

SUMMARY
BACKGROUND
TDP-43 is a major component of the ubiquitinated inclusions that characterise amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) with ubiquitin inclusions (FTLD-U). TDP-43 is an RNA-binding and DNA-binding protein that has many functions and is encoded by the TAR DNA-binding protein gene (TARDBP) on chromosome 1. Our aim was to investigate whether TARDBP is a candidate disease gene for familial ALS that is not associated with mutations in superoxide dismutase 1 (SOD1).
METHODS
TARDBP was sequenced in 259 patients with ALS, FTLD, or both. We used TaqMan-based SNP genotyping to screen for the identifi ed variants in control groups matched to two kindreds of patients for age and ethnic origin. Additional clinical, genetic, and pathological assessments were made in these two families.
FINDINGS
We identified two variants, p.Gly290Ala and p.Gly298Ser, in TARDBP in two familial ALS kindreds and we observed TDP-43 neuropathology in the CNS tissue available from one family. The variants are considered pathogenic mutations because they co-segregate with disease in both families, are absent in ethnically-matched controls, and are associated with TDP-43 neuropathology in several family members.
INTERPRETATION
The p.Gly290Ala and p.Gly298Ser mutations are located in the glycine-rich domain that regulates gene expression and mediates protein-protein interactions; in particular TDP-43 binds to heterogeneous ribonucleoproteins (hnRNPs) via this domain. We postulate that due to the varied and important cellular functions of TDP-43, these mutations may cause neurodegeneration through both gains and losses of function. The finding of TARDBP mutations implicates TDP-43 as an active mediator of neurodegeneration in a novel class of disorders, TDP-43 proteinopathies, a class of disorder that includes ALS and FTLD-U.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease (MND) and is characterized by relentless degeneration of upper and lower motor neurons leading to progressive weakness and eventually death within 3-5 years.1 Most cases are sporadic, but ~10% of cases are familial.1 Familial ALS (FALS) is genetically heterogeneous. The most common FALS gene is Cu/Zn superoxide dismutase 1 (SOD1). Mutations in this gene predominantly cause autosomal dominant disease and account for ~20% of FALS cases.2 Mutations in other genes cause rarer forms of ALS. These genes include Sentaxin, a DNA/RNA helicase that causes juvenile ALS,3 Alsin,4,5 Dynactin,6,7 angiogenin,8 and synaptobrevin associated membrane protein B.9 Sporadic ALS, which accounts for more than 90% of ALS cases, has been more difficult to genetically solve. In addition, a locus on chromosome 9p21, for which the gene has not yet been identified, is responsible for a form of ALS with frontotemporal lobar degeneration (FTLD).10-12 FTLD is a heterogeneous group of neurodegenerative disorders that have in common behavioral and/or language dysfunction and are now considered to be the most common non-motor deficit in ALS patients.12-14 There is a growing body of evidence that a spectrum of frontal lobe dysfunction is present in up to 50% of ALS patients, with as many as 20% showing abnormalities meeting Neary criteria for clinical FTLD.15-17
This clinical overlap between ALS and FTLD is particularly interesting because, neuropathologically, both ALS and the most common FTLD subtype known as FTLD-U are characterized by ubiquitin-positive and tau and α-synuclein negative neuronal and glial cytoplasmic CNS inclusions. The major protein in both ALS and FTLD-U inclusions was recently identified and is the ubiquinated TAR-DNA binding protein 43 known as TDP-43.18 This finding was rapidly confirmed by others.19 Interestingly, TDP-43 pathology is not found in SOD1-associated FALS suggesting that mechanistically ALS is heterogeneous.20 TDP-43 is a 414-amino acid nuclear protein encoded by the TARDBP gene on chromosome 1p36.2. It was originally identified as a 43 kd transcription repressor that binds to the TAR-DNA element of HIV, and, because of its molecular weight of 43 kDa, it was named TDP-43.21 Functionally, TDP-43 is involved in regulating gene expression and spicing, and in particular, is part of a complex involved in splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene.22-24
The observed TDP-43 inclusions, in both ALS and FTLD-U, puts these two disorders in a class of neurodegenerative diseases where abnormal protein aggregation is observed.25 Protein aggregation disorders include Alzheimer’s disease (AD), Parkinson’s disease (PD), prion disorders, tauopathies, trinucleotide repeat disorders, and other rare brain amyloidoses. In each example listed, dominant mutations are known in the gene encoding the deposited protein, and these mutations accounts for at least a rare fraction of the disease. For example, rare mutations in APP that encodes the Aβ peptide found in AD amyloid plaques is a very rare cause of AD.25 Likewise mutations in the gene encoding α-synuclein, the protein found in Lewy body deposits in PD, are a very rare cause of PD.25 Mutations in MAPT, the gene that encodes tau, the protein component of neurofibrillary tangles, cause some cases of FTD without ubiquinated TDP-43 deposits.26 Thus, TARDBP is an obvious candidate gene for FALS and other familial FTD syndromes not caused by MAPT or progranulin mutations. It is not surprising therefore, that while this manuscript was in preparation, several mutations in TARDBP were identified in ALS cases by two groups, though no associated neuropathology has been reported.27,28 The mutations all cluster in exon 6 of TARDBP and segregate with disease in two FALS kindred (A315T and M337V), while two mutations were identified in sporadic cases (Q331K and G294A). However, several other published studies found no TARDBP mutations in FTLD (214 cases29 plus 173 cases30) or sporadic ALS (237 cases)30. In this study we present the results of DNA sequence analysis of 259 patients with ALS and/or FTLD in order to further define the spectrum of TARDBP mutations. We report here the identification of two novel genetic variants in the C-terminal glycine-rich domain of TARDBP and provide evidence for their pathogenic role in ALS. In addition, because affected members of one of our familial ALS kindreds had neuropathological evaluation, this is the first report to directly link a TARDBP mutation with TDP-43 pathology to autosomal dominantly inherited ALS. This finding further implicates TDP-43 as an active participant rather than innocent bystander in the novel class of neurodegenerative disorders, the TDP-43 proteinopathies, which includes ALS and FTLD-U.
METHODS
Participants
The TARDBP gene was screened for mutations in 259 patients with clinical ALS (n=148) or FTLD/ALS (n=20), as well as in autopsy cases with confirmed TDP-43 pathology and neuropathological diagnoses of ALS (n=44) and FTLD-U or FTLD plus MND (n=47). Autopsy cases with mutations in the progranulin gene were excluded. The characteristics of these cases are given in Table 1. In particular, 65 (25.1%) of the 259 cases were familial and of these, 39 (60%) were patients with ALS and no dementia. Dementia was present overall in 64/259 (24.7%) cases and in 26 of the 65 familial cases (40%). These cases originated from neurodegenerative disease clinics or brain banks at the University of Pennsylvania (UPenn), University of Washington (UW), University of San Diego, and Center for Neuropathology and Prion Research (ZNP) in Munich, Germany. The majority of cases in the cohort were Caucasian (237/259, 91.5%), although three families (1.2%) were of Asian descent and five (5%) originated from Guam Chammoros (Table 1). In addition, members of two families with identified variants were tested. All samples were collected with institutional review board approval and informed consent was obtained from all participants. Pedigrees were constructed using Progeny software (Progeny Software, LLC, South Bend, IN).
Table 1.
Demographic, Autopsy and Family History Characteristics of Cases Studied
| Mean AOO* (years +/- 1SD) | 57.2 +/- 12.8 (range 8-82) | |
|
| ||
| Mean AOD* (years +/- 1SD) | 63.0 +/- 11.8 (range 37-92) | |
|
| ||
| M:F (% Male) | 1.24:1 (55.4%) | |
|
| ||
| N | Percent | |
|
| ||
| Total cases evaluated | 259 | |
|
| ||
| Dementia | ||
| Present# | 64 | 24.7% |
| Absent | 188 | 72.6% |
| Unknown | 7 | 2.7% |
|
| ||
| Autopsy diagnosis | ||
| Total | 91 | 35.0% |
| ALS | 44 of 91 | 48.3% |
| FTLD-U or FTLD-MND | 47 of 91 | 51.6% |
|
| ||
| Ethnicity | ||
| Caucasian | 237 | 91.5% |
| Guam Chammoro | 5 | 5.0% |
| Asian | 3 | 1.2% |
| Black | 1 | 0.4% |
| Unknown | 13 | 1.9% |
|
| ||
| Family History | ||
| Familial ALS (# without dementia) | 65 (39)¥ | 25.1% (60%)¥ |
| Family history of NDD other than ALS | 41 | 15.8% |
| Sporadic | 86 | 33.2% |
| Unknown or unavailable history | 67 | 25.9% |
Mean age of onset (AOO) of 149 clinical cases and mean age of death (AOD) of 88 autopsy cases with available information from total of 259 cases.
Dementia was predominantly of the frontotemporal dementia type
The number of familial cases without any dementia is given in parenthesis as well as the percentage of the total familial cases represented by this subgroup.
NDD, neurodegenerative disorder; SD, standard deviation
Genetic Analysis
Genomic DNA was extracted from blood (patients) or brain (autopsy cases) using standard methods (Qiagen Inc., Valencia, CA). For one sample, DNA was obtained from paraffin-embedded tissue also with Qiagen reagents. The entire coding region of TARDBP, consisting of exons 2-5 and the first 531 nucleotides (nt) of exon 6 as well as an average of 100 nt of flanking 3’ and 5’ intronic regions of each exon were fully sequenced in both directions. Variants identified were confirmed with repeat sequencing of new amplicons. Primers used to amplify part or all of each exon were selected using Primer3 software and are available upon request.
At UPenn, amplification reactions (50 μl) were performed with 200 ng DNA using AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA) and 0.8 μM (final concentration) each primer. Touchdown amplification protocol used consisted of 95°C (10 min), followed by 14 cycles of 95°C (30 s), 60°C (exons 4-6) or 58°C (exons 2, 3) with a reduction of 0.5°C per cycle (30 s), 72°C (1 min), and 20 cycles of 95°C (30 s), 53°C (30 s), 72°C (1 min), with a final 72°C extension (5 min). Amplification products were purified using AMPure (Agencourt Bioscience, Beverly, MA) followed by single pass bidirectional sequencing performed by Agencourt Bioscience or with a CEQ8000 (Beckman Coulter, Fullerton, CA) with Beckman Coulter sequencing reagents. Results were analyzed using Mutation Surveyor software (SoftGenetics, PA).
At UW and ZNP, amplification reactions of 50 ng genomic DNA included 20 pmol primers and 10 μl HotStarTaq DNA Polymerase master mixture (Qiagen Inc., Valencia, CA) in a final volume of 20 μl. Amplification conditions (except for exon 6) were as follows: 1 cycle of 95°C for 15 min, 32 cycles of 98°C (20 s), 60°C (30 s), 72°C (2 min), and a final 5 min extension at 72°C. Upon completion of amplification, 1 μl of ExoSAP-IT (USB Corp, Cleveland, OH) was added to the PCR products and digested overnight at 37°C to remove the residual primers and dNTPs. ExoSAP-treated PCR products (4 μl) were sequenced (30 cycles) using BigDye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) in a final volume of 10 μl. The sequence data were collected and analyzed on a 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA), and the presence of nucleotide variants was identified by sequence alignment using SEQUENCHER software (Gene Codes Corp, Ann Arbor, MI).
SNP Genotyping of TARDBP Variants
We genotyped control samples for TARDBP variants NM_007375:c.869G>C (p.Gly290Ala) and c.892G>A (p.Gly298Ser) using a TaqMan chemistry-based allelic discrimination assay with “Assay by Design” (Applied Biosystems, Foster City, CA) probes using an Applied Biosystems 7900 followed by analysis with Sequence Detection System 2.2.1 software (Applied Biosystems). Control samples were obtained from the following sources: 276 controls from the Coriell Institute (Neurologically Normal Caucasian control panels, Camden, NJ), 48 clinical controls (mean age 76) from the Alzheimer Disease Center at UPenn, 42 brain autopsy samples (mean age 69) without evidence of neurodegenerative diseases from UPenn, and 381 controls from elderly normal controls from the UW. The c.892G>A variant was also assessed in 380 Chinese control subjects (mean age 72) from National Taiwan University. The DNA samples with the variants identified by DNA sequencing served as positive controls for each assay. An upper confidence bound (p) of a mutation frequency when none are observed in a control population is computed using the equation (1-p)N = α where N is the number of chromosomes tested, and α is the significance level.
Neuropathological Analysis
Three cases from the family with c.892G>A (p.Gly298Ser) had post-mortem evaluation and standard neuropathological evaluation (II-5, II-7, and III-4 in Figure 1C) including histologic staining with Luxol Fast Blue, Periodic Acid Schiff, and Hematoxylin and Eosin. Two cases (II-7 and III-4) had remaining paraffin-embedded tissue available for immunostaining. TDP-43 (Protein Tech, 1:2000) and α-synuclein (LB509, 1:200) immunostaining were performed using previously published methodology.31,32
Figure 1. FALS pedigrees.
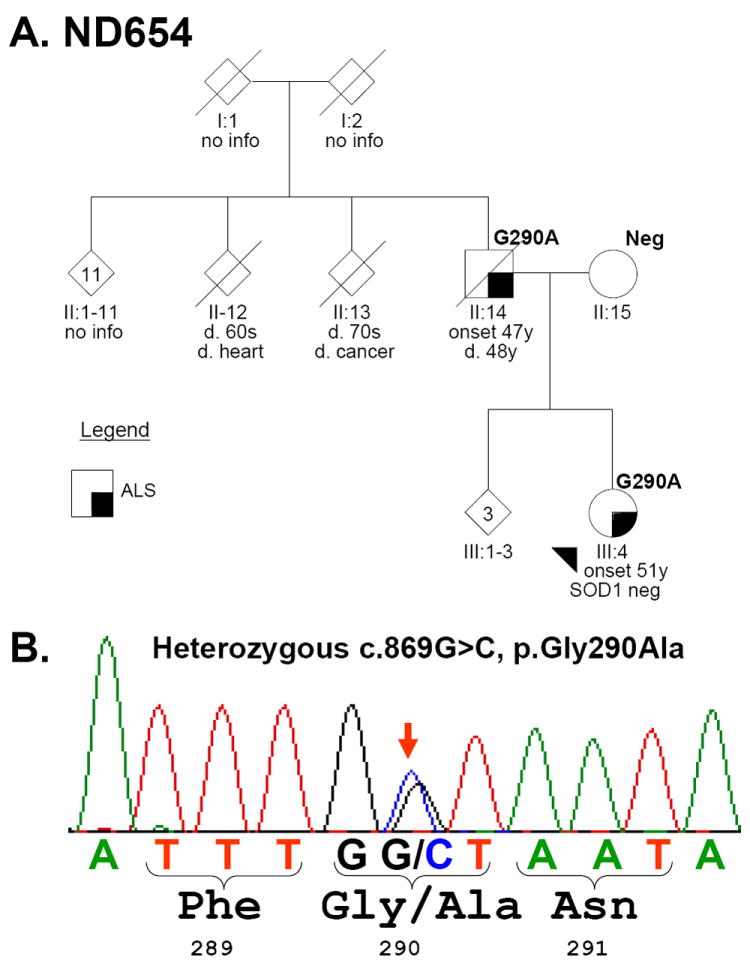
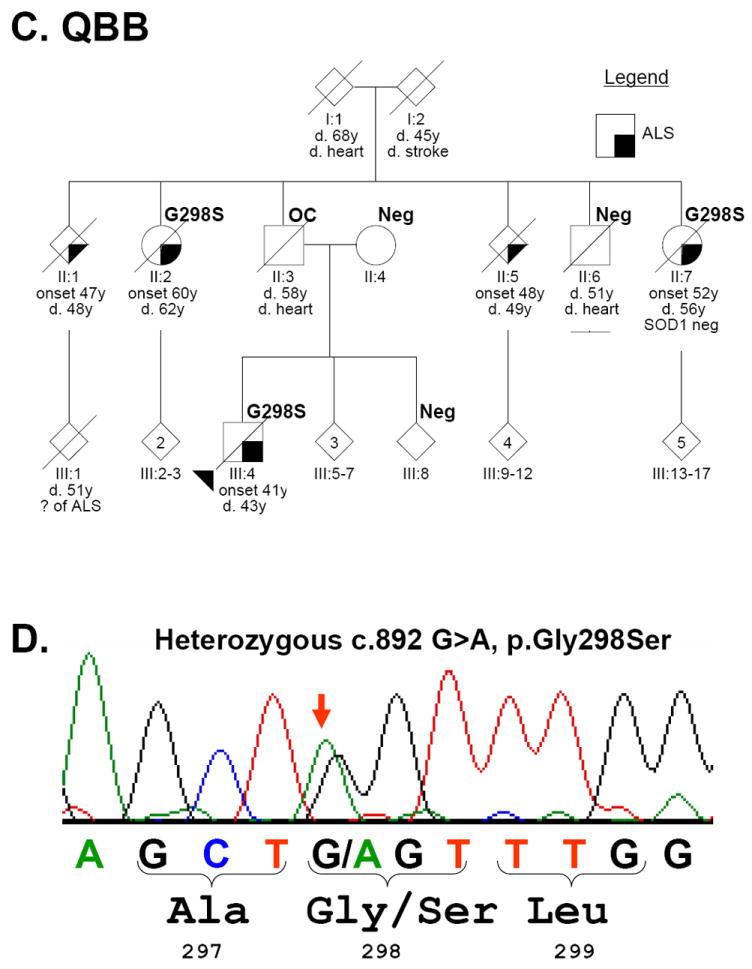
(A). Family ND654 with TARDBP p.Gly290Ala (G290A) and (C). QBB with p.Gly298Ser (G290S). The sequencing electropherograms with heterozygous peaks marked with a red arrow are shown in B and D, respectively. Each individual is marked with generation and individual numbers. The proband in each family is marked with an arrowhead. Gender was removed from many individuals (diamonds) to protect privacy. Multiple siblings are indicated with a number inside the symbol. Clinical status is in the legends and TARDBP genetic testing results are indicated at upper right the symbol as “Neg” (negative) or with the mutation. OC: obligate carrier.
Role of the funding source
The study sponsors had no role in the study design, data collection, data analysis, data interpretation, or writing of this report. The corresponding author had full access to all data in this study and had the final responsibility for the decision to submit for publication.
RESULTS
We identified two heterozygous missense variants, NM_007375:c.869G>C (p.Gly290Ala) (Figure 1B) and c.892G>A (p.Gly298Ser) (Figure 1D), in exon 6 of TARDBP in two families with apparent autosomal dominant FALS (Figure 1). Family ND654 (Figure 1A) is Caucasian (maternal side of Polish ancestry and paternal side of Austrian and Ukrainian heritage) and family QBB (Figure 1C) is Chinese. To evaluate pathogenicity, we tested all additional available family members and controls for these mutations. Controls screened consisted of 747 elderly Caucasians for both mutations and 380 elderly Chinese for the c.892G>A mutation. Neither mutation was found in the controls tested. For Caucasians, the 95% upper confidence limit for the frequency of each of the mutations is 0.002 based on neither mutation occurring in 747 Caucasian controls. For the p.Gly298Ser mutation in Chinese, the 95% upper confidence limit for the frequency is 0.004 based on no examples of the mutation in 380 Chinese controls.
The p.Gly290Ala mutation was found in the two affected members of family ND654; clinical characteristics are summarized in Table 2. The proband in family ND654 (III:4 in Figure 1A) presented with dysarthria and dysphagia at age 51 and was diagnosed with ALS 8 months after the onset of symptoms. Electrophysiological studies showed active denervation; SOD1 testing was negative for a mutation. Over the course of the next 13 months she rapidly developed limb weakness, became anarthric, required a feeding tube for nutrition as well as non-invasive ventilatory support and subsequently died in hospice care. An autopsy was not performed. Her father (II:14) presented with arm weakness at age 47 before rapid progression to death 16 months after onset with a clinical diagnosis of ALS. An autopsy was performed which showed degeneration of anterior horn cells, most prominently in the cervical cord, consistent with ALS. At the time of this genetic analysis (~40 years after the autopsy) CNS tissue was not available for study, but DNA testing of non-CNS paraffin-embedded tissue confirmed the presence of the p.Gly290Ala mutation in the father, while the unaffected mother (II:15) was negative. No other affected or unaffected older individuals were available. The siblings (III:1-3), who are similar in age to the proband, have not been clinically evaluated, but are alive and well.
Table 2.
Clinical Characteristics of Families with TARDBP Mutations
| Family | ND654 | QBB | |||||
|---|---|---|---|---|---|---|---|
| Individual | II:14 | III:4 | II:1 | II:2 | II:5 | II:7 | III:4 |
| TARDBP | G290A | G290A | ND | G298S | ND | G298S | G298S |
| Age of onset | 47 | 51 | 47 | 60 | 48 | 52 | 41 |
| Age of death | 48 | 52 | 48 | 62 | 49 | 56 | 43 |
| Site of onset | Spinal | Bulbar | ND | Bulbar | ND | Spinal | Spinal |
| Dementia | ND | No | No | No | No | No | No |
| UMN signs | ↑DTRs, Spastic gait | ↑DTRs | ND | ↑DTRs, Spastic dysarthria | ND | No | ↑DTRs |
| LMN signs | Fasciculations, weakness, atrophy | Fasciculations, weakness, atrophy | ND | Fasciculations, weakness, atrophy | ND | Fasciculations, weakness, atrophy | Fasciculations, weakness, atrophy |
| Electrophysiology | ND | Active denervation | ND | Fibrillations, fasciculations, positive sharp waves | ND | Fibrillations, fasciculations, positive sharp waves | Fibrillations, fasciculations, positive sharp waves |
| Labs/imaging | Normal myelogram | Normal MRI brain/spine | Normal CSF, normal MRI brain | Normal MRI brain, normal myelogram | |||
| Genetic testing | SOD1 neg | SOD1 neg | |||||
Clinical characteristics of FALS families with TARDBP mutations.
ND = no data
DTRs = deep tendon reflexes
In the QBB family, there are five cases of rapidly progressive ALS in two generations (Figure 1C). Clinical characteristics for all five cases are summarized in Table 2; detailed clinical information was available for three family members. Briefly, the onset of disease consisted of left hand weakness in III:4, right leg weakness in II:7 and dysarthria in II:2. Electrophysiological studies in these three cases were consistent with ALS, showing fasciculations, fibrillations, and positive sharp waves. In addition, SOD1 testing in individual II:7 was negative for a mutation. In all instances, the disease rapidly progressed to quadraparesis and compromised respiratory function. The TARDBP p.Gly298Ser mutation was present in all three affected individuals (II:2, II:7, and III:4) with available DNA, while it was absent in two unaffected individuals (II:6, III:8). In addition, p.Gly298Ser was absent in II:4 (the mother of proband III:4) thus making the father (II:3) an obligate carrier. Individual III-1 died at age 51 and by report of the family was said to have ALS.
CNS autopsies were performed for QBB subjects II:5, II:7 and III:4 (Figure 1B) and all three cases had neuropathologic changes consistent with a diagnosis of ALS including neuronal loss, gliosis and Bunina bodies in the anterior horns of the spinal cord and pallor of the corticospinal tracts. CNS tissues from II-7 and III-4 were available for further analyses and Betz cell loss was observed in the primary motor cortex of both cases. Sections of medulla revealed pallor of the pyramidal tracts, but only case III:4 showed hypoglossal neuron loss. Significantly, immunohistochemistry performed using previously described methods, revealed the presence of TDP-43 inclusions in upper and lower motor neurons and elsewhere in the CNS of both cases (Figures 2 and 3).18,32
Figure 2. Immunohistochemical detection of TDP-43 inclusions in Family QBB with p.Gly298Ser.
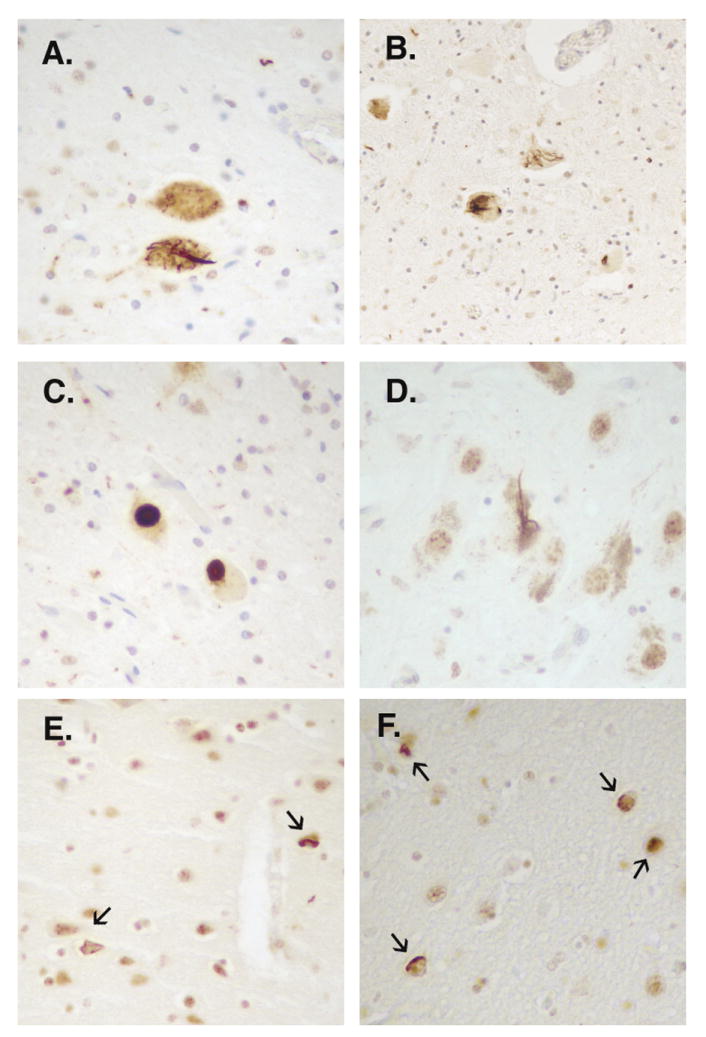
TDP-43 immunohistochemistry conducted in cases II-7 (A, C, E) and III-4 (B, D, F) using a rabbit polyclonal antibody to TDP-43 (Protein Science, Chicago, IL). Filamentous and round TDP-43 immunopositive inclusions were observed in anterior horn cells (A, B, C) of the spinal cord and in substantia nigra neurons (D) of both cases. Additional TDP-43 inclusions were detected in the cingulate gyrus of case II-7 (E) and in the amygdala of both cases (F).
Figure 3. Other examples of TDP-43 pathology.
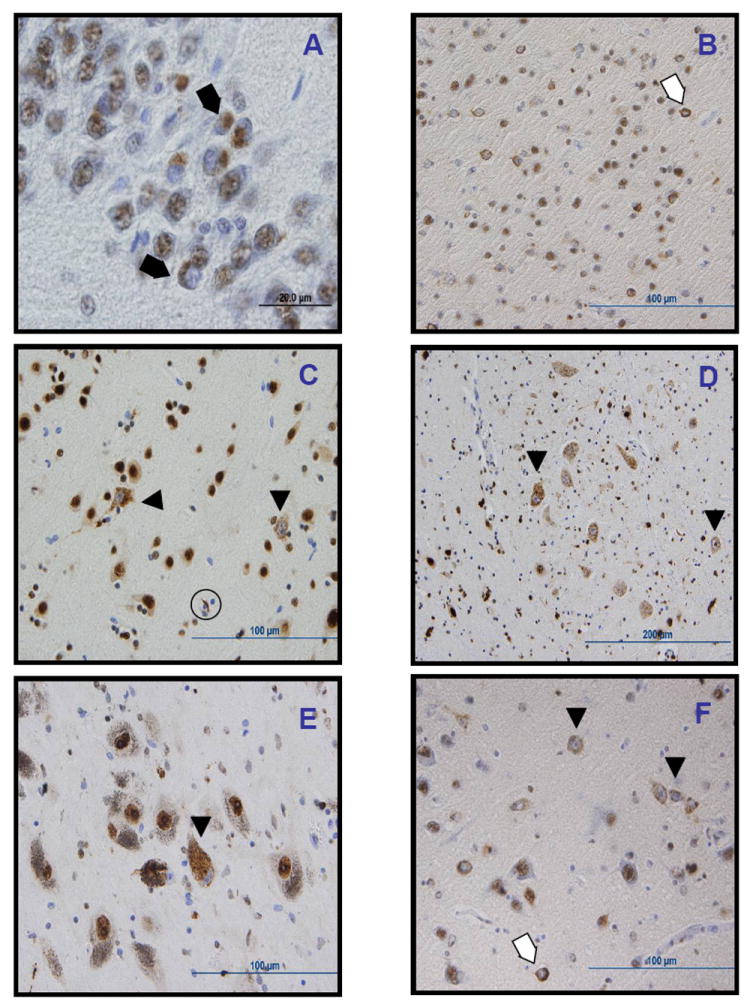
Other TDP-43 positive lesions included dense round inclusions in the dentate gyrus of the hippocampus in case III-4 (A); ring-like dense cytoplasmic inclusions (thin arrows) and diffuse stippled cytoplasmic staining with nuclear clearing or “pre-inclusions” (arrowheads) in the entorhinal cortex (B), temporal cortex (C), hyppoglossal nucleus (D), substantia nigra (E), and amygdala (F), as well as a glial inclusions (thick arrow, C). All images obtained from case III-4, except for C, from case II-7.
For the QBB family, we TDP-43 immunostained cases II-7 and III-4 and observed filamentous and round immunopositive inclusions in anterior horn cells (Figure 2A, B, C), as well as abundant diffuse granular cytoplasmic staining with nuclear clearing of remaining motor neurons, which may reflect pre-inclusions and will be referred to here as such. These pre-inclusions were also found in the hypoglossal nuclei of both cases (Figure 3D). The round inclusions observed in case II-7 were not demonstrated by hematoxylin and eosin staining nor with α-synuclein immunostaining. TDP-43 immunopositive neuronal and neuritic inclusions and pre-inclusions, together with glial inclusions also were observed in diverse distributions and frequencies in the substantia nigra (Figures 2D and 3E), dentate gyrus of the hippocampus (Figure 3A), cingulate gyrus of case II-7 (Figure 2E), frontal cortex of case III-4, the amygdala (Figure 2F and 3F), and frontal and temporal cortices of both cases (Figure 3B, C). Overall, in both cases, there was a significant proportion of pre-inclusions among the TDP-43 pathological findings, higher than observed in other sporadic and familial cases of ALS, which might reflect a unique characteristic pattern of neuropathology for the TDP-43 TARDBP mutation herein described; however, further analysis of other cases will be needed to confirm this hypothesis. Alpha-synuclein immunostaining of spinal cord, medulla, substantia nigra, and amygdala was negative for neuronal lesions but positive for gliosis in spinal cord anterior horns.33
DISCUSSION
We report the identification of two new TARDBP mutations associated with FALS in 2 separate kindreds with apparent autosomal dominant inheritance. These mutations are absent in controls and segregate with disease in two pedigrees described. Furthermore, abnormal aggregated TDP-43 was present in two members of the QBB family that had the Gly298Ser mutation. While the family data and pathological evidence presented in support of a pathogenic role are stronger for Gly298Ser, the similarity and close proximity of the Gly290Ala variant in another family support its pathogenic association with FALS. In addition, these mutations are clustered in the same exon 6 region as previously identified TARDBP mutations, in particular the Gly294Ala mutation, which further supports their pathogenicity.27,28
How might these TARDBP variants cause disease? TDP-43 is a ubiquitously expressed, highly conserved protein with several functional domains. These domains include a nuclear localization signal (amino acids (aa) 82-98), two RNA-recognition motifs (RRM1, aa ~106-175 and RRM2, aa ~191-262), and a C-terminal glycine-rich domain (aa ~274-413). This glycine-rich domain, in which both of our mutations are found, regulates the alternative splicing of several genes. These include the CFTR gene,22,34 that is altered in cystic fibrosis, and the apolipoprotein IIa gene.35 In addition, the C-terminal domain functions in the transcriptional repression of the mouse testis-specific SP-10 gene, regulating the tissue-specific expression of this gene.36 Finally, the glycine-rich C-terminal domain binds several heterogeneous nuclear ribonucleoproteins involved in the biogenesis of mRNA.37,38 Thus, while the physiological functions of TDP-43 are as yet incompletely characterized, TDP-43, and in particular the C-terminal domain, appears to function in the regulation of gene expression.34
The fact that both the Gly298Ser and Gly290Ala mutations identified here, as well as the Gly294Ala TARDBP mutation recently identified27,28 all involve Gly residues within 10 aa of each other in the C-terminal domain of TDP-43, suggests that all three variants may cause disease through similar mechanisms. Specifically, we propose that these variants may alter the normal function(s) of this domain in regulating gene expression.
While the TARDBP mutations reported here occurred in approximately 1.1% (2/188) of non-dementia ALS cases and approximately 0.8% (2/259) of all cases in this study, this is not unprecedented since mutations in the SNCA gene encoding α-synuclein, the major protein component of Lewy bodies in PD, are a rare cause of familial PD. However, familial cases without dementia (39/65, 60%) only represented 15.1% (39/259) of total cases (Table 1), thus, mutations were identified in 5.1% of non-dementia FALS (2/39) and 3.1% (2/65) of all familial cases tested, including FTLD-U. This is similar to the Gitcho et al. study identifying Ala315Thr among 38 familial cases (~2.6%) with ALS, FTLD-MND, and FTLD-U.27 However, it is higher than the rate reported by Sreedharan et al. in which TARDBP mutations were identified in 0.6% (1/154) FALS cases and 0.5% (2/372) sporadic ALS cases.28
In addition to linking variations in the TARDBP gene to clinical FALS, our study demonstrates for the first time the presence of TDP-43 neuropathology in affected family members bearing the TARDBP mutations. Specifically, cases with the Gly298Ser mutation in TARDBP have TDP-43-positive inclusions in many areas of the CNS, including remaining anterior horn cells in diseased spinal cord.
The identification of three separate mutations in TARDBP associated with FALS, with confirmed TDP-43 positive pathology in at least one FALS kindred bearing one of these mutations, provides a direct link between TARDBP nucleotide substitution, TDP-43 neuropathology and ALS neurodegeneration. Further studies will help to better elucidate the pathomechanisms underlying diseases characterized by TDP-43 proteinopathy such as ALS and FTLD-U.
Acknowledgments
The authors would thank Hillary Lipe for clinical assistance. In addition, we extend our appreciation to the patients and families who made this research possible. This work was funded by the National Institutes of Health (AG10124, AG17586, AG005136-22, PO1 AG14382), the Department of Veterans Affairs Funds, the Friedrich-Baur Stiftung (0017/2007), the US Public Health Service, the ALS Association, and a fellowship from Fundació ’la Caixa’, Spain.
Footnotes
AUTHORS’ CONTRIBUTIONS
VMV wrote the manuscript with assistance from JBL, TDB, LBE, DC, EMW, ASCP, MML, MN, JQT, VMYL, GDS, and CY. Genetic analyses of cohort and data were contributed by VMV, LMB, WY, DC, EMW, ES, GDS, and CY. Neuropathological analysis and resources were provided by JBL, MML, TJM, JQT, and VMYL. Cases were contributed to the study by LBE, DC, EMW, ASCP, ES, LM, MG, MN, RK, and DRG. Controls were contributed by DC, EMW, ES, ILW, and WSY. All authors participated in the review of the manuscript and approved the final draft.
References
- 1.Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 2.Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11-12):956–72. doi: 10.1016/j.bbadis.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74(6):1128–35. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadano S, Hand CK, Osuga H, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29(2):166–73. doi: 10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29(2):160–5. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- 6.Munch C, Sedlmeier R, Meyer T, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63(4):724–6. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- 7.Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–6. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 8.Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and ’sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38(4):411–3. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75(5):822–31. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66(6):839–44. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 11.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129(Pt 4):868–76. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 12.Valdmanis PN, Dupre N, Bouchard JP, et al. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007;64(2):240–5. doi: 10.1001/archneur.64.2.240. [DOI] [PubMed] [Google Scholar]
- 13.Kumar-Singh S, Van Broeckhoven C. Frontotemporal lobar degeneration: current concepts in the light of recent advances. Brain Pathol. 2007;17(1):104–14. doi: 10.1111/j.1750-3639.2007.00055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elman LB, McCluskey L, Grossman M. Motor Neuron Disease and Frontotemporal Lobar Degeneration: A Tale of Two Disorders Linked To TDP-43. Neurosignals. 2008;16(1):85–90. doi: 10.1159/000109762. [DOI] [PubMed] [Google Scholar]
- 15.Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64(4):530–4. doi: 10.1001/archneur.64.4.530. [DOI] [PubMed] [Google Scholar]
- 16.Murphy J, Henry R, Lomen-Hoerth C. Establishing subtypes of the continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64(3):330–4. doi: 10.1001/archneur.64.3.330. [DOI] [PubMed] [Google Scholar]
- 17.Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60(7):1094–7. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- 18.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 19.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–34. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 21.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–96. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ayala YM, Pantano S, D’Ambrogio A, et al. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348(3):575–88. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 23.Buratti E, Brindisi A, Pagani F, Baralle FE. Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet. 2004;74(6):1322–5. doi: 10.1086/420978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. Embo J. 2001;20(7):1774–84. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trojanowski JQ, Lee VM. “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer’s disease and other neurodegenerative disorders. Ann N Y Acad Sci. 2000;924:62–7. doi: 10.1111/j.1749-6632.2000.tb05561.x. [DOI] [PubMed] [Google Scholar]
- 26.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64(10):1388–94. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 27.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008 doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science. 2008 doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rollinson S, Snowden JS, Neary D, Morrison KE, Mann DM, Pickering-Brown SM. TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci Lett. 2007;419(1):1–4. doi: 10.1016/j.neulet.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 30.Gijselinck I, Sleegers K, Engelborghs S, et al. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Leverenz JB, Fishel MA, Peskind ER, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol. 2006;63(3):370–6. doi: 10.1001/archneur.63.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leverenz JB, Yu CE, Montine TJ, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130(Pt 5):1360–74. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- 33.Doherty MJ, Bird TD, Leverenz JB. Alpha-synuclein in motor neuron disease: an immunohistologic study. Acta Neuropathol. 2004;107(2):169–75. doi: 10.1007/s00401-003-0790-2. [DOI] [PubMed] [Google Scholar]
- 34.Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–78. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- 35.Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33(18):6000–10. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abhyankar MM, Urekar C, Reddi PP. A Novel CpG-free Vertebrate Insulator Silences the Testis-specific SP-10 Gene in Somatic Tissues: ROLE FOR TDP-43 IN INSULATOR FUNCTION. J Biol Chem. 2007;282(50):36143–54. doi: 10.1074/jbc.M705811200. [DOI] [PubMed] [Google Scholar]
- 37.Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572–84. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 38.Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83(1):130–9. doi: 10.1016/s0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]