

Abstract
The 26S proteasome is a 2.5 MDa, 32-subunit ATP-dependent protease that is responsible for the degradation of ubiquitinated protein targets in all eukaryotic cells. This proteolytic machine consists of a barrel-shaped peptidase capped by a large regulatory particle, which contains a heterohexameric AAA+ unfoldase as well as several structural modules of previously unknown function. Recent electron microscopy studies have allowed major breakthroughs in understanding the architecture of the regulatory particle, revealing that the additional modules provide a structural framework to position critical, ubiquitin-interacting subunits and thus allow the 26S proteasome to function as a universal degradation machine for a wide variety of protein substrates. The electron microscopy studies have also uncovered surprising asymmetries in the spatial arrangement of proteasome subunits, yet the functional significance of these architectural features remains unclear. This review will summarize the recent findings on 26S proteasome structure and discuss the mechanistic implications for substrate binding, deubiquitination, unfolding, and degradation.
Introduction
Energy-dependent protein degradation is required for the specific and irreversible removal of critical regulatory proteins, as well as the general control of protein content and quality in the cell. This highly regulated form of proteolysis is performed by ATP-dependent proteases, a family of multi-subunit protein complexes that share the common architecture of a barrel-shaped compartmental peptidase topped by a hexameric AAA+ unfoldase (ATPases Associated with various cellular Activities) 1.
Protein peptide bonds are metastable. Their hydrolysis is exergonic and can be accelerated by peptidases, but in folded proteins their exposure to solvent or proteolytic active sites is strongly restricted by higher-order structures. ATP-dependent proteases utilize the input of energy to make their protein substrates selectively accessible for degradation by mechanically unfolding and translocating them into a self-compartmentalized degradation chamber. The compartmental peptidases of these proteases are formed from two or four stacked rings of six or seven subunits, with proteolytic active sites sequestered on the internal surface of the barrel-shaped cavity. Access to this cavity is restricted by narrow axial pores that are only wide enough to accommodate an unfolded polypeptide chain 2. Protein substrates must therefore be threaded into this proteolytic chamber by AAA+ unfoldases that bind and gate the entryway. These unfoldases are hexameric rings of ATPase subunits, which consist of a small and a large AAA+ sub-domain and contain conserved loops that line the central pore of the ring. The loops directly contact the substrate protein, transduce the ATP-hydrolysis-induced conformational changes of individual ATPase subunits, and thereby exert a pulling force to unfold and translocate the substrate through the narrow central pore and into the peptidase 3, 4, 5. Each subunit in the hexamer can communicate with its neighbor, allowing the ring to coordinate activities and work as a single motor 6, 7, 8.
Prokaryotes and eukaryotes have evolved different approaches to meet the proteolytic requirements of the cell. Prokaryotes, and bacteria in particular, contain many different ATPdependent proteases that recognize distinct sets of substrate targeting signals and thus regulate different cellular processes 9, 10. These targeting signals are typically short peptide sequences that are integral parts of the substrate polypeptide. They can either be recognized directly by the unfoldase or first interact with adaptor proteins that subsequently bind the unfoldase and deliver substrates for degradation 9, 10. The bacterial ClpXP protease, for example, recognizes the so-called ssrA tag, an 11-amino-acid sequence that is added to the C-terminus of incompletely translated proteins at stalled ribosomes. The last three amino acids of this tag bind directly to the ClpX unfoldase pore, but the starvation-induced adaptor protein SspB can enhance substrate affinity by interacting with both ClpX and the N-terminal portion of the tag 11. Another bacterial protease, ClpAP, collaborates with the adaptor protein ClpS to identify N-end rule substrates through their N-terminal amino acid 12. And the protease Lon performs its role in bacterial protein quality control by recognizing specific sequences that are rich in aromatic amino acids and become exposed from the hydrophobic core when a protein is damaged or unfolded. An exception from the general rule of integral targeting signals in bacteria is the Pup system, which is present in the actinomycetes branch (including Mycobacterium tuberculosis, the infectious pathogen of tuberculosis) as the result of horizontal gene transfer from eukaryotes. In this system, post-translational modification of substrates with the small protein Pup (prokaryotic ubiquitin-like protein) targets them for degradation by the homomeric unfoldase Mpa bound to a compartmental peptidase 13, 14. However, besides this rare Pup system, the many different bacterial ATP-dependent proteases recognize a variety of substrate targeting signals that are typically included in the primary sequence of substrate proteins.
Energy-dependent protein degradation in archaea is catalyzed by Lon proteases and the PAN (proteasome-activating nucleotidase)-20S or Cdc48-20S proteasomes 15, 16, 17, but the degradation signals for these proteases are still poorly understood 18. Similar to ubiquitin in eukaryotes, small archaeal modifier proteins (SAMPs) have been found attached to proteins, but whether they function as targeting signals for proteasomal degradation has yet to be determined 19, 20. Interestingly, in-vitro studies using non-endogenous, bacterial sequences have shown that the archaeal proteasomes can efficiently recognize intrinsic substrate targeting signals, similar to their bacterial counterparts 21.
Eukaryotes, on the other hand, have evolved to rely on only one major cytosolic ATP-dependent protease, the 26S proteasome. Along with damaged and truncated proteins, this universal protease degrades hundreds of regulatory proteins and thus controls a myriad of essential cellular processes, including the cell cycle, DNA replication, transcription, and stress responses 22. The proteasome specifically recognizes substrates by one versatile, small protein tag called ubiquitin, which is post-translationally attached to substrate proteins by ubiquitin ligases (see Box 1: the Ubiquitin Proteasome System). Ubiquitin ligases facilitate the transfer of ubiquitin to lysine side-chains on a target protein as well as lysines on ubiquitin itself, resulting in the formation of ubiquitin chains. These ubiquitin chains can then bind to the proteasome and tether substrates for engagement by the proteasomal AAA+ unfoldase.
BOX 1: The Ubiquitin Proteasome System.
Ubiquitination occurs by way of a three-enzyme cascade. Ubiquitin is first activated by an ubiquitin-activating protein (E1), which couples ATP hydrolysis to the formation of a thioester bond between its active-site cysteine and the C-terminus of ubiquitin. E1 then transfers the activated ubiquitin to the active-site cysteine of an ubiquitin-conjugating enzyme (E2). Finally an ubiquitin-protein ligase (E3) facilitates the transfer of the ubiquitin from an E2 to a lysine on specific target proteins83. This final step can be repeated several times with the same substrate, resulting in ubiquitination of multiple lysines and/or the assembly of polyubiquitin chains84. It is thought that chain formation can occur on any of the seven lysines of ubiquitin, with specificity for a particular lysine conferred by the E2. Finally these ubiquitinated substrates may be targeted for degradation or signal other types of cellular regulation, depending on the length and linkage of the ubiquitin chain. The best-understood ubiquitin chains are K48-linked, which confer proteasomal degradation, and K63-linked, which are typically involved in non-proteolytic endocytic signaling and DNA repair85. Alternatively, substrates may have their ubiquitin chains removed by cellular deubiquitinating enzymes.
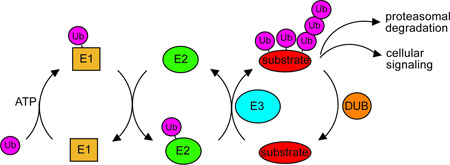
The ubiquitin tag may carry signaling information in its length, linkage, and placement on a substrate. Ubiquitin contains seven lysines (K6, K11, K27, K29, K33, K48, and K63) and a free N-terminal amino group, all of which can be used to link consecutive ubiquitin molecules. Different ubiquitin linkages result in polyubiquitin chains with diverse quaternary structures that likely alter proteasome affinity and may affect substrate degradation efficiency 23, 24. In addition, substrate-attached ubiquitin chains vary in length. While it has been found with K48-linked chains that a substrate must carry a chain of at least four ubiquitins for efficient degradation, the detailed effects of chain length on substrate recognition and degradation efficiency has yet to be determined 25. Finally, the location of the ubiquitin chain on a substrate may play an important role in degradation. After a substrate has been tethered to the proteasome by its ubiquitin chain, it must be engaged by the AAA+ unfoldase. This engagement requires an unstructured initiation site in the substrate protein, either a flexible terminus or internal loop, which extends long enough beyond the ubiquitin tether to enter the unfoldase pore 26. Thus, the position of the ubiquitin chain relative to the unstructured initiation site is likely to affect degradation efficiency. Progress towards understanding the information contained in ubiquitin chains and the impact on substrate degradation rates has been limited by a lack of structural information on the 26S proteasome and insufficient quantitative analyses of ubiquitin binding and substrate processing. However, it is evident that the post-translational ubiquitin tagging system of eukaryotes has the potential to carry far more information than the intrinsic substrate targeting mechanisms of prokaryotes.
Also contrary to prokaryotic systems, ubiquitin targeting signals must be removed by an essential deubiquitinating enzyme (DUB) on the proteasome before a substrate can be efficiently degraded 13, 27. Cleaved ubiquitin chains are then hydrolyzed into ubiquitin monomers for reuse in the cell 28. Ubiquitin chain removal is likely to be particularly important because ubiquitin is very thermodynamically stable and may be a significant unfolding challenge for the proteasome. Furthermore, even after unfolding, the branched polypeptide chains of uncleaved polyubiquitin may not be easily accommodated by the proteasome pore and represent a considerable obstacle for fast degradation. Indeed, the degradation rate of a substrate with an attached, uncleavable ubiquitin is lowered dramatically 29. Thus a key difference between the eukaryotic and simpler prokaryotic systems is that the ubiquitin targeting signal is added to substrates post-translationally and must be subsequently removed before degradation.
The 26S proteasome contains several large structural modules in addition to the basic AAA+ unfoldase and compartmental peptidase that all ATP-dependent proteases share. The compartmental peptidase of the proteasome, called the core particle, is capped by a 19-subunit assembly called the regulatory particle, which contains the AAA+ unfoldase as well as all of the subunits necessary to bind and remove ubiquitin chains. These ubiquitin-interacting modules are absent from proteasomal ancestors like the archaeal PAN-20S protease, which consists of four stacked homoheptameric peptidase rings capped on either end by a homohexameric AAA+ unfoldase (Figure 1). The additional structures became a part of the 26S proteasome at the prokaryotic to eukaryotic transition and were likely added to accommodate the ubiquitin signaling system that evolved around the same time 30, 31. Indeed, studies over the past decade have shown that individual subunits of these modules function in ubiquitin binding and deubiquitination, but the role of the majority of extra subunits and how they are positioned to receive, interpret, and remove ubiquitin signals has remained unclear. Furthermore, even the 26S proteasome’s basic ATP-dependent degradation machinery is more complicated than its prokaryotic counterparts. The eukaryotic proteasome is the only ATP-dependent protease in which all subunits in the AAA+ unfoldase and the stacked rings of the compartmental peptidase are distinct, resulting in a fully heteromeric machine. Together, these embellishments make the 26S proteasome a significantly larger and more complicated enzyme than its prokaryotic precursors, but the functional importance of this complexity has been poorly understood (Figure 1).
Figure 1. The 26S proteasome is a heteromeric and asymmetric ATP-dependent protease with higher complexity than its prokaryotic ancestors.

A) Model of the homo-hexameric archaeal unfoldase PAN (cyan) bound to the core particle (grey). The separate crystal structures of the N-ring (3H43), the ATPases (3H4M), and core particle (3H4P) 38 were docked together and rendered with a surface resolution of 4 Å to match more closely the EM structure of the 26S proteasome. B) Cryo-EM reconstruction of the 26S proteasome at subnanometer resolution 33. The core particle (grey) and AAA+ unfoldase subunits (cyan) resemble the archaeal PAN-20S protease in size and shape, but bear distinct asymmetries due to their heteromeric ring architectures. The additional structural modules that do not share homology with other compartmental peptidases and have likely been added to accommodate ubiquitin signaling are shown in orange. They include the lid sub-complex, the torroidal subunits Rpn1 and Rpn2, and the ubiquitin receptors.
A previous lack of structural information on the 26S proteasome, and the regulatory particle in particular, has strongly limited our knowledge of the mechanisms for substrate recognition and degradation. However, numerous recent electron microscopy (EM) and X-ray crystallography studies have helped to shed light on how this complicated enzyme processes its substrates. Cryo-EM reconstructions of the 26S proteasome from three different organisms (S. cerevisiae 32, 33, S. pombe 34, and human 35) at sub-nanometer resolution, together with subunit localizations and crystal structures of isolated subunits, have dramatically improved our understanding of the architecture of the regulatory particle and the spatial arrangement of key subunits for ubiquitin interaction. These novel structural data have offered important explanations for the ubiquitinchain and substrate-geometry requirements previously established based on biochemical experiments, and have allowed the generation of plausible hypotheses for the mechanisms of ubiquitin binding, deubiquitination, substrate unfolding and translocation by the proteasome. While these structural data have provided insight into how the 26S proteasome has adapted from simpler ATP-dependent proteases to accommodate the ubiquitin-targeting signal, they have also identified numerous novel questions. The structure of the eukaryotic proteasome displays surprising asymmetries, whose functional roles remain to be investigated. This review will summarize the recent advances in proteasome structure and the associated implications for substrate recognition and processing, as well as outline future directions for mechanistic and structural studies.
Proteasome components and structure
The 26S proteasome contains a fully diversified set of AAA+ unfoldase and compartmental peptidase subunits (Table 1). It is these differentiated subunits that allow the specific and asymmetric placement of the additional structural modules required to accommodate ubiquitin signaling. The proteasome core particle is made up of seven different alpha subunits (α1–7) and seven different β subunits (β1–7) that form four stacked, seven-membered rings 36. This peptidase is capped by the regulatory particle, which consists of two stably associated subcomplexes, the lid and the base (Figure 2). The base is a 10-subunit sub-complex that contains the heterohexameric AAA+ unfoldase (Rpt1-6), two large structural proteins (Rpn1 and Rpn2), one intrinsic ubiquitin receptor (Rpn13), and a non-essential DUB (Ubp6). The base also provides binding sites for the three transiently associated substrate-recruiting subunits, the shuttle factors Rad23, Ddi1, and Dsk2. The lid is a 10-subunit sub-complex that includes six PCI (proteasome-CSN-eIF3)-domain containing subunits (Rpn3, Rpn5, Rpn6, Rpn7, Rpn9, and Rpn12), two Jab1/MPN-domain proteins (Rpn8 and the essential DUB, Rpn11), the second intrinsic ubiquitin receptor (Rpn10), and a small structural protein (Sem1). While the structure of the core particle has been well studied 36, the overall architecture of the regulatory particle and the spatial relationship between the lid and base sub-complexes has been elucidated only recently. Surprisingly, the new structural data show that the lid binds to the side of the base and even contacts the core particle.
Table 1.
Proteasome subunits
| Subunits | Structure and Function |
|---|---|
| Base sub-complex: | |
| Rpt1 | AAA+ domain subunit |
| Rpt2 | AAA+ domain subunit |
| Rpt6 | AAA+ domain subunit |
| Rpt3 | AAA+ domain subunit |
| Rpt4 | AAA+ domain subunit |
| Rpt5 | AAA+ domain subunit |
| Rpn1 | PC-repeat solenoid, binds Ubp6 and shuttle factors Rad23, Ddi1, and Dsk2 |
| Rpn2 | PC-repeat solenoid, binds Rpn13 |
| Rpn13 | Ubiquitin binding by Pru domain |
| Lid sub-complex: | |
| Rpn9 | PCI-domain subunit |
| Rpn5 | PCI-domain subunit |
| Rpn6 | PCI-domain subunit |
| Rpn7 | PCI-domain subunit |
| Rpn3 | PCI-domain subunit |
| Rpn12 | PCI-domain subunit |
| Rpn8 | Inactive Jab1/MPN domain |
| Rpn11 | Active Jab1/MPN domain with JAMM motif deubiquitination active site |
| Rpn10 | Ubiquitin binding by UIM, lid binding by VWA domain |
| Core Particle: | |
| α1–7 | gating subunits |
| β1–7 | β1, β2, β5 contain proteolytic active sites |
Figure 2. The 26S proteasome can be separated into three stably associated subcomplexes.

A) Cryo-EM reconstruction of the eukaryotic proteasome holoenzyme with the core particle in grey and the regulatory particle multicolored 33. B) Individual sub-complexes of the 26S proteasome. The regulatory particle can be separated into two stable sub-complexes, the lid and the base. The base consists of the AAA+ ATPase subunits Rpt1-6, (cyan), two large torroidal subunits, Rpn1 (purple) and Rpn2 (light blue), and the ubiquitin receptor Rpn13 (orange). The lid contains six PCI-domain subunits (multicolored) along with the essential DUB Rpn11 (green) and the ubiquitin receptor Rpn10 (yellow). A central cross-section of the core particle (grey) is shown to allow visualization of the barrel-shaped central cavity.
The structure of the base sub-complex
The AAA+ unfoldase of the base sub-complex shares overall homology with other hexameric unfoldases, but in contrast to its prokaryotic counterparts, all of its subunits are distinct. This heterohexameric set of ATPase subunits is observed even in the most primitive eukaryotic organisms (like the amitochondriate diplomonad Giardia), suggesting that eukaryotes evolved these distinct ATPases immediately following their divergence from archaea, when ubiquitin and the additional regulatory particle subunits also evolved 30, 31. Disulfide engineering and crosslinking has been used to show that the ATPase subunits arrange in a particular order: Rpt1-Rpt2-Rpt6-Rpt3-Rpt4-Rpt5 37. This ordering of unique Rpt subunits within the heterohexameric ring creates the specific attachment points necessary for the asymmetric placement of the additional base subunits and the lid sub-complex (Figure 3A).
Figure 3. Subunit architecture of the lid and base sub-complexes.

A) Multiple views of the base sub-complex. A molecular model based on the PAN crystal structure has been docked into the EM density for Rpt1-6 to highlight each subunit of the heterohexamer 38, 86. The N-terminal segment of Rpt1 (dark blue) forms a minimal coiled coil with Rpt2 (light blue) that provides a docking site for Rpn1 (purple) at the periphery of the unfoldase. Rpt 6 (red) forms a long coiled coil with Rpt3 (green) that positions Rpn2 (light blue) above the AAA+ ring. Rpn2 in turn holds Rpn13 (orange) high above the pore of the unfoldase. The coiled coil formed by the N-terminal segments of Rpt4 (yellow) and Rpt5 (orange) does not appear to interact with any other proteasome subunits, but may stabilize the UIM of Rpn10. B) Multiple views of the lid subcomplex in its holoenzyme-bound conformation. The first view (far left) shows the base-facing, inner surface of the lid. The PCI-domain subunits in this orientation, from left to right, are Rpn12 (light green), Rpn3 (yellow), Rpn7 (purple), Rpn6 (blue), Rpn5 (light yellow) and Rpn9 (pink). The central green density was initially identified as Rpn11, but may be an MPN domain dimer of Rpn11 and Rpn8. In turn, the central density depicted in red, initially proposed to be Rpn8, may be a helix bundle formed by the C-termini of all lid subunits 32. The ubiquitin receptor Rpn10 (yellow, right) is positioned high above the unfoldase pore through interactions with Rpn11 and Rpn9 33.
Each ATPase subunit consists of an AAA+ domain and an N-terminal domain, which contains a variable N-terminal sequence followed by a conserved OB (oligomer binding)-fold. Previous insight into the architecture of the N-terminal domains had come from a structure of PAN, the homohexameric archaeal ancestor of the eukaryotic proteasomal unfoldase 38. The six OB-fold sub-domains of PAN form a ring-shaped structure (the N-ring), while the N-terminal sequences adopt alpha-helical conformations and pair into three coiled coils. A conserved proline residue at the base of the N-terminal helix adopts a cis-conformation in every other PAN subunit around the hexameric ring, introducing a sharp kink of the helix that allows coiled-coil formation with its neighbor. It seemed likely that the eukaryotic proteasomal ATPases would form similar N-terminal coiled-coils, as Rpt2, Rpt3, and Rpt5 contain a conserved proline residue that would allow the kinks required for specific subunit pairing. In fact, recent EM reconstructions of the 26S proteasome revealed such coiled coils, but they differ significantly in length and therefore introduce a break in the unfoldase symmetry that may give them unique structural roles 32, 33. In EM structures of the S. cerevesiae holoenzyme, the N-terminal sequences of Rpt3 and Rpt6 form a long coiled-coil that creates a structural beam, allowing Rpn2 and the lid sub-complex to attach to the base high above the central pore of the unfoldase. In contrast, the N-terminal sequences of Rpt1 and Rpt2 form a much shorter and less-defined coiled coil that appears to bind tightly to the side of Rpn1, positioning this large subunit at the periphery of the base. Similar to Rpt3 and Rpt6, the N-terminal sequences of Rpt4 and Rpt5 form a long coiled coil, but it is not observed to interact with any other proteasome subunit. Notably however, the ubiquitin-binding subunit Rpn10 is localized in close proximity to this Rpt4/5 coiled coil. Rpn10 consists of two domains, a globular von-Willebrand-A (VWA) domain and a small ubiquitininteracting motif (UIM) that is connected via a long flexible linker and thus may not be visible by EM. Unpublished EM data show increased flexibility and a consequent lack of defined electron density for the Rpt4/5 coiled coil if Rpn10 is deleted. While Rpn10’s VWA domain clearly does not interact with Rpt4/5, its UIM may contact the tip of the coiled coil, allowing mutual stabilization and potential communication between the ubiquitin receptor and the unfoldase. An asymmetry in the three coiled coils, as observed for the S. cerevisiae proteasome, may be present and functionally relevant for other eukaryotes as well, even though it was not clearly resolved in the EM reconstructions of the human and S. pombe proteasomes 34, 35.
The heterohexameric AAA+ unfoldase creates the specific docking sites for the large torroidal subunits Rpn1 and Rpn2, which themselves provide the binding surfaces for many of the ubiquitin-interacting subunits of the proteasome. As predicted based on their numerous PC (proteasome/cyclosome) repeats, both Rpn1 and Rpn2 have been shown in recent EM and crystallography studies to each form double solenoid structures that transition into an extended arm 32, 33, 35, 39, 40, 41. Rpn1 docks to the base by interacting with the N-terminal helix of Rpt1 as well as the AAA+ domains of Rpt1 and Rpt2. It appears to be particularly flexible, based on its more variable electron density and consequent lower resolution in the EM reconstructions, even though individual PC repeats are still clearly discernable 33. This flexible Rpn1 binds the intrinsic DUB Ubp6, which is therefore also poorly defined in the reconstruction 33. Still, Ubp6 can be generally positioned at the outer surface of Rpn1 and relatively far away from the central processing pore. This may be an ideal placement, given the role of this DUB in internal cleavage or trimming of ubiquitin chains from their distal end 33, 42, 43. Rpn1 also provides binding sites for the transiently associated ubiquitin shuttle factors Rad23, Ddi1, and Dsk2 44. These shuttle factors likely bind to the surface of the solenoid domain of Rpn1 45, which thus creates a large landing platform to receive ubiquitinated substrates and position them near the unfoldase pore for engagement by the translocation machinery. Rpn1’s intrinsic flexibility may therefore play an important role in proper substrate delivery, allowing the enzyme to accommodate substrates of highly variable size, shape, and ubiquitin-chain length.
Rpn2 is positioned high above the unfoldase pore through tight interactions with the Rpt3/6 coiled coil. The crystal structure of the isolated Rpn2 at 2.7 Å resolution shows an extended N-terminal arm, followed by the large toroidal PC domain and a small, globular C-terminal domain 41. It docks nicely into all three holoenzyme EM structures, which unambiguously confirms the localization of Rpn2 within the proteasome complex. This docking reveals that the extended N-terminal arm of Rpn2 provides a strong attachment point for the lid sub-complex. Furthermore, it verifies that the intrinsic ubiquitin receptor Rpn13 binds to the C-terminal region of Rpn2, as had been shown in biochemical experiments 46.
The recent structural data allow us for the first time to localize the ubiquitin receptors within the context of the holoenzyme. Rpn13 is positioned high above the pore of the unfoldase through interactions with the C-terminus and the top of the solenoid domain of Rpn2. Docking the crystal structure of Rpn13 into the EM density of the holoenzyme shows that its ubiquitin-binding pru (pleckstrin-like receptor for ubiquitin) domain faces outward and would therefore position a bound ubiquitin chain above the unfoldase pore. The location of the second intrinsic ubiquitin receptor, Rpn10, is surprising, as this subunit had been previously considered a component of the base 22, 47. However, EM data on proteasomes purified from Rpn10 deletion strains as well as the unambiguous docking of an Rpn10 crystal structure into the holoenzyme electron density indicate that its globular VWA domain interacts exclusively with subunits of the lid, which will be discussed below.
The structure of the lid sub-complex
The lid is a stably associated PCI-domain-containing sub-complex of 10 subunits (Figure 3B). The recent EM reconstructions have elucidated that it binds to the side of the base and core particle, providing a structural scaffold to position the essential deubiquitinating enzyme Rpn11 and the ubiquitin receptor Rpn10. The lid subunits Rpn9, 5, 6, 7, 3, and 12 interact with each other through their C-terminal PCI domains to form a horseshoe-shaped structure 33. The N-termini of these subunits extend radially outwards from the PCI horseshoe, with elongated helixturn- helix domains creating the fingers of an overall hand-shaped arrangement. The crystal structure of the isolated Rpn6 fits well into the segmented EM density and confirms the localization of this subunit in the center of the lid sub-complex 48. Besides the six PCI-containing subunits, the lid includes two Jab1/MPN-domain proteins, Rpn8 and the DUB Rpn11. EM density across the top of the PCI horseshoe, bridging Rpn3 and Rpn9, had originally been assigned to Rpn8 33. However, based on flexible fitting of a MPN domain crystal structure from human Rpn8, part of this density near Rpn3 has recently been suggested to represent a helix bundle formed by the C-termini of all lid subunits 32. For both models, the MPN domain of Rpn11 is held in the palm of the hand-shaped lid structure through extensive contacts with the MPN domain of Rpn8. Thus, the key deubiquitination active site of Rpn11 is exposed and located just above the entrance to the pore of the unfoldase. Rpn11, together with Rpn9, also interacts with the ubiquitin receptor Rpn10, which is therefore located above the pore and nearby the DUB active site. Overall, this lid architecture agrees well with previous data from native-state mass spectrometry studies that demonstrated the existence of two sub-assemblies within the lid, Rpn5/6/8/9/11 and Rpn3/7/12 49.
The recent EM reconstructions of the 26S proteasome provided the first insight into the structural role of PCI domains in multi-subunit assemblies. The lid sub-complex shows high homology and one-to-one subunit correspondence with the other two known PCI-containing protein complexes, the CSN and eIF3. Evolutionary analysis of these complexes in early eukaryotes suggests that the proteasome lid may have formed first and thus may be the precursor of CSN or eIF3 22. Docking of a PCI-domain crystal structure into the lid EM density reveals that the proteasomal PCI domains interact laterally with each other 33. Although the structures of CSN and eIF3 have not yet been solved at higher resolution, it is likely that the PCI domains in those complexes exhibit a very similar architecture 50. The lid, CSN, and eIF3 each include one or two Jab1/MPN-domain proteins, which often (but not always) contain a zinc metalloprotease active site, termed a JAMM motif (defined as EXnHXHX10D). Therefore, the main function of PCI-domain protein complexes may be to properly position MPN-domain subunits for catalysis. Of the two MPN-domain proteins in the lid, only Rpn11 has a JAMM motif that catalyzes the essential cleavage of ubiquitin chains from proteasome substrates, while Rpn8 lacks that motif and thus has no enzymatic activity 22, 27, 51. In CSN, the subunit Csn5 contains a JAMM-motif active site and is responsible for removing the small ubiquitin-like modifier Nedd8 from the SCF ubiquitin ligase to regulate its activity 52. Like the lid, CSN from many organisms contains a second, inactive MPN-domain protein. Inactive MPN domain subunits likely have scaffolding functions that help position and stabilize the catalytically active subunits. For example, Rpn8 and Rpn11 directly contact each other in the lid, which may be a conserved architectural feature also for CSN. In eIF3, both MPN-domain subunits appear to be inactive and therefore play solely structural roles.
The hand-shaped lid docks onto the base subcomplex and core particle through numerous contacts that stabilize the base-core interaction (our unpublished data). Rpn3 and Rpn12 make tight contacts with the extended N-terminal arm of Rpn2, while the alpha-helical bundle of the lid C-termini anchors to the Rpt3/Rpt6 coiled coil 32. Comparing EM structures of the lid in its isolated and holoenzyme-bound form revealed significant conformational changes in lid subunits upon binding to the base and core particle 33. Rpn11, for example, is re-positioned such that it extends away from the PCI horseshoe to reach across the pore of the unfoldase and contact the solenoid portion of Rpn2. In the isolated lid, the curled-up N-terminal finger of Rpn5 seems to block access to the catalytic domain of Rpn11, whereas upon incorporation into the holoenzyme, it swings down to contact the alpha-1 subunit of the core particle, potentially opening access to the Rpn11 active site. Such auto-inhibition would explain the previously described lack of DUB activity by isolated lid 27, which may prevent the futile deubiquitination of substrates by lid that is not properly assembled with the degradation machinery.
In addition to contacts with Rpn2 and the Rpt3/6 coiled coil, lid subunits also interact with the AAA+ domains of the base sub-complex. The broad contacts of Rpn5, 6, and 7 with the AAA+ domains of Rpt4, 3, and 6 are surprising, especially given that the EM structure was determined with fully functional proteasomes in the presence of saturating ATP. Recent crystal structures comparing the nucleotide bound and unbound conformations of a related unfoldase, ClpX from E. coli, show the entire ring flexing as subunits transition through different states of the ATP-hydrolysis cycle 53, 54. The contacts between the lid and AAA+ domains in the proteasome imply that either the heterohexameric unfoldase is rather static or the structure reflects a default state adopted in the absence of substrate and the observed contacts are broken during active protein translocation. In fact, structural studies on the isolated Rpn6 revealed a surface conservation that extends upwards towards the PCI horseshoe, beyond the observed contacts with the AAA+ domain of the base. This additional region of conservation on Rpn6 may be indicative of alternative modes of binding to Rpt3 as the ATPase ring transitions through different nucleotide states 48.
Also surprising are the previously unknown interactions between the lid and the core particle. The N-terminal portions of Rpn5 and Rpn6 reach down beyond the base to contact the coreparticle subunits α1 and α2, respectively 33, 48. These contacts may thus allow direct communication between the lid and core. Importantly, Rpn5 and Rpn6 might function as a clamp that bridges the static lid and core in order to accommodate the conformational changes of the base moving through different nucleotide states while holding ubiquitin receptors and Rpn11 in place. Suspension of the Rpn11 active site above the pore may be critical for allowing substrate access to the translocation machinery and proper positioning of the substrate-bound ubiquitin chain for DUB cleavage, which may also explain the translocation dependence of deubiquitination, as discussed below.
The lid positions the ubiquitin receptor Rpn10 above the pore and next to Rpn11. The Rpn10 VWA domain solely contacts Rpn9 and the Rpn8/11 MPN dimer, which is contrary to previous models that suggested Rpn10 as an intrinsic base subunit or a stabilizing link between base and lid 47. However, Rpn10 might indirectly stabilize the lid-base complex, as it interacts with the extended, Rpn2-bound conformation of the MPN dimer. Docking of the Rpn10 VWA domain crystal structure indicates that Rpn10’s UIM, and therefore also a bound ubiquitin chain, would likely be positioned below the VWA domain, at approximately the same level as Rpn11 and above the unfoldase pore.
The structure of the core particle
The core particle is the structurally and functionally best-understood sub-complex of the 26S proteasome 36. It consists of 28 subunits arranged in four stacked heptameric rings that create a barrel-shaped structure with a sequestered internal chamber. The rings consist of either all-α- or all-β-type subunits (α1–7 and β1–7), stacked in an α-β-β-α pattern. α- and β-type subunits share a similar fold, in which two antiparallel β sheets are sandwiched between two layers of α helices, but differences in their N-termini result in distinct functions 55. The α-type subunits have N-terminal sequences that project into the center of the heptameric ring and form an interdigitated network, restricting access to the internal cavity. The β-type subunits instead have N-termini that contain the key proteolytic residues of the N-terminal nucleophile (Ntn) family of protein hydrolases. In contrast to prokaryotes, which have homomeric peptidase rings, eukaryotes have evolved seven distinct α and β subunits, which occupy unique positions in the heptameric rings and contribute specific regulatory and proteolytic advantages. For instance, only three of the seven β subunits have a functional active site, and each has a different endopeptidase specificity: β1 selectively cleaves after acidic residues, β2 after tryptic residues, and β5 after hydrophobic residues. These differentiated active sites likely facilitate the complete destruction of diverse protein substrates into short peptides. The arrangement of active and inactive subunits, with β1 and β2 next to each other and adjacent to their second copies in the stacked β-ring, but separated from β5 across the ring, may allow the creation of unique substrate binding pockets within the internal degradation chamber of this peptidase 55.
Substrate entry into the internal chamber of the core particle requires opening of the access portal and thus substantial structural rearrangements of the α-subunit N-termini. This gate opening is triggered by binding of the regulatory particle, which docks with the C-terminal extensions, or tails, of its ATPase subunits into the hydrophobic pockets located at the interfaces of α-subunits around the top of the α-ring. A specific C-terminal HbYX motif (hydrophobic/tyrosine/unspecified residue) in the tails is critical for inducing the conformational changes of the α-subunits and opening of the channel 56, 57. Only three of the ATPases in the heterohexameric unfoldase of the 26S proteasome, Rpt2, Rpt3, and Rpt5, contain HbYX sequences. Interestingly, in the EM reconstruction of the yeast holoenzyme only the tails of those three subunits are visible, bound to their respective α-pockets in the core particle (α3–α4, α1–α2, and α5–α6) 33. However, the tails of Rpt1, 4, and 6 might interact with the core as well, but simply be too dynamic to result in a defined electron density. In fact, recent crosslinking studies suggested that all tails, including the ones lacking the HbYX motif, dock into α-pockets, some of them even sampling two nearby pockets 58. The ATPase C-terminal tails might thus be rather promiscuous and dynamic 59.
Numerous EM studies have noted that the AAA+ unfoldase binds the core particle slightly offset from a coaxial alignment 60, 61. More recent EM reconstructions that distinguish between the lid and base sub-complexes have revealed that the base is shifted by approximately 10 Å towards the lid, creating a kink in the central processing channel of the proteasome 33. Whether this angled channel for substrate entry into the core particle has any functional relevance remains unknown, but it may help to guide unfolded polypeptides along certain grooves of the inner surface of the peptidase, through the ante-chamber, and towards specific active sites 55, 62. The offset alignment of base and core may be primarily induced by the lateral attachment of the lid sub-complex, however, specific contacts between base and core themselves might also contribute to this asymmetry. The C-terminal α-helix of the core subunit α4, for instance, extends upwards above the top of the α-ring to interact with Rpn1 of the base. In addition, Ubp6 is bound to Rpn1 and therefore localized nearby the α4 extension 33. This proximity of Ubp6 to the core particle may explain the role of this DUB in stimulating gate opening upon ubiquitin-chain binding to the regulatory particle 63.
Implications of the 26S proteasome architecture for substrate degradation mechanism
In contrast to the more simple prokaryotic AAA+ proteases, substrate degradation by the 26S proteasome requires several regulatory-particle mediated processing steps prior to protein unfolding and translocation. Subunits of the regulatory particle must tether a substrate by its ubiquitin chain, position its unstructured initiation region for engagement by the unfoldase, and cleave off its ubiquitin chain before degradation can commence. Consequently, the subunits of the regulatory particle must be spatially arranged to efficiently facilitate these initial processing steps, but retain enough flexibility to accommodate the large variety of proteasome substrates that may differ in the length, location and number of ubiquitin chains, the position and length of the unstructured initiation site, and general protein size and domain architecture 64, 65.
Both of the proteasome’s intrinsic ubiquitin receptors are positioned high above the unfoldase, with the DUB Rpn11 located approximately equidistant between them and directly above the entrance to the central pore. Provided a sufficient length, ubiquitin chains might bind to multiple receptors simultaneously, which could enhance binding affinity or modulate substrate positioning 66. However, knockout studies have shown that efficient substrate delivery to the proteasome does not depend on the simultaneous interaction of ubiquitin chains with multiple receptors 65, 67. Therefore, the simplest degradation model would include the following steps: 1) a substrate is tethered by its ubiquitin chain to one of the ubiquitin receptors, 2) its flexible degradation initiation site is engaged by the subjacent pore of the AAA+ unfoldase, 3) translocation commences and potentially helps position the ubiquitin-modified lysine in the active site of Rpn11 for ubiquitin-chain removal, 4) as translocation by the base continues, the globular parts of the substrate are mechanically unfolded and the unstructured polypeptide is threaded into the core particle for proteolysis (Figure 4).
Figure 4. Mechanistic model for substrate degradation by the 26S proteasome.

1) A chain of four or more ubiquitins (purple) binds to an ubiquitin receptor (orange) on the proteasome, tethering the substrate (red). 2) Structures in the subjacent pore of the AAA+ unfoldase (cyan) engage the flexible initiation site of the substrate. 3) Translocation commences and helps to position the isopeptide bond of the ubiquitin-modified lysine in the active site of Rpn11 (green) for deubiquitination. 4) The substrate is unfolded and translocated through the AAA+ ring and into the core particle.
Ubiquitin binding
Elegant biochemical studies using K48-linked chains have shown that a minimum length of four ubiquitins is required for efficient substrate degradation, with no improvement in rate for longer chains 25. However, the mechanistic basis for tetra-ubiquitin being the canonical proteasome targeting signal had remained elusive. The recent EM reconstructions of the proteasome now provide a plausible model for this chain-length dependence. Rpn11-mediated cleavage of the isopeptide bond at the proximal ubiquitin is likely more efficient and energetically favorable if the ubiquitin chain is not yet released from the receptor. Thus, an ubiquitin chain would have to be long enough to span the distance between the DUB and either Rpn13 or the UIM of Rpn10, both of which are located 70 – 80 Å from the predicted position of the Rpn11 active site. Since both ubiquitin receptors bind to the hydrophobic patches of ubiquitin, a bound ubiquitin chain would need to be in an extended, open conformation with hydrophobic patches exposed. While solution NMR studies of K48-linked tetra-ubiquitin suggest that these chains may spend more time in a closed conformation, they also show that chains are highly dynamic and constantly sampling the open state, providing little barrier for binding to a receptor 68. In this open conformation, a single ubiquitin moiety contributes ~30 Å in length, and it would take three linked ubiquitins to span the distance between Rpn10 or 13 and Rpn11. Moreover, structural and quantitative binding studies using isolated Rpn10 and Rpn13 have shown that both receptors bind ubiquitin chains between two consecutive ubiquitin moieties. Hence, at least a tetra-ubiquitin chain would be necessary on a substrate to allow the simultaneous interaction with an intrinsic receptor and deubiquitination by Rpn11. Shuttle receptors seem to provide an alternative path for substrate recruitment to the 26S proteasome. The receptors Rad23, Ddi1, and Dsk2 bind to Rpn1, which positions them ~80–120 Å away from Rpn11 33. The inherent flexibility of Rpn1 may allow the receptors to adjust to substrates of variable size and ubiquitin-chain length for optimal deubiquitination and engagement.
In general, the number of ubiquitins required for efficient degradation may vary with the linkage type of the ubiquitin chain. Depending on the lysine residue used to link consecutive ubiquitin moieties, chains differ significantly in their extended length and adopt distinct quaternary structures. While K48- and K11-linked ubiquitin chains are the best-understood proteasometargeting signals, quantitative mass spectrometry has shown that all ubiquitin linkages, except K63, can target substrates for proteasomal degradation in vivo 69. K63-linked chains are typically involved in non-proteolytic endocytic and DNA-damage signaling, although they are able to efficiently deliver proteins for proteasomal degradation in vitro 70 (our unpublished work). Substrates modified with K63-linked chains may be protected from degradation by the 26S proteasome in the cell due to K63-specific binding proteins, K63-specific DUBs, or a sequestered localization. When studied in isolation, Rpn10 does appear to display a 10-fold preference for K48-linked versus K63-linked di-ubiquitin 71. However, aside from this study, little binding preference for K11, K48, and K63 linkages has been observed, and even less is known about ubiquitin chains linked through K6, K27, K29, K33. The intrinsic ubiquitin receptors, the shuttle factors, or either of the DUBs could display a preference for these alternative ubiquitin chains, allowing the proteasome to interpret the potentially intricate ubiquitin code.
Ubiquitin chains on cellular substrates can vary significantly in length, and it is still unclear how chain length beyond the required four ubiquitins and the potential multivalent binding to several ubiquitin receptors might affect degradation 72. While longer chains do not seem to enhance substrate recruitment in vitro 25, in vivo they would better withstand any trimming deubiquitination from the distal end, and thus be more likely to allow a substrate to arrive at the proteasome with a sufficiently long targeting signal. Analyses of substrate levels in cell lysates have suggested that a high processivity of ubiquitination correlates with more rapid degradation by the proteasome 73, which may be indicative of a resistance to trimming deubiquitination in the cell. However, it appears that the most-essential degradation targets in the cell do not necessarily receive the longest ubiquitin chains 74, 75.
Engagement
To avoid dissociation from the proteasome upon deubiquitination, a substrate polypetide likely must be engaged with the unfolding machinery of the base before the removal of its ubiquitin chain. This engagement requires the substrate to have an unstructured initiation site, which must be long enough to reach through the narrow N-ring of the unfoldase and into the AAA+ pore. In addition, this tail must be sufficiently spaced from the ubiquitin attachment point on the substrate to allow deubiquitination by Rpn11 even after engagement. Biochemical experiments have shown that the tail length required for efficient degradation is around 30 amino acids, which could easily span the distance of 60 Å between Rpn11 and the AAA+ pore 26, 33. However, this minimal tail length also depends on the receptor utilized for substrate delivery and the length of the attached ubiquitin chain 26.
Deubiquitination
Interestingly, deubiquitination has been shown to require ATP-hydrolysis, which may indicate dependence on initial substrate translocation by the unfoldase. Despite sufficient length, a chain of four or more ubiquitins bound to a receptor may still require the translocation of the attached substrate in order to pull the proximal ubiquitin moiety into the Rpn11 active site, especially because Rpn11 appears to lack significant intrinsic affinity for ubiquitin 76 (our unpublished data). In some instances, however, mono- or di-ubiquitination has been reported to be sufficient for degradation 77, 78, 79. In those cases, deubiquitination may depend on substrate translocation by the base because the ubiquitin has to be pulled off the receptor and into the Rpn11 active site.
The second DUB of the yeast proteasome, Ubp6, appears to interact with the outer surface of Rpn1 and is thus, compared to other ubiquitin-interacting subunits, located the furthest away from the entrance to the pore. This location might enable Ubp6 to trim extended or unnecessary ubiquitin chains from their distal end. Because Ubp6 is positioned closer to Rad23, Dsk2, and Ddi1 than to Rpn10 or 13, it may preferentially act on substrates delivered by these shuttle receptors.
Substrate unfolding and translocation
The asymmetric placement of ubiquitin receptors and DUBs relative to the pore may necessitate a corresponding asymmetry in the unfolding and translocation machinery. Interestingly, the heterohexameric base sub-complex shows strong intrinsic asymmetry, with different Rpt subunits in distinct orientations. Current mechanistic models for AAA+ unfoldases predict that individual subunits in the hexamer are at different stages in the ATPase cycle and undergo coordinated conformational changes in response to ATP binding or hydrolysis 53, 54, 59, 80. Surprisingly, the recent EM reconstruction of proteasome in the absence of substrate but with saturating ATP shows the AAA+ domains of all six Rpts well resolved and in a particular orientation, indicating that the base sub-complex was in an identical and well-defined conformation for the thousands of averaged proteasome particles 33. In this conformation, the large AAA sub-domains of Rpt1-6 adopt a spiral-staircase arrangement, with Rpt3 at the highest and Rpt2 at the lowest position relative to the 20S core peptidase, whereas the C-terminal small AAA sub-domains of all subunits lie basically in one plane above the peptidase.
Subunit staggering and partial or complete staircase arrangements around the hexameric ring have been previously observed for prokaryotic protein unfoldases and helicases of the RecA or AAA+ superfamilies 53, 81, 82. It had been suggested that individual subunits in these homohexameric enzymes consecutively progress through the different conformational stages of the spiral staircase, and thereby translocate substrate through the central pore. However, the six-fold symmetry of the motors makes it impossible to know whether the observed staircase geometries are in fact snapshots of molecules transitioning through different stages or reflect more static subunit arrangements that are identical for all particles. For the proteasome with its hetero-hexameric ATPase ring, it is conceivable that the observed spiral staircase persists even in the presence of protein substrates, which may get translocated by local pore-loop motions in individual Rpt subunits rather than larger-scale, quaternary conformational changes. Alternatively, the defined staircase orientation of Rpt1-6 might represent an apo state of the proteasome that is adopted only in the absence of substrate to allow the optimal engagement of an incoming polypeptide. Given the asymmetric spatial arrangement of ubiquitin receptors and DUBs above the unfoldase, substrate polypeptides may approach the central processing pore from particular angles, and a spiral staircase with the lid-facing Rpt3 subunit in the top position might play a special role in making first contact with the substrate and handing it over to Rpt subunits below. As substrate translocation commences, individual Rpts may then undergo larger motions that change their vertical positions within the spiral staircase arrangement of the hetero-hexameric ring, similar to the models previously proposed for AAA+ helicases 81. Future biochemical and structural studies will be required to distinguish between the two models for proteasomal substrate translocation, which rely on either static or dynamic spiral staircase arrangements.
Future directions
While recent electron microscopy studies of the proteasome have provided an important structural context for previous findings of biochemical analyses, numerous new questions have emerged and remain unanswered. Many of these questions involve the architectural asymmetries of the proteasome, for instance whether the different locations of Rpn10, Rpn13, and the ubiquitin shuttle receptors relative to the processing pore or DUBs have functional consequences for degradation or determine substrate preferences. Furthermore, the EM reconstructions of the proteasome in the presence of ATP revealed a specific spiral staircase arrangement of the six AAA+ subunits, but whether this arrangement is maintained during substrate degradation remains unclear. Future studies will also have to address how proteasome sub-complexes communicate with each other and respond to substrate binding, whether the DUB Rpn11 is in fact auto-inhibited in the isolated lid, and what functional role the significant offset from coaxial alignment between base and core particle may play for substrate processing. Finally, we still know very little about the potentially complex information hidden in the ubiquitin code and how proteasome degradation may be fine-tuned by variations in ubiquitin chain length, linkage, and location on a protein substrate. We look forward to the answers of these and many more questions in the near future.
Highlights.
-
-
Asymmetric architecture of the 26S proteasome regulatory particle revealed by cryo-EM
-
-
Novel insights into spatial arrangements of Ub receptors, DUBs, and the AAA unfoldase
-
-
Proteasome architecture provides a glimpse into mechanisms for protein degradation
-
-
Processing of Ubiquitin-tagged substrates requires additional structural complexity
Acknowledgements
We thank the members of the Martin laboratory for helpful discussions, and José Ramon López- Blanco and Pablo Chacón (IQFR- Rocasolano Physical Chemistry Institute, Madrid, Spain) for flexible fitting of EM densities. M.E.M. acknowledges support by the American Cancer Society (grant 121453-PF-11-178-01-TBE), and A.M. thanks the National Institute of Health (grant R01- GM094497-01A1) and the Searle Scholars Program for funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 2.Sauer RT, et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin A, Baker TA, Sauer RT. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 2008;15:1147–1151. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maillard RA, et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 2011;145:459–469. doi: 10.1016/j.cell.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell. 2011;145:257–267. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Augustin S, et al. An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol Cell. 2009;35:574–585. doi: 10.1016/j.molcel.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hishida T, Han YW, Fujimoto S, Iwasaki H, Shinagawa H. Direct evidence that a conserved arginine in RuvB AAA+ ATPase acts as an allosteric effector for the ATPase activity of the adjacent subunit in a hexamer. Proc Natl Acad Sci U S A. 2004;101:9573–9577. doi: 10.1073/pnas.0403584101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol. 2004;146:106–112. doi: 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Baker TA, Sauer RT. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci. 2006;31:647–653. doi: 10.1016/j.tibs.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirstein J, Moliere N, Dougan DA, Turgay K. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol. 2009;7:589–599. doi: 10.1038/nrmicro2185. [DOI] [PubMed] [Google Scholar]
- 11.Flynn JM, et al. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc Natl Acad Sci U S A. 2001;98:10584–10589. doi: 10.1073/pnas.191375298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt R, Zahn R, Bukau B, Mogk A. ClpS is the recognition component for Escherichia coli substrates of the N-end rule degradation pathway. Mol Microbiol. 2009;72:506–517. doi: 10.1111/j.1365-2958.2009.06666.x. [DOI] [PubMed] [Google Scholar]
- 13.Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup's N-terminus. Embo J. 29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bremm A, Komander D. A further case of Dop-ing in bacterial pupylation. EMBO Rep. 11:722–723. doi: 10.1038/embor.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zwickl P, Ng D, Woo KM, Klenk HP, Goldberg AL. An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26 S proteasome, activates protein breakdown by 20 S proteasomes. J Biol Chem. 1999;274:26008–26014. doi: 10.1074/jbc.274.37.26008. [DOI] [PubMed] [Google Scholar]
- 16.Barthelme D, Sauer RT. Identification of the Cdc48*20S proteasome as an ancient AAA+ proteolytic machine. Science. 337:843–846. doi: 10.1126/science.1224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maupin-Furlow JA, et al. Archaeal proteasomes and other regulatory proteases. Curr Opin Microbiol. 2005;8:720–728. doi: 10.1016/j.mib.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Maupin-Furlow J. Proteasomes and protein conjugation across domains of life. Nat Rev Microbiol. 10:100–111. doi: 10.1038/nrmicro2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maupin-Furlow JA, et al. Proteasomes from structure to function: perspectives from Archaea. Curr Top Dev Biol. 2006;75:125–169. doi: 10.1016/S0070-2153(06)75005-0. [DOI] [PubMed] [Google Scholar]
- 20.Humbard MA, et al. Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii. Nature. 463:54–60. doi: 10.1038/nature08659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benaroudj N, Goldberg AL. PAN the proteasome-activating nucleotidase from archaebacteria, is a protein-unfolding molecular chaperone. Nat Cell Biol. 2000;2:833–839. doi: 10.1038/35041081. [DOI] [PubMed] [Google Scholar]
- 22.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bremm A, Freund SM, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol. 2010;17:939–947. doi: 10.1038/nsmb.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fushman D, Wilkinson KD. Structure and recognition of polyubiquitin chains of different lengths and linkage. F1000 Biol Rep. 3:26. doi: 10.3410/B3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. Embo J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inobe T, Fishbain S, Prakash S, Matouschek A. Defining the geometry of the two-component proteasome degron. Nat Chem Biol. 2011;7:161–167. doi: 10.1038/nchembio.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 28.Wilkinson KD, et al. Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry. 1995;34:14535–14546. doi: 10.1021/bi00044a032. [DOI] [PubMed] [Google Scholar]
- 29.Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 30.Volker C, Lupas AN. Molecular evolution of proteasomes. Curr Top Microbiol Immunol. 2002;268:1–22. doi: 10.1007/978-3-642-59414-4_1. [DOI] [PubMed] [Google Scholar]
- 31.Cavalier-Smith T. Rooting the tree of life by transition analyses. Biol Direct. 2006;1:19. doi: 10.1186/1745-6150-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck F, et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1213333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lander GC, et al. Complete subunit architecture of the proteasome regulatory particle. Nature. 482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lasker K, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci U S A. 109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.da Fonseca PC, He J, Morris EP. Molecular model of the human 26S proteasome. Mol Cell. 46:54–66. doi: 10.1016/j.molcel.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Groll M, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 37.Tomko RJ, Jr, Funakoshi M, Schneider K, Wang J, Hochstrasser M. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol Cell. 2010;38:393–403. doi: 10.1016/j.molcel.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F, et al. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34:473–484. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kajava AV. What curves alpha-solenoids? Evidence for an alpha-helical toroid structure of Rpn1 and Rpn2 proteins of the 26 S proteasome. J Biol Chem. 2002;277:49791–49798. doi: 10.1074/jbc.M204982200. [DOI] [PubMed] [Google Scholar]
- 40.Effantin G, Rosenzweig R, Glickman MH, Steven AC. Electron microscopic evidence in support of alpha-solenoid models of proteasomal subunits Rpn1 and Rpn2. J Mol Biol. 2009;386:1204–1211. doi: 10.1016/j.jmb.2009.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He J, et al. The structure of the 26S proteasome subunit Rpn2 reveals its PC repeat domain as a closed toroid of two concentric alpha-helical rings. Structure. 20:513–521. doi: 10.1016/j.str.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 42.Hanna J, et al. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006;127:99–111. doi: 10.1016/j.cell.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 43.Leggett DS, et al. Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- 44.Elsasser S, et al. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat Cell Biol. 2002;4:725–730. doi: 10.1038/ncb845. [DOI] [PubMed] [Google Scholar]
- 45.Gomez TA, Kolawa N, Gee M, Sweredoski MJ, Deshaies RJ. Identification of a functional docking site in the Rpn1 LRR domain for the UBA-UBL domain protein Ddi1. BMC Biol. 2011;9:33. doi: 10.1186/1741-7007-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schreiner P, et al. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature. 2008;453:548–552. doi: 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glickman MH, et al. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94:615–623. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- 48.Pathare GR, et al. The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc Natl Acad Sci U S A. 109:149–154. doi: 10.1073/pnas.1117648108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol. 2006;4:e267. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Enchev RI, et al. Structural Basis for a Reciprocal Regulation between SCF and CSN. Cell Rep. doi: 10.1016/j.celrep.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rinaldi T, et al. Participation of the proteasomal lid subunit Rpn11 in mitochondrial morphology and function is mapped to a distinct C-terminal domain. Biochem J. 2004;381:275–285. doi: 10.1042/BJ20040008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cope GA, et al. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–611. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 53.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139:744–756. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glynn SE, Nager AR, Baker TA, Sauer RT. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol. 19:616–622. doi: 10.1038/nsmb.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gallastegui N, Groll M. The 26S proteasome: assembly and function of a destructive machine. Trends Biochem Sci. 35:634–642. doi: 10.1016/j.tibs.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 56.Smith DM, et al. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim YC, DeMartino GN. C termini of proteasomal ATPases play nonequivalent roles in cellular assembly of mammalian 26 S proteasome. J Biol Chem. 286:26652–26666. doi: 10.1074/jbc.M111.246793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tian G, et al. An asymmetric interface between the regulatory and core particles of the proteasome. Nat Struct Mol Biol. 2011;18:1259–1267. doi: 10.1038/nsmb.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith DM, Fraga H, Reis C, Kafri G, Goldberg AL. ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell. 144:526–538. doi: 10.1016/j.cell.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bohn S, et al. Structure of the 26S proteasome from Schizosaccharomyces pombe at subnanometer resolution. Proc Natl Acad Sci U S A. 2010;107:20992–20997. doi: 10.1073/pnas.1015530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nickell S, et al. Insights into the molecular architecture of the 26S proteasome. Proc Natl Acad Sci U S A. 2009;106:11943–11947. doi: 10.1073/pnas.0905081106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell. 41:8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peth A, Besche HC, Goldberg AL. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol Cell. 2009;36:794–804. doi: 10.1016/j.molcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim W, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mayor T, Graumann J, Bryan J, MacCoss MJ, Deshaies RJ. Quantitative profiling of ubiquitylated proteins reveals proteasome substrates and the substrate repertoire influenced by the Rpn10 receptor pathway. Mol Cell Proteomics. 2007;6:1885–1895. doi: 10.1074/mcp.M700264-MCP200. [DOI] [PubMed] [Google Scholar]
- 66.Sakata E, et al. Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. Proc Natl Acad Sci U S A. 109:1479–1484. doi: 10.1073/pnas.1119394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Husnjak K, et al. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. 2008;453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eddins MJ, Varadan R, Fushman D, Pickart CM, Wolberger C. Crystal structure and solution NMR studies of Lys48-linked tetraubiquitin at neutral pH. J Mol Biol. 2007;367:204–211. doi: 10.1016/j.jmb.2006.12.065. [DOI] [PubMed] [Google Scholar]
- 69.Xu P, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saeki Y, et al. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. Embo J. 2009;28:359–371. doi: 10.1038/emboj.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Riedinger C, et al. Structure of Rpn10 and its interactions with polyubiquitin chains and the proteasome subunit Rpn12. J Biol Chem. 2010;285:33992–34003. doi: 10.1074/jbc.M110.134510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang D, et al. Together, Rpn10 and Dsk2 can serve as a polyubiquitin chain-length sensor. Mol Cell. 2009;36:1018–1033. doi: 10.1016/j.molcel.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rape M, Reddy SK, Kirschner MW. The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell. 2006;124:89–103. doi: 10.1016/j.cell.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 74.Matyskiela ME, Morgan DO. Analysis of activator-binding sites on the APC/C supports a cooperative substrate-binding mechanism. Mol Cell. 2009;34:68–80. doi: 10.1016/j.molcel.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carroll CW, Enquist-Newman M, Morgan DO. The APC subunit Doc1 promotes recognition of the substrate destruction box. Curr Biol. 2005;15:11–18. doi: 10.1016/j.cub.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 76.Sato Y, et al. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358–362. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- 77.Dimova NV, et al. APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat Cell Biol. 14:168–176. doi: 10.1038/ncb2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shabek N, et al. The Size of the Proteasomal Substrate Determines Whether Its Degradation Will Be Mediated by Mono- or Polyubiquitylation. Mol Cell. doi: 10.1016/j.molcel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 79.Kirkpatrick DS, et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 80.Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–1120. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 81.Thomsen ND, Berger JM. Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–534. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 83.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 84.Matyskiela ME, Rodrigo-Brenni MC, Morgan DO. Mechanisms of ubiquitin transfer by the anaphase-promoting complex. J Biol. 2009;8:92. doi: 10.1186/jbiol184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 18:520–528. doi: 10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- 86.Lopez-Blanco JR, Garzon JI, Chacon P. iMod: multipurpose normal mode analysis in internal coordinates. Bioinformatics. 27:2843–2850. doi: 10.1093/bioinformatics/btr497. [DOI] [PubMed] [Google Scholar]