

Abstract
Iron–sulfur clusters are ubiquitous electron transfer cofactors in hydrogenases. Their types and redox properties are important for H2 catalysis, but, recently, their role in a protection mechanism against oxidative inactivation has also been recognized for a [4Fe-3S] cluster in O2-tolerant group 1 [NiFe] hydrogenases. This cluster, which is uniquely coordinated by six cysteines, is situated in the proximity of the catalytic [NiFe] site and exhibits unusual redox versatility. The [4Fe-3S] cluster in hydrogenase (Hase) I from Aquifex aeolicus performs two redox transitions within a very small potential range, forming a superoxidized state above +200 mV vs. standard hydrogen electrode (SHE). Crystallographic data has revealed that this state is stabilized by the coordination of one of the iron atoms to a backbone nitrogen. Thus, the proximal [4Fe-3S] cluster undergoes redox-dependent changes to serve multiple purposes beyond classical electron transfer. In this paper, we present field-dependent 57Fe-Mössbauer and EPR data for Hase I, which, in conjunction with spectroscopically calibrated density functional theory (DFT) calculations, reveal the distribution of Fe valences and spin-coupling schemes for the iron–sulfur clusters. The data demonstrate that the electronic structure of the [4Fe-3S] core in its three oxidation states closely resembles that of corresponding conventional [4Fe-4S] cubanes, albeit with distinct differences for some individual iron sites. The medial and distal iron–sulfur clusters have similar electronic properties as the corresponding cofactors in standard hydrogenases, although their redox potentials are higher.
Catalysis of hydrogen conversion in [NiFe] hydrogenases relies on electron transfer between the active site and a chain of iron–sulfur clusters (1). The most abundant components of such chains are low-potential tetranuclear [4Fe-4S] centers of the ferredoxin type (2). In the so-called group I hydrogenases, such [4Fe-4S] clusters are found close to the active [NiFe] site and near the enzyme’s surface (in the so-called proximal and distal positions, respectively) (1). A third cluster in medial position, typically of the [3Fe-4S] type, completes the electron transfer pathway.
Recently, a subclass of O2-tolerant, membrane-bound hydrogenases (MBH) was recognized to possess an uncommon proximal iron–sulfur cluster with unique redox properties that enable a protection mechanism against oxidative inactivation of the [NiFe] site. Typical representatives of such hydrogenases are those from Aquifex aeolicus (3), Hydrogenovibrio marinus (4), Ralstonia eutropha (5), and Escherichia coli (6). Three independent crystal structure investigations (4, 5, 7) revealed that the proximal cluster in these enzymes is not a classical [4Fe-4S] but, instead, a [4Fe-3S] cluster with an unusual six-cysteine binding motif (CXCCX94CX4CX28C) that is unique for the subclass of O2-tolerant enzymes (8, 9) (SI Appendix, Fig. S1). Four of the six cysteine residues (C) coordinate the cluster in the usual manner as terminal ligands to the [4Fe-3S] core, whereas the sulfur (S−) atom of one of the supernumerary cysteines (C) substitutes for an inorganic sulfide (S2−) in the cubane core, and the other serves as a second terminal ligand to one of the Fe atoms (4, 5, 7) (Fig. 1). In contrast, the structures of the medial and distal iron–sulfur clusters and their arrangement relative to the [NiFe] site are almost identical to those of standard hydrogenases (1, 4, 5).
Fig. 1.
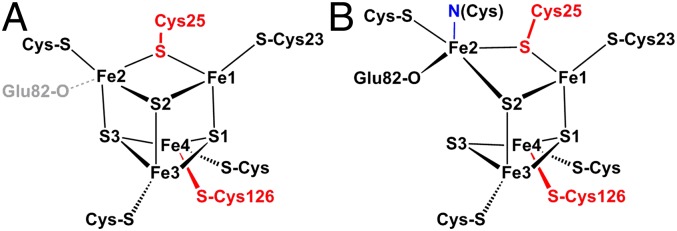
Structures of the [4Fe-3S] cluster of H. marinus hydrogenase in the reduced (3+) state (A) and in the superoxidized (5+) state (B). (Labeling of iron sites is like in ref. 4; conversion to the alternative scheme of refs. 5 and 7 is achieved by mapping Fe1→1, Fe2→4, Fe3→2, Fe4→3.) The two “supernumerary” cysteines, Cys25 and Cys126, are indicated in red, Glu82 is sketched in gray (A) or black (B). The cysteine closest to the [NiFe] site (Cys23), and the bond to the backbone nitrogen in the superoxidized state (blue), are also indicated. The distance between Fe2 and S3 varies from 240 picometers (pm) in the reduced-state (A) to 401 pm in the superoxidized state (B).
Conventional [4Fe-4S] clusters perform either a single-electron 1+/2+ redox transition (in low-potential ferredoxins) or a 2+/3+ transition in high-potential iron-sulfur proteins (HiPIPs) (2). Both forms share the same isoelectronic 2+ oxidation state with spin S = 0, albeit with some minor differences in structures and electron delocalization (10, 11). In contrast, the proximal [4Fe-3S] cluster in hydrogenase (Hase) I from A. aeolicus can attain three redox states within a redox potential span of only 150 mV, two of which are paramagnetic with S = 1/2, as we could show by EPR-detected potentiometric titrations (3). Particularly, a superoxidized state, not known for low-potential [4Fe-4S] clusters (12), manifests at high potentials (3). According to the crystal structure of H. marinus hydrogenase (4) and EPR-detected electrochemical titrations on A. aeolicus Hase I (3), this superoxidized [4Fe-3S]5+ state is stabilized by structural changes arising from deprotonation of a backbone-nitrogen atom and concomitant nitrogen coordination to one of the Fe sites (3, 4). Additionally, a glutamate group was found within bonding distance to the cluster (7).
In this report, we present a detailed electronic structural investigation of the iron–sulfur cofactors in A. aeolicus Hase I based on EPR and applied-field Mössbauer spectroscopy in conjunction with quantum-chemical calculations. In particular, the proximal [4Fe-3S] cluster has been thoroughly examined based on the results of the crystallographic studies. Local properties of the Fe atoms involved in the redox-induced structural changes have been identified and their spin and charge distribution has been compared with that of conventional [4Fe-4S] clusters (13–15) in view of the crucial role of the [4Fe-3S] cluster toward O2-tolerance within this class of enzymes.
Results
Hase I from A. aeolicus grown on 57Fe-enriched media (3) was treated with hydrogen, ascorbate, and porphyrexide or air to obtain fully reduced, oxidized, and superoxidized samples, as listed in Table 1 and SI Appendix, Table S1. The redox states of the cofactors were assigned according to our previous titration data (3), whereby EPR measurements on aliquots of the Mössbauer samples were used to verify the spin counts of the (paramagnetic) cofactors and EPR silence of the [4Fe-3S]4+ cluster in the ascorbate-treated sample (SI Appendix, Fig. S2).
Table 1.
FeS cofactors and redox states of Hase I
| Preparation/site | Proximal | Medial | Distal | [NiFe] |
| H2-reduction* | [4Fe-3S]3+ | [3Fe-4S]0 | [4Fe-4S]1+ | Ni-R |
| S = 1/2 | S = 2 | S = 1/2 | S = 0 | |
| Oxidation† | [4Fe-3S]4+ | [3Fe-4S]1+ | [4Fe-4S]2+ | Ni-B |
| S = 0 | S = 1/2 | S = 0 | S = 1/2 | |
| Superoxidation‡ | [4Fe-3S]5+ | [3Fe-4S]1+ | [4Fe-4S]2+ | Ni-B |
| S = 1/2 | S = 1/2 | S = 0 | S = 1/2 |
Cluster oxidation numbers are obtained from the spectroscopically derived oxidation numbers of Fe with the core sulfides taken in their closed shell form (S2−).
*Fully reduced with H2 gas.
†Partially reduced with ascorbate (SI Appendix).
‡Full superoxidation with porphyrexide; alternative preparation in air yields ∼20% contamination with the proximal [4Fe-3S]4+ cluster, accounted for in Mössbauer simulations.
The UV-visible spectra of superoxidized Hase I prepared in air show broad bands around 400 nm, arising from S-to-Fe charge transfer transitions (16) (SI Appendix, Fig. S3). The spectra resemble those of other oxidized O2-tolerant group I enzymes (17), but they cannot be distinguished from those of standard [NiFe] hydrogenases without superoxidized [4Fe-3S] clusters. Interestingly, however, these features could not be discriminated from those of oxidized HiPIP proteins (18) or alkylated ferredoxin:thioredoxin reductase (NEM-FTR) with cubane clusters in the corresponding 3+ state (19). Likewise, EPR spectra of the [4Fe-3S] cluster in 3+ and 5+ states (3) resemble those of [4Fe-4S] centers in the corresponding 1+ and 3+ states (19–22). This is not completely unexpected, because the 4Fe cores are formally isoelectronic and the modified [4Fe-3S] cores differ from classical cubanes only by an inorganic S2− ligand. Although the cluster oxidation numbers are no observables, they can be substantiated from electric and magnetic Mössbauer hyperfine interactions, probing the local oxidation and spin states of the metal ions.
Electric Mössbauer Parameters and Valence States.
Mössbauer spectra were measured with reduced, oxidized, and superoxidized samples of Hase I. The zero-field spectra recorded at 160 K could be decomposed in a number of quadrupole doublets without paramagnetic broadening because of fast-spin relaxation (Fig. 2 and SI Appendix, Tables S2–S4). Five or six distinct subspectra could be identified that arise from [NiFe] and distinct groups of the 11 iron sites within the valence-delocalized FeS cofactors. Primarily, we used three subspectra with constrained relative intensities for the iron sites, which have the formal oxidation states “Fe2+,” “Fe2.5+,” and “Fe3+” (a detailed list of formal valences is found in SI Appendix, Table S1). In addition, a subspectrum with 8% intensity was adopted for the low-spin Fe(II) site in the [NiFe] cluster (1/12 of the total iron content).
Fig. 2.

Zero-field Mössbauer spectra of Hase I recorded at 160 K. The subspectra are Lorentzian doublets [yellow, Fe2+; green, Fe2.5+; blue, Fe3+; purple, special site (S) of [4Fe-3S] clusters; orange, ferrous site (F) of reduced [4Fe-3S] cluster in the H2-reduced sample; light-blue, low-spin Fe(II) in [NiFe]; red, sum of subspectra]. The dotted vertical lines represent the ferric character increasing with oxidation state.
Moreover, a unique feature of the Hase I spectra in all three different preparations are resolved lines at the wings of the absorptions, which reveal an additional special subspectrum (S) with moderate isomer shift but, remarkably large quadrupole splitting (δ/|ΔEQ| = 0.51/2.61 mm·s−1, 0.48/2.23 mm·s−1, and 0.46/2.42 mm·s−1 for reduced, oxidized, and superoxidized samples). Its relative intensity, ∼7.3–8.6%, accounts for a fully occupied, genuine iron site. Because this subspectrum was not observed for standard hydrogenases (23–25) (although the distal cluster has an uncommon histidine-coordinated iron corner), we assign it to the modified [4Fe-3S] cluster, present only in O2-tolerant hydrogenases. This is corroborated by the applied-field spectra given below, because such a large quadrupole splitting could be assigned neither to a part of the diamagnetic distal cluster in oxidized samples nor to the integer-spin S = 2 component of the medial 3Fe cluster.
In addition to the five subspectra introduced so far (Fe2+, Fe2.5+, Fe3+, special site “S,” and Fe(II) low-spin), we had to add a sixth subspectrum with markedly larger isomer shift (δ/ΔEQ = 0.72/1.56 mm·s−1) to account for a second distinct site with rather localized ferrous character, occurring only in the spectrum of the H2-reduced enzyme (Fig. 2). This component (F) was also required for consistent simulation of the magnetic Mössbauer spectra and was likewise attributed to a site of the reduced [4Fe-3S]3+ cluster (see below).
In summary, the zero-field Mössbauer spectra of Hase I obtained under three different redox conditions could be well simulated by using a scheme of subspectra for one 3Fe and two 4Fe clusters, of which the proximal [4Fe-3S] cluster can be superoxidized and occur in three oxidation states. These states are isoelectronic to the formal [3Fe2+, Fe3+], [2Fe2+, 2Fe3+], and [1Fe2+, 3Fe3+] motifs recognized for classical [4Fe-4S] centers (although physiologically these three never occur in one system). The overall increase of ferric character in the series is reflected in the Mössbauer subspectra by a decrease of the average isomer shifts (<δ>[4Fe-3S] = 0.52, 0.43, 0.36 mm·s−1).
Magnetic Mössbauer Spectra and Spin Distribution.
Applied-field Mössbauer spectra of fully reduced and superoxidized Hase I recorded at 4.2, 80, and 160 K are shown in Fig. 3. Since the magnetic spectra exhibit higher site selectivity than the zero-field spectra, not least because the signs of electric field gradients (efg) and induced internal fields can be resolved from the field dependence of the spectra (24–26), separate subspectra had to be introduced for all 12 iron sites. To this end, the five to six collective subspectra introduced above for the different classes of mixed-valence irons sites seen in the zero-field spectra were broken up into their cluster contributions (SI Appendix, Tables S2–S4). For the sake of clarity, we present the result in four collective subspectra corresponding to the three FeS clusters and the [NiFe] center.
Fig. 3.

Magnetic Mössbauer spectra of Hase I reduced with H2 (A) and superoxidized with air (B). Fields of 0.1–7 T were applied perpendicular to the γ-ray beam. The solid lines represent global simulations (SI Appendix, Tables S6 and S7) with the following contributions: proximal [4Fe-3S]3+ (purple), medial [3Fe-4S]0 (light green), distal [4Fe-4S]1+ (blue) (A); and proximal [4Fe-3S]5+ (purple), medial [3Fe-4S]1+ (light green), distal [4Fe-4S]2+ (blue) (B). Fe(II) of the [NiFe] site (orange) occurs in both A and B.
Spin relaxation was found to be slow for the paramagnetic centers at 4.2 K and fast at 80 and 160 K, which means that the internal fields virtually average to zero at the high temperatures. The condition is valuable for assigning efg parameters, i.e., the sign of ΔEQ and the asymmetry parameter η. The zero-field values of δ and ΔEQ were taken in this program as starting points and valuable constraints for global optimization of the magnetic spectra (allowing for only the usual temperature variations).
In contrast, the large magnetic splitting observed at 4.2 K indicates strong internal fields at some iron nuclei attributable to strong magnetic hyperfine coupling with static spin expectation values, 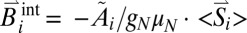 (26). The temperature and field dependence of the hyperfine coupling could be simulated well with
(26). The temperature and field dependence of the hyperfine coupling could be simulated well with 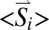 values derived from the usual spin Hamiltonian for clusters with strong intrinsic spin coupling (SI Appendix). However, weak isotropic coupling between the proximal cluster, Ni-B, and the medial cluster had to be introduced with J values up to ∼0.02 cm−1 for the superoxidized enzyme. This coupling, which causes reduction of magnetic splittings at weak fields, was established first by X- and W-band EPR studies (SI Appendix, Figs. S4 and S5 and Table S5) (3).
values derived from the usual spin Hamiltonian for clusters with strong intrinsic spin coupling (SI Appendix). However, weak isotropic coupling between the proximal cluster, Ni-B, and the medial cluster had to be introduced with J values up to ∼0.02 cm−1 for the superoxidized enzyme. This coupling, which causes reduction of magnetic splittings at weak fields, was established first by X- and W-band EPR studies (SI Appendix, Figs. S4 and S5 and Table S5) (3).
The signs of the local magnetic hyperfine coupling constants found for the 57Fe-nuclei are consistent with the usual spin distribution known for 3Fe and 4Fe clusters in their different magnetic ground states and spin-coupling schemes (a survey is given in SI Appendix, Table S1). Iron sites with majority spin density (↓) have negative A tensors with respect to the cluster spin, whereas minority spin sites (↑) have positive A-values (26). Interestingly, the classical spin-distribution pattern also holds for the proximal [4Fe-3S] cluster, which, in this respect, resembles its isoelectronic unmodified analogs.
In the H2-reduced enzyme, all iron–sulfur centers are paramagnetic and exhibit appreciable hyperfine splitting at 4.2 K (Fig. 3A). Only the low-spin Fe(II) site of the [NiFe] center shows weak magnetic splitting from the applied field only, as expected for the diamagnetic Ni-R state (orange trace). The distal [4Fe-4S]1+ cluster (blue) may be described by a delocalized mixed-valence pair Fe2.5+-Fe2.5+ with subspin (S* = 9/2) and a ferrous pair with (S* = 4) (14, 15). Accordingly, the spin minority ferrous pair has positive hyperfine parameters A, whereas the Fe2.5+-Fe2.5+ pair has negative A values (SI Appendix, Table S6). Although the distal cluster is known to have a modified Fe subsite with a fifth Nε nitrogen ligand from a histidine (1, 4, 5, 7), the spectra do not exhibit unusually large δ or ΔEQ. Similar to previous studies on [NiFe] hydrogenases (23–25), the effect may be small and not resolved in the present spectra.
The medial [3Fe-4S]0 cluster (light green, Fig. 3A) was simulated with S = 2, arising from antiferromagnetic coupling of a delocalized mixed-valence Fe2.5+-Fe2.5+ pair (S* = 9/2) and a ferric ion (S = 5/2). The effective hyperfine values resemble those of other [3Fe-4S] clusters (26, 27), with negative A for the mixed valence pair (majority spin) and positive A for the ferric site. The zero-field splitting parameters D = –2.20 cm−1, E/D = 0.26 are typical for [3Fe-4S]0 clusters in hydrogenases (23).
The g values for the reduced proximal [4Fe-3S] cluster, as taken from previous EPR work (3), are similar to those of the isoelectronic conventional [4Fe-4S]1+ clusters (20, 28). Correspondingly, the magnetic Mössbauer spectra (purple trace, Fig. 3A) could be fitted with the same spin-coupling scheme found for reduced, low-potential ferredoxins; a delocalized mixed-valence pair Fe2.5+-Fe2.5+ (S* = 9/2) is antiferromagnetically coupled to a ferrous Fe2+-Fe2+ pair (S* = 4). Based on the signs of the A values, the subspectra with δ = 0.50 mm·s−1/ΔEQ = +2.60 mm·s−1 and with δ = 0.44 mm·s−1/ΔEQ = +1.23 mm·s−1 are ascribed to the ferrous pair (positive A). The result corroborates the oxidation state +3 for the reduced [4Fe-3S] core (spin-coupling scheme and Mössbauer parameters as for 3 Fe2+, 1 Fe3+, completed with 3 S2-; Fig. 1 and SI Appendix, Table S1). The A values for the [4Fe-3S]3+ cluster are generally similar to those of [4Fe-4S]1+ clusters (27), except that site (F) has relatively large A values, in accordance with a rather localized ferrous valence (δ = 0.72 mm·s−1) and relatively low covalency (SI Appendix, Table S6).
In superoxidized Hase I, the medial and proximal FeS clusters are paramagnetic (Table 1), whereas the distal [4Fe-4S]2+ cluster is diamagnetic and shows only weak magnetic splitting from the external magnetic field (blue trace in Fig. 3B). Likewise, the ferrous low-spin site of the [NiFe] center has a vanishing hyperfine field (orange trace; Fig. 3B), which shows that covalently transferred spin density from nickel is negligible. In contrast, the [3Fe-4S]1+ cluster is an all-ferric center with S = 1/2 and small g anisotropy arising from spin frustration (23, 28, 29). This is seen from the moderately strong overall splitting in the magnetic Mössbauer spectrum (light green trace; Fig. 3B). The A values are characteristic of three-iron sulfur clusters as found previously, with one of the A tensors being characteristically small (SI Appendix, Table S7) (23).
The proximal cluster in its superoxidized state is oxidized by two electrons more than in the H2-reduced enzyme. Paramagnetism of the resulting [4Fe-3S]5+ state is supported by the magnetic splitting in the field-dependent Mössbauer spectra. However, predicting a spin-coupling scheme is difficult because of the irregular structure of the cluster (Fig. 1B), which lacks a direct bridge between Fe2 and Fe4. We adopted the general pattern of all mixed-valence four-Fe clusters, that pairs of irons harbor minority and majority spin populations (positive and negative A values). The best fits performed with this assumption required that the special subspectrum (S) with the largest quadrupole splitting (δ/ΔEQ = 0.46/2.45 mm·s−1) and a site with small isomer shift (0.28 mm·s−1) have positive A values. This appears to be formally consistent with the presence of a mixed-valence localized pair (S* = 9/2), antiparallel coupled to a ferric pair (2× S = 5/2), as recently proposed by a theoretical/crystallographic study (7). However, the assignment of subspectra to iron sites, obtained from density functional theory (DFT) calculations, rules out this simple scheme, as is discussed below.
Electronic Structure Calculations.
Detailed interpretation of the Mössbauer data were obtained from DFT calculations of the electronic structure of the proximal [4Fe-3S] cluster in all three oxidation states. To this end, cluster models have been constructed on the basis of the crystal structures reported for the membrane-bound hydrogenases of H. marinus and R. eutropha (4, 5). Constrained optimizations have been performed with C-α carbons of coordinating amino acids fixed to their crystallographic positions (SI Appendix, Fig. S8). Potential binding of Glu82 to Fe2 and its protonation state has been probed systematically. In general, all possible broken-symmetry (BS) solutions have been calculated for each structure. The Mössbauer parameters for each of the four iron sites have been used as criteria for the selection of the best BS solution (isomer shift δ, quadrupole splitting ΔEQ with sign, asymmetry parameter η of the efg, and the sign of the isotropic A value, Aiso). Calculations were carried out with standard functionals (SI Appendix) without adjustments of fractional Hartree–Fock exchange or other ad hoc modifications. The agreement between calculated and experimental metric details, as well as Mössbauer spectroscopic parameters is reasonable (Table 2 and SI Appendix, Tables S8–S14). Calculated anisotropic A tensors and relations of the efg- and A-tensor axes is found in SI Appendix, Table S15.
Table 2.
Calculated Mössbauer parameters for the iron sites of the proximal [4Fe-3S] cluster
| δ, mm·s−1 | ΔEQ, mm·s−1 | η | Aiso/gNµN,* T | Spin population | Charge | |
| Reduced [4Fe-3S]3+ | ||||||
| Fe1 (F) | 0.60 (0.71) | +1.41 (+1.52) | 0.28 (0.3) | −45.8 (−31.3) | +3.5 | −0.15 |
| Fe2 | 0.35 (0.44) | +1.25† (+1.23) | 0.99 (0.9) | +31.6 (+20.0) | −3.1 | −0.38 |
| Fe3 | 0.36 (0.42) | +0.93 (+0.84) | 0.3 (0.3) | −36.2 (−25.0) | +3.1 | −0.38 |
| Fe4 (S) | 0.46 (0.50) | +2.13 (+2.60) | 0.51 (0.1) | +19.2 (+14.0) | −2.7 | −0.41 |
| Superoxidized [4Fe-3S]5+ | ||||||
| Fe1 | 0.39 (0.40) | (−)1.56† (−1.00) | 0.83 (0.7) | −43.5 (−24.3) | 3.3 | −0.33 |
| Fe2 (S) | 0.38 (0.46) | +2.25 (+2.45) | 0.4 (0.5) | +35.4 (+18.6) | −2.4 | −0.09 |
| Fe3 | 0.30 (0.39) | (+)0.81† (+0.70) | 0.74 (1.0) | −41.8 (−34.7) | 3.3 | −0.37 |
| Fe4 | 0.22 (0.28) | +0.74 (+0.60) | 0.56 (0.3) | +42.5 (+24.2) | −3.4 | −0.35 |
Calculated values (bold) are tentatively assigned to the experimental values given in parentheses (see SI Appendix, Tables S6 and S7). For the assignment of special sites, see Fig.1. (F), ferrous subspectrum; (S), special subspectrum.
*Isotropic experimental values are the average of the anisotropic values given in SI Appendix, Tables S6 and S7.
†The efg tensor has two components of similar size but different sign because of the large asymmetry parameter η close to 1. For details of tensor orientations with respect to the magnetic axes see SI Appendix.
Based on the ΔEQ values alone, the best BS solution for the superoxidized cluster is labeled Ox2_14 (SI Appendix, Table S9, spin populations of Fe1 and Fe4 flipped) and corresponds to BS13 in ref. 7. However, this solution does not properly reproduce the experimental δ values or the sign of the A values, of which the latter reveals a basically wrong spin-coupling scheme. Instead, our best solution, which is found within 4 kcal/mol of the energy minimum, has the spin populations of Fe2 and Fe4 flipped (Ox2_24), yielding proper values for δ, ΔEQ, η, and the A values. In both cases, Glu82 is required to be unprotonated.
On the basis of the calculations, the special subspectrum (S) of the superoxidized [4Fe-3S]5+cluster with the large quadrupole splitting (ΔEQ = 2.45 mm·s−1) is assigned to the nitrogen-coordinated Fe2. This agrees with chemical intuition, where a large efg is expected because of the anisotropic covalency that results from asymmetric coordination with a unique, short N bond (4). The result is in contrast to the assignment of subspectrum (S) to Fe1 (7), which has quasitetrahedral symmetry. Note that our suggested spin distribution (αβαβ spin alignment for Fe1, Fe2, Fe3, Fe4, respectively) does not correspond to a standard coupling scheme observed for iron–sulfur clusters. The standard spin-coupling scheme involving a delocalized mixed-valence pair and two “ferric” ions of the type [2Fe2.5+, 2Fe3+] does not apply because Fe2 and Fe4 with minority spin cannot form a strongly coupled mixed-valence pair with S* = 9/2 since they are not connected by bridges that would transmit strong intersite coupling. Consequently, the total spin S = 1/2 cannot arise from simple antiparallel coupling of S* = 9/2 to two ferric sites with S = 5/2 each. Weak bonding interactions found in the DFT model, by contrast, indicate that Fe1 and Fe3 may be regarded as the valence-delocalized “Fe2+/Fe3+” pair (Mayer bond order 0.6, SI Appendix, Fig. S9). This makes it more difficult to explain why the remaining ferric sites do not determine the majority spin. Apparently, the asymmetric topology of the [4Fe-3S]5+ cluster leads to a more complex spin-density distribution pattern compared with standard cubanes, such that the resulting charge and spin distribution is energetically more favorable than in HiPIP clusters, as can be suspected on the basis of the redox potentials.
Selection of the best BS solution for the reduced [4Fe-3S]3+ cluster is less clear cut than for the superoxidized state because several solutions exist (SI Appendix, Table S10). Importantly, all reasonable choices imply that Glu82 is unprotonated. This raises some concerns with respect to its suspected role as a base (7). Our best result (Red2_24) shows the spin populations on Fe2 and Fe4 to be flipped. Based on this solution, Fe4 and Fe1 are assigned to the special subspectra (S) and (F) found for the reduced proximal cluster, i.e., those with largest ΔEQ and highest δ, respectively (Table 2). Interestingly, the iron site with the largest quadrupole splitting (subspectrum S) has changed from Fe2 in the superoxidized structure to Fe4 in the reduced structure, in which Fe4 has the unusual two-cysteine coordination in an otherwise rather cuboidal cluster. Unlike for the superoxidized case, the optimized structure for Red2_24 forms a tightly closed cubane with a long Fe4–S-Cys25 separation. The spin distribution obtained from the calculations, in agreement with the experimental A values, is best explained by the presence of a mixed-valence pair (Fe1 and Fe3) and two ferrous sites (Fe2 and Fe4), which render the special subspectrum (F) with the highest isomer shift part of the mixed-valence pair.
Discussion
Tetranuclear iron–sulfur centers are common in biology, exhibiting great diversity in binding motifs, redox properties, and catalytic roles (2, 15, 21, 30). The proximal [4Fe-3S] cluster found in Hase I from A. aeolicus and other organisms (4, 5, 7) differs from these conventional cubanes by having a distorted core structure, as well as a unique six-cysteine coordination mode, common for all group I O2-tolerant hydrogenases (9). Magnetic Mössbauer data presented in this study, in conjunction with our previous EPR results, substantiate the notion that the [4Fe-3S] cluster can attain three redox states with formal charges 5+, 4+, and 3+, which are isoelectronic to the 3+, 2+, and 1+ states of conventional [4Fe-4S] clusters (SI Appendix, Table S1). The oxidation numbers in this formal concept are obtained from spectroscopically probed oxidation states of the iron atoms with the inorganic sulfur ligands taken in their usual closed shell configuration (S2−). Our data suggest that the valence-delocalization pattern and the spin-coupling schemes for the [4Fe-3S] cluster are similar to those of conventional [4Fe-4S] clusters, at least for the reduced and oxidized state. The superoxidized [4Fe-3S]5+ cluster may also be regarded as comprising a mixed-valence Fe2.5+/Fe2.5+ and a diferric pair, comparable with classical oxidized HiPIP-type clusters (19, 21, 22, 28), but the spin interactions appear to be more complex than in HiPIP clusters. The reduced [4Fe-3S]3+ cluster shares the pattern of a mixed-valence and a diferrous pair with the reduced cubanes in bacterial ferredoxins (14, 15). Consistent with these extreme oxidation states, the [4Fe-3S]4+ state appears to be well described by two mixed-valence pairs.
The Mössbauer spectra of the proximal [4Fe-3S] center exhibit throughout a particular subspectrum (S) with large quadrupole splitting, which indicates unusual coordination of a distinct Fe site (31) in all three oxidation states. Although analogous subspectra are known for [4Fe-4S] clusters participating in chemical reactions, site-differentiated Fe atoms with partially trapped valences are usually observed only in one of the two accessible redox states. The perturbations are mostly associated with substrate binding (30) and arise from five-coordination or otherwise perturbed protein ligation (31). Interestingly, the majority of “perturbed” clusters show signs of a “special” Fe subsite only in their diamagnetic [4Fe-4S]2+ state. Some notable examples are alkylated ferredoxin:thioredoxin reductase (NEM-FTR) (19), the CCG cluster in succinate:quinone oxidoreductase (SQR) (21), the S-adenosyl methionine (SAM) bound cluster in pyruvate formate lyase (PFL) (30), and the cluster in the LytB protein from E. coli (32). Although the presence of trapped-valence sites disturbs the spin-dependent delocalization (33), the diamagnetic state (S = 0) of these clusters and of the [4Fe-3S]4+ cluster in A. aeolicus is most likely stabilized through vibronic-coupling and/or unequal J-coupling constants.
Spectroscopy-oriented quantum chemical calculations provided a detailed description of the electronic structure of the [4Fe-3S] cluster and correlated the spin and valence distribution derived from EPR and Mössbauer spectra to the recently reported crystallographic data (4, 5, 7). In particular, it could be shown that the unusual Mössbauer subspectra (S) and (F) found for the H2-reduced state of the [4Fe-3S] cluster can be assigned to Fe4 and Fe1, respectively. Whereas three Fe sites in the reduced structure retain quasitetrahedral FeS4 coordination, Fe4 is unusual in that it is ligated by two terminal cysteine ligands and comes out of the core of the pseudocube (Fig. 1), which predestines it as the special site (S) with large quadrupole splitting, corroborated by the calculation.
In contrast, the most unusual site in the superoxidized structure is Fe2. Accordingly, the DFT calculations clearly assign (S) to that iron, although this may have essentially ferric character (if Fe1-Fe3 is regarded as mixed-valence pair). Although ferric compounds are typically characterized by small ΔEQ because of the vanishing valence contribution of the 3d5 configuration to the efg, large ΔEQ are encountered for such a site if charge asymmetry is caused by a strongly covalent bond, such as with oxoligands, for example (34, 35). Thus, the DFT result that Fe2 provides subspectrum (S) in the [4Fe-3S]5+ cluster agrees with the crystallographic data, which show that superoxidation is accompanied by deprotonation of a backbone amide nitrogen of the tandem CC motif and nitrogen coordination to Fe2, forming a tight nitrogen bond (209 pm). The recent crystal structure and theoretical study for the E. coli Hyd-1 hydrogenase has shown that Fe2 may be five-coordinate with a nearby deprotonated glutamate. Our DFT results support this interpretation, particularly for the superoxidized state, because the interpretation of the Mössbauer data appears to be very difficult on the basis of a different model.
We note in passing that the spectroscopic properties of the other clusters, i.e., the medial [3Fe-4S] and the distal [4Fe-4S] centers, are in the range of those reported for other hydrogenases (23–25), which is in agreement with the preserved structural features (4, 5).
Superoxidation of the proximal [4Fe-3S]4+ cluster to the 5+ state, stabilized by deprotonation and binding of the amide nitrogen of the CC tandem motif and the incorporation of the nitrogen as a ligand to Fe2, is comparable to that of the P cluster in the A. vinelandii nitrogenase upon its two-electron oxidation from the native (PN) to the oxidized state (POX) (35). In this step, one of the Fe loses a sulfide bond and binds to the backbone amide nitrogen of an Sγ-coordinated cysteine group. In addition, a second Fe coordinates to the Oγ atom of a serine, thereby avoiding disruption of the cluster. A second case of a backbone nitrogen coordinating a Fe atom is that of nitrile dehydratase (36). Notably, nitrile dehydratase also yields a large ΔEQ (37), however, as expected for ferric low-spin ions.
The presence of supernumerary cysteines in the coordination sphere of the [4Fe-3S] cluster appears to be essential for its ability to trigger deprotonation and binding of a peptide amide nitrogen to tune its electronic properties for two redox transitions within 150 mV (3). Exchange of the two “additional” cysteines, Cys25 and Cys126, in R. eutropha MBH, with the usual glycine residues found in standard hydrogenases, has shown that the [4Fe-3S] cluster may be involved in a protection mechanism against inactivation of the [NiFe] center by molecular oxygen. The distance and orientation of the [4Fe-3S] cluster with respect to the [NiFe] site in the respective crystal structures (4) are essentially identical in the 3+ and 5+ states, where the ligand of the [4Fe-3S] cluster closest to the [NiFe] site is the conserved Cys23 (Fig. 1). It is tempting to assume that electron transfer proceeds via covalent bonds from this cysteine to the active site. The exchange-coupling constant J between the proximal [4Fe-3S]5+ and the [NiFe] site is 209 × 10−4 cm−1, whereas that between the reduced [4Fe-3S]3+ and the [NiFe] site is only 36 × 10−4 cm−1 (3). Electron-transfer rates are proportional to the magnitude of the exchange interaction and this significant difference in J values could affect these rates (38).
The [4Fe-3S] cluster uses surplus-cysteine ligation to stabilize two redox couples unprecedented in biological [4Fe-4S] cubanes. Our Mössbauer and DFT results show a distribution of Fe valences and local spin populations that basically resemble those of classical cubanes, especially in lower oxidation states. In detail, however, the electronic properties of the sites participating in the chemistry are distinct, particularly in the superoxidized state. The redox transitions are clearly metal-centered. Due to its proximity to the [NiFe] center, the proximal [4Fe-3S] cluster has in fact been discussed in the past as being, to some degree, part of the catalytic core of the enzyme (39). Therefore, considering its very positive redox potential and its appreciable electronic coupling to the [NiFe] site, it is not surprising that the [4Fe-3S] cofactor is able to perform at least two functions through redox-dependent structural changes; to mediate catalytic electron transfer and to serve as an electron reservoir for the efficient reductive removal of O2.
Materials and Methods
The enzyme Hase I was isolated under semianaerobic conditions from the hyperthermophile Aquifex aeolicus grown in 57Fe enriched media. The theoretical methods are cited in the text. Details of the experimental procedures, and spectroscopic and theoretical descriptions, including sequence alignments and EPR and Mössbauer spectra, as well as tables with simulation parameters and DFT calculations are available in SI Appendix.
Supplementary Material
Acknowledgments
This work was supported by European Union/Energy Network SOLAR-H2 (Seventh Framework Programme Contract 212508) and Région Provence-Alpes-Côte d’Azur.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1202575110/-/DCSupplemental.
References
- 1.Volbeda A, et al. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature. 1995;373(6515):580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 2.Beinert H. Iron-sulfur proteins: Ancient structures, still full of surprises. J Biol Inorg Chem. 2000;5(1):2–15. doi: 10.1007/s007750050002. [DOI] [PubMed] [Google Scholar]
- 3.Pandelia ME, et al. Characterization of a unique [FeS] cluster in the electron transfer chain of the oxygen tolerant [NiFe] hydrogenase from Aquifex aeolicus. Proc Natl Acad Sci USA. 2011;108(15):6097–6102. doi: 10.1073/pnas.1100610108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shomura Y, Yoon KS, Nishihara H, Higuchi Y. Structural basis for a [4Fe-3S] cluster in the oxygen-tolerant membrane-bound [NiFe]-hydrogenase. Nature. 2011;479(7372):253–256. doi: 10.1038/nature10504. [DOI] [PubMed] [Google Scholar]
- 5.Fritsch J, et al. The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature. 2011;479(7372):249–252. doi: 10.1038/nature10505. [DOI] [PubMed] [Google Scholar]
- 6.Lukey MJ, et al. Oxygen-tolerant [NiFe]-hydrogenases: The individual and collective importance of supernumerary cysteines at the proximal Fe-S cluster. J Am Chem Soc. 2011;133(42):16881–16892. doi: 10.1021/ja205393w. [DOI] [PubMed] [Google Scholar]
- 7.Volbeda A, et al. X-ray crystallographic and computational studies of the O2-tolerant [NiFe]-hydrogenase 1 from Escherichia coli. Proc Natl Acad Sci USA. 2012;109(14):5305–5310. doi: 10.1073/pnas.1119806109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goris T, et al. A unique iron-sulfur cluster is crucial for oxygen tolerance of a [NiFe]-hydrogenase. Nat Chem Biol. 2011;7(5):310–318. doi: 10.1038/nchembio.555. [DOI] [PubMed] [Google Scholar]
- 9.Pandelia ME, Lubitz W, Nitschke W. Evolution and diversification of Group 1 [NiFe] hydrogenases. Is there a phylogenetic marker for O(2)-tolerance? Biochim Biophys Acta. 2012;1817(9):1565–1575. doi: 10.1016/j.bbabio.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Dey A, et al. Solvent tuning of electrochemical potentials in the active sites of HiPIP versus ferredoxin. Science. 2007;318(5855):1464–1468. doi: 10.1126/science.1147753. [DOI] [PubMed] [Google Scholar]
- 11.Niu SQ, Ichiye T. Cleavage of [4Fe-4S]-type clusters: Breaking the symmetry. J Phys Chem A. 2009;113(19):5710–5717. doi: 10.1021/jp900402y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heering HA, Bulsink YB, Hagen WR, Meyer TE. Reversible super-reduction of the cubane [4Fe-4S](3+;2+;1+) in the high-potential iron-sulfur protein under non-denaturing conditions. EPR spectroscopic and electrochemical studies. Eur J Biochem. 1995;232(3):811–817. [PubMed] [Google Scholar]
- 13.Solomon EI, Xie XJ, Dey A. Mixed valent sites in biological electron transfer. Chem Soc Rev. 2008;37(4):623–638. doi: 10.1039/b714577m. [DOI] [PubMed] [Google Scholar]
- 14.Noodleman L, Peng CY, Case DA, Mouesca JM. Orbital interactions, electron delocalization and spin coupling in iron-sulfur clusters. Coord Chem Rev. 1995;144:199–244. [Google Scholar]
- 15.Beinert H, Holm RH, Münck E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science. 1997;277(5326):653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 16.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. Mill Valley, CA: University Science Books; 1994. [Google Scholar]
- 17.Yoon KS, Fukuda K, Fujisawa K, Nishihara H. Purification and characterization of a highly thermostable, oxygen-resistant, respiratory [NiFe]-hydrogenase from a marine, aerobic hydrogen-oxidizing bacterium Hydrogenovibrio marinus. Int J Hydrogen Energy. 2011;36:7081–7088. [Google Scholar]
- 18.Moulis JM, Lutz M, Gaillard J, Noodleman L. Characterization of [4Fe-4Se]2+/3+ high-potential iron-sulfur protein from Chromatium vinosum. Biochemistry. 1988;27:8712–8719. [Google Scholar]
- 19.Walters EM, et al. Role of histidine-86 in the catalytic mechanism of ferredoxin:thioredoxin reductase. Biochemistry. 2009;48(5):1016–1024. doi: 10.1021/bi802074p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mouesca JM, Lamotte B. Iron-sulfur clusters and their electronic and magnetic properties. Coord Chem Rev. 1998;178:1573–1614. [Google Scholar]
- 21.Hamann N, et al. The CCG-domain-containing subunit SdhE of succinate:quinone oxidoreductase from Sulfolobus solfataricus P2 binds a [4Fe-4S] cluster. J Biol Inorg Chem. 2009;14(3):457–470. doi: 10.1007/s00775-008-0462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Middleton P, Dickson DPE, Johnson CE, Rush JD. Interpretation of the Mössbauer spectra of the high-potential iron protein from Chromatium. Eur J Biochem. 1980;104(1):289–296. doi: 10.1111/j.1432-1033.1980.tb04427.x. [DOI] [PubMed] [Google Scholar]
- 23.Teixeira M, et al. Redox intermediates of Desulfovibrio gigas [NiFe] hydrogenase generated under hydrogen. Mössbauer and EPR characterization of the metal centers. J Biol Chem. 1989;264(28):16435–16450. [PubMed] [Google Scholar]
- 24.Huynh BH, et al. On the active sites of the [NiFe] hydrogenase from Desulfovibrio gigas. Mössbauer and redox-titration studies. J Biol Chem. 1987;262(2):795–800. [PubMed] [Google Scholar]
- 25.Surerus KK, et al. Further characterization of the spin coupling observed in oxidized hydrogenase from Chromatium vinosum. A Mössbauer and multifrequency EPR study. Biochemistry. 1994;33(16):4980–4993. doi: 10.1021/bi00182a029. [DOI] [PubMed] [Google Scholar]
- 26.Schünemann V, Winkler H. Structure and dynamics of biomolecules studied by Mössbauer spectroscopy. Rep Prog Phys. 2000;63:263–353. [Google Scholar]
- 27.Mouesca JM, Noodleman L, Case DA, Lamotte B. Spin-densities and spin coupling in iron-sulfur clusters - a new analysis of hyperfine coupling-constants. Inorg Chem. 1995;34:4347–4359. [Google Scholar]
- 28.Cammack R, Patil DS, Fernandez VM. Electron-spin-resonance/electron-paramagnetic-resonance spectroscopy of iron-sulphur enzymes. Biochem Soc Trans. 1985;13(3):572–578. doi: 10.1042/bst0130572. [DOI] [PubMed] [Google Scholar]
- 29.Kent TA, Huynh BH, Münck E. Iron-sulfur proteins: Spin-coupling model for three-iron clusters. Proc Natl Acad Sci USA. 1980;77(11):6574–6576. doi: 10.1073/pnas.77.11.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. Coordination of adenosylmethionine to a unique iron site of the [4Fe-4S] of pyruvate formate-lyase activating enzyme: A Mössbauer spectroscopic study. J Am Chem Soc. 2002;124(6):912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 31.Ciurli S, et al. Subsite-differentiated analogs of native [4Fe-4S]2+ clusters - preparation of clusters with 5-coordinate and 6-coordinate subsites and modulation of redox potentials and charge-distributions. J Am Chem Soc. 1990;112:2654–2664. [Google Scholar]
- 32.Seemann M, et al. Isoprenoid biosynthesis via the MEP pathway: In vivo Mössbauer spectroscopy identifies a [4Fe-4S]2+ center with unusual coordination sphere in the LytB protein. J Am Chem Soc. 2009;131(37):13184–13185. doi: 10.1021/ja9012408. [DOI] [PubMed] [Google Scholar]
- 33.Belinskii M, Bertini I, Galas O, Luchinat C. The electronic-structure of the Fe4S43+ cluster in proteins - the importance of double exchange parameter. Z Naturforsch A. 1995;50:75–80. [Google Scholar]
- 34.Kurtz DM. Oxo-bridged and hydroxo-bridged diiron complexes - A chemical perspective on a biological unit. Chem Rev. 1990;90:585–606. [Google Scholar]
- 35.Peters JW, et al. Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry. 1997;36(6):1181–1187. doi: 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- 36.Huang WJ, et al. Crystal structure of nitrile hydratase reveals a novel iron centre in a novel fold. Structure. 1997;5(5):691–699. doi: 10.1016/s0969-2126(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 37.Popescu VC, et al. Mössbauer and EPR studies of the photoactivation of nitrile hydratase. Biochemistry. 2001;40(27):7984–7991. doi: 10.1021/bi010198f. [DOI] [PubMed] [Google Scholar]
- 38.More C, et al. Study of the spin-spin interactions between the metal centers of Desulfovibrio gigas aldehyde oxidoreductase: Identification of the reducible sites of the [2Fe-2S]1+,2+ clusters. Biochemistry. 2005;44(34):11628–11635. doi: 10.1021/bi0510025. [DOI] [PubMed] [Google Scholar]
- 39.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.