Arrowheads indicate detected HVS. ECoG traces in A–C have the same voltage scale. (D and E) HVS comparison in all experiments. Each line represents a single experiment, and different colors represent different animals. CP55940 (CP, 0.3 mg/kg) reliably increases HVS, and this effect was reversed (D) and abolished (E) by injection of the CB1R antagonist AM251 (AM, 3 mg/kg). (F and G) Similar to D and E, but the amplitude of ECoG oscillations faster than 12 Hz is compared. Same experiments and color code as in D and E. P values in D–G are from Wilcoxon paired two-sided signed rank test. Bold green traces in D and F show the quantification of the illustrative experiment shown in A–C.
CB1R Activation at GABAergic Striatonigral Synapses Increases the Incidence of Thalamocortical HVS.
We next set out to understand how CP55940 i.p. injections increase HVS. Although rodent HVS function is still a matter of debate [analog to human μ rhythm (29) or model for absence-like seizure actvity (30)], there is ample evidence that they share mechanistic similarities with the well-studied thalamocortical sleep spindle oscillations (31–35). On the one hand, the basal ganglia is a major site controlling thalamocortical oscillations (28, 36) via giant inhibitory nigrothalamic synapses (37). Specifically, potentiation or blockade of GABAergic transmission in the substantia nigra pars reticulata (SNr) decreases or increases HVS incidence, respectively (36, 38). On the other hand, the highest density of CB1R in the brain is found in the SNr on striatal inhibitory terminals (1, 20), where CB1R activation potently decreases GABA release (39). To test the hypothesis that CB1R expressed by striatonigral neurons (D1-positive medium spiny neurons forming the basal ganglia direct pathway) mediate the increase in HVS observed after systemic injection of CP55940, we took advantage of conditional mutant mice lacking CB1R expression in the striatal neurons projecting to the SNr (D1-CB1R−/− mice) (19). In WT mice, CP55940 systemic injections induced HVS (Fig. 2A), but no such effect was seen in the D1-CB1R−/− mice (Fig. 2 B and C). CP55940 effects on HVS were also abolished in GABA-CB1R−/− and CaMK-CB1R−/− mice (Fig. S4), which lack CB1R expression in, respectively, GABAergic and principal forebrain neurons (24). In GABA-CB1R−/− and CaMK-CB1R−/− mice, striatal neurons projecting to the SNr lack CB1R expression. Thus, this result (Fig. S4) confirms that CB1R expressed by striatonigral neurons is required for the CP55940-induced increase in HVS. Importantly, the ECoG oscillatory content during baseline recordings was similar across all types of conditional mutant mice (Fig. S5), ruling out the possibility that the differential effects of CP55940 in WT and CB1R mutant mice originate from alterations in baseline cortical network activity.
Fig. 2.
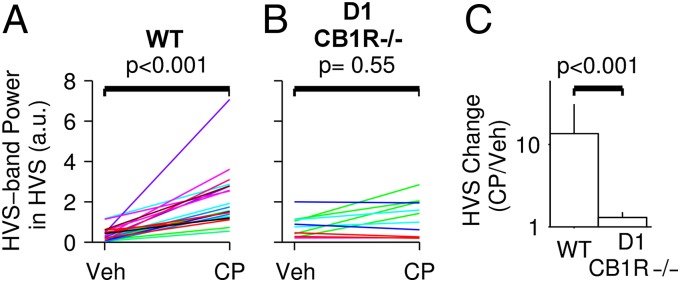
CB1R expressed by D1-positive striatal neurons are required for the CP55940-induced increase in HVS. (A and B) Group effect of vehicle and CP55940 (CP, 0.3 mg/kg) injections on HVS in WT animals (A) and mice lacking CB1R expression in D1-positive striatal neurons (D1-CB1R−/−; B). (C) Comparison of the effects of CP55940 on HVS between WT and D1-CB1R−/− mice. Histograms show mean changes in HVS power and incidence. Error bars are SEM. P value is from the Wilcoxon rank sum test.
The lack of cannabinoid-induced enhancement in HVS in D1-CB1R−/− mice suggested that, in the SNr, activation of CB1R expressed by striatonigral neurons is necessary to increase HVS. To test more directly this hypothesis, CP55940 was locally injected in the SNr of WT and D1-CB1R−/− mice. In WT mice, injections of CP55940 in the SNr induced HVS, an effect that could be reversed by a subsequent injection of the GABA-A receptor agonist muscimol (Fig. 3 A and B). Conversely, in D1-CB1R−/− mice, intra-SNr injections of CP55940 failed to increase HVS (Fig. 3 C and D; P < 0.001 vs. WT data), whereas subsequent injections of the GABA-A antagonist bicuculline increased HVS (Fig. 3C). Altogether these results are consistent with a scenario in which CP55940 enhances HVS incidence by decreasing GABA release from direct pathway striatal neurons projecting to the SNr.
Fig. 3.

Intra-SNr activation of CB1R expressed by D1-positive striatal neurons induces HVS. (A and C) Single experiments in WT (A) and D1-CB1R−/− (C) mice. (Left) Track of the injection cannula. Black traces are ECoG during baseline and after injections (500 nL) of vehicle, CP55940 (20 mg/mL), and muscimol (0.5 mg/mL; A) or bicuculline (0.2 mg/mL; C). Rasters show detected HVS. Colored histograms show HVS power and incidence quantification for these two experiments. (B and D) Group effect of intra-SNr CP55940 (CP) injections on HVS in WT (B) and D1-CB1R−/− (D) mice. (Scale bars, 5 min and 1 mV.)
Activation of CB1R Expressed by Cortical Glutamatergic Neurons Decreases Fast Neocortical Oscillations and Limits Cannabinoid-Induced Increase in HVS.
Systemic injection of CP55940 enhances HVS while decreasing the power of fast (>12 Hz) ECoG oscillations in mice (Fig. 1). To test whether these effects shared the same mechanism, we quantified the effect of CP55940 on the amplitude of fast ECoG oscillations in CB1R conditional mutant mice and their WT littermates. The CP55940-induced decrease in fast ECoG oscillations power was intact in D1-CB1R−/− mice (Fig. 4), suggesting that the cannabinoid-induced increase in HVS and decrease in fast ECoG oscillations have distinct mechanisms. In contrast, the decrease in fast ECoG oscillations was significantly reduced in Glu-CB1R−/− and CaMK-CB1R−/− mice compared with WT littermates (Fig. 4). These results show that activation of CB1R expressed by glutamatergic cortical neurons is, at least in part, responsible for the reduced neuronal network synchrony observed in vivo after cannabinoids injections. Additionally, CP55940-induced decrease in fast ECoG oscillations was stronger in GABA-CB1R−/− mice than in WT littermates (Fig. 4), suggesting that activations of CB1R expressed on cortical GABAergic and glutamatergic neurons exert a bidirectional control over fast ECoG oscillations.
Fig. 4.

Cannabinoid-induced decrease in fast ECoG oscillations power is reduced in mice lacking CB1R in glutamatergic cortical neurons. (A) Effects of vehicle and CP55940 (CP) injections on the amplitude of fast (>12 Hz) ECoG oscillations in, from left to right, WT, D1-CB1R−/− mice, mice lacking CB1R in glutamatergic cortical neurons (Glu-CB1R−/−), in all principal forebrain neurons (CaMK-CB1R−/−), and in all GABAergic forebrain neurons (GABA-CB1R−/−). (B) Mean changes in HVS and SEM for WT and mutant mice. P values are from Wilcoxon rank sum test vs. WT data. Dotted lines represent mean change in the WT group.
A desynchronization function of CB1R expressed by glutamatergic cortical neurons is additionally supported by the observation that the cannabinoid-induced increase in HVS was stronger in Glu-CB1R−/− mice than in WT animals (Fig. 5 A–C). Indeed, CP55940 induced prominent HVS and high-amplitude isolated spike-and-wave complexes in the mutant mice (Fig. 5 D and E). This result suggests that, in WT animals, activation of CB1R expressed by glutamatergic cortical neurons limits the increase in HVS observed after CP55940 injection.
Fig. 5.

CB1R expressed by glutamatergic cortical neurons limit the cannabinoid-induced increase in HVS. (A and B) Effects of vehicle and CP55940 (CP) injections on HVS incidence and power in WT (A) and Glu-CB1R−/− (B) mice. (C) Comparison of the mean changes in HVS for these mice. (D) Single experiment in which ECoG (black) was recorded from a Glu-CB1R−/− mouse after vehicle (Upper) and CP55940 (Lower) i.p. injections. Colored rasters show detected HVS. (Scale bar, 5 min.) (E) Upper trace is taken from the rectangle in D. Arrowheads show two HVS. The two lower traces are taken from the upper trace (1, 2) and show, respectively, a strong HVS and isolated spike-and-wave seizure-like discharges. (Scale bars, 1 s and 0.75 mV.)
Discussion
Here we report that cannabinoids have contrasting effects on neocortical network synchrony, characterized by the appearance of hypersynchronous thalamocortical oscillations and decreased amplitude of fast ECoG oscillations. Taking advantage of conditional mice lacking CB1R expression in specific neuronal populations, we broke down these effects into region- and cell-specific CB1R activation: CB1R at striatonigral inhibitory synapses are responsible for the cannabinoid-induced thalamocortical hypersynchrony, whereas CB1R activation at cortical excitatory synapses reduces synchrony of fast neocortical oscillations.
We found that in immobile mice, systemic CB1R activation generated highly synchronous oscillatory bursts of the ECoG with a frequency, duration, and amplitude characteristic of thalamocortical HVS (25, 26, 40). We further showed that this hypersynchronous effect was mediated by CB1R located in the SNr on striatal GABAergic synaptic terminals. Finally, intra-SNr injections of GABA-A agonist and antagonist, respectively, reversed and mimicked the effects of CP55940 on HVS. Altogether, our results are best explained by the following mechanism: systemic CB1R activation induces thalamocortical HVS by decreasing GABA release at striatonigral synapses (Fig. 6). This hypothesis is supported by previous studies on CB1R synaptic physiology in the basal ganglia and the known role of the SNr in controlling thalamocortical oscillations. First, the highest level of CB1R immunoreactivity in rodent is found in the SNr (18). In this nucleus, CB1R is localized on axons and terminals of striatal neurons (20), and its activation potently inhibits striatonigral GABAergic transmission (20, 39). Second, the basal ganglia are known to modulate thalamocortical oscillations (28, 36). Specifically, hyperpolarization of thalamocortical relay cells is a condition sine qua non for the emergence of thalamocortical oscillations (13, 31, 34), and the giant inhibitory synapses between GABAergic neurons of the SNr and thalamocortical relay cells have been proposed to contribute to this hyperpolarization (37). In agreement with an oscillation-promoting role of nigrothalamic cells, pharmacological excitation or inhibition of SNr neurons increased or decreased HVS incidence, respectively (38, 41). Finally, dopamine depletion or blockade of dopamine receptors in the striatum, which decreased the activity of direct pathway striatal neurons (42), also enhanced the incidence of HVS (28, 43). Altogether these studies suggest that the cannabinoid-induced increase in HVS we report is best explained by the disinhibition of nigrothalamic GABAergic neurons (Fig. 6).
Fig. 6.

Hypothesized mechanism responsible for the cannabinoid-induced thalamocortical HVS. (A and B) Schematic representation of the circuit responsible for HVS control during vehicle (A) or CB1R agonist (B) injections. During vehicle injection the oscillation-promoting nigrothalamic GABAergic neurons are kept inhibited by striatonigral inhibitory neurons. After cannabinoid injection, the nigrothalamic oscillation-promoting neurons are disinhibited after CB1R activation at striatonigral synapses.
In mice lacking CB1R on glutamatergic cortical neurons (Glu-CB1R−/−), the cannabinoid-induced increase in HVS was stronger than in their WT littermates, and CP55940 generated a seizure-like pattern of activity. This result suggests that in WT mice, striatonigral and cortical CB1R compete and modulate thalamocortical oscillations in opposite directions. The partial counterbalancing effect of “cortical” CB1R could be due to a decrease in release of glutamate in the neocortex, in the thalamus (from corticothalamic excitatory neurons), or in both areas. This is in agreement with a role of cortical input on the thalamic reticular neurons to control thalamocortical HVS oscillations (34, 44). A role for CB1R expressed by cortical glutamatergic neurons in reducing network synchrony is further supported by the observation that cannabinoid-induced decrease in fast neocortical oscillations was reduced in Glu- and CaMK-CB1R−/− mice. This function is in line with studies showing that “glutamatergic cortical” CB1R provide protection against kainic acid-induced seizures (23, 24) and with recent works suggesting that cannabinoids decrease hippocampal γ and ripple oscillations by decreasing excitatory transmission (6, 11). CB1R expressed by brain astroglial cells have been recently shown to participate in the control of glutamatergic synaptic transmission and plasticity (45–47). Our results suggest that the effects of CP55940 on fast ECoG oscillations are mostly independent from astroglial CB1R: in fact, at odds with astroglial CB1R-dependent long-term depression of excitatory synaptic transmission (47), CP55940 effects were reversed by injections of a CB1R antagonist and were significantly reduced in Glu-CB1R−/− mice. Still, at this stage, we cannot entirely exclude that astroglial CB1R contribute to the decrease in fast ECoG oscillations, and future investigation will address directly this possibility.
There are several possible specific behavioral implications of our findings. First, the reported dual pro- and antioscillatory functions of CB1R expressed in, respectively, subcortical and cortical regions might be related to the complex control of network excitability exerted by CB1R in physiopathological conditions, such as during epileptiform seizures (15–17, 23, 48). Additionally, we believe that the increase in thalamocortical HVS after CB1R activation in the SNr offers a perspective to understand the psychoactive effects associated with marijuana consumption. In this context, it is important to emphasize that thalamocortical HVS have been proposed to constitute a brain state that facilitates detection of weak sensory stimulation (29, 49). More generally, the activity of the thalamocortical system controls vigilance states and gates the perception of sensory stimulation (50). The recreational consumption of marijuana is well known to produce a “high” characterized by an altered consciousness and an intensification of sensory perceptions (51). Strikingly, in both human and rodent brains, the highest expression of CB1R is found in the SNr on striatonigral synapses (18, 20, 52, 53). Therefore, an exciting hypothesis for future investigation is that the sensory/behavioral “high” experienced during marijuana consumption is due to an aberrant thalamocortical synchrony via massive CB1R activation in the SNr.
Experimental Procedures
Experimental procedures are described in SI Experimental Procedures. This section describes the conditional CB1R mutant mice used, electrophysiological recording methods, local and systemic pharmacological injections, and data analysis. All animal procedures were conducted in accordance with standard ethical guidelines (European Communities Directive 86/60-EEC) and were approved by the local ethical committee (Comité d'Experimentació Animal, Universitat de Barcelona, Ref 520/08).
Supplementary Material
Acknowledgments
We thank O. Manzoni, I. Bureau, H. Martin, and D. Jercog for excellent discussions and careful reading of the manuscript; D. Gonzales, N. Aubailly, M. Metna, D. Terrier, and T. Wiesner for mouse care and genotyping; and A. Cei for help in early experiments on this project. This work was funded by Ministerio de Ciencia e Innovación Grant BFU2008-03946 (to D.R.), Marie Curie International Reintegration Grant IRG230976 (to D.R.), Institut National de la Santé et de la Recherche Médicale (INSERM) (G.M.), Region Aquitaine (E.S.-G. and G.M.), and the European Research Council (ERC-2010-StG-260515, to G.M.). D.R. was supported by a Ramon-Y-Cajal fellowship from the Spanish Ministerio de Ciencia e Innovación and the Avenir program from INSERM. P.E.R.-O. was supported by Marie Curie International Incoming Fellowship IIF253873. E.S.-G. was supported by the Fyssen Fondation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217144110/-/DCSupplemental.
References
- 1.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83(3):1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 2.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4(11):873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 3.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89(1):309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 4.Robbe D, et al. Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nat Neurosci. 2006;9(12):1526–1533. doi: 10.1038/nn1801. [DOI] [PubMed] [Google Scholar]
- 5.Robbe D, Buzsáki G. Alteration of theta timescale dynamics of hippocampal place cells by a cannabinoid is associated with memory impairment. J Neurosci. 2009;29(40):12597–12605. doi: 10.1523/JNEUROSCI.2407-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maier N, et al. Cannabinoids disrupt hippocampal sharp wave-ripples via inhibition of glutamate release. Hippocampus. 2012;22(6):1350–1362. doi: 10.1002/hipo.20971. [DOI] [PubMed] [Google Scholar]
- 7.Kucewicz MT, Tricklebank MD, Bogacz R, Jones MW. Dysfunctional prefrontal cortical network activity and interactions following cannabinoid receptor activation. J Neurosci. 2011;31(43):15560–15568. doi: 10.1523/JNEUROSCI.2970-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hajós M, Hoffmann WE, Kocsis B. Activation of cannabinoid-1 receptors disrupts sensory gating and neuronal oscillation: Relevance to schizophrenia. Biol Psychiatry. 2008;63(11):1075–1083. doi: 10.1016/j.biopsych.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Goonawardena AV, Riedel G, Hampson RE. Cannabinoids alter spontaneous firing, bursting, and cell synchrony of hippocampal principal cells. Hippocampus. 2011;21(5):520–531. doi: 10.1002/hipo.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hájos N, et al. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12(9):3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 11.Holderith N, et al. Cannabinoids attenuate hippocampal γ oscillations by suppressing excitatory synaptic input onto CA3 pyramidal neurons and fast spiking basket cells. J Physiol. 2011;589(Pt 20):4921–4934. doi: 10.1113/jphysiol.2011.216259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsicano G, Kuner R. In: Cannabinoids and the Brain. Köfalvi A, editor. New York: Springer; 2008. pp. 161–201. [Google Scholar]
- 13.Steriade M, Contreras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol. 1998;80(3):1439–1455. doi: 10.1152/jn.1998.80.3.1439. [DOI] [PubMed] [Google Scholar]
- 14.Vergnes M, Boehrer A, Simler S, Bernasconi R, Marescaux C. Opposite effects of GABAB receptor antagonists on absences and convulsive seizures. Eur J Pharmacol. 1997;332(3):245–255. doi: 10.1016/s0014-2999(97)01085-6. [DOI] [PubMed] [Google Scholar]
- 15.Turkanis SA, Karler R. Central excitatory properties of delta 9-tetrahydrocannabinol and its metabolites in iron-induced epileptic rats. Neuropharmacology. 1982;21(1):7–13. doi: 10.1016/0028-3908(82)90204-0. [DOI] [PubMed] [Google Scholar]
- 16.Fish BS, Consroe P, Fox RR. Convulsant-anticonvulsant properties of delta-9-tetrahydrocannabinol in rabbits. Behav Genet. 1983;13(2):205–211. doi: 10.1007/BF01065669. [DOI] [PubMed] [Google Scholar]
- 17.Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J Neurosci. 2003;23(9):3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herkenham M, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87(5):1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monory K, et al. Genetic dissection of behavioural and autonomic effects of Delta(9)-tetrahydrocannabinol in mice. PLoS Biol. 2007;5(10):e269. doi: 10.1371/journal.pbio.0050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mátyás F, et al. Subcellular localization of type 1 cannabinoid receptors in the rat basal ganglia. Neuroscience. 2006;137(1):337–361. doi: 10.1016/j.neuroscience.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Meier MH, et al. Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc Natl Acad Sci USA. 2012;109(40):E2657–E2664. doi: 10.1073/pnas.1206820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bossong MG, et al. Effects of δ9-tetrahydrocannabinol on human working memory function. Biol Psychiatry. 2012;71(8):693–699. doi: 10.1016/j.biopsych.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Marsicano G, et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302(5642):84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- 24.Monory K, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51(4):455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arain FM, Boyd KL, Gallagher MJ. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor α1 subunit. Epilepsia. 2012;53(8):e161–e165. doi: 10.1111/j.1528-1167.2012.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ernst WL, Zhang Y, Yoo JW, Ernst SJ, Noebels JL. Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J Neurosci. 2009;29(6):1615–1625. doi: 10.1523/JNEUROSCI.2081-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryan LJ. Characterization of cortical spindles in DBA/2 and C57BL/6 inbred mice. Brain Res Bull. 1984;13(4):549–558. doi: 10.1016/0361-9230(84)90037-6. [DOI] [PubMed] [Google Scholar]
- 28.Buzsáki G, Smith A, Berger S, Fisher LJ, Gage FH. Petit mal epilepsy and parkinsonian tremor: Hypothesis of a common pacemaker. Neuroscience. 1990;36(1):1–14. doi: 10.1016/0306-4522(90)90345-5. [DOI] [PubMed] [Google Scholar]
- 29.Wiest MC, Nicolelis MA. Behavioral detection of tactile stimuli during 7-12 Hz cortical oscillations in awake rats. Nat Neurosci. 2003;6(9):913–914. doi: 10.1038/nn1107. [DOI] [PubMed] [Google Scholar]
- 30.Shaw F-Z. Is spontaneous high-voltage rhythmic spike discharge in Long Evans rats an absence-like seizure activity? J Neurophysiol. 2004;91(1):63–77. doi: 10.1152/jn.00487.2003. [DOI] [PubMed] [Google Scholar]
- 31.Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: Is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62(5):612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kandel A, Buzsáki G. Cellular-synaptic generation of sleep spindles, spike-and-wave discharges, and evoked thalamocortical responses in the neocortex of the rat. J Neurosci. 1997;17(17):6783–6797. doi: 10.1523/JNEUROSCI.17-17-06783.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCormick DA, Bal T. Sleep and arousal: Thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- 34.Timofeev I, Steriade M. Neocortical seizures: Initiation, development and cessation. Neuroscience. 2004;123(2):299–336. doi: 10.1016/j.neuroscience.2003.08.051. [DOI] [PubMed] [Google Scholar]
- 35.Buzsáki G. The thalamic clock: Emergent network properties. Neuroscience. 1991;41(2-3):351–364. doi: 10.1016/0306-4522(91)90332-i. [DOI] [PubMed] [Google Scholar]
- 36.Deransart C, Vercueil L, Marescaux C, Depaulis A. The role of basal ganglia in the control of generalized absence seizures. Epilepsy Res. 1998;32(1-2):213–223. doi: 10.1016/s0920-1211(98)00053-9. [DOI] [PubMed] [Google Scholar]
- 37.Bodor AL, Giber K, Rovó Z, Ulbert I, Acsády L. Structural correlates of efficient GABAergic transmission in the basal ganglia-thalamus pathway. J Neurosci. 2008;28(12):3090–3102. doi: 10.1523/JNEUROSCI.5266-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deransart C, Lê-Pham BT, Hirsch E, Marescaux C, Depaulis A. Inhibition of the substantia nigra suppresses absences and clonic seizures in audiogenic rats, but not tonic seizures: Evidence for seizure specificity of the nigral control. Neuroscience. 2001;105(1):203–211. doi: 10.1016/s0306-4522(01)00165-8. [DOI] [PubMed] [Google Scholar]
- 39.Wallmichrath I, Szabo B. Cannabinoids inhibit striatonigral GABAergic neurotransmission in the mouse. Neuroscience. 2002;113(3):671–682. doi: 10.1016/s0306-4522(02)00109-4. [DOI] [PubMed] [Google Scholar]
- 40.Radek RJ, Curzon P, Decker MW. Characterization of high voltage spindles and spatial memory in young, mature and aged rats. Brain Res Bull. 1994;33(2):183–188. doi: 10.1016/0361-9230(94)90249-6. [DOI] [PubMed] [Google Scholar]
- 41.Paz JT, Chavez M, Saillet S, Deniau JM, Charpier S. Activity of ventral medial thalamic neurons during absence seizures and modulation of cortical paroxysms by the nigrothalamic pathway. J Neurosci. 2007;27(4):929–941. doi: 10.1523/JNEUROSCI.4677-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mallet N, Ballion B, Le Moine C, Gonon F. Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J Neurosci. 2006;26(14):3875–3884. doi: 10.1523/JNEUROSCI.4439-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dejean C, Gross CE, Bioulac B, Boraud T. Dynamic changes in the cortex-basal ganglia network after dopamine depletion in the rat. J Neurophysiol. 2008;100(1):385–396. doi: 10.1152/jn.90466.2008. [DOI] [PubMed] [Google Scholar]
- 44.Polack P-O, Mahon S, Chavez M, Charpier S. Inactivation of the somatosensory cortex prevents paroxysmal oscillations in cortical and related thalamic neurons in a genetic model of absence epilepsy. Cereb Cortex. 2009;19(9):2078–2091. doi: 10.1093/cercor/bhn237. [DOI] [PubMed] [Google Scholar]
- 45.Navarrete M, Araque A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron. 2010;68(1):113–126. doi: 10.1016/j.neuron.2010.08.043. [DOI] [PubMed] [Google Scholar]
- 46.Min R, Nevian T. Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat Neurosci. 2012;15(5):746–753. doi: 10.1038/nn.3075. [DOI] [PubMed] [Google Scholar]
- 47.Han J, et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell. 2012;148(5):1039–1050. doi: 10.1016/j.cell.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 48.Lutz B. On-demand activation of the endocannabinoid system in the control of neuronal excitability and epileptiform seizures. Biochem Pharmacol. 2004;68(9):1691–1698. doi: 10.1016/j.bcp.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Nicolelis MAL, Fanselow EE. Thalamocortical optimization of tactile processing according to behavioral state. Nat Neurosci. 2002;5:517–523. doi: 10.1038/nn0602-517. [DOI] [PubMed] [Google Scholar]
- 50.Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101(2):243–276. doi: 10.1016/s0306-4522(00)00353-5. [DOI] [PubMed] [Google Scholar]
- 51.Iversen L. The Science of Marijuana. New York: Oxford Univ Press; 2000. [Google Scholar]
- 52.Mailleux P, Vanderhaeghen JJ. Localization of cannabinoid receptor in the human developing and adult basal ganglia. Higher levels in the striatonigral neurons. Neurosci Lett. 1992;148(1-2):173–176. doi: 10.1016/0304-3940(92)90832-r. [DOI] [PubMed] [Google Scholar]
- 53.Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77(2):299–318. doi: 10.1016/s0306-4522(96)00428-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.