

Abstract
NEDD8 (neural precursor cell expressed, developmentally down-regulated 8) is a ubiquitin-like molecule whose action on modifying protein substrates is critical in various cellular functions but whose importance in the immune system is not well understood. Here we investigated the role of protein neddylation in regulating T-cell function using an in vivo knockdown technique. We found that reduced expression of Ubc12 in CD4+ T cells led to impaired T-cell receptor/CD28-induced proliferation and cytokine production both in vitro and in vivo, accompanied by reduced Erk activation. These findings were recapitulated by treatment with MLN4924, an inhibitor of NEDD8-activating enzyme. Furthermore, Shc, an adaptor molecule between antigen receptors and the Ras/Erk pathway, was identified as a target for neddylation. Importantly, mice adoptively transferred with Ubc12 knockdown CD4+ T cells showed markedly ameliorated allergic responses. This study thus identifies an important role for protein neddylation in T-cell function, which may serve as a therapeutic target for inflammatory diseases.
Keywords: allergy, signal transduction, posttranslational modification
Engagement of T-cell antigen receptor (TCR) and costimulatory molecules leads to the activation of CD4+ T cells through several signaling pathways, which ultimately induces proliferation, cytokine production, and differentiation into different subsets of T helper type (Th) cells and memory cells (1). Notably, posttranslational modification by ubiquitin and ubiquitin-like proteins (UBLs) has emerged as a critical mechanism regulating T-cell function (2, 3). UBLs compose a diverse group of evolutionarily conserved small proteins, and attachment of different UBLs to a target has different biological consequences (4). Among the UBLs, NEDD8 (neural precursor cell expressed, developmentally down-regulated 8) is covalently attached to lysines in substrate proteins by a series of enzymatic reactions similar to those involved in ubiquitination, including the E1 (Nae1/Uba3) and E2 (Ubc12) reactions (5). The well-characterized substrates of NEDD8 modification are the cullin subunits of cullin-RING ubiquitin E3 ligases, and this modification of cullins is critical for the transfer of ubiquitin from recruited E2-ubiquitin to the substrate (6, 7). The importance of this pathway was further underscored by the recent development of a pharmacologic inhibitor of Nae1, MLN4924, which inhibits cullin neddylation, resulting in substrate accumulation and cancer cell apoptosis by the deregulation of S-phase DNA synthesis, and offers great promise for the treatment of cancer (8–10).
In the present study, we investigated whether the NEDD8 pathway is involved in CD4+ T-cell function. Using siRNA-mediated depletion of the NEDD8 system in vivo and the NEDD8-activating enzyme inhibitor MLN4924, we found that the NEDD8 pathway is required for TCR-induced proliferation and activation. Interestingly, our results demonstrate that Erk activation by TCR stimulation requires an intact neddylation system. Differentiation of CD4+ T cells from Ubc12 knockdown bone marrow chimeric mice into Th1 and Th2 subsets was markedly decreased. Mice transferred with Ubc12 knockdown CD4+ T cells exhibited a deficiency in the ability to develop T-cell–driven airway inflammation. Thus, the NEDD8 pathway appears to play an important role in CD4+ T-cell–mediated inflammatory responses.
Results and Discussion
NEDD8 Pathway Is Required for CD4+ T-Cell Function.
To explore the function of the NEDD8 pathway in the immune system, particularly in CD4+ T cells, we generated retroviral vectors expressing both GFP and an shRNA targeting Ubc12. Bone marrow cells isolated from SJL B6 mice (CD45.1+) were transduced with the retrovirus bearing Ubc12 shRNA or control shRNA, then transplanted into lethally irradiated C57BL/6 mice (CD45.2+). After 8 wk, newly reconstituted GFP+CD4+ T cells were sorted from the spleen and lymph nodes of bone marrow chimeric mice. Western blot analysis showed a significant decrease in Ubc12 protein expression in the sorted GFP+ T-cell population (Fig. 1A).
Fig. 1.

Ubc12 is required for TCR-induced proliferation and activation. (A) Immunoblot analysis of Ubc12 was performed in sorted GFP+ control (ctl) or GFP+ shUbc12 CD4+ T cells (Inset). Peripheral naïve CD4+ T cells sorted from shRNA expressing chimeric mice were stimulated with various concentration of anti-CD3 and anti-CD28 mAb for 3 d and then pulsed for an additional 8 h with 3H-thymidine. Cell proliferation was measured by 3H-thymidine uptake. (B) Cell cycle analysis of control (Left) and shUbc12 CD4+ (Right) T cells. The cells were activated for 23 h with anti-CD3/CD28 mAbs, then pulsed for 1 h with EdU and analyzed by flow cytometry after staining for EdU and 7-AAD. (C) Control (Left) and shUbc12 CD4+ (Right) violet-labeled CD4+ T cells were stimulated with anti-CD3/CD28 mAbs for 2 d, after which cell division was analyzed by flow cytometry. (D) Flow cytometry for cell surface expression of CD69 (Left) and CD25 (Right) in sorted control and shUbc12 CD4+ T cells stimulated for 16 h with anti-CD3/CD28 mAbs. The results are representative of three repeated experiments. (E) IL-2 (Left) and IFN-γ (Right) production in control and shUbc12 CD4+ T cells stimulated for 48 h with anti-CD3 and anti-CD28, measured by ELISA. (F) Control (Left) and Ubc12 shRNA-expressing (Right) OT-II TCR transgenic mice were generated by reconstitution of retrovirally transduced bone marrow cells from OT-II mice into lethally irradiated recipient mice (n = 3). After 8 w, isolated CD4+ T cells from OT-II control or OT-II Ubc12 knockdown chimeric mice were adoptively transferred into C57BL/6J recipient mice, followed by immunization with OVA plus CFA. At 5 d after immunization, splenocytes and lymph node cells were stimulated with OVA323–339 peptide for 24 h and analyzed by flow cytometry using anti–IL-2 and anti–IFN-γ antibodies. (G) Sorted naïve CD4+ T cells were stimulated with anti-CD3/CD28 mAbs and differentiated in vitro under Th1-inducing (Upper) or Th2-inducing (Lower) conditions. Results are from three repeated experiments.
We next analyzed T-cell development in the thymuses of Ubc12 knockdown chimeric mice. Decreased expression of Ubc12 had no obvious effect on the development of CD4+, CD8+, or CD4+CD8+ thymocytes, and similarly, the proportion of CD4+ and CD8+ T cells in the spleens of these mice was comparable to that in control mice (Fig. S1 A and B). We quantified memory and naive T cells on the basis of surface expression of the activation markers CD44 and CD62L. Compared with control mice, the Ubc12 knockdown chimeric mice had a significantly lower frequency of memory CD4+ T cells with a concomitantly higher frequency of naive CD4+ T cells (Fig. S1C).
To assess the function of Ubc12 in T-cell proliferation, we stimulated control and Ubc12 knockdown naïve CD4+ T cells in vitro with anti-CD3 and anti-CD28. Proliferation of the stimulated knockdown T cells was substantially reduced as measured by 3H-thymidine incorporation (Fig. 1A). Staining with 5-ethynyl-2′-deoxyuridine (EdU) and 7-amino-actinomycin D (7-ADD) revealed that Ubc12 knockdown T cells were blocked in the G0/G1 phase of the cell cycle and did not progress efficiently through the S phase (Fig. 1B). Analysis of individual cell division of violet dye-labeled CD4+ T cells by flow cytometry showed that Ubc12 knockdown T cells divided much more slowly than control T cells (Fig. 1C) and also failed to up-regulate the activation markers CD69 and CD25 to the same extent as control cells (Fig. 1D). Evaluation of cell death by a 7-AAD exclusion assay showed similar 7-AAD staining in control and Ubc12 knockdown T cells after 24 h of TCR stimulation (Fig. S2), suggesting that Ubc12 knockdown does not affect the survival of CD4+ T cells in response to TCR activation. Taken together, these results demonstrate a critical role for the NEDD8 pathway in the positive regulation of CD4+ T-cell activation.
To further test whether other components of the NEDD8 pathway also affect T-cell function, we designed a single retroviral vector bearing three shRNA cassettes targeting the neddylation components of Nae1, Ubc12, and NEDD8 (shTriple) or individually (Fig. S3A). The knockdown efficiencies of various hairpin constructs were measured by Western blot analysis (Fig. S3B). Inhibition of the NEDD8 pathway by the shRNA-mediated simultaneous knockdown of the neddylation components also led to impaired proliferation and division of CD4+ T cells (Fig. S3 C and D). In addition, IL-2 production was substantially reduced in the triple-knockdown T cells (Fig. S3E). Furthermore, Nae1-only knockdown CD4+ T cells also displayed a decrease in proliferation and IL-2 production in response to TCR activation (Fig. S3 F and G).
Defective T-Cell Differentiation by Ubc12 Knockdown.
We next investigated the function of the NEDD8 pathway in TCR-induced cytokine production. We found that the secretion of IL-2 and IFN-γ was markedly impaired in both shUbc12- and shTriple-expressing cells compared with control cells (Fig. 1E and Fig. S3E). We further examined the in vivo function of the neddylation pathway in CD4+ T-cell–mediated immune responses using the OT-II transgenic TCR adoptive transfer system. For this, purified CD4+ T cells from OT-II control or OT-II Ubc12 knockdown chimeric mice were adoptively transferred into C57BL/6J recipient mice. These mice were then immunized with ovalbumin (OVA) emulsified in complete Freund’s adjuvant (CFA) at 1 d after adoptive transfer. Splenocytes obtained from OVA-immunized mice were cultured in the presence of OVA, and IFN-γ and IL-2 production was analyzed by intracellular cytokine staining. Splenic CD4+ T cells from the mice transferred with Ubc12 knockdown OT-II CD4+ T cells exhibited impaired ability to produce IFN-γ and IL-2 in response to a secondary antigen challenge in vitro (Fig. 1F). These data suggest that the neddylation pathway is involved in antigen-driven cytokine production by CD4+ T cells.
We then examined whether Ubc12 knockdown affects the differentiation of CD4+ T cells into effector Th cell subsets. We stimulated naïve CD4+ T cells from control and Ubc12 knockdown chimeric mice with anti-CD3 and anti-CD28 mAbs, cultured them under either Th1- or Th2-polarizing conditions, and then analyzed IFN-γ and IL-4 production. Ubc12 knockdown CD4+ T cells produced less IFN-γ and IL-4 compared with control cells under Th1 and Th2 culture conditions, respectively (Fig. 1G), indicating that the NEDD8 pathway is required for efficient Th1 and Th2 differentiation.
MLN4924 Inhibits T-Cell Function.
We further examined the effects of blocking the NEDD8 pathway on T-cell activation using the Nae1-specific inhibitor MLN4924 (8). CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAbs in the presence of indicated concentrations of MLN4924 for 16 h. Consistent with the shRNA results, MLN4924 suppressed the anti-CD3/CD28-induced proliferation of CD4+ T cells as assessed by EdU/7-AAD staining (Fig. 2A). The inhibitor also blocked T-cell cycle progression from G0/G1 to S phase after activation, as well as up-regulation of the activation markers CD69 and CD25 in response to TCR stimulation (Fig. 2B). We next examined the effect of MLN4924 on IL-2 production. Treatment with the inhibitor markedly decreased IL-2 production in T cells (Fig. 2C). Quantitative RT-PCR analysis revealed less IL-2 mRNA in MLN4924-treated T cells (Fig. 2D). It should be noted that MLN4924-treated CD4+ T cells did not show a substantial increase in cell death under low-concentration treatment, as measured by 7-AAD staining (Fig. 2E), indicating that the inhibitory effect of MLN4924 on T-cell activation is not due to an increase in cell death. Together, the use of MLN4924 recapitulated the siRNA knockdown results, supporting a potent positive function of the NEDD8 pathway in T-cell activation.
Fig. 2.
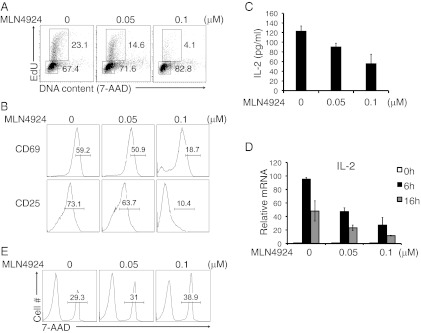
MLN4924 treatment blocks TCR-induced proliferation and activation. (A) Flow cytometry of the cell cycle status of CD4+ T cells stimulated with anti-CD3 and anti-CD28 for 16 h in the presence of the indicated concentrations of MLN4924 diluted in DMSO or equivalent volumes of DMSO alone. (B) CD4+ T cells were analyzed for surface expression of CD69 (Upper) and CD25 (Lower) after stimulation with anti-CD3 and anti-CD28 in the presence of the indicated concentrations of MLN4924. (C) IL-2 production by CD4+ T cells stimulated with anti-CD3/CD28 mAbs in the absence or presence of MLN4924, measured by ELISA. (D) Quantitative RT-PCR analysis of IL-2 mRNA. (E) Cell death of stimulated CD4+ T cells examined by 7-AAD staining and FACS analysis.
NEDD8 Pathway Is Required for Erk Activation in T Cells.
To understand the molecular mechanism by which NEDD8 pathway regulates CD4+ T-cell function, we analyzed several TCR-induced signaling events in Ubc12 knockdown CD4+ T cells. Knockdown of Ubc12 reduced the neddylation of cullin-1 (Fig. 3A), suggesting the efficient blockage of the neddylation pathway by Ubc12 ablation. Activation of the Erk1/2 by TCR/CD28 stimulation was profoundly impaired in the Ubc12 knockdown CD4+ T cells, whereas phosphorylation of p38 was slightly decreased (Fig. 3A). To confirm this result, we next pretreated CD4+ T cells for 16 h with MLN4924 before subjecting these cells to TCR stimulation with anti-CD3/CD28 mAbs. Pretreatment with 0.1 μM MLN4924 reduced the activation of Erk, whereas the phosphorylation of PLC-γ1 was unchanged by treatment with the inhibitor (Fig. 3B). As expected, MLN4924 pretreatment efficiently blocked the neddylation of cullin-1 (Fig. 3B). Taken together, these data indicate that the neddylation pathway is involved in TCR-stimulated Erk activation.
Fig. 3.

The neddylation pathway is involved in Erk activation. (A) Immunoblot analysis of control (Left) and shUbc12 (Right) CD4+ T cells. The CD4+ T cells sorted from the chimeric mice were stimulated with anti-CD3 and anti-CD28 for the indicated time periods. Cell lysates were separated by SDS/PAGE and immunoblotted with the indicated antibodies. (B) CD4+ T cells were preactivated with anti-CD3 in the presence of the indicated concentrations of MLN4924 for 16 h and then restimulated with a combination of anti-CD3 and anti-CD28. (C) Jurkat E6.1 cells stably expressing FLAG-NEDD8 were treated with MLN4924 for 12 h or left untreated before being collected. Cell lysates were boiled in 1% SDS-containing lysis buffer, then diluted to 0.1% SDS and immunoprecipitated with anti-FLAG–conjugated beads, followed by elution with triple FLAG peptide. The eluates were subjected to Western blot analysis using the indicated antibodies. (D) The three isoforms of Shc. All isoforms contain an N-terminal PTB domain, a CH domain, and a C-terminal SH2 domain. (E) Jurkat cells were transfected with FLAG-tagged p66, p52, or p46 with HA-tagged NEDD8. The samples were processed for immunoprecipitation as described in C. (F) The peptide sequence at the top is derived from MS analysis of p66Shc; an asterisk indicates the modified lysine residue. Jurkat cells were transfected with expression vector encoding WT (wt) p52Shc or p52Shc lysine mutant (K3R). Where indicated, combinations of plasmids expressing HA-NEDD8 and/or Ubc12 were cotransfected. (G) CD4+ T cells were stimulated with anti-CD3/CD28 antibodies and retrovirally transduced with Myc-tagged p52Shc WT-IRES-GFP (WT) or Myc-tagged p52Shc K3R-IRES-GFP (K3R). Whole-cell lysates were immunoprecipitated with anti-ZAP70 antibody and blotted with the indicated antibodies. (H) CD4+ T cells were treated with MLN4924 for 16 h or left untreated. After TCR stimulation, the cell lysates were subjected to coimmunoprecipitation with anti-ZAP70 antibody.
Given that NEDD8 modification of cullin-1 has been demonstrated to be necessary for the ubiquitination of IκB-α and subsequent activation of NF-κB (11, 12), we also examined IκB-α degradation by TCR stimulation. Ubc12 knockdown T cells showed an only slight delay in IκB-α degradation in response to TCR stimulation compared with control T cells (Fig. 3A). We also examined phorbol 12-myristate 13-acetate/ionomycin- or TNF-α–induced IκB-α degradation in the NEDD8 inhibitor-treated T cells, and found that treatment of T cells with MLN4924 led to retardation of the stimulus-induced IκB-α degradation, suggesting that this pharmacologic inhibitor indeed functions in the NF-κB activation process in T cells (Fig. S4). Ubc12 knockdown did not lead to effective blockage of this pathway, however.
Adaptor Protein Shc Is a Target for Neddylation.
We asked whether other NEDD8-modified proteins may be responsible for the observed Erk activation on TCR stimulation. In an effort to identify currently unknown NEDD8 substrates, we generated Jurkat E6.1 cells stably expressing N-terminal 3× FLAG-tagged NEDD8. The cells were lysed under strong denaturing conditions to block noncovalent protein–protein interactions, and FLAG-NEDD8–conjugated species were subjected to anti-FLAG immunoaffinity purification. Proteomic analysis by MS identified the adaptor protein Shc as a possible target (Fig. S5). We confirmed this observation by an immunoprecipitation experiment. As a control, we treated the stable cell lines with 1 μM MLN4924 for 12 h. As expected, we detected the NEDD8-modified forms of cullin-1 and Ubc12 in the immunoprecipitates from the stable cells, but not in immunoprecipitants from MLN4924-treated cells (Fig. 3C). The molecular mass of immunoprecipitated proteins increased by ∼20 kDa, consistent with a 3× FLAG-tagged NEDD8 conjugation. Western blot analysis with anti-Shc antibodies revealed a slower-migrating anti-Shc reactive band of ∼70 kDa. Treatment of cells with MLN4924 caused the disappearance of this band in the immunoprecipitated samples.
Shc is expressed as three isoforms (p66, p52, and p46) encoded by the same genetic locus. All three isoforms have an SH2 domain, a proline-rich CH1 domain, and a phospho-tyrosine binding (PTB) domain (Fig. 3D) (13, 14). Given that Jurkat E6.1 cells did not express p66Shc at detectable levels, the slow-migrating band could represent an NEDD8-modified form of either p52Shc or p46Shc. To examine which isoforms of Shc can be modified with NEDD8, we generated expression vectors encoding FLAG-tagged Shc isoforms. Jurkat cells were transfected with plasmids encoding FLAG-tagged p66Shc, p52Shc, or p46Shc isoforms along with a vector expressing HA-tagged NEDD8. Cells were lysed and immunoprecipitated with anti-FLAG antibody, and then immunoblotted with anti-HA or anti-FLAG antibody. Whereas p66Shc and p52Shc were conjugated to NEDD8, no conjugation was observed with p46Shc (Fig. 3E). Because the sole difference between p52Shc and p46Shc is the presence of an additional 45 amino acids at the N terminus of p52Shc, this finding suggests that the first 45 amino acids contain a potential NEDD8 conjugation site.
To identify a lysine residue in p52Shc that acts as the site for neddylation, we performed MS analysis on NEDD8-conjugated p66Shc immunopurified from cells expressing FLAG-p66Shc and HA-NEDD8. The analysis revealed that p66Shc was conjugated with NEDD8 at lysine 113, which is located at position 3 in p52Shc (Fig. 3F). To convincingly prove that the identified lysine residue is a NEDD8 acceptor site for p52Shc, we constructed a mammalian expression vector carrying a mutation of the lysine residue to an arginine residue (K3R). Plasmids encoding the K3R mutant were transfected into Jurkat cells in combination with HA-NEDD8. Here p52Shc (K3R) failed to become neddylated even in the presence of Ubc12 overexpression, suggesting that lysine 3 is the NEDD8 acceptor site of p52Shc (Fig. 3F).
To further explore the potential mechanisms of Shc neddylation on Erk activation, we examined the complex formation of ZAP70 with Shc and Grb2, and found that p52Shc interacted with ZAP70 and Grb2 in TCR-stimulated CD4+ T cells, but that the Shc K3R mutation impaired the recruitment of Grb2 to the ZAP70-Grb2 complex (Fig. 3G). Treatment of CD4+ T cells with MLN4924 also resulted in perturbed TCR-induced complex formation of Shc-Grb2 with ZAP70 (Fig. 3H). These findings suggest that neddylation of Shc may facilitate the formation of a ZAP70-Shc-Grb2 signaling complex and affect downstream Erk activation.
NEDD8 Pathway Is Important for Airway Inflammation.
We next examined whether the NEDD8 pathway in CD4+ T cells may influence the development of airway inflammatory responses. We used an adoptive transfer model of experimental allergy (Fig. 4A). GFP+ OT-II T cells were isolated from the mice transplanted with control or Ubc12 shRNA-transduced bone marrow cells and then transferred i.v. into recipient B6 mice. At 1 d after transfer, recipient mice were immunized i.p. with OVA in alum adjuvant and then repeatedly challenged with OVA for 10 d after immunization. At 24 h after the last OVA challenge, the lungs were lavaged, and the bronchoalveolar lavage (BAL) fluid was examined. OVA sensitization and challenge resulted in an increased number of leukocytes in BAL fluid in the mice transferred with control OT-II T cells; however, mice transferred with Ubc12 knockdown T cells showed a substantially reduced number of infiltrating inflammatory cells in the lungs (Fig. 4C). Histological analysis revealed prominent cellular infiltration around the bronchioles in control mice, but significantly more attenuated cell infiltration in the mice transferred with Ubc12 knockdown T cells (Fig. 4B). Consistent with a defective allergic response, the mice transferred with Ubc12 knockdown T cells also had significantly lower levels of the Th2 cytokines IL-4 and IL-5 in the BAL fluid compared with control mice (Fig. 4D). Taken together, these results suggest that the NEDD8 pathway is important for efficient Th2-mediated lung inflammation.
Fig. 4.

Role pf the NEDD8 pathway in the development of allergic inflammation. (A) GFP+CD4+ T cells from OT-II control or OT-II Ubc12 knockdown chimeric mice were adoptively transferred into C57BL/6J recipient mice, followed by immunization with OVA plus alum (n = 3). Starting at 10 d after immunization, the mice were challenged intranasally with OVA protein for the next 4 d. The mice were examined at 24 h after the last challenge. (B) Lungs from control mice (Left) and shUbc12 knockdown chimeric mice (Right) were sectioned and stained with H&E to visualize inflammatory infiltrates. (C) Total numbers of eosinophils (Eos), neutrophils (Neu), monocytes (Mon), and lymphocytes (Lym) were calculated by FACS analysis. (D) IL-4 (Left) and IL-5 (Right) concentrations in BAL fluid were measured by ELISA at 24 h after the final OVA challenge. The results are representative of two repeated experiments.
The data presented here demonstrate that the NEDD8 system regulates various aspects of CD4+ T-cell function, including proliferation, cytokine production, and Th cell differentiation. Inhibition of the NEDD8 pathway by shRNA-mediated knockdown of neddylation components or MLN4924 led to impaired proliferation and IL-2 production of CD4+ T cells. Interestingly, the neddylation pathway seemed to have a more dominant effect on Erk activation in TCR-stimulated CD4+ T cells. Using a combination of molecular and proteomics approaches, we have identified a target Shc for neddylation, suggesting the possibility that neddylation of p52Shc might affect the formation of ZAP70-Shc-Grb2 signaling complexes; however, more detailed studies are needed to clarify the exact molecular mechanisms as a future direction of investigation. Importantly, we have shown that the NEDD8 pathway is critical for the development of allergic responses, as demonstrated in adoptive transfer experiments. Recent preclinical trials of MLN4924 for various cancers have yielded promising results for the manipulation of the neddylation pathway in treating human diseases (10). Our data on the knockdown of Ubc12 in attenuating airway inflammation suggest that this pathway may represent a potentially important molecular target for therapeutic intervention of T-cell–mediated inflammatory and autoimmune diseases.
Materials and Methods
Retroviral Transduction and Bone Marrow Reconstitution.
C57BL/6 mice (CD45.2), B6.SJL mice (CD45.1), and OT-II mice (OVA-specific TCR Tg mice) were obtained from Jackson Laboratories. All mice were housed under specific pathogen-free conditions. To generate shRNA-expressing bone marrow chimeric mice, Phoenix-Eco packaging cells were transfected with 3 µg of LMP vector with 9 µl of TransIT-LT1 (Mirus). At 48 h, the culture supernatant containing retrovirus was collected. Mature T-cell–depleted bone marrow cells from B6.SJL mice (CD45.1) were cultured for 24 h in 10 ng/mL IL-3, 10 ng/mL IL-6, and 100 ng/mL SCF (all from Peprotech) containing complete DMEM before initial retroviral infection. Mature T-cell–depleted bone marrow cells were infected with retrovirus along with 5 µg/mL polybrene (Sigma-Aldrich) by spin inoculation (420 × g for 1 h). At 2 d after infection, retroviral transduced bone marrow cells were injected into lethally irradiated (900 rad) C57BL/6 recipient mice. Recipient mice were killed at 8 wk after reconstitution for further analysis. All animal experiment protocols were approved by the Institutional Animal Care and Use Committee of the La Jolla Institute for Allergy and Immunology.
Adoptive Transfer of OT-II T Cells and Immunization.
Bone marrow cells from TCR transgenic OT-II mice were transduced with control or Ubc12 shRNA-expressing retroviruses, which were then transplanted into lethally irradiated mice. After 8 wk, isolated CD4+ T cells from OT-II control or OT-II Ubc12 knockdown chimeric mice were adoptively transferred into C57BL/6J recipient mice. At 1 d after transfer, the recipient mice were immunized i.p. with OVA (50 μg, Grade V; Sigma-Aldrich) emulsified in complete Freund’s adjuvant (BD Diagnostics) by s.c. injection. At 10 d after immunization, cells were collected from spleen and lymph nodes and cultured with OVA323–339 peptide (10 μg/mL; AnaSpec) for 8 h at 37 °C in the presence of GolgiStop protein inhibitor (BD Biosciences).
Antigen-Induced Airway Inflammation.
GFP+CD4+ T cells were sorted from OT-II control and OT-II Ubc12 knockdown chimeric mice, and 1 × 106 GFP+CD4+ T cells were injected i.v. into C57BL/6 recipient mice. At 1 d after transfer, the recipient mice were sensitized by i.p. injection of 20 μg of OVA protein (Grade V; Sigma-Aldrich) adsorbed to 2 mg of aluminum hydroxide (alum) gel (Imject Alum; Pierce). At 10 d after immunization, the mice were challenged intranasally with OVA protein for 4 consecutive days. Mice were killed at 24 h after the last aerosol challenge and assessed for lung inflammation. BAL fluid was collected for assessment of cytokine levels and histological analysis.
More detailed information on the experiments is provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank J. Lopez and C. Elly for mouse breeding and K. Iwanami for technical help. This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases Grants R01 AI62969 and R01 AI78272. H.-s.J. was supported in part by National Research Foundation of Korea Grant NRF-2009-352-C00098.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213819110/-/DCSupplemental.
References
- 1.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: A reversible process of modification. Nat Rev Immunol. 2005;5(12):941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun SC. Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008;8(7):501–511. doi: 10.1038/nri2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 5.Harper JW, Schulman BA. Structural complexity in ubiquitin recognition. Cell. 2006;124(6):1133–1136. doi: 10.1016/j.cell.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32(1):21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duda DM, et al. Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell. 2008;134(6):995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 9.Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70(24):10310–10320. doi: 10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer. 2010;1(7):708–716. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Read MA, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20(7):2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amir RE, Haecker H, Karin M, Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: Identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene. 2004;23(14):2540–2547. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Lorenz U, Ravichandran KS. Role of Shc in T-cell development and function. Immunol Rev. 2003;191:183–195. doi: 10.1034/j.1600-065x.2003.00025.x. [DOI] [PubMed] [Google Scholar]
- 14.Finetti F, Savino MT, Baldari CT. Positive and negative regulation of antigen receptor signaling by the Shc family of protein adapters. Immunol Rev. 2009;232(1):115–134. doi: 10.1111/j.1600-065X.2009.00826.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.