

Abstract
Pathological conditions, such as cancers, viral infections, and autoimmune diseases, are associated with abnormal cytokine production, and the morbidity associated with many medical disorders is often directly a result of cytokine production. Because of the absence of negative feedback control occurring in some pathophysiologic situations, a given cytokine may flood and accumulate in the extracellular compartment of tissues or tumors thereby impairing the cytokine network homeostasis and contributing to local pathogenesis. To evaluate whether the rise of anti-cytokine Abs by vaccination is an effective way to treat these pathological conditions without being harmful to the organism, we have analyzed each step of the cytokine process (involving cytokine production, target response, and feedback regulation) and have considered them in the local context of effector–target cell microenvironment and in the overall context of the macroenvironment of the immune system of the organism. In pathologic tissues, Abs of high affinity, as raised by anti-cytokine vaccination, should neutralize the pool of cytokines ectopically accumulated in the extracellular compartment, thus counteracting their pathogenic effects. In contrast, the same Abs should not interfere with cytokine processes occurring in normal tissues, because under physiologic conditions cytokine production by effector cells (induced by activation but controlled by negative feedback regulation) does not accumulate in the extracellular compartment. These concepts are consistent with results showing that following animal and human anti-cytokine vaccination, induction of high-affinity Abs has proven to be safe and effective and encourages this approach as a pioneering avenue of therapy.
Cytokines normally exert their biological activities within the context of a cytokine network to maintain homeostatic mechanisms particularly relevant to the generation of immune reactions, inflammatory processes, remodeling of tissues, hemopoiesis, and angiogenesis. Imbalance in cytokine production or cytokine receptor expression and/or dysregulation of a cytokine process provide the basis for generating pathological disorders. This study focuses on the rationale of using vaccines against different cytokines in an attempt to control these pathologic events.
Cytokines and Cytokine Network: Pivotal in the Functioning of the Immune System
Parenchymal and stromal cells of various tissues communicate by means of cell-membrane contacts but also by means of soluble factors, especially cytokines. These signals released by the cells are short-range ones and control cell growth, differentiation cell death, and effector functions, including the secretion of other cytokines, as is evident for immune cells, T and B lymphocytes, and antigen-presenting cells, for which cytokines represent the key signals ruling over the various steps of immune reactions (1).
Cytokines comprise a family of proteins that include those called ILs, lymphokines, monokines, IFNs, and chemokines (2). Cytokines are characterized by a short half-life, local (autocrine/paracrine) signaling activity, and rarely systemic signaling [tumor necrosis factor (TNF)-α, IL-1, and IL-6, as observed in septic shock]. Like polypeptide hormones, cytokines are recognized by specific receptors present on the surface of target cells. In contrast with hormones, however, their targets and effects are highly pleiotropic and redundant (3). Moreover, although specific hormones are secreted by specialized cell types, each of these being anatomically localized in one endocrine gland, individual cytokines are produced by different types of effector cells. These include blood cells such as lymphocytes, natural killer cells, and antigen-presenting cells. Cytokine-producing blood cells circulate from one tissue to another. The cell migration in part accounts for the overall interdependency of cytokines. The interdependency of these regulatory signals is displayed also at the level of individual tissues, because the release of a particular cytokine by effector cells depends on microenvironmental stimuli among which signals are mediated by other cytokines. Consequently, the complex physiologic homeostasis taking place in the tissues is coordinated by the cytokine network, which is a set of interdependent regulatory cytokines and their corresponding receptors (3).
Cytokine Process: Cytokine Production, Effects, and Regulation
Production of an individual cytokine by an effector cell is usually not constitutive but is rather elicited by various stimuli such as viral or bacterial components, protein antigens, mitogens, and other cytokines. Under physiological conditions, activated effector cells trigger a transient but composite reaction. Viewed at a single effector (E) cell level, the composite reaction taking place in a tissue includes the activated E cell, the surrounding extracellular matrix (ECM), and target cells. The reaction consists of three successive stages (Fig. 1).
Figure 1.

Cytokine process in normal (A) and vaccinated (B) subjects under physiological condition. Cytokines (●) produced by E cells after activation (↘) initially may form immune complexes with ECM Abs (t1) and trigger a target cell (T; t2 and t'2). After T-cell response (✖), negative feedback down-regulates the cytokine process (cytokine synthesis and target receptor expression; t3 and t'3). Note that natural Abs (Y) or vaccine-induced Abs of high affinity (YY), complexed with the cytokine, delay the process for only a few minutes.
Step 1: Cytokine Production by Activated Effector Cells.
On activation, an effector cell is the site of cytokine gene transcription and cytokine synthesis. The cytokine transported to the cell membrane via Golgi microvesicles (4) is not stored into granules but continuously released into the juxtaposed ECM as long as it is synthesized. Indeed, cytokine release is transient. For example, in vitro release of IL-2 by phytohemagglutinin (PHA)-activated human T cells starts a few hours after stimulation and may last 48 ± 24 h, depending on the experimental conditions. In mice, after (bacterial) lipopolysaccharide (LPS) activation, Kupffer cells coating the liver sinusoids transiently release inflammatory cytokines. TNF-α is produced first after 1 h, IL-1β is produced after 3 h, and IL-1α is produced after 6 h. The release drops very rapidly for TNF-α; it drops after 6 h for IL-1β and after 18 h for IL-1α (5).
Step 2: Cytokine–Receptor (C–R) Binding and Target-Cell Response.
The half-life of a released cytokine is very brief, generally lasting only minutes. During this period, the cytokine crosses the effector–target (E–T) microinterval (made of the ECM) and binds to specific membrane receptors constituted most often by di- or trimeric polypeptide chains borne by neighboring competent target cells. Competence is related to expression of receptors that depend on genetic and microenvironmental factors. For instance, in Leishmania major-infected C3H mice (H2k), the production of IL-12 by natural killer cells triggers a T cell helper (TH)1 response on antigen-activated CD4 cell targets that express IL-12 β1 and β2 receptors. By contrast, infected BALB/c mice (H2d)—whose Leishmania antigen-activated CD4 cell targets do not respond to IL-12 because of different receptor regulation—develop a TH2 response (6).
C–R binding chiefly depends on the binding affinity value, which is generally high (see Turnover Rates and Binding Affinity of Target Receptors vs. Lymph Abs). The kinetics of C–R binding can be influenced by other factors also, including ECM components such as fibronectin, collagen IV, laminin, glycosaminoglycan, and other matrix substrates and serum proteins such as soluble receptors, receptor antagonists, immunoglobulins (including specific Abs), immune complexes, macroglobulins, and proteases—all of which may be present in the slow-circulating scarce lymph that moistens the ECM microinterval.
The target-cell response is initiated when a sufficient amount (a quantum) of cytokines has bound to the receptors. C–R complexes activate a cascade of metabolic processes in the competent target cell(s), involving cell membrane modulation with C–R complex internalization and receptor amplification (7, 8), transducing signals, transcription, protein synthesis, and, ultimately, changes in target-cell physiology. As long as C–R binding stimuli are produced, target cells develop their response.
Step 3: Down-Regulation of Cytokine Production and Receptor Expression.
Cytokine production is discontinued by negative feedback mechanisms after the target-cell response. In most cases, cytokine synthesis is switched off at the transcriptional level as a result of a cascade of as yet ill-characterized cellular endogenous reactions, but initiated by autocrine/paracrine target signals. These signals include the cytokine itself, acting in a negative feedback autocrine loop, as for IFN-α (9) and IL-10 (10) or, alternatively, paracrine inhibitory factors, among which are other cytokines triggered by or derived from the cytokine process itself. Thus, IL-4 turns off IL-12 production by means of antigen-presenting cells or natural killer cells (11) and, reciprocally, IL-12 turns off IL-4 secretion by means of TH2 cells (12).
Parallel with the cytokine negative feedback control, there is usually a down-regulation of the corresponding receptors most likely resulting from the disappearance of C–R-binding stimuli. Cytokines are released no longer by E cells into the ECM, and residual cytokines rapidly lose their stimulating capacity because of their short half-lives. It is noteworthy that in some instances, other factors contribute to the negative regulatory process in the target cell such as in soluble receptors shed by the target cells found for IL-1-, IL-2-, IL-4-, IFN-γ-, and TNF-α-induced reactions (13), and these receptors compete with membrane receptors for cytokine binding. Receptor antagonists may be released by effector cells also, such as IL-1Ra (14). The latter are devoid of agonistic effects but compete with effective cytokines for binding to target receptors.
Natural Anti-Cytokine Abs
Circulating anti-cytokine natural Abs are generally present in the sera of healthy individuals, although at low levels. Although they are detected most often at background levels by ELISA, they can be purified by affinity chromatography. In a study of 200 healthy individuals, anti-IFN-α autoAbs were isolated from the sera of all subjects (15). Natural Abs directed to other cytokines, including IL-6, IL-1, and IFN-γ, were isolated also (16, 17). These results are not surprising because, in contrast to T cells, anti-cytokine Ab-producing B cells have not been selected negatively and are still present albeit silenced (self-ignorance; ref. 18). The capacity to raise anti-cytokine Abs in response to pathophysiologic events is restricted given the absence of corresponding TH cells.
In patients undergoing treatment with cytokines, the specific Ab levels are increased only slightly. This is the case, for example, in patients receiving IFN-α therapy. Nonetheless, in these subjects, the presence of autoAbs was not associated with any notable effect (19). Also in patients infected with HIV-1, higher than normal circulating anti-IFN-α Abs are found (20). In these patients, the increase of autoAbs is likely related to the B cell polyclonal activation associated with HIV-1 infection, and they are at low levels and again without apparent notable effects.
Reasons for the Negligible Effects of Anti-Cytokine Abs on Cytokine Processes Under Physiologic Conditions
Negative Feedback Regulation of Cytokine Production in ECM Environment with Slow Lymph Flow.
During a cytokine process occurring in a tissue, anti-cytokine Abs in the stroma may be locally antagonistic to cytokines. Although competing with target receptors for binding cytokines, these Abs do not actually affect the local reactions under physiologic conditions. This conclusion is documented by in vivo experiments (21, 22). Anti-epidermal growth factor (EGF)-immunized adult rats were unaffected by the induction and presence of high-affinity autoAbs, and the EGF-containing organs did not show any histologic signs of inflammation or tissue damage (21). Also, high affinity neutralizing anti-IL-1 autoAbs elicited by active immunization proved to be fully innocuous in mice (22).
Free Abs are renewed poorly in the stromal compartment of a tissue under physiologic conditions because of the negligible lymph turnover flow, and they do not exercise any effect on cytokine processes because of the negative feedback control of cytokine production, which is turned off when the target-cell reaction has been effective (Fig. 1). As discussed in Turnover Rates and Binding Affinity of Target Receptors vs. Lymph Abs, the poorly renewed lymph Abs, by binding cytokines (Fig. 1A), may minimally delay (a few minutes) but do not hamper the homeostatically regulated cytokine processes that last for hours (5). This also seems to be the case for high-affinity Abs (Fig. 1B) that were raised after vaccination (21, 22).
Turnover Rates and Binding Affinity of Target Receptors vs. Lymph Abs.
The competition between receptors (R) and Abs for binding the same cytokine (C) in the ECM microenvironment is governed by the mass action law according to the following double equilibrium:
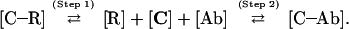 |
1 |
During a cytokine process, the equilibrium (step 1) [C–R] ⇄ [R] + [C] at the target membrane side is likely to be displaced toward C–R complex formation because of the rapid turnover of free receptors and the rapid internalization of C–R complexes. On the other hand, the equilibrium (step 2) [C] + [Ab] ⇄ [C–Ab] is likely reached in minutes and should change only slightly because of the slow turnover of the scarce lymph moistening of the ECM (Fig. 1). As a consequence of the different turnover rates of receptors and Abs, the equilibrium is shifted to the left along the course of the cytokine reaction, favoring cytokine binding to membrane receptors.
The equilibrium shift to C–R complexes is promoted further by the higher cytokine-binding affinity of receptors compared with that of Abs. During the course of an immunization, the dissociation constant (Kd) of Abs, as measured by immunoprecipitation of radiolabeled antigen, fluorescence transfer, or ELISA, varies from 10−7 to 10−10 M (23, 24), whereas that of receptors during cytokine processes varies from 10−10 to 10−12 M (5, 25–33). More precisely, the Kd of high-affinity receptors are in the range of 1–10 pM for vascular endothelial growth factor (25), 10–50 pM for IL-2 (26), IL-12 (27), TNF-α (28), IL-6 (29) and transforming growth factor (TGF)-β (30), and 100–500 pM for IL-1 (5), IL-4 (31), IL-10 (32), and IFN-α (33), vs. 100–100,000 pM for Abs (23, 24).
Dysregulation of Cytokine Production
Cytokines may be produced continuously when regulatory feedback circuits are not functioning in E cells, as may occur in cancer- or virus-infected cells, when genetic changes vicariously turn on cytokine-promoter genes (Fig. 2). For example, the immunosuppressive TGF-β cytokine is expressed abnormally by freshly isolated cells (34–36) and cell lines (37, 38) from tumors including ovarian cancer and uterine cervix carcinoma. The potentially immunosuppressive IL-10 is overexpressed likewise by freshly isolated cells (36, 39) and cell lines (40) from various cancers. The proinflammatory IL-6 is produced aberrantly by fresh tumor cells also (41, 42). Fas ligand is produced by isolated primary cells from human colon, lung, and esophageal cancers (43–45). Finally, angiogenic vascular endothelial growth factor is released in culture supernatants of freshly isolated primary cells from malignant tumors (46–48). In patients infected with HIV-1, fresh cells from Kaposi sarcoma tumors overexpress proinflammatory cytokines including IL-6, IL-1, TNF-α, and IFN-γ (49, 50).
Figure 2.

Effects of vaccination against immunosuppressive cytokines on tumor cell growth. Cancer cells may constitutively release immunosuppressive cytokines (●), including TGF-β (34–36) or IL-10 (39), which accumulate in the extracellular compartment. (A) Natural Abs (Y) present in ECM cannot contain the accumulated cytokine (no. 1), stromal T lymphocytes are anergized, (no. 2), and cancer cell growth occurs (no. 3). (B) High-titer and high-affinity Abs (YY), triggered by an appropriate anti-cytokine vaccine, neutralize the suppressive signal and block its effects (no. 1). T lymphocytes may be activated spontaneously by either a tumor cell antigen or better by a conventional anti-tumor vaccine (no. 2). As a result, effector cytotoxic T lymphocytes (CTLs) directed against cancer cells control tumor growth (no. 3).
Continuous release of a cytokine in the absence of controlled regulation results in a flood of the signal molecule and its subsequent accumulation in the ECM (Fig. 2). Under impaired negative feedback, several cytokines [including IFN-α (51)] that are barely detectable in normal serum given their short half-lives, untimely may be overproduced by a tissue or a tumor and found at abnormally high concentrations in the serum. This is the case met with patients with AIDS in whom HIV-1-induced dysregulation of IFN-α production (52) is associated with high titers of the circulating cytokine (53).
The dysregulated cytokine overproduction may lead to a cytokine network homeostatic imbalance in the local concerned tissue, resulting in absence or excess of other regulatory signals. This seems to be the case in various pathologies including AIDS and cancer. Uncontrolled release by cancer cells of signaling factors could untimely activate immune and/or endothelial stromal cells in the malignant tissue. The resulting induction of undesirable cytokines by these stromal cells may locally impair the cytokine network in the tumor and favor the establishment of immune suppression and neoangiogenesis, features characterizing the cancer cell's microenvironment. Angiogenic processes may be triggered by vascular endothelial growth factor (54) and by inflammatory cytokines (TNF-α, IL-1, IL-6, or IFN-γ) that are overproduced by cancer or stromal cells in malignant tumors (55, 56). Overproduction of TGF-β by tumor cells (34–38) may result in local T cell anergy and inhibition of cellular immune response, i.e., immune escape of cancer cells (Fig. 2A). Also, apoptosis of immune T cells occurs as a consequence of the abnormal production of TNF-α or Fas ligand (57) from malignant tumors, which may diminish further the immune control of the tumor cells.
Anti-Cytokine Abs and Cytokine Processes in Pathologic Tissues
Considering their low levels and the inability of organisms to mount an effective anti-cytokine immune response under natural (nonimmunized) conditions (see Natural Anti-Cytokine Abs), circulating anti-cytokine autoAbs seem to play a negligible role in the regulation of cytokine overproduction. However, vaccination may induce higher levels of long-lasting circulating Abs that, under pathophysiologic conditions consisting of an ectopic accumulation of cytokines in the extracellular compartment, could effectively bind the corresponding cytokine and counteract the pathogenic effects of its overproduction or transiently hamper the composite reaction it induces.
Inhibition by Anti-Cytokine Abs of the Pathogenic Effects of Accumulated Cytokine Released from Cells Deprived of Negative Feedback Regulation.
When homeostatic negative regulation is impaired or absent, the cytokine may be produced continuously and released locally, as if its tap remained open. The “flood” resulting from this overproduction and the accumulation of the cytokine in the ECM compartment may trigger inappropriate stimulation of nearby immune or endothelial stromal cells, resulting in local pathophysiologic changes in the concerned tissue or tumor as depicted for cancer cells secreting IL-10 (39) or TGF-β (refs. 34–36; Fig. 2A). In this instance, high titers of specific anti-cytokine Abs induced by effective immunization (see Safe and Effective Anti-Cytokine Therapeutic Vaccination) should neutralize dysregulated cytokine overproduction locally and inhibit its pathogenic effects (Fig. 2B).
Transient Inhibition by Specific Abs of a Cytokine Process Occurring in Pathologic Tissues Containing Accumulated Extracellular Lymph.
Microbial infection of a tissue may trigger an immune reaction characterized first by a local inflammation leading to a flooding of body fluid (lymph) and its accumulation in the extracellular compartment (edema), followed by a specific immune response. Both processes depend on cytokines and the reactions these signals induce (3). After passive anti-cytokine Ab administration, the lymph accumulated locally in the ECM as a result of inflammation is likely to represent a permanent source that continuously supplies high-affinity Abs. These Abs are capable of binding to and removing the cytokine, thus inhibiting the cytokine process that involves the target-cell response and its subsequent negative feedback. The studies of Hondowicz et al. (6, 58) and of Constantinescu et al. (59) on mice infected with L. major support these concepts.
C3H mice infected in the footpad with L. major resolve their lesions by induction of a TH1 immune response. The specific TH1 response resulted from the cytokine process triggered on antigen-activated CD4 targets by the release of IL-12 from natural killer cells. By contrast, infected BALB/c mice, which display a TH2 response to L. major (see Cytokine Process: Cytokine Production, Effects, and Regulation; Step 2: Cytokine–Receptor (C–R) Binding and Target-Cell Response), fail to heal and ultimately succumb to infection after a few weeks (6, 58, 59). When massive administration of high-affinity anti-IL-12 Abs were administered repeatedly for 3 weeks to infected C3H mice, the inflammatory process increased because of the absence of a TH1 response and the footpad lesion developed, whereas in infected BALB/c mice, the IL-12-induced reaction in antigen-activated CD4 targets leading to a TH1-effective response could not occur for genetic reasons (6). In Ab-treated C3H mice, the IL-12-triggered process was blocked as long as massive anti-IL-12 Abs of high affinity (as a result of repeated administration) were loading the lymphatic fluid and interfered locally with the binding of IL-12 to its specific receptors on CD4 cell targets. However, after interruption of anti-IL-12 Ab injections, the mice immune reaction switched from a TH2 to TH1 response with IFN-γ production (59), and the footpad lesion healed and resistance to L. major occurred albeit with delay.
These experiments showed that lymph Abs directed against a given cytokine transiently interfere with the cytokine reaction under a specific histopathologic microenvironment, namely one with accumulated lymph. Here, the lymph acts as a reservoir to deliver continuously free Abs that entrap, crossreact with, and remove the released cytokine. In contrast, under physiologic conditions, lymph Abs rapidly convert into immune complexes but are renewed only negligibly (see Reasons for the Negligible Effects of Anti-Cytokine Abs on Cytokine Processes Under Physiologic Conditions) and do not interfere with cytokine processes, as observed experimentally for anti-Il-1 mice (22) and for anti-EGF-immunized rats (21, 60).
Of interest, anti-EGF- (60) and anti-nerve growth factor (NGF; refs. 61 and 62)-immunized female rats were mated with nonimmunized males. Although immunized mothers remained healthy, the offspring exhibited teratogenic tissue lesions (60–62). Thus, in developing offspring of anti-NGF-immunized female rats, maternal Abs of high affinity could neutralize NGFs acting on immature neurons (63) and hamper nerve tissue development (62).
Safe and Effective Anti-Cytokine Therapeutic Vaccination
Vaccination Objectives.
The objective of an anti-cytokine therapeutic vaccine is to raise Ab levels over the background levels of natural Abs while enhancing their affinity and neutralizing activity. Thus, in altered tissue or in a tumor, neutralizing Abs may block the accumulation of an overproduced cytokine, thereby inhibiting its pathogenic effects. An anti-cytokine T cell response is neither required nor likely beneficial for this purpose, and it is not expected because in contrast to still-existing anti-cytokine B cells, T cells have undergone a thymic-negative clonal selection against self-cytokines.
Vaccination Procedures.
To mount a high-affinity autoAb response against self cytokines in patients, two immunizing procedures are currently available to break immunological tolerance (22). The first procedure can be applied to subjects exhibiting B cell polyclonal activation, as is the case of patients infected with HIV. B cell polyclonal activation also can be achieved experimentally after (bacterial) lipopolysaccharide (LPS) treatment (5). The vaccine consists of an inactivated but immunogenic cytokine. Absence of toxicity may be obtained by various cytokine-inactivation procedures such as the chemical formaldehyde treatment similar to that used for converting bacterial toxins into toxoids (64). These treatments have the advantage of stabilizing the antigen. An example is the formalinated human IFN-α vaccine that has been used safely in patients infected with HIV-1 (65–67). In the second procedure, the nontoxic immunogenic cytokine is coupled chemically to a foreign TH protein such as tetanus toxoid, purified protein derivative (PPD), or keyhole limpet hemocyanin (KLH), to promote the T-dependent B cell response. This procedure has been applied successfully to the preparations of anti-TNF-α (68, 69), anti-IL-9 (70), anti-EGF (71), and anti-IL-1 (22) vaccines. Taking into account the relative short lives of the memory B cells (72) activated through either of these two immunizing procedures, boosting 2–4 times per year may be necessary to maintain substantial high-affinity Ab levels (66, 67).
Animal Experimentation.
Over the last decade, anti-cytokine vaccinations (both passive and active) have been performed to combat different pathogenic disorders originating from cytokine dysregulation (Table 1 Animal experimentation). Although massive doses of specific Abs (mg) have been administered, in one inoculation no complications related to inhibition of the corresponding cytokine process have been reported, and disease improvement was observed generally. As an example, Sadick et al. (73) succeeded in curing L. major with passive administration of anti-IL-4 mAbs to sensitive mice. Also, active immunization with an anti-IL-9 vaccine eliciting high titer of specific Abs proved to be safe and effective in preventing worm expulsion and blood eosinophilia in Trichuris muris-infected mice (70).
Table 1.
Anticytokine therapeutic vaccination
| Cytokine target | Vaccine procedure | Preparation | Therapeutic indication | Refs. |
|---|---|---|---|---|
| Animal experimentation | ||||
| EGF | Active | EGF in CFA | Cancers | 21 |
| IL-1 | Active | PPD–IL-1α | Inflammation | 22 |
| TNF-α | Active | OVA–peptide–TNF-α | Murine polyarthritis | 68, 69 |
| IL-9 | Active | OVA–IL-9 | Murine parasitasis and eosinophilia | 70 |
| IL-4 | Passive | Murine mAb | Allergy to L. major | 73 |
| IL-12 | Passive | Murine mAbs | Colitis | 74 |
| TGF-β | Passive | Monospecific rabbit Abs | Immune-induced lung fibrosis | 75 |
| Clinical trials | ||||
| IFN-α | Active | Inactivated IFN-α | AIDS | 65–67 |
| EGF | Active | Tetanus toxoid–EGF | Colon, lung, stomach, and prostate cancers | 71 |
| IL-4 | Passive | Humanized mAb | Allergy | 76 |
| IL-6 | Passive | mAb | Castleman disease | 77 |
| IL-8 | Passive | Humanized mAb | Psoriasis | 78 |
| TNF-α | Passive | Human, humanized, chimeric mAb | Rheumatoid arthritis, Crohn's disease | 78 |
| VEGF | Passive | Humanized mAb | Colorectal cancer | 78 |
| TGF-β2 | Passive | Human Ab | Glaucoma | 78 |
VEGF, vascular endothelial growth factor.
Human Trials.
Passive and active vaccinations also have been performed in humans (Table 1 Clinical trials). Passive immunization used human Abs or humanized mAbs for treatment of allergy (76), cancers (77, 78), rheumatoid arthritis, and autoimmune diseases including psoriasis and Crohn's disease (78). Here again, massive doses of Abs in the range of milligrams were administered repeatedly without undesirable reactions and were shown to be beneficial. A limitation of passive immunization is the possibility of production of anti-anti-cytokine Abs as a result of repeated injections of heterogenous Abs with risk of disease recurrence. Further limitation may result from restricted affinity of presently available humanized mAbs. Treatment of Castleman disease with anti-IL-6 Abs was transiently successful, but in this study, recurrence occurred after a few boosting injections (77). Active immunization against anti-IFN-α (65, 66) and EGF (71) phase I trials, respectively, carried out in subjects infected with HIV-1 and in patients with malignant carcinomas including colon, lung, and prostate cancer, provided evidence for compliance, safety, and immunogenicity of the preparation. The immunogens used were formaldehyde-inactivated IFN-α and EGF coupled to tetanus toxoid and led to a dramatic rise of specific Abs. Further, an anti-IFN-α multicentric double-blind trial (European/Israeli II/III) was carried out in HIV-1-infected patients with or without retroviral therapy in Italy, Belgium, and Israel. The trial confirmed safety and immunogenicity of the previous anti-IFN-α vaccine trials and proved to be significantly beneficial to the vaccines compared with placebo-treated patients (67).
Conclusions
In this report, we provide rationale, feasibility, and therapeutic use for active immunization against cytokines. We stressed that cytokines released from homeostatically dysregulated effector cells, accumulating in the extracellular compartment of an abnormal tissue or a tumor, can be neutralized by specific Abs and their pathogenic effects can be counteracted. These basic considerations are supported by animal and human vaccine trials that trigger anti-cytokine B but not T cell memory response, thus exhibiting high Ab titers over a limited period [around 12 weeks (72)]. Vaccine effects being transient, boosting injections have to be repeated. Anti-cytokine vaccinations seemed to be safe, devoid of side effects in normal tissues, and effective, as long as high Ab levels were maintained. These data together with the lack of problems in patient compliance (66, 67) prompt their extended use in human trials to combat cytokine-induced immunopathogenesis. Anti-cytokine vaccines, however, are contraindicated in pregnant women because of a risk of teratogenic lesions in the offspring (60–62).
Current vaccines have shown limitations in their use for diseases such as AIDS (65–67), allergies (76), and cancer (71, 78). In these diseases, investigators have targeted exclusively the antigenic aggressor, be it a microbe, a cell, or an allergen, but not the cytokine immune dysregulation that the aggressor triggers. We think that the cytokine dysregulation contributes to the impairment of the vaccine immune reaction. In particular, current cancer vaccines do not combat immunosuppression surrounding cancer cells in malignant tumors; AIDS vaccines do not as yet counter the HIV-1-induced cellular immunosuppression, and specific vaccines against allergic disorders do not control the immune dysregulation induced by the allergens. We strategize that in all these instances, additional anti-cytokine vaccination may allow a conventional vaccine to mount an adequate immune reaction against the antigenic aggressor.
To have high-affinity Abs neutralizing an overproduced cytokine in patients presenting these chronic diseases, both vaccine and Ab therapy might be used. Given their ephemeral effects (72), vaccines will require repeated booster injections (2–4 per year) to be effective, whereas Ab therapy, lacking B cell memory, may be limited by (i) the needs for massive Ab administration at a time (which could generate transient undesirable reactions, local and systemic); (ii) short duration of the protective effects because of a lack of immune B cell memory, thereby requiring frequent repetition of administration; and (iii) the use either of humanized Abs, which are usually of low affinity, or of heterologous Abs, whose repeated administration will likely result in an antiAb response (79). Nevertheless, in a combined passive and active vaccination, the initial administration of specific Abs should confer an immediate helpful effect. Further, a prior passive immunization could be used as a tool to assess whether anti-cytokine vaccine is beneficial in the treatment of a chronic disease, including AIDS, cancer, autoimmunity, or allergy.
Abbreviations
- ECM
extracellular matrix
- C–R binding
cytokine–receptor binding
- TNF
tumor necrosis factor
- EGF
epidermal growth factor
- E
effector
- TH
T cell helper
- TGF
transforming growth factor
References
- 1.Cohen S, Bigazzi P E, Yoshida T. Cell Immunol. 1974;12:150–159. doi: 10.1016/0008-8749(74)90066-5. [DOI] [PubMed] [Google Scholar]
- 2.Aarden L A. J Immunol. 1979;123:2298–2299. [Google Scholar]
- 3.Thomas A, editor. The Cytokine Handbook. 3rd Ed. San Diego-London: Academic; 1998. pp. 1–20. [Google Scholar]
- 4.Zagury D, Uhr J W, Jamieson J D, Palade G E. J Cell Biol. 1970;46:52–63. doi: 10.1083/jcb.46.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chensue S W, Terebuh P D, Remick D G, Scales W E, Kunkel S L. Am J Pathol. 1991;13:395–402. [PMC free article] [PubMed] [Google Scholar]
- 6.Hondowicz B D, Scharton-Kersten T M, Jones D E, Scott P. J Immunol. 1997;159:5024–5031. [PubMed] [Google Scholar]
- 7.Zhang Y, Pilon G, Marette A, Baracos V E. Am J Physiol. 2000;279:E196–E205. doi: 10.1152/ajpendo.2000.279.1.E196. [DOI] [PubMed] [Google Scholar]
- 8.Grenfell S J, Smithers N, Solari R. Cytokine. 1992;4:114–124. doi: 10.1016/1043-4666(92)90046-t. [DOI] [PubMed] [Google Scholar]
- 9.Chany C. J Interferon Res. 1990;10:453–459. doi: 10.1089/jir.1990.10.453. [DOI] [PubMed] [Google Scholar]
- 10.Knolle P A, Uhrig A, Protzer U, Trippler M, Duchmann R, Meyer zum Buschenfelde K H, Gerken G. Hepatology. 1998;27:93–99. doi: 10.1002/hep.510270116. [DOI] [PubMed] [Google Scholar]
- 11.Bonder C S, Finlay-Jones J J, Hart P H. Immunology. 1999;96:529–536. doi: 10.1046/j.1365-2567.1999.00711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeKruyff R H, Fang Y, Wolf S F, Umetsu D T. J Immunol. 1995;154:2578–2587. [PubMed] [Google Scholar]
- 13.Fernandez-Botran R. Crit Rev Clin Lab Sci. 1999;36:165–224. doi: 10.1080/10408369991239196. [DOI] [PubMed] [Google Scholar]
- 14.Arend W P. J Clin Invest. 1991;88:1445–1451. doi: 10.1172/JCI115453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross C, Hansen M B, Schyberg T, Berg K. Clin Exp Immunol. 1990;82:57–62. doi: 10.1111/j.1365-2249.1990.tb05403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svenson M, Hansen M B, Ross C, Diamant M, Rieneck K, Nielsen H, Bendtzen K. Blood. 1998;91:2054–2061. [PubMed] [Google Scholar]
- 17.Caruso A, Turano A. Biotherapy. 1997;10:29–37. doi: 10.1007/BF02678215. [DOI] [PubMed] [Google Scholar]
- 18.Theofilopoulos A N. Immunol Today. 1995;16:90–98. doi: 10.1016/0167-5699(95)80095-6. [DOI] [PubMed] [Google Scholar]
- 19.Bonino F, Baldi M, Negro F, Oliveri F, Colombatto P, Bellati G, Brunetto M R. J Interferon Cytokine Res. 1997;17, Suppl 1:35–38. [PubMed] [Google Scholar]
- 20.Fall L S, Chams V, Le Coq H, Fouchard M, M'Bika J-P, Gringeri A, Santagostino E, Bizzini B. Cell Mol Biol. 1995;41:409–416. [PubMed] [Google Scholar]
- 21.Raaberg L, Nexo E, Poulsen S S, Jorgensen P E. Pediatr Res. 1995;37:169–174. doi: 10.1203/00006450-199502000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Svenson M, Hansen M B, Thomsen A R, Diamant M, Nansen A, Rieneck K, Otterness I G, Bendtzen K. J Immunol Methods. 2000;236:1–8. doi: 10.1016/s0022-1759(99)00232-x. [DOI] [PubMed] [Google Scholar]
- 23.Foote J, Eisen H N. Proc Natl Acad Sci USA. 1995;92:1254–1256. doi: 10.1073/pnas.92.5.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friguet B, Chaffotte A F, Djavadi-Ohaniance L, Goldberg M E. J Immunol Methods. 1985;77:305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 25.Vaisman N, Gospodarowicz D, Neufeld G. J Biol Chem. 1990;265:19461–19466. [PubMed] [Google Scholar]
- 26.Minamoto S, Itoh S, Kono T, Doi T, Hatakeyama M. Immunol Lett. 1989;20:139–147. doi: 10.1016/0165-2478(89)90099-0. [DOI] [PubMed] [Google Scholar]
- 27.Chizzonite R, Truitt T, Desai B B, Nunes P, Podlaski F J, Stern A S. J Immunol. 1992;148:3117–3124. [PubMed] [Google Scholar]
- 28.Grell M, Wajant H, Zimmermann G, Scheurich P. Proc Natl Acad Sci USA. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi M, Tanaka J, Imamura M, Maeda S, Iwasaki H, Tanaka M, Tsudu Y, Sakurada K, Miyazaki T. Br J Haematol. 1993;83:535–538. doi: 10.1111/j.1365-2141.1993.tb04687.x. [DOI] [PubMed] [Google Scholar]
- 30.Tripathi R C, Borisuth N S, Kolli S P, Tripathi B J. Invest Ophthalmol Visual Sci. 1993;34:260–263. [PubMed] [Google Scholar]
- 31.Obiri N I, Siegel J P, Varricchio F, Puri R K. Clin Exp Immunol. 1994;95:148–155. doi: 10.1111/j.1365-2249.1994.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurlander J, Lai C F, Tan J, Chou C C, Geisler C H, Schriber J, Blumenson L E, Narula S K, Baumann H, Caligiuri M A. Blood. 1997;89:4146–4152. [PubMed] [Google Scholar]
- 33.Lau J Y, Sheron N, Morris A G, Bomford A B, Alexander G J, Williams R. Hepatology. 1991;13:332–338. [PubMed] [Google Scholar]
- 34.Santin A D, Hiserodt J C, DiSaia P J, Pecorelli S, Granger G A. Gynecol Oncol. 1996;61:403–408. doi: 10.1006/gyno.1996.0164. [DOI] [PubMed] [Google Scholar]
- 35.Santin A D, Hermonat P L, Hiserodt J C, Fruehauf J, Schranz V, Barclay D, Pecorelli S, Parham G P. Gynecol Oncol. 1997;64:477–480. doi: 10.1006/gyno.1996.4579. [DOI] [PubMed] [Google Scholar]
- 36.Karcher J, Reisser C, Daniel V, Herold-Mende C. HNO. 1999;47:879–884. doi: 10.1007/s001060050528. [DOI] [PubMed] [Google Scholar]
- 37.Bequet-Romero M, Lopez-Ocejo O. Biochem Biophys Res Commun. 2000;277:55–61. doi: 10.1006/bbrc.2000.3628. [DOI] [PubMed] [Google Scholar]
- 38.Teicher B A, Kakeji Y, Ara G, Herbst R S, Northey D. In Vivo. 1997;11:453–461. [PubMed] [Google Scholar]
- 39.Nakagomi H, Pisa P, Pisa E K, Yamamoto Y, Halapi E, Backlin K, Juhlin C, Kiessling R. Int J Cancer. 1995;63:366–371. doi: 10.1002/ijc.2910630311. [DOI] [PubMed] [Google Scholar]
- 40.Gastl G A, Abrams J S, Nanus D M, Oosterkamp R, Silver J, Liu F, Chen M, Albino A P, Bander N H. Int J Cancer. 1993;55:96–101. doi: 10.1002/ijc.2910550118. [DOI] [PubMed] [Google Scholar]
- 41.Woods K V, El-Naggar A, Clayman G L, Grimm E A. Cancer Res. 1998;58:3132–3141. [PubMed] [Google Scholar]
- 42.Kyo S, Kanaya T, Takakura M, Inoue M. Gynecol Oncol. 2000;78:383–387. doi: 10.1006/gyno.2000.5904. [DOI] [PubMed] [Google Scholar]
- 43.Shiraki K, Tsuji N, Shioda T, Isselbacher K J, Takahashi H. Proc Natl Acad Sci USA. 1997;94:6420–6425. doi: 10.1073/pnas.94.12.6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niehans G A, Brunner T, Frizelle S P, Liston J C, Salerno C T, Knapp D J, Green D R, Kratzke R A. Cancer Res. 1997;57:1007–1012. [PubMed] [Google Scholar]
- 45.Bennett M W, O'Connell J, O'Sullivan G C, Brady C, Roche D, Collins J K, Shanahan F. J Immunol. 1998;160:5669–5675. [PubMed] [Google Scholar]
- 46.Hussong J W, Rodgers G M, Shami P J. Blood. 2000;95:309–313. [PubMed] [Google Scholar]
- 47.Akagi K, Ikeda Y, Miyazaki M, Abe T, Kinoshita J, Maehara Y, Sigumachi K. Br J Cancer. 2000;83:887–891. doi: 10.1054/bjoc.2000.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park Y N, Kim Y B, Yang K M, Park C. Arch Pathol Lab Med. 2000;124:1061–1065. doi: 10.5858/2000-124-1061-IEOVEG. [DOI] [PubMed] [Google Scholar]
- 49.Ensoli B, Nakamura S, Salahuddin S Z, Biberfeld P, Larsson L, Beaver B, Wong-Staal F, Gallo R C. Science. 1989;243:223–226. doi: 10.1126/science.2643161. [DOI] [PubMed] [Google Scholar]
- 50.Samaniego F, Markham P D, Gendelman R, Watanabe Y, Kao V, Kowalski K, Sonnabend J A, Pintus A, Gallo R C, Ensoli B. Am J Pathol. 1998;152:1433–1443. [PMC free article] [PubMed] [Google Scholar]
- 51.Ambrus J L, Poiesz B J, Lillie M A, Stadler I, Di Berardino L A, Chadha K C. Am J Med. 1989;87:405–407. doi: 10.1016/s0002-9343(89)80822-8. [DOI] [PubMed] [Google Scholar]
- 52.Popik W, Pitha P M. Virology. 1992;189:435–447. doi: 10.1016/0042-6822(92)90567-9. [DOI] [PubMed] [Google Scholar]
- 53.Francis M L, Meltzer M S, Gendelman H E. AIDS Res Hum Retroviruses. 1992;8:199–207. doi: 10.1089/aid.1992.8.199. [DOI] [PubMed] [Google Scholar]
- 54.Martiny-Baron G, Marme D. Curr Opin Biotechnol. 1995;6:675–680. doi: 10.1016/0958-1669(95)80111-1. [DOI] [PubMed] [Google Scholar]
- 55.Folkman J. Semin Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 56.Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. J Leukocyte Biol. 1994;55:410–422. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- 57.Owen-Schaub L, Chan H, Cusack J C, Roth J, Hill L L. Int J Oncol. 2000;17:5–12. [PubMed] [Google Scholar]
- 58.Hondowicz B D, Park A Y, Elloso M M, Scott P. Eur J Immunol. 2000;30:2007–2014. doi: 10.1002/1521-4141(200007)30:7<2007::AID-IMMU2007>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 59.Constantinescu C S, Hondowicz B D, Elloso M M, Wysocka M, Trinchieri G, Scott P. Eur J Immunol. 1998;28:2227–2233. doi: 10.1002/(SICI)1521-4141(199807)28:07<2227::AID-IMMU2227>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 60.Raaberg L, Nexo E, Jorgensen P E, Poulsen S S, Jakab M. Pediatr Res. 1995;37:175–181. doi: 10.1203/00006450-199502000-00009. [DOI] [PubMed] [Google Scholar]
- 61.Gorin P D, Johnson E M. Proc Natl Acad Sci USA. 1979;76:5382–5386. doi: 10.1073/pnas.76.10.5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson E M, Gorin P D, Brandeis L D, Pearson J. Science. 1980;210:916–918. doi: 10.1126/science.7192014. [DOI] [PubMed] [Google Scholar]
- 63.Müller W A. Developmental Biology. New York: Springer; 1996. pp. 248–249. [Google Scholar]
- 64.Ramon G. Compt Rend Hebdomadaires Seances Acad Sci. 1923;177:1338–1340. [Google Scholar]
- 65.Zagury D, Bernard J, Halbreich A, Bizzini B, Carelli C, Achour A, Defer M C, Bertho J M, Lanneval K, Zagury J F, et al. J Acquired Immune Defic Syndr. 1992;5:676–681. [PubMed] [Google Scholar]
- 66.Gringeri A, Santagostino E, Mannucci P M, Tradati F, Cultraro D, Buzzi A, Criscuolo M, David A, Guillemot L, Barre-Sinoussi F, et al. J Acquired Immune Defic Syndr. 1994;7:978–988. [PubMed] [Google Scholar]
- 67.Gringeri A, Musicco M, Hermans P, Bentwich Z, Cusini M, Bergamasco A, Santagostino E, Burny A, Bizzini B, Zagury D. J Acquired Immune Defic Syndr. 1999;20:358–370. doi: 10.1097/00042560-199904010-00006. [DOI] [PubMed] [Google Scholar]
- 68.Dalum I, Butler D M, Jensen M R, Hindersson P, Steinaa L, Waterston A M, Grell S N, Feldmann M, Elsner H I, Mouritsen S. Nat Biotechnol. 1999;17:666–669. doi: 10.1038/10878. [DOI] [PubMed] [Google Scholar]
- 69.Dalum I, Jensen M R, Hindersson P, Elsner H I, Mouritsen S. J Immunol. 1996;157:4796–4804. [PubMed] [Google Scholar]
- 70.Richard M, Grencis R K, Humphreys N E, Renauld J C, Van Snick J. Proc Natl Acad Sci USA. 2000;97:767–772. doi: 10.1073/pnas.97.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonzalez G, Crombet T, Catala M, Mirabal V, Hernandez J C, Gonzalez Y, Marinello P, Guillen G, Lage A. Ann Oncol. 1998;9:431–435. doi: 10.1023/a:1008261031034. [DOI] [PubMed] [Google Scholar]
- 72.Gray D, Skarvall H. Nature (London) 1988;336:70–73. doi: 10.1038/336070a0. [DOI] [PubMed] [Google Scholar]
- 73.Sadick M D, Heinzel F P, Holaday B J, Pu R T, Dawkins R S, Locksley R M. J Exp Med. 1990;171:115–127. doi: 10.1084/jem.171.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fuss I J, Marth T, Neurath M F, Pearlstein G R, Jain A, Strober W. Gastroenterology. 1999;117:1078–1088. doi: 10.1016/s0016-5085(99)70392-6. [DOI] [PubMed] [Google Scholar]
- 75.Denis M. Immunology. 1994;82:584–590. [PMC free article] [PubMed] [Google Scholar]
- 76.Heusser C. Arb Paul Ehrlich Inst Bundesamt Sera Impfstoffe Frankfurt A M. 1994;87:283–291. [PubMed] [Google Scholar]
- 77.Beck J T, Hsu S M, Wijdenes J, Bataille R, Klein B, Vesole D, Hayden K, Jagannath S, Barlogie B. N Engl J Med. 1994;330:602–605. doi: 10.1056/NEJM199403033300904. [DOI] [PubMed] [Google Scholar]
- 78.Glennie M J, Johnson P W. Immunol Today. 2000;21:403–410. doi: 10.1016/s0167-5699(00)01669-8. [DOI] [PubMed] [Google Scholar]
- 79.Clark M. Immunol Today. 2000;21:397–402. doi: 10.1016/s0167-5699(00)01680-7. [DOI] [PubMed] [Google Scholar]