

Abstract
Streptococcus pyogenes (group A streptococcus, GAS) is a human bacterial pathogen of global significance, causing severe invasive diseases associated with serious morbidity and mortality. To survive within the host and establish an infection, GAS requires essential nutrients, including iron. The streptococcal hemoprotein receptor (Shr) is a surface-localized GAS protein that binds heme-containing proteins and extracellular matrix components. In this study, we employ targeted allelic exchange mutagenesis to investigate the role of Shr in the pathogenesis of the globally disseminated serotype M1T1 GAS. The shr mutant exhibited a growth defect in iron-restricted medium supplemented with ferric chloride, but no significant differences were observed in neutrophil survival, antimicrobial peptide resistance, cell surface charge, fibronectin-binding or adherence to human epithelial cells and keratinocytes, compared with wild-type. However, the shr mutant displayed a reduction in human blood proliferation, laminin-binding capacity and was attenuated for virulence in in vivo models of skin and systemic infection. We conclude that Shr augments GAS adherence to laminin, an important extracellular matrix attachment component. Furthermore, Shr-mediated iron uptake contributes to GAS growth in human blood, and is required for full virulence of serotype M1T1 GAS in mouse models of invasive disease.
Keywords: hemoprotein, iron acquisition, iron starvation, laminin-binding, Shr, Streptococcus pyogenes, virulence, whole blood
Introduction
Streptococcus pyogenes, commonly referred to as group A streptococcus (GAS), is one of the world’s leading human pathogens, responsible for significant morbidity and economic loss worldwide. GAS is a β-hemolytic Gram-positive bacterium that causes a wide spectrum of diseases, ranging from superficial skin infections (pharyngitis, impetigo and cellulitis), to severe life-threatening invasive diseases (bacteremia, streptococcal toxic shock-like syndrome and necrotizing faciitis), and immunologically-mediated postinfection sequelae (rheumatic fever, rheumatic heart disease and acute poststreptococcal glomerulonephritis).1 Most invasive GAS diseases are associated with the M1 serotype, and a marked increase in GAS invasive disease severity over the past three decades has been attributed to a single globally disseminated clone of serotype M1T1.2 This clone is distinguished from related M1 serotypes by two bacteriophage-encoded virulence factors, designated extracellular streptodornase D (Sda1) and streptococcal exotoxin type A (SpeA).2 The acquisition of the Sda1 and SpeA prophages is proposed to enhance the fitness and virulence potential of M1T1 GAS relative to the ancestral M1 serotype.1
With the exception of the Lyme disease pathogen Borrelia burgdorferi3 and certain Lactococcus species,4 almost all microorganisms require iron to support growth and essential cellular processes. For the majority of bacterial pathogens, the acquisition of iron from host sources is essential for survival and the establishment of infection.5 While iron uptake has been studied extensively in Gram-negative bacteria, only recently has progress been made to identify the iron transport mechanisms in Gram-positive pathogens, including Staphylococcus aureus6 and Corynebacterium species.7 Estimates indicate that pathogenic bacteria require a minimum iron concentration of 10−8 M to support growth;8 however, most free extracellular iron is complexed by iron-binding proteins circulating in blood,9 such as lactoferrin and transferrin, severely limiting the amount of free iron available to the pathogen (~10−18 M).10 GAS cannot survive on the amount of free iron available in the human host11 and utilize heme-containing proteins such as hemoglobin, myoglobin, the hemoglobin-haptoglobin complex and heme-albumin as alternative iron sources during the course of infection.12 However, neither the mechanism(s) of GAS iron acquisition from hemoproteins, nor the role of iron in GAS pathogenicity, are fully understood.
Bates et al.12 identified the ATP-binding cassette transporter siaABC (streptococcal iron acquisition), also known as hts (heme transport S. pyogenes),13 which shares significant sequence homology to bacterial transporters involved in iron uptake. The siaABC transporter is part of the highly conserved 10-gene sia operon, an iron-regulated operon involved in heme acquisition and transport in GAS. The first gene in the sia operon, shr (streptococcal hemoprotein receptor), encodes for a large (145 kDa) surface-associated hydrophilic protein, Shr, with no significant sequence homology to known heme or hemoprotein receptors.12 This membrane-bound surface receptor lacks a C-terminal LPXTG cell wall anchoring motif, but has a functional N-terminal signal peptide and is secreted into the extracellular milieu.12,14 Shr binds hexacoordinate heme and heme-containing proteins (hemoglobin, myoglobin, heme-albumin and hemoglobin-haptoglobin complex)12 and directly transfers the heme group to Shp (streptococcal heme-binding protein), encoded by the second gene in the sia operon, shp.15 The two NEAr transporter (NEAT) domains of Shr play a central role in heme binding, while a unique N-terminal domain interacts with methemoglobin.16 In addition to iron acquisition, Shr is reported be an adhesin that mediates bacterial attachment to extracellular matrix components fibronectin and laminin.14 Shr is expressed in vivo during the course of GAS infection, and is required for full virulence of serotype M49 GAS in an intramuscular zebrafish model of necrotizing fasciitis.14
Previous studies on Shr have focused primarily on GAS strains SF370 (M1 serotype lacking speA and sda1), MGAS5005 (M1T1 serotype harboring a mutation in the covRS two-component regulator),17 JRS4 (M6 serotype)10 and NZ131 (M49 serotype).14 In this investigation, we evaluate for the first time the role of Shr in the pathogenesis of the globally disseminated wild-type (covRS intact) serotype M1T1 GAS. We demonstrate that Shr facilitates GAS adherence to the laminin glycoprotein family, and that Shr-mediated iron uptake contributes to growth in human blood. Finally, we assess for the first time the role of Shr in a mammalian in vivo system, demonstrating a contribution to virulence in two murine models of infection.
Results
Exogenously supplied iron supports in vitro GAS growth under iron-starvation conditions
Previous studies have shown that iron-depleted media reduces the growth rate of several different GAS serotypes, including M1,12,14,18,19 M610,20 and M49.16 Here, we characterized the growth of serotype M1T1 GAS in an iron-restricted medium comprised of RPMI 1640 with 20% heat-inactivated fetal bovine serum (FBS) to support GAS growth. RPMI alone does not permit growth (data not shown), and GAS cannot utilize lactoferrin or transferrin, the major ferric carrier proteins in FBS.10 Overnight stationary phase cultures of wild-type (WT) 5448, the isogenic mutant 5448Δshr, and the complementation strain 5448Δshr pShr, were washed with PBS and inoculated into fresh medium to a starting OD600 of 0.1. In iron-free media, all strains grew at similar rates (Fig. 1A). The addition of ferric chloride to a final concentration of 250 μM did not restore 5448Δshr growth kinetics to WT levels (Fig. 1B). Complementation of 5448Δshr with a plasmid overexpressing Shr (5448Δshr pShr) restored growth to WT levels in the presence of iron (Fig. 1B). Quantitative real-time PCR analyses verified that shr gene expression was abrogated in 5448Δshr, and upregulated in 5448Δshr pShr, compared with WT (Fig. 1C). Moreover, all strains expressed equivalent levels of shp (Fig. 1C), the gene immediately downstream of shr in the M1T1 GAS genome, confirming that the growth defect of 5448Δshr is due to a specific shr mutation rather than a pleiotropic or polar effect on gene expression levels. Overall, these data suggest that Shr plays a pivotal role in M1T1 GAS iron acquisition and growth under iron-starvation conditions.
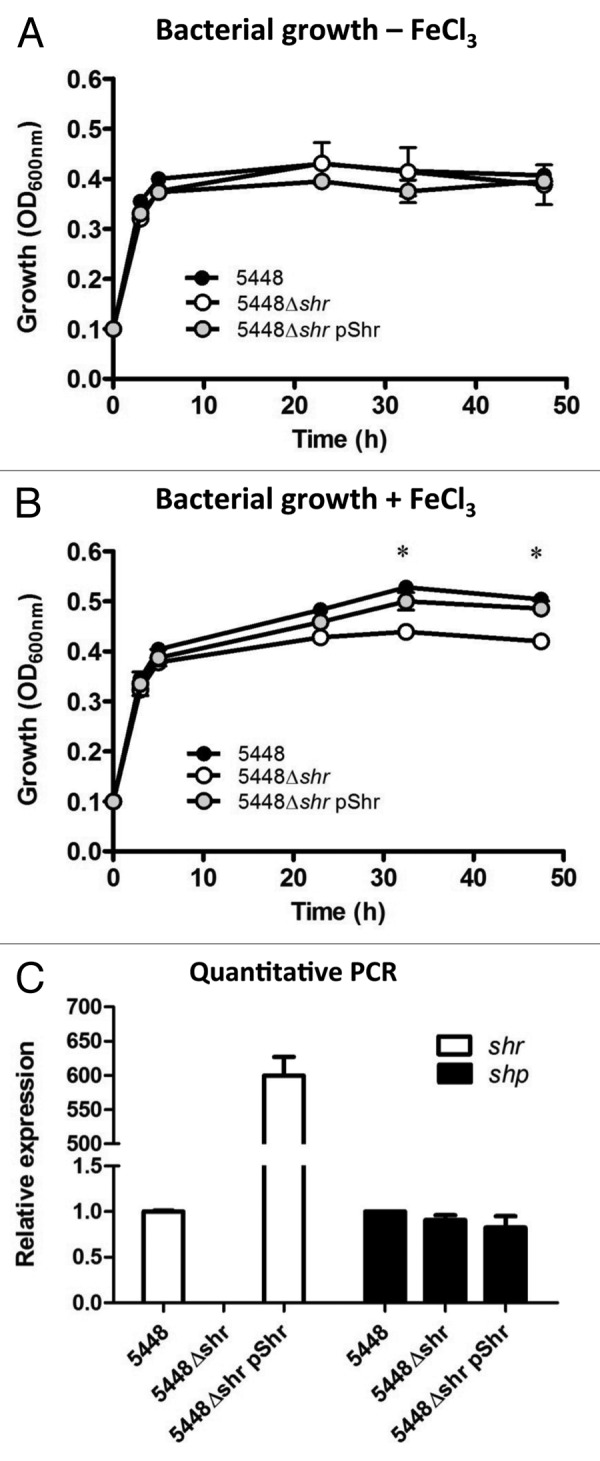
Figure 1. Shr enhances bacterial growth in the presence of free iron. Growth of WT M1T1 GAS isolate 5548, the isogenic 5448Δshr mutant, and the complemented strain 5448Δshr pShr were assessed under iron-depleted conditions (A) and in the presence of 250 μM ferric chloride (B) by measuring the turbidity of cultures at OD600. Data points denote the arithmetic mean ± SEM and are representative of two independent experiments, each performed in duplicate. The asterisk indicates values that are statistically significantly different (p < 0.01) from WT strain 5448. (C) Quantitative real-time PCR analysis of shr and shp expression levels in stationary phase cultures of WT, 5448Δshr and 5448Δshr pShr. Data were normalized to the housekeeping gene gyrA and relative expression was compared with WT.
Shr promotes GAS growth in whole blood, but does not enhance neutrophil resistance
Next we examined whether Shr contributes to M1T1 GAS proliferation in whole blood. While the growth kinetics of WT and 5448Δshr were initially comparable in human blood with orbital rotation, at 90 and 120 min post-inoculation the shr mutant demonstrated a significant impairment in growth (Fig. 2A). Similar results were observed under static assay conditions (data not shown). The complemented strain 5448Δshr pShr displayed equivalent growth to WT at all time points, confirming the growth defect of the mutant is specifically caused by the deletion of shr (Fig. 2A). Human neutrophils are one of the principal mediators of opsonophagocytic bacterial killing in human blood.21 To test the potential role of Shr in GAS resistance to host innate immune clearance, we co-incubated bacteria with freshly isolated human neutrophils. The loss of Shr did not influence neutrophil clearance, in comparison to WT (Fig. 2B). Preincubation of bacteria in human serum did not enhance neutrophil opsonophagocytic killing of 5448Δshr, compared with WT and 5448Δshr pShr (data not shown). These data suggest that while Shr is necessary to support M1T1 GAS growth in human blood, the principle mechanism is not increased resistance to neutrophil-mediated killing.
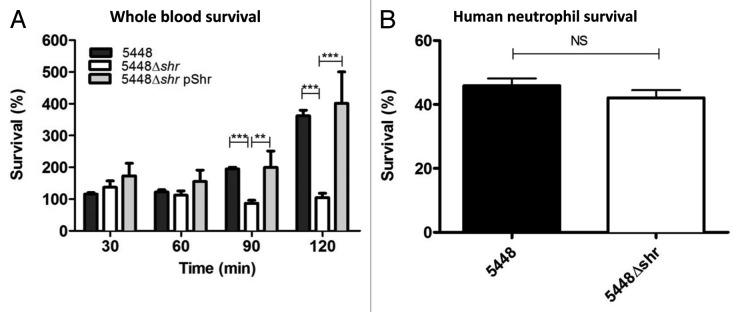
Figure 2. Shr enhances bacterial growth in human blood, but does not contribute to neutrophil resistance. (A) Mid-logarithmic phase bacteria were incubated with whole human blood and plated at different time points for enumeration. Each bar denotes the arithmetic mean ± SEM. The asterisks indicate values that are statistically significantly different (p < 0.001). Percent survival was calculated as the ratio of surviving CFU after incubation vs. the initial inoculum. (B) Shr does not contribute to GAS survival after 30 min incubation with purified human neutrophils. Data represent the arithmetic mean ± SEM. Percent survival of the bacteria was calculated relative to control wells.
Shr does not influence GAS antimicrobial peptide resistance or surface charge
Human cathelicidin antimicrobial peptide LL-37, a short positively charged peptide expressed by epithelial cells and phagocytes, serves as the first-line defense against invasive bacterial infections, including GAS.20 LL-37 expression is induced by tissue damage, inflammation, or infection, but it is expressed at very low or undetectable levels at baseline. In the Gram-positive human pathogen S. aureus, the iron acquisition protein IsdA, which shares homology with Shr, was shown to contribute to cathelicidin resistance by decreasing the hydrophobicity of the bacterial cell wall.22,23 Therefore, we investigated whether Shr expression influences bacterial resistance to LL-37 killing. Following 2 h incubation in the presence of LL-37 (16 μM), no significant differences were observed between WT and 5448Δshr survival (Fig. 3A), suggesting that Shr does not contribute to antimicrobial peptide resistance. Moreover, loss of Shr did not have an impact on the surface hydrophobicity of M1T1 GAS, as determined by the N-hexadecane partition assay (Fig. 3B). Finally, the 5448Δshr mutant bound cytochrome c at comparable levels to the WT parent strain (Fig. 3C), indicating that the absence of Shr does not perturb M1T1 GAS cell surface charge, another factor often implicated in antimicrobial peptide susceptibility/resistance.
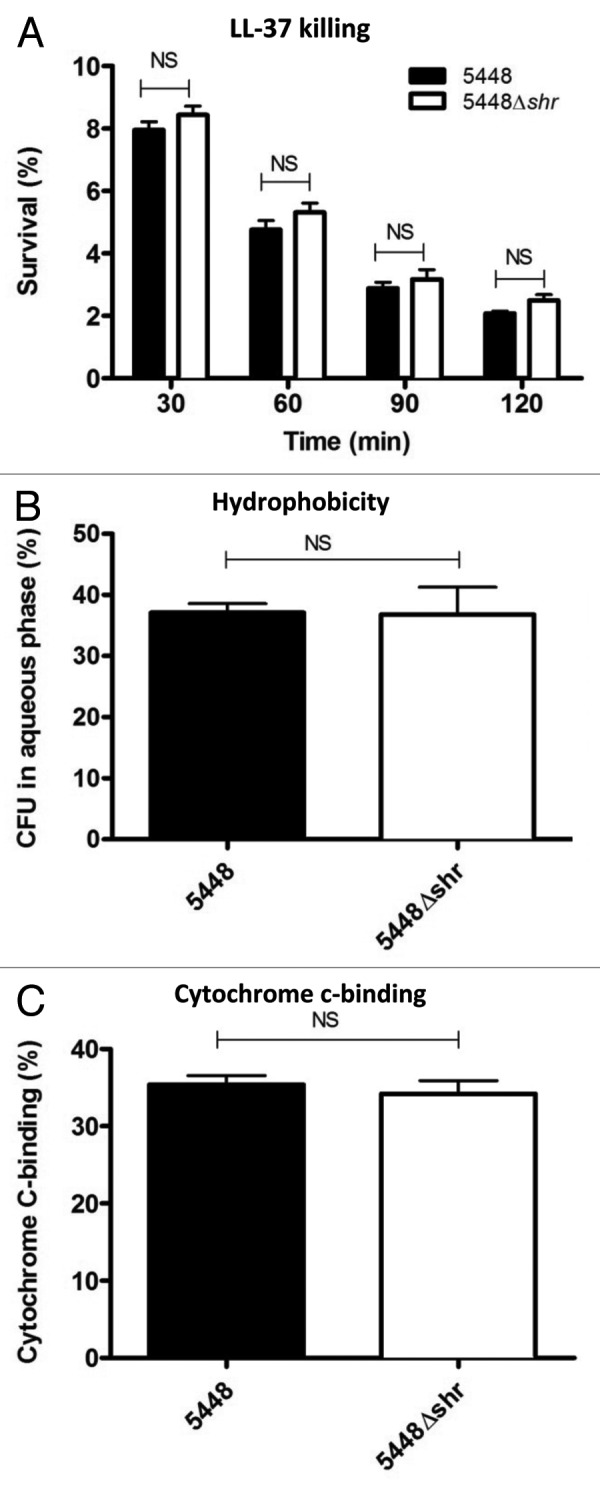
Figure 3. Shr does not interfere with human cathelicidin antimicrobial peptide LL-37 or cell surface charge. (A) Mid-logarithmic phase bacterial cultures were incubated with LL-37 (16 μM), serially diluted and plated onto agar at different time points for enumeration of surviving bacteria. Percent survival was determined by dividing surviving CFU by the inocula and multiplying by 100%. Values denote the arithmetic mean ± SEM. (B) The degree of surface hydrophobicity was determined by incubation with hexadecane and quantification of the bacteria in the aqueous phase. Data represent the mean ± SEM. (C) For cell surface charge assays, overnight stationary phase bacterial pellets were washed and incubated with cytochrome c at final concentration of 0.5 mg/ml. The percent cytochrome c-binding was determined by measuring the absorbance at 530 nm.
Shr enhances GAS binding to extracellular matrix glycoprotein laminin, but not fibronectin
In addition to iron uptake, Shr is a surface-localized and exposed adhesin with a proposed role in adherence to extracellular matrix components fibronectin and laminin.14 Here, we demonstrate that in comparison to WT M1T1 GAS, 5448Δshr showed a significant reduction in laminin-binding (Fig. 4A). The complemented strain, 5448Δshr pShr, bound laminin more efficiently than WT (Fig. 4A), most likely due to the overexpression of Shr (Fig. 1C). Although there was a trend toward reduced adherence of 5448Δshr to fibronectin, this difference did not reach statistical significance under our assay conditions (Fig. 4B). These data imply a role for Shr in the binding of M1T1 GAS to laminin but not fibronectin.

Figure 4. Shr enhances binding to extracellular matrix glycoprotein laminin, but not fibronectin. After incubation of mid-logarithmic phase bacteria (2 × 107 CFU) in 96-well plates coated with laminin (A) or fibronectin (B) at final concentration of 20 μg/ml, the unattached bacteria were removed by washing and the bound bacteria released with trypsin prior to serial dilution and plating for enumeration. Data represent the arithmetic mean ± SEM. The asterisks indicate values that are statistically significantly different (p < 0.0001).
Adherence of GAS to human epithelial cells and keratinocytes is not influenced by Shr
Previously, Fisher and coworkers demonstrated that a shr mutant in serotype M49 GAS exhibited reduced adherence to HEp-2 cells.14 In this investigation, we interrogated the role of Shr in the adherence of WT M1T1 GAS and the isogenic 5448Δshr to HEp-2 epithelial cells (Fig. 5A) and HaCaT keratinocytes (Fig. 5B), and observed no significant differences in either cell line. Therefore, Shr does not play an essential role in serotype M1T1 GAS adherence to epithelial cells or keratinocytes.
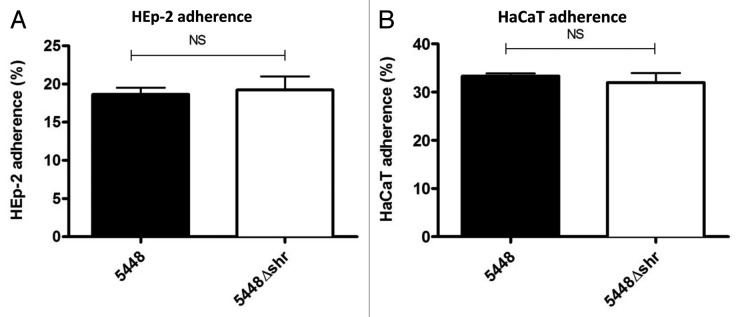
Figure 5. Adherence of WT 5448 and 5448Δshr to HEp-2 and HaCaT cells. Confluent HEp-2 (A) and HaCaT (B) monolayers were incubated with mid-log phase bacterial cultures at 37°C and 5% CO2 for 30 min. After washing unattached bacteria and lysing the cells, the percent binding was determined by the dividing the number of bound bacteria per well by the starting inoculum and multiplying by 100%. Each bar shows the mean ± SEM.
Shr promotes virulence in mouse models of skin and systemic infection
In a zebrafish model of invasive infection, Shr enhances the virulence of serotype M49 GAS.14 To determine whether Shr contributes to in vivo virulence in a mammalian model, mice were subcutaneously infected with non-lethal doses WT M1T1 GAS or 5448Δshr mutant, and lesion size measured daily for 3 d. At 24, 48 and 72 h post infection (Fig. 6A), lesion size was significantly reduced for 5448Δshr-infected mice. Extending this analysis to a systemic infection model, mice were injected intraperitoneally with WT or 5448Δshr, and the mortality rate monitored twice daily for 6 d. WT infection resulted in rapid death (100% mortality at day 1.5), while the mortality rate was significantly reduced for 5448Δshr (Fig. 6B). For the first time, this study shows that Shr is required for full virulence of M1T1 GAS in subcutaneous and systemic mouse models of infection.
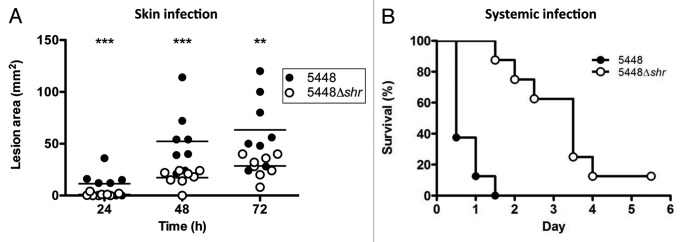
Figure 6. Shr promotes GAS virulence in murine models of infection. (A) Separate cohorts of female CD-1 mice 10 weeks of age (n = 8) were subcutaneously challenged with non-lethal doses of WT 5448 or 5448Δshr (~1 × 107 CFU/100 μl) and lesion size monitored daily. The asterisks indicate values that are statistically significantly different (p < 0.05). (B) Shr is required for full virulence in a mouse model of systemic infection. CD-1 female mice (10 weeks of age; n = 8) were injected intraperitoneally with either WT 5448 or 5448Δshr (~4 × 106 CFU/200 μL PBS with 5% gastric mucin) and monitored twice daily for mortality (p < 0.0001).
Discussion
Iron is an essential nutrient for most bacterial pathogens, functioning as a catalyst in electron transport systems and an important cofactor in many essential enzymes. The inactivation of genes involved in heme acquisition in Vibrio cholerae,24 serotype M49 GAS,14 Bordetella pertussis25 and S. aureus26 attenuates virulence in animal infection models. The heme acquisition machinery of GAS parallels that of S. aureus.12 The GAS Shr protein is reminiscent of NEAT-containing receptors, such as the heme-binding Isd proteins in S. aureus, and related proteins from other Gram-positive bacteria.27 However, relatively little is known about the role that iron plays in the physiology and virulence of GAS. The major objective of this study was to explore the role of Shr in the pathogenesis of serotype M1T1 GAS, one of the leading causes of severe invasive infections worldwide annually.28 Here, for the first time, we use precise targeted mutagenesis to demonstrate that Shr contributes to M1T1 GAS growth in iron-restricted media and human blood, adherence to laminin glycoprotein and virulence in mouse models of skin and systemic infection.
GAS is an obligate human pathogen that requires an iron source for growth and essential metabolic processes. Previously, it was demonstrated that Shr does not bind lactoferrin or transferrin,10 the principal ferric carrier proteins in serum. In this study, growth assays were performed in iron-limited RPMI 1640 medium supplemented with 20% FBS to support bacterial growth. In the absence of iron, the growth rates were similar for WT, 5448Δshr and the complemented strain 5448Δshr pShr. Upon supplementation of the medium with elemental iron FeCl3, 5448Δshr maintained its original growth rate, while the growth kinetics of the WT and complemented strain displayed a small but statistically significant enhancement (Fig. 1B). An M1 SF370 strain with a polar mutation in shr did not affect growth in iron-restricted medium supplemented with hemoglobin, whole blood or ferric citrate.12 Here, by using precise in-frame allelic exchange mutagenesis and single-gene complementation analysis, we demonstrate that iron is required for optimal GAS growth, and that Shr is important for iron uptake in the invasive M1T1 serotype, as shown previously for serotype M6 GAS.10
GAS is a β-hemolytic bacterium that may use hemolysis as a means of acquiring iron from hemoglobin and other heme-containing proteins circulating within the human host.10,29 GAS secrete streptolysin S (SLS), a potent membrane-active hemolysin that efficiently lyses a broad spectrum of host cells including red blood cells.30 This study demonstrates that Shr promotes GAS growth in whole human blood, as evidenced by the reduced growth rate of 5448Δshr relative to WT. We hypothesize that 5448Δshr is unable to utilize the hemoproteins released by SLS-mediated lysis for growth support and metabolic functions. In support of this hypothesis, iron restriction increases the hemolytic activity of GAS, and induces the expression of SLS and other genes encoding putative hemolysins.31 Further, the reduced growth rate of 5448Δshr in iron-limited media and human blood correlates with reduced virulence in mouse models of systemic and skin infection. This is the first report to demonstrate that Shr is a virulence factor of serotype M1T1 GAS in mouse infection models. Laminin is abundant in the skin,32 and the diminished laminin-binding of 5448Δshr may contribute to a colonization defect resulting in reduced skin lesion sizes in our subcutaneous model of mouse infection. These mammalian virulence data validate an earlier report conducted with a shr deletion-insertion mutant in serotype M49 GAS, which exhibited a 50-fold increase in LD50 compared with WT, and reduced mortality in a zebrafish intramuscular infection model.14
In addition to iron uptake, the two NEAT domains of Shr protein have been implicated in GAS adherence to ECM ligands.14,16 Purified recombinant Shr has been shown to specifically interact with fibronectin and laminin immobilized on microtiter plates.14 Lactococcus lactis heterologously expressing GAS Shr on the bacterial surface gained the capacity to bind fibronectin, compared with WT, suggesting a possible adhesin role for Shr.14 Here, we demonstrate that Shr contributes to M1T1 GAS binding to laminin, but not fibronectin, most likely due to the expression of multiple fibronectin-binding proteins on the GAS cell surface. In addition, we found that Shr does not mediate binding to HEp-2 cells, contrary to a previous report showing a 40% reduction in HEp-2 adherence for a shr mutant in serotype M49 GAS.14 To account for this discrepancy, we hypothesize that additional adhesins expressed on the M1T1 GAS cell surface that mediate binding to HEp-2 cells, such as the M1 protein,33,34 may compensate for the loss Shr.
In contrast to findings reported with a S. aureus mutant lacking the homologous iron acquisition protein IsdA,22 we did not detect an effect of Shr deletion on cell wall hydrophobicity, sensitivity to cathelicidin antimicrobial peptides, nor sensitivity to killing by neutrophils, which utilize cathelicidin for killing in the phagolysosome and extracellular traps. M1T1 GAS express a large array of cathelicidin resistance factors, including dlt operon-encoded cell wall-modifying lipoteichoic acids,35 surface hyaluronic acid capsule,36 secreted streptococcal inhibitor of complement (SIC)37 and the N-terminal domain of the surface-anchored M1 protein,38 which together may provide a functional redundancy sufficient to obscure any independent contribution of Shr to the cathelicidin resistance phenotype.
Further biochemical and biophysical studies are required to fully elucidate the iron uptake mechanisms of GAS, including Shr. Convalescent sera from GAS infected mice exhibit high titer Shr antibodies, demonstrating that Shr is immunogenic and expressed in vivo during the course of GAS infection.14 More recently, Huang et al.39 demonstrated that the highly conserved Shr protein could serve as a vaccine candidate antigen since it conferred partial protection against systemic GAS infection in passive and active immunization models. Thus, our demonstration of a virulence role for Shr in mouse infection models with invasive M1T1 GAS perhaps provides added rationale for inclusion of this protein in GAS subunit vaccine formulations.
Materials and Methods
Bacterial strains
M1T1 GAS strain 5448 was originally isolated from a patient with necrotizing fasciitis and streptococcal toxic shock-like syndrome.40 For routine growth, GAS was propagated at 37°C on Todd-Hewitt agar (THA), or in liquid cultures of Todd-Hewitt broth (THB, Hardy Diagnostics) without shaking. When required, the growth medium was supplemented with erythromycin (Erm) 5 μg/ml or chloramphenicol (Cm) 2 μg/ml.
Precise in-frame allelic exchange mutagenesis of shr
To construct the 5448Δshr allelic exchange mutant, the upstream (824 bp) and downstream (835 bp) regions of the shr gene (GenBank Gene ID: 902026) were PCR amplified from the 5448 chromosome using the following primer pairs: (1) shr-xho1_upF (5′-CGACTCGAGCTTGGGGACAATGCACTTG-3′, XhoI restriction site underlined) + shr-upR [5′-CCAGTGATTTTTTTCTCCATTTTCAATCAACTTTCTAAAAATTTTATGAATAAGCTAAC-3′ with the 20 bp 5′ extension matching 5′ end of the chloramphenicol acetyltransferase (cat) gene]; (2) shr-downF (5′-TGGCAGGGCGGGGCGTAAGAAACTTAGTACCAAGTGAAATAACAAAAAAGCTAGTC-3′ with the 18 bp 5′ extension matching 3′ end of the cat gene) + shr-xbaI-downR (5′-CGCTCTAGACATAGCACCTGTGTGAGACTCATCTTG-3′, XbaI site underlined). PCR was performed with PfuUltra II (Stratagene) using the respective upstream and downstream fragments and a cat gene amplicon to yield a fusion product in which cat precisely replaced the gene of interest. This fusion product was recovered by TA cloning into pCR2.1-TOPO (Invitrogen), and subsequently cloned into the temperature-sensitive erythromycin (Erm) resistant plasmid pHY304. The resulting knockout plasmid was transformed into WT 5448 by electroporation and transformants plated on THA-Erm 5 μg/ml. Single recombination events were identified by shifting to the non-permissive temperature (37°C) while maintaining Erm selection. Selective pressure was relaxed by serial passage at 30°C without antibiotics, and double-crossover events identified by screening for colonies exhibiting a Cm-resistant and Erm-sensitive phenotype. Precise, in-frame allelic exchange of shr with the cat gene in the 5448 chromosome (5448Δshr) was confirmed by PCR using primers specific for cat and shr. For complementation of 5448Δshr, the shr gene was PCR amplified from 5448 using primers Shr_For_EcoRI (5′-AGCGAATTCATGAAAAAAATCAGCAAATG-3′) and Shr_Rev_BamHI (5′-AGCGGATCCTTATTTAAATAATGTCTTTCG-3′). The PCR product was cloned into pCR2.1-TOPO (Invitrogen) and subcloned into the Erm-resistant expression vector pDCerm.41 The resulting plasmid, pShr, was introduced via electroporation into GAS 5448Δshr to construct complemented strain 5448Δshr pShr.
RNA isolation and quantitative PCR
Total RNA was extracted from overnight GAS cultures using the RNeasy Mini Kit (Qiagen). Bacteria were mechanically disrupted in a MagNA Lyser (Roche) with 1.0 mm diameter zirconia glass beads (BioSpec). RNA was treated with DNase (Turbo DNase, Ambion) and 1 μg total RNA reverse transcribed to cDNA (iScript, BioRad). Real-time PCR was performed using iQ SYBR Green Supermix (BioRad) with the following primer sets at 1 μM final concentration: shr forward 5′-TGGGACTGAACGTCTTGAAA-3′ and shr reverse 5′-ACTCGCCTCGAATCTGTTCT-3′; shp forward 5′-GGGACAACACTAGATATTGC-3′ and shp reverse 5′-GACCACTTCTCTGCCCATAG-3′; gyrA forward 5′-GAAGTGATCCCTGGACCTGA-3′ and gyrA reverse 5′-CCCGACCTGTTTGAGTTGTT-3′. Data were normalized to gyrA and relative expression was compared with WT by the ΔΔCt method.42
Bacterial growth under iron starvation conditions
Test tubes were soaked overnight in 1 M HCl and rinsed 10 times with Milli-Q water prior to use. Overnight stationary phase cultures of GAS in 4 ml THB were centrifuged at 3,220× g for 10 min, washed three times with PBS, then resuspended in 300 μl RPMI 1640 medium (Invitrogen) supplemented with 20% fetal bovine serum (FBS; Gibco) heat-inactivated (HI) at 70°C for 30 min. The bacterial concentrate was used to adjust 10 ml of RPMI + 20% HI FBS to a starting optical density at 600 nm (OD600) of 0.1. When required, ferric chloride (Sigma) was added to a final concentration of 250 μM from a sterile-filtered 0.1 M stock solution of FeCl3.6H2O in 0.1 M HCl. Growth at 37°C under static conditions was monitored by measuring the OD600 for 2 d. Data were pooled and normalized from two independent experiments, each performed in triplicate.
Bacterial growth in whole human blood
Blood was drawn from healthy volunteers into heparinized syringes. One hundred microliters of mid-logarithmic phase bacteria in PBS (2 × 104 colony forming units, CFU), was mixed with 400 μl of whole human blood in siliconized tubes and incubated at 37°C with orbital rotation or under static conditions. Aliquots (25 μl) were removed, 10-fold serially diluted and plated onto THA for enumeration of surviving CFU at different time points. Data were pooled and normalized from three independent experiments, each performed in triplicate.
Neutrophil isolation and killing assay
Human neutrophils were isolated from the blood of healthy donors using the PolymorphPrep system (Axis-Shield). Neutrophils were seeded into 96-well plates (Costar) at a final concentration of 2 × 105 cells/well in RPMI 1640 + 2% HI FBS (30 min at 56°C). Bacteria were grown overnight in THB, diluted 1:10 into fresh medium, and grown to mid-log phase, corresponding to an OD600 of 0.4 (~2 × 108 CFU/ml). Bacteria were resuspended in RPMI + 2% HI FBS and added to each well containing neutrophils at a multiplicity of infection of 0.1. For opsonophagocytic assays, washed mid-log phase bacteria were pre-incubated in 80% human serum for 1 h at 37°C with orbital rotation, then diluted in RPMI to final 2% serum concentration and a multiplicity of infection of 0.1. An aliquot from each well (25 μl) was 10-fold serially diluted in PBS and immediately plated on THA for counting (time = 0). The assay plate was placed at 37°C + 5% CO2 for 30 min and 25 μl aliquots diluted in PBS and plated for enumeration of CFU. Percent survival of the bacteria was calculated in comparison to bacterial control wells grown under the same conditions in the absence of neutrophils. Data were pooled and normalized from three independent experiments, each performed in triplicate.
LL-37 killing kinetics
GAS were grown to mid-log phase (OD600 = 0.4) in DMEM (Invitrogen) + 10% THB and seeded into 96-well plates (Costar) at a concentration 105 CFU/well in 225 μl final volume. DMEM contains carbonate to enhance bacterial susceptibility to LL-37, while 10% THB supports GAS growth.43 LL-37 (American Peptide Company) was added to each well at final concentration of 16 μM. Aliquots of 25 μl were 10-fold serially diluted in PBS and plated onto THA at each time point for enumeration. Percent survival was determined as the number of CFU recovered divided by number of initial CFU, multiplied by 100%. Data were pooled and normalized to WT from three independent experiments, each performed in triplicate.
Cell surface hydrophobicity assay
Hydrophobicity was determined by the hexadecane method as previously described,44 with the following modifications. Overnight cultures were inoculated into THB (1:10) and grown at 37°C to mid-log phase (OD600 = 0.4). GAS were centrifuged at 3,220× g for 10 min, and the pellet resuspended in 3 ml PBS. Sterile tubes containing 300 μl of hexadecane (Sigma) were overlaid with 900 μl of bacterial culture, vortexed vigorously for 2 min and incubated for 10 min at room temperature to allow phase separation. Aliquots (25 μl) of the lower aqueous phase were serially diluted in PBS and plated onto THA for enumeration. Percent hydrophobicity was determined as the ratio of bacteria in the aqueous phase to the starting inoculum, multiplied by 100%. Data were pooled and normalized from five independent experiments, each performed in triplicate.
Cytochrome c-binding
Bacterial cultures (20 ml) were grown overnight at 37°C. In the morning, the pellet was washed twice with 20 mM MOPS (4-morpholinepropanesulfonic acid) buffer (pH 7.0) and adjusted to an OD600 = 0.7 in 10 ml of buffer. The cultures were centrifuged at 3,200× g for 10 min and resuspended in 1 ml of buffer. In a 96-well V-bottom plate (Costar), 190 μl of bacterial culture was mixed with cytochrome c at a final concentration of 0.5 mg/ml. The plate was incubated at room temperature for 10 min and centrifuged at 3,200× g for 10 min. Supernatant (100 μl) was transferred to a flat-bottom 96-well plate (Costar) and the absorbance measured at 530 nm with the SpectraMax 250 (Molecular Devices). The percent binding was determined by measuring the amount of bacteria bound to cytochrome c at OD530. Data were pooled and normalized from three independent experiments, each performed in triplicate.
Laminin and fibronectin adherence assays
High-binding EIA/RIA 96-well microtiter plates (Costar) were coated overnight at 4°C with 40 μg of murine laminin or human fibronectin (Sigma) in 200 μl 50 mM Na2CO3 (pH 9.5). The wells were washed three times with PBS and blocked with 1% bovine serum albumin in PBS for 2 h at 37°C. GAS was grown to mid-logarithmic phase, washed with PBS and adjusted to an OD600 = 0.4 (~2 × 108 CFU/ml). The plates were washed five times with PBS, seeded with 100 μl (2 × 107 CFU) of bacterial solution, centrifuged at 500× g for 10 min, incubated at 37°C for 1 h and then washed five times with PBS to remove non-adherent bacteria. Adherent bacteria were released by the addition of 100 μl 0.25% trypsin/1 mM EDTA (Gibco) to each well for 10 min at 37°C. After adding 100 μl PBS to each well, bacteria were 10-fold serially diluted in PBS and plated onto THA for enumeration. Adherence was calculated as a percentage of the initial inoculum. Data were pooled and normalized from three independent experiments, each performed in triplicate.
Adherence to epithelial cells and keratinocytes
Assays were performed using human laryngeal epithelia (HEp-2) and human keratinocyte (HaCaT) cells, as described previously.45 Briefly, both cell lines were propagated as monolayers in RPMI 1640 (Invitrogen) supplemented with 2% HI FBS at 37°C with 5% CO2. A tissue culture treated 24-well plate (Costar) was seeded with 2 × 105 cells/well and grown overnight. Immediately prior to the assay, the culture media for both cell lines was replaced with fresh RPMI + 2% FBS. GAS cultures were grown overnight and inoculated into fresh media in the morning and grown to an OD600 = 0.4. Bacteria were diluted in PBS and added at a multiplicity of infection of 10:1. The plates were centrifuged at 500× g for 5 min and incubated at 37°C in 5% CO2. After 30 min, the monolayers were washed 5 times with PBS to remove unattached bacteria, and 100 μl of trypsin added to release the cells. After incubation for 5 min at 37°C, the cells were lysed by addition of Triton X-100 to each well at a final concentration of 0.025%. Bacteria were serially diluted and plated on THA for enumeration. Bacterial adherence was calculated as a percentage of the initial inoculum. Bacterial strains grew equally in RPMI + 2% FBS for the 30 min duration of the experiment. Trypan blue staining showed HEp-2 and HaCaT cell viability ≥ 95%. Data were pooled and normalized from three independent experiments, each performed in triplicate.
Mouse infection models
Mice were subcutaneously (s.c.) inoculated with non-lethal doses essentially as previously described.36 Briefly, 4 ml overnight stationary phase GAS cultures were diluted 1:10 in fresh THB and grown to mid-logarithmic phase (OD600 ~0.4). Cultures were centrifuged at 3,220× g for 10 min, washed once with 20 ml PBS, and bacterial pellets resuspended in 300 μl PBS final volume. The bacterial concentrate was used to adjust 10 ml PBS to OD600 = 0.4 (~2 × 108 CFU/ml), prior to centrifugation at 3,220× g for 10 min and 10-fold concentration in PBS. Cohorts (n = 10) of 10-week-old female CD-1 mice (Charles River Laboratories) were inoculated s.c. with 100 μl of bacteria and lesion size measured daily for 3 d. For intraperitoneal (i.p.) infections, bacterial cultures were adjusted to OD600 = 0.4 as described above, mixed with an equal volume of 10% gastric mucin (Sigma) in PBS and 200 μl injected i.p. into 10-week-old female CD-1 mice (n = 10). Survival was monitored twice daily for 6 d. Inocula were 10-fold serially diluted in PBS, plated onto THA and incubated overnight at 37°C for enumeration.
Statistical analyses
Growth assays and LL-37 killing kinetics were compared by 2-way ANOVA. Laminin-binding assays were analyzed by 1-way ANOVA. Human neutrophil, hydrophobicity, cytochrome c-binding, fibronectin-binding, mammalian cell adherence assays and skin lesion sizes were assessed using two-tailed unpaired t-tests. Kaplan-Meier survival curves were compared using the log-rank test. At a significance level of 0.05, differences were considered significantly different at p < 0.05. All statistical analyses were performed using GraphPad Prism version 5 (GraphPad Software Inc.).
Ethical approval
Permission to collect human blood under informed consent was approved by the UCSD Human Research Protection Program. Procedures used for all animal experiments were approved by the UCSD Institutional Animal Care and Use Committee.
Acknowledgments
J.N.C. was supported by an Overseas Biomedical Training Fellowship (514639) and Project Grant (APP1033258) from the National Health and Medical Research Council of Australia. S.D. and V.N. were supported by NIH grants AI077780 and AI48176. The authors wish to thank Fred Beasley for technical advice regarding the iron growth assays.
Glossary
Abbreviations:
- GAS
group A streptococcus
- Shr
streptococcal hemoprotein receptor
- Sia
streptococcal iron acquisition
- WT
wild-type
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/virulence/article/21933
References
- 1.Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–36. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 2.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, et al. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis. 2005;192:771–82. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 3.Posey JE, Gherardini FC. Lack of a role for iron in the Lyme disease pathogen. Science. 2000;288:1651–3. doi: 10.1126/science.288.5471.1651. [DOI] [PubMed] [Google Scholar]
- 4.Archibald F. Lactobacillus plantarum, an organism not requiring iron. FEMS Microbiol Lett. 1983;19:29–32. doi: 10.1111/j.1574-6968.1983.tb00504.x. [DOI] [Google Scholar]
- 5.Payne SM. Iron acquisition in microbial pathogenesis. Trends Microbiol. 1993;1:66–9. doi: 10.1016/0966-842X(93)90036-Q. [DOI] [PubMed] [Google Scholar]
- 6.Maresso AW, Schneewind O. Iron acquisition and transport in Staphylococcus aureus. Biometals. 2006;19:193–203. doi: 10.1007/s10534-005-4863-7. [DOI] [PubMed] [Google Scholar]
- 7.Drazek ES, Hammack CA, Schmitt MP. Corynebacterium diphtheriae genes required for acquisition of iron from haemin and haemoglobin are homologous to ABC haemin transporters. Mol Microbiol. 2000;36:68–84. doi: 10.1046/j.1365-2958.2000.01818.x. [DOI] [PubMed] [Google Scholar]
- 8.Ge R, Sun X, He Q. Iron acquisition by Streptococcus species: An updated review. Front Biol China. 2009;4:392–401. doi: 10.1007/s11515-009-0035-4. [DOI] [Google Scholar]
- 9.Mietzner TA, Morse SA. The role of iron-binding proteins in the survival of pathogenic bacteria. Annu Rev Nutr. 1994;14:471–93. doi: 10.1146/annurev.nu.14.070194.002351. [DOI] [PubMed] [Google Scholar]
- 10.Eichenbaum Z, Muller E, Morse SA, Scott JR. Acquisition of iron from host proteins by the group A streptococcus. Infect Immun. 1996;64:5428–9. doi: 10.1128/iai.64.12.5428-5429.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinberg ED. Iron and infection. Microbiol Rev. 1978;42:45–66. doi: 10.1128/mr.42.1.45-66.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bates CS, Montañez GE, Woods CR, Vincent RM, Eichenbaum Z. Identification and characterization of a Streptococcus pyogenes operon involved in binding of hemoproteins and acquisition of iron. Infect Immun. 2003;71:1042–55. doi: 10.1128/IAI.71.3.1042-1055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei B, Liu M, Voyich JM, Prater CI, Kala SV, DeLeo FR, et al. Identification and characterization of HtsA, a second heme-binding protein made by Streptococcus pyogenes. Infect Immun. 2003;71:5962–9. doi: 10.1128/IAI.71.10.5962-5969.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher M, Huang YS, Li X, McIver KS, Toukoki C, Eichenbaum Z. Shr is a broad-spectrum surface receptor that contributes to adherence and virulence in group A streptococcus. Infect Immun. 2008;76:5006–15. doi: 10.1128/IAI.00300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu H, Liu M, Lei B. The surface protein Shr of Streptococcus pyogenes binds heme and transfers it to the streptococcal heme-binding protein Shp. BMC Microbiol. 2008;8:15. doi: 10.1186/1471-2180-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ouattara M, Cunha EB, Li X, Huang YS, Dixon D, Eichenbaum Z. Shr of group A streptococcus is a new type of composite NEAT protein involved in sequestering haem from methaemoglobin. Mol Microbiol. 2010;78:739–56. doi: 10.1111/j.1365-2958.2010.07367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2006;2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lei B, Smoot LM, Menning HM, Voyich JM, Kala SV, Deleo FR, et al. Identification and characterization of a novel heme-associated cell surface protein made by Streptococcus pyogenes. Infect Immun. 2002;70:4494–500. doi: 10.1128/IAI.70.8.4494-4500.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eichenbaum Z, Green BD, Scott JR. Iron starvation causes release from the group A streptococcus of the ADP-ribosylating protein called plasmin receptor or surface glyceraldehyde-3-phosphate-dehydrogenase. Infect Immun. 1996;64:1956–60. doi: 10.1128/iai.64.6.1956-1960.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Froehlich BJ, Bates C, Scott JR. Streptococcus pyogenes CovRS mediates growth in iron starvation and in the presence of the human cationic antimicrobial peptide LL-37. J Bacteriol. 2009;191:673–7. doi: 10.1128/JB.01256-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voyich JM, Musser JM, DeLeo FR. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect. 2004;6:1117–23. doi: 10.1016/j.micinf.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 22.Clarke SR, Mohamed R, Bian L, Routh AF, Kokai-Kun JF, Mond JJ, et al. The Staphylococcus aureus surface protein IsdA mediates resistance to innate defenses of human skin. Cell Host Microbe. 2007;1:199–212. doi: 10.1016/j.chom.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Zinkernagel AS, Nizet V. Staphylococcus aureus: a blemish on skin immunity. Cell Host Microbe. 2007;1:161–2. doi: 10.1016/j.chom.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Henderson DP, Payne SM. Vibrio cholerae iron transport systems: roles of heme and siderophore iron transport in virulence and identification of a gene associated with multiple iron transport systems. Infect Immun. 1994;62:5120–5. doi: 10.1128/iai.62.11.5120-5125.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brickman TJ, Vanderpool CK, Armstrong SK. Heme transport contributes to in vivo fitness of Bordetella pertussis during primary infection in mice. Infect Immun. 2006;74:1741–4. doi: 10.1128/IAI.74.3.1741-1744.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pishchany G, Dickey SE, Skaar EP. Subcellular localization of the Staphylococcus aureus heme iron transport components IsdA and IsdB. Infect Immun. 2009;77:2624–34. doi: 10.1128/IAI.01531-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu H, Xie G, Liu M, Olson JS, Fabian M, Dooley DM, et al. Pathway for heme uptake from human methemoglobin by the iron-regulated surface determinants system of Staphylococcus aureus. J Biol Chem. 2008;283:18450–60. doi: 10.1074/jbc.M801466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aziz RK, Kotb M. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis. 2008;14:1511–7. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Francis RT, Jr., Booth JW, Becker RR. Uptake of iron from hemoglobin and the haptoglobin-hemoglobin complex by hemolytic bacteria. Int J Biochem. 1985;17:767–73. doi: 10.1016/0020-711X(85)90262-9. [DOI] [PubMed] [Google Scholar]
- 30.Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, et al. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol Microbiol. 2005;56:681–95. doi: 10.1111/j.1365-2958.2005.04583.x. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths BB, McClain O. The role of iron in the growth and hemolysin (Streptolysin S) production in Streptococcus pyogenes. J Basic Microbiol. 1988;28:427–36. doi: 10.1002/jobm.3620280703. [DOI] [PubMed] [Google Scholar]
- 32.Sugawara K, Tsuruta D, Ishii M, Jones JC, Kobayashi H. Laminin-332 and -511 in skin. Exp Dermatol. 2008;17:473–80. doi: 10.1111/j.1600-0625.2008.00721.x. [DOI] [PubMed] [Google Scholar]
- 33.Courtney HS, Hasty DL, Dale JB. Molecular mechanisms of adhesion, colonization, and invasion of group A streptococci. Ann Med. 2002;34:77–87. doi: 10.1080/07853890252953464. [DOI] [PubMed] [Google Scholar]
- 34.Molinari G, Chhatwal GS. Streptococcal invasion. Curr Opin Microbiol. 1999;2:56–61. doi: 10.1016/S1369-5274(99)80010-1. [DOI] [PubMed] [Google Scholar]
- 35.Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, et al. D-alanylation of teichoic acids promotes group A streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol. 2005;187:6719–25. doi: 10.1128/JB.187.19.6719-6725.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cole JN, Pence MA, von Kockritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, et al. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus mBio 2010; 1; [DOI] [PMC free article] [PubMed]
- 37.Pence MA, Rooijakkers SH, Cogen AL, Cole JN, Hollands A, Gallo RL, et al. Streptococcal inhibitor of complement promotes innate immune resistance phenotypes of invasive M1T1 group A Streptococcus. J Innate Immun. 2010;2:587–95. doi: 10.1159/000317672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lauth X, von Köckritz-Blickwede M, McNamara CW, Myskowski S, Zinkernagel AS, Beall B, et al. M1 protein allows group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J Innate Immun. 2009;1:202–14. doi: 10.1159/000203645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang YS, Fisher M, Nasrawi Z, Eichenbaum Z. Defense from the group A Streptococcus by active and passive vaccination with the streptococcal hemoprotein receptor. J Infect Dis. 2011;203:1595–601. doi: 10.1093/infdis/jir149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatellier S, Ihendyane N, Kansal RG, Khambaty F, Basma H, Norrby-Teglund A, et al. Genetic relatedness and superantigen expression in group A streptococcus serotype M1 isolates from patients with severe and nonsevere invasive diseases. Infect Immun. 2000;68:3523–34. doi: 10.1128/IAI.68.6.3523-3534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeng A, Sakota V, Li Z, Datta V, Beall B, Nizet V. Molecular genetic analysis of a group A Streptococcus operon encoding serum opacity factor and a novel fibronectin-binding protein, SfbX. J Bacteriol. 2003;185:1208–17. doi: 10.1128/JB.185.4.1208-1217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dorschner RA, Lopez-Garcia B, Peschel A, Kraus D, Morikawa K, Nizet V, et al. The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J. 2006;20:35–42. doi: 10.1096/fj.05-4406com. [DOI] [PubMed] [Google Scholar]
- 44.Courtney HS, Ofek I, Penfound T, Nizet V, Pence MA, Kreikemeyer B, et al. Relationship between expression of the family of M proteins and lipoteichoic acid to hydrophobicity and biofilm formation in Streptococcus pyogenes. PLoS One. 2009;4:e4166. doi: 10.1371/journal.pone.0004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Timmer AM, Kristian SA, Datta V, Jeng A, Gillen CM, Walker MJ, et al. Serum opacity factor promotes group A streptococcal epithelial cell invasion and virulence. Mol Microbiol. 2006;62:15–25. doi: 10.1111/j.1365-2958.2006.05337.x. [DOI] [PubMed] [Google Scholar]