

Abstract
Although the protein native state is a Boltzmann conformational ensemble, practical applications often require a representative model from the most populated region of that distribution. The acidity of the backbone amides, as reflected in hydrogen exchange rates, is exquisitely sensitive to the surrounding charge and dielectric volume distribution. For each of four proteins, three independently determined X-ray structures of differing crystallographic resolution were used to predict exchange for the static solvent-exposed amide hydrogens. The average correlation coefficients range from 0.74 for ubiquitin to 0.93 for Pyrococcus furiosus rubredoxin, reflecting the larger range of experimental exchange rates exhibited by the latter protein. The exchange prediction errors modestly correlate with the crystallographic resolution. MODELLER 9v6-derived homology models at ~60% sequence identity (36% identity for chymotrypsin inhibitor CI2) yielded correlation coefficients that are ~0.1 smaller than for the cognate X-ray structures. The most recently deposited NOE-based ubiquitin structure and the original NMR structure of CI2 fail to provide statistically significant predictions of hydrogen exchange. However, the more recent RECOORD refinement study of CI2 yielded predictions comparable to the X-ray and homology model-based analyses.
Keywords: hydrogen exchange, NMR, continuum dielectric, protein electrostatics, structural analysis
1. Introduction
Significant conformational flexibility occurs even in the comparatively well-ordered proteins that have proven to be more amenable to detailed structural characterization. As a result, the accurate prediction of any conformation-dependent observable will formally require the correct modeling of the conformational distribution of that protein, if not the rate of conformational interchange as well. In practice, the field of structural biology has long achieved major successes by interpreting protein reactivities and interactions based on single structures obtained from crystallographic analysis. Although the continual expansion of computational resources has greatly stimulated efforts to model the conformational distribution of the protein native state, the need to determine a “most representative” structure remains a critical issue in numerous applications.
With the advances in computational capacity, the field of molecular docking/virtual screening has progressed from placing rigid ligands into rigid protein binding sites [1] to docking of flexible ligands into rigid protein sites, docking of flexible ligands onto proteins with rigid backbones and flexible sidechains, and most recently the incorporation of limited protein backbone flexibility as well [2]. The latter two stages are well typified by the original formulation [3] of the popular ROSETTALIGAND program of Baker and colleagues and the subsequent extension of this program [4] in which energy minimization of the backbone conformation is applied with harmonic constraints placed on the protein Cα atoms. In the latter case, ROSETTALIGAND required 2–4 hr on a 20 processor cluster for each ligand. As many of the structure libraries currently being used in virtual screening studies contain millions of compounds, such a detailed analysis of ligand-receptor flexibility is only viable in later stages of optimization.
Similar considerations apply to QM-MM calculations of enzymatic reactivity in which the protein-ligand complex is divided between a set of atoms in the primary subsystem that is represented quantum mechanically, while the remainder of the system is modeled classically. Due to the high computational demands of the quantum mechanical calculations, conformational sampling for the protein structure is severely limited as compared to a standard classical MD simulation. As a result, significant inaccuracies in the initial structural model used to set up the QM/MM calculation can not be readily overcome.
Optimal utilization of single structure-based analysis serves a major conceptual role as well. An analysis that requires the averaging of characteristics over two or more distinct protein conformations is invariably severely underdetermined by experimental structural constraints. To establish a physical significance to any such underdetermined comparison between predicted data and experimental measurement necessarily assumes that the model conformations represent a proper sampling of the Boltzmann ensemble [5].
Although molecular dynamics or Monte Carlo sampling approaches can be used to generate a set of energetically plausible protein conformations, their correspondence to an accurate Boltzmann sampling of the native state distribution is generally untested and in some cases have been found to be seriously in error [5,6]. Furthermore, the added computational demands of carrying out parallel calculations initiated from differing starting conformations will generally necessitate a reduction in flexibility testing in molecular docking studies or a restriction of the basis set used in the quantum mechanical calculations in enzymatic modeling studies.
Crystallographic studies generally provide a quite accurate representation of the predominant protein conformation within a crystal lattice. However, demonstration of the degree to which this detailed conformation is preserved in solution is often problematic. NMR solution structure determinations account for ~10% of the recent depositions in the Protein Data Bank [7], the great majority of which are based primarily on NOE distance constraints. Various comparisons between NMR and X-ray structures of the same protein have nearly always yielded differences that are substantially larger than the estimated uncertainties within each individual structure determination [8,9]. However, the degree to which these differences reflect actual variations in structure between the crystal and solution state remains an open question.
On occasion, protein NMR structures have been found to be sufficiently similar to the corresponding crystal structure so that these NMR structures can be used as molecular replacement models for solution of the crystallographic phase problem [10,11]. A standard rule of thumb holds that molecular replacement is generally feasible using initial structural models within 1.5 Å rmsd for the heavy atoms [11], and indeed in favorable cases, even de novo model folding predictions are sufficiently accurate for that purpose [12,13].
For protein functions, such as enzymatic catalysis and intermolecular recognition, a more stringent criterion for structural accuracy may be required to obtain quantitative predictions. It is commonly held that crystallographic analysis treats a protein as a single static structure, while in contrast NMR solution structure determinations treat proteins as dynamic conformational ensembles. In reality, these roles are largely reversed. In addition to the familiar isotropic and anisotropic atomic B factors that nominally model high frequency vibrations and librations, larger scale collective motions have been integrated into protein crystallographic refinement by normal mode [14,15] and TLS (translation, rotation, screw-rotation) analysis [16,17]. However, it should be noted that modeling of such larger scale motion in crystallographic refinement is not without its challenges [18].
In contrast, nearly all NMR protein structure determinations reported to date are based on the assumption that a single fixed conformation is able to simultaneously satisfy all experimental constraints. In the standard protocol, a stochastic simulation is carried to generate such a single conformation. Multiple repetitions of this procedure provide a set of views for a single structure solution to the problem of simultaneous satisfaction of all experimental constraints. This set can not represent an ‘ensemble’ in the thermodynamic sense of the term.
In lieu of a suitably accurate de novo determination of the solution structure for a given protein, utility can be obtained by establishing an experimental method that robustly tests whether a given model conformation is consistent with an optimal representative structure. The wide range of reactivities exhibited by enzymes is understood to reflect an exquisite sensitivity to the detailed structural and electrostatic environment of the protein catalytic active site. This sensitivity to the chemical accuracy of the protein structure can be extended across its entire solvent interface by consideration of the mechanistically simple hydroxide-catalyzed amide hydrogen exchange reaction. Conventionally, protein hydrogen exchange has long been interpreted in terms of a steric blocking paradigm [19–21] so that, in stark contrast to the basic tenets of chemical enzymology, the exchange reactivity of a protein amide is determined by its exposure to solvent and is effectively independent of the conformation of the surrounding protein structure. In reality, the reactivity of individual backbone amides varies markedly as a function of electrostatic interactions ranging from the local geometry of the peptide backbone [22–24] to interactions between formal charge interactions separated by as much as 12–14 Å [25,26].
We have recently reported that backbone amides which are solvent-exposed in the corresponding X-ray structures exhibit a billion-fold range in hydroxide-catalyzed exchange rates [26,27]. Furthermore, for the four model proteins considered in these studies, this billion-fold range in exchange rates was found to be predictable to within a factor of 7 using individual models based on the highest resolution X-ray structures available [27]. In the case of ubiquitin, the use of a model ensemble based on a uniform sampling of trajectory frames from standard unconstrained molecular simulations reduced the variation in the accuracy of hydrogen rate predictions for nearly all of the well-exposed amide hydrogens to a factor of 4 [5]. Slightly better predictions were obtained when this same set of well-exposed amides were analyzed using previously reported model ensembles of ubiquitin (PBD codes 2NR2, 2K39 and 2KOX) that were systematically constrained to match the extensive set of experimentally-derived NOE distance bounds [5].
Given the appreciable success in predicting a wide range of amide hydrogen exchange rates for several different proteins using the highest resolution X-ray structure to model the predominant conformation in solution giving rise to hydrogen exchange, the present study examines the degree to which the ability to achieve single conformation-based predictions of hydrogen exchange reactivity depends upon the resolution of the X-ray model. We then examine the utility of crystallographically-derived homology models and NMR solution structure models in yielding reliable single conformation-based predictions.
2. Computational methods
2.1. Protein structures and modeling
For each X-ray crystallographically-derived structure, hydrogens were added using the program REDUCE [28]. For the NMR structures, hydrogen atoms were stripped and then added back with REDUCE. Electron density was not reported for several C-terminal residues in either the ubiquitin X-ray structure pdb code 1YIW [29] or the 1.00 Å resolution structure of the K29Q variant (kindly provided by S. Ramaswamy (U. of Iowa) and A. D. Robertson (National Psoriasis Foundation), citations of this structure have been previously published [27,30]). We computationally terminated the truncated C-terminus with an N-methylamide cap. For this study, the Gln 29 sidechain of the K29Q variant was truncated to alanine for the calculation of peptide acidity for the amide of Glu 16, as described previously [27]. The original CI2 X-ray structure (pdb code 2CI2 [31]) incorrectly identified residue 78 (or residue 59 in the N-terminal truncated form as numbered in this manuscript [32]) as glutamic acid. This sidechain was computationally transformed into glutamine based on the higher resolution 1LW6 structure [33]. In contrast to our earlier study [27], we did not computationally transform Cys 22 of the various FK506 binding protein structures into the more base-stable valine variant which corresponds to the protein sample used in the experimental hydrogen exchange measurements. Among the static solvent-exposed amides, Lys 47 is closest to the Cβ atom of residue 22 (7.1 Å), and it exhibits the largest change in predicted acidity (0.11 pH units) when this computational mutation is performed.
For each protein, the X-ray structure of an evolutionarily related homolog was identified by the BLASTP program to yield an ungapped alignment across the protein sequence. Using only this sequence alignment as input, a single round of homology modeling was carried out with the MODELLER 9v6 program [34] to generate five models of each target protein. The model yielding the best molpdf fit score was utilized for continuum dielectric calculations.
2.2. Continuum dielectric calculations
Nonlinear Poisson-Boltzmann calculations were carried out using the DelPhi algorithm [35] with a grid spacing of 0.25 Å, a 50% fill value using an ionic strength of 150 mM and internal and solvent dielectric values of 3 and 78.5, respectively. The internal dielectric value is derived from correlation between predicted and observed hydrogen exchange rates in proteins [26,27,36] and peptides [24,37]. The CHARMM22 [38] atomic charge and atomic radius parameters were supplemented with a density functional theory-derived charge distribution of the peptide anion [27]. The protein termini and the lysine, arginine, aspartate, and glutamate residues were set to the charged state. As earlier reported [26,27], nearly all of the monitored amides exhibit linear log exchange rate vs. pH dependencies with unit slope from below pH 6 to above pH 10, indicative of simple hydroxide catalysis. The log kex rates for amides near the sidechains of His 68 in ubiquitin and His 87 and His 94 in FKBP12 exhibit deviations from simple linearity, consistent with the known pK values for these sidechains [39,40], such that the kOH− rate constants for the neutral imidazole form of the proteins could be determined [27].
For each protein conformation, the electrostatic potential was calculated for the individual peptide anions that are formed by removal of the amide hydrogen from each of the solvent exposed residues. Peptide acidity predictions were carried out on every amide hydrogen with a solvent-accessible surface area of at least 0.5 Å2, as determined by the SURFV program [41] using the default set of atomic radii [42]. To facilitate comparisons between protein amide anions in differing conformations, an N-methylacetamide (or N-methylacetamide anion) molecule was added to the continuum dielectric lattice volume in each calculation such that the distance between the N-methylacetamide nitrogen and the nearest formal charge was at least 16 Å and such that no intermolecular atomic distance was less than 8 Å, resulting in negligible intermolecular electrostatic interactions [24]. The differential electrostatic potentials for the various peptide anions were placed onto the pH scale noting that the acidity of water (pKa of 15.7) and the diffusion limit of hydroxide reactivity (2 × 1010 M−1s−1 [43,44]) at 25 °C predict that an amide with a pKa of 26 will have an exchange rate constant kOH− of 1.0 M−1s−1.
When an aspartic acid sidechain has a χ1 torsion angle near −60° (g−) or +60° (g+), the carboxylate group is oriented near to the amide nitrogen of that residue, suppressing the predicted exchange reactivity. Unhindered rotation to the trans χ1 rotamer is used to model the conformer that dominates the exchange reactivity [26]. Similarly, other residues with a g+ χ1 rotamer were rotated to alternate unhindered rotamers [27]. The 1YIW X-ray structure of ubiquitin contains three nonequivalent monomers in the asymmetric unit. When a given amide hydrogen is solvent-accessible by at least 0.5 Å2 in all three monomers, the most acidic conformer was used in the subsequent analysis. The analogous logic was applied to sidechains that adopt dual conformations in other X-ray structures.
To account for the potentially rapid dielectric response of the sidechain hydroxyl hydrogens, when serine and threonine residues containing gauche χ1 sidechain torsion angles have solvent-exposed amides, the peptide conformer acidity for such residues was calculated according to the water dielectric equivalence assumption, in which the serine sidechain is truncated to alanine, and the threonine side chain is truncated to α-aminobutyrate [27].
3. Results and discussion
3.1. Structural dependence of peptide hydrogen exchange reactivity
As first demonstrated by Molday and Kallen [43], amides behave as normal Eigen [45] acids so that, for the weakly acidic protein backbone amides, the kinetic acidities reflected in their hydrogen exchange rates are essentially equal to their thermodynamic acidities. More precisely, the hydrogen exchange rate is attenuated from the diffusion limit by a factor of Ki/(Ki + 1), where Ki is the equilibrium constant for transfer of a proton from the amide to an hydroxide ion. The brief lifetime of the strongly basic peptide anion that is inferred from its diffusion-limited reaction rate is directly supported by the ~5 ps residence lifetime of an hydroxide ion in water [46] and the similar lifetimes that are observed for photoactivated strong acids and bases [47,48].
The brief lifetime of the peptide anion charge state severely limits the shielding contributions from protein conformational transitions, since reorganizations that occur on a longer timescale can not effectively shield that charge state [26,49]. As a result, electronic polarizability effects dominate the dielectric shielding. This, in turn, implies that the assumption of a uniform internal volume dielectric provides a reasonably robust approximation for modeling peptide ionization behavior. In marked contrast, the less robustly predictable ionizable protein sidechains have charge state lifetimes in the s-ms timeframe near neutral pH, so that any assumption of a uniform dielectric approximation necessarily treats the dielectric shielding effects from a wide range of protein conformational dynamics in a globally averaged fashion.
The Curtin-Hammett principle of conformationally dependent reactivity [50] argues that the product ratio depends only upon the difference in the transition state free energies of the reactive species which, in turn, corresponds to the sum of differential free energies among the conformers and the differential free energy of activation for each of those conformers [6]. If the electrostatic interactions surrounding a given backbone amide predict a strong suppression of its ionization for the crystallographically determined structure and transition to a conformation which reduces this suppression is energetically accessible, this alternate conformation will generally dominate the exchange reactivity. The most widely observed local interaction of this nature occurs for aspartate residues in which the sidechain carboxylate is oriented gauche to its own backbone nitrogen [26,27,36,37]. Continuum dielectric calculations applying the finite difference solver DelPhi [35] to reported X-ray structures indicate as much as 105 to 106-fold suppression of ionization for these conformations [26,37]. In such instances, even a modest population of a trans conformation for the aspartate sidechain would be predicted to dominate the exchange reactivity of this residue. In the single conformation-based hydrogen exchange predictions considered in this study, unhindered rotation of the aspartate sidechain to the trans χ1 rotamer is analyzed [26,27]. Such an effect applies to Asp 39 in ubiquitin in which the three X-ray structures of differing crystal forms that are further considered in this study each vary in the sidechain orientation of this residue (Fig. 1).
Fig. 1.
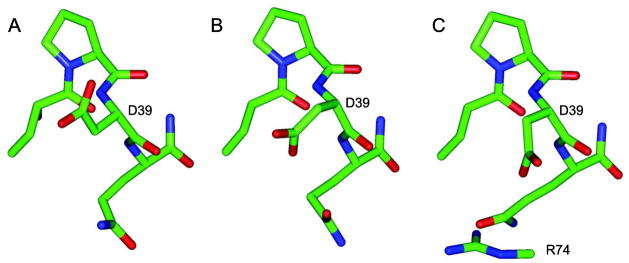
Orientational variations of the Asp 39 sidechain in ubiquitin X-ray structures from differing crystal forms. The sidechain χ1 torsion angle varies from g− to g+ to an eclipsed orientation near +120° for the 1.00 Å (A [27,30]), 1.39 Å (B [29]) and 1.80 Å (C [84]) structures, respectively. Only the lowest resolution structure presents a potential interresidue interaction for the Asp 39 sidechain, a salt bridge to Arg 74 in the mobile C-terminal tail. None of the structures exhibit interactions that would inhibit a transition to the trans rotamer state.
A qualitatively similar effect occurs when the orientation of the sidechain Cγ atom in a gauche+ χ1 rotamer results in decreased solvation of the peptide anion. This much smaller predicted suppression of peptide ionization can also be usefully compensated for by rotating the sidechain to an unhindered χ1 rotamer [27]. Application of these two systematic dominant acidic conformer transitions provide marked improvements in the predictive power of single protein structure-based predictions of protein hydrogen exchange [26,27].
The accuracy with which experimental hydrogen exchange rate constants kOH− can be determined for backbone amides in small proteins [27] and the precision with which continuum dielectric algorithms predict differences in acidity for those sites [24,37] are both at least an order of magnitude better than the current ability to predict experimental protein exchange rates from conformational analysis (errors in log kOH− > ~0.5 [36]). To further assess the statistical errors that arise from lattice grid placement for calculations on an individual protein structure as carried out in this study, 30 independent peptide acidity analyses of Pf rubredoxin were carried out with the lattice position of the 1BQ8 X-ray structure [51] randomly shifted. The mean rmsd value for the predicted acidities of the solvent-exposed amides was 0.046 pH units.
3.2. Testing the dominant acidic conformer analysis in the NMR-restrained model ensemble of ubiquitin 2NR2
To apply the simplifying assumption of a dominant acidic conformer of the protein, local interactions that strongly suppress peptide ionization are considered. As illustration, in each X-ray structure of ubiquitin the Asp 52 sidechain is in a g− χ1 rotamer, forming a salt bridge with the sidechain of Lys 27. Using these conformations to predict the acidity of the Asp 52 peptide yields exchange reactivities that are up to 105 lower than the experimentally observed exchange rate constant [36]. Rotation of the Asp 52 sidechain to the trans χ1 rotamer yields a hydrogen exchange rate prediction that closely matches the experimental data [27].
As noted above, the Asp 39 sidechain of ubiquitin is oriented in a different χ1 conformation in each of the three crystal structures. Regarding the weaker suppression of peptide ionization that results from other sidechain types adopting a g+ χ1 conformation, Lys 63 adopts such a g+ conformation in each of these three X-ray structures (in the 1YIW structure Lys 63 is trans in molecule C, but it is g+ in molecules A and B). Gln 62 adopts a g+ conformation in molecule A of 1YIW and a g− rotamer in all of the other crystal structures. No other Cγ-bearing sidechain of ubiquitin in these three X-ray structures exhibits a g+ χ1 conformation in addition to offering a seemingly unhindered transition to either a g− or trans χ1 rotamer.
The fact that applying χ1 rotamer transitions of the dominant acidic conformer analysis to the sidechains of Asp 52, Asp 39 and Lys 63 yield robust predictions of the experimental exchange rates for each of these three residues [27] does not demonstrate that the reactivity within the experimental Boltzmann distribution is achieved by this means. To gain further insight into the physical plausibility of this analysis, exchange reactivity predictions were carried out for each of the 144 conformations in the 2NR2 ubiquitin ensemble [52]. With regards to the well-exposed peptide hydrogens of ubiquitin, the NOE-restrained 2NR2 ensemble provides more accurate hydrogen exchange predictions than did either the NOE- and residual dipolar coupling-restrained 2K39 [53] or 2KOX [54] ensembles or an unconstrained MD simulation-based ensemble [5,6,36].
A 0.5 Å2 peptide hydrogen exposure criterion was used to select residues for the single structure-based hydrogen exchange rate predictions. In the ensemble averaging analyses, for each amide hydrogen the number of conformations exhibiting at least 0.5 Å2 exposure to solvent was found to provide a useful criterion for estimating the adequacy of the statistical sampling for the chemically reactive conformations of that amide [36], although all solvent-exposed conformations are utilized in ensemble-averaged reactivity calculations [5,6,36]. An area of 0.5 Å2 corresponds to approximately the maximum solvent exposure for any peptide hydrogen involved in a intramolecular hydrogen bond [6].
In the hydrogen exchange analysis of the individual conformations of the 2NR2 ensemble, Asp 52 was excluded. The salt bridge between Asp 52 and Lys 27 is strongly preserved among the models in this ensemble. The markedly depressed reactivity predicted for this residue indicates that the kinetically significant conformations at this site are inadequately sampled in this ensemble. The 144 models of the 2NR2 ensemble yield log kOH− predictions with a median rmsd value of 1.05. The central half of this distribution of log kOH− predictions yields rmsd values between 0.90 and 1.20. Model 92 was found to best predict the experimental log kOH− values with an rmsd of 0.58 and a correlation coefficient of 0.88 (Fig. 2), which is only slightly worse than the values obtained via ensemble averaging [5]. The 2NR2 ensemble was derived by restrained molecular dynamics simulations initiated from the 1UBQ crystallographic coordinates. In generating model 92 the initial g+ rotamer of Lys 63 evolved to a g− rotamer, and the initial eclipsed sidechain conformation of Asp 39 was transformed to a fully trans orientation. These two sidechain transitions that evolve in the derivation of model 92 directly reproduce the specific sidechain rotations predicted from the dominant acidic conformer analysis [27] applied to this crystal structure.
Fig. 2.

Correlation of hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann (PB)-derived pK values predicted from model 92 of the 2NR2 ubiquitin ensemble [52] with the use of CHARMM22 electrostatic parameters [38] and an internal dielectric constant of 3 at 25°C. Excluding Glu 2 and the four conformationally disordered residues at the C-terminus, thermodynamic acidities were predicted for all backbone amide hydrogens that are exposed to solvent by at least 0.5 Å2. As previously discussed [36], the sampling of sidechain rotamers of Asp 52 is restricted in the 2NR2 ensemble by a salt bridge interaction with Lys 27. The rmsd (σ) and correlation coefficient (r) values are indicated, excluding Asp 52.
Only 10 of the 144 models in the 2NR2 ensemble have undergone a similar pair of transitions to a trans χ1 rotamer for Asp 39 and to either a g− or trans χ1 rotamer for Lys 63. Of these ten models, only six (including model 92) have χ1 rotamers for each of the solvent-exposed residues that are consistent with the simple sidechain rotation protocol proposed for identifying more highly acidic conformers. Thus, among the structural variations represented in the 2NR2 ensemble that might yield more accurate predictions of the experimental hydrogen exchange data, the simple modeling approach proposed for the dominant acidic conformer analysis [27] successfully identified the most effective transitions.
3.3. Predicting hydrogen exchange from protein X-ray structures
For each of four proteins, Pyrococcus furiosus (Pf) rubredoxin, human FK506 binding protein (FKBP12), barley chymotrypsin inhibitor 2 (CI2) and human ubiquitin, three crystal structures of differing resolution were used to predict the hydrogen exchange reactivities for the static solvent-exposed peptide hydrogens. With the exception of Pf rubredoxin, each of the three crystal structures for each protein was derived from a different crystal form. In the case of Pf rubredoxin (Fig. 3), the 1.10 Å resolution crystal form (pdb 1BQ8 [51]) and the two lower resolution structures 1VCX (1.50 Å resolution neutron diffraction study [55]) and 1CAA (1.80 Å resolution [56]) yield similar quality hydrogen exchange rate predictions (rmsd values for log kOH− of 0.96, 0.97 and 1.03, respectively). The prediction for Ile 12 is significantly improved over our previous analysis [26], in part, due to the somewhat larger log kOH− value that applies to the charge state of the protein near physiological conditions. Furthermore, in the present study the nearby phenolic hydroxyl hydrogen of Tyr 11 is modeled to be oriented toward the aqueous phase, as reported in the high resolution neutron diffraction analysis of this protein [55,57] in contrast to being oriented toward the interior as predicted by the Reduce program [28] used in our previous analysis.
Fig. 3.

Correlation of Pf rubredoxin hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann-derived pK values. Predictions were derived from X-ray structures at 1.10 Å resolution (PDB code 1BQ8 [51] in red), 1.80 Å resolution (PDB code 1CAA [56] in blue), and the 1.50 Å resolution neutron diffraction structure (PDB code 1VCX [55] in green). Residue identification for the highest resolution data has been previously reported [26].
In Fig. 4A is illustrated the correlation between predicted peptide acidities and experimental hydrogen rates for the three available X-ray structures of the unligated form of human FKBP12. The deviation of the predicted peptide pK values from the observed log kOH− values increases from 0.82 to 1.07 to 1.14, as the resolution limit increases from 0.92 Å (pdb code 2PPN [58]) to 1.85 Å (pdb code 1D6O [59]) to 2.20 Å (pdb code 1FKK, a M49V, H94N, V98I variant [60]).
Fig. 4.

Correlation of hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann-derived pK values predicted from X-ray structures. In panel A are given the predictions derived from X-ray structures of human FKBP12 at 0.92 Å resolution (PDB code 2PPN [58] in red), 1.85 Å resolution (PDB code 1D6O [59] in green) and 2.20 Å resolution (PDB code 1FKK [60] in blue). In panel B are given the predictions derived from X-ray structures of CI2 at 1.50 Å resolution (PDB code 1LW6 [33] in red), 2.00 Å resolution (PDB code 2CI2 [31] in green) and 2.10 Å resolution (PDB code 2SNI [61] in blue) is given in panel B. In panel C are given the predictions derived from X-ray structures of ubiquitin at 1.00 Å resolution (K29Q variant [27,30] in red), 1.39 Å resolution (PDB code 1YIW [29] in green) and 1.80 Å resolution (PDB code 1UBQ [84] in blue). Amides exposed to solvent > 0.5 Å2 that are solvent inaccessible in the highest resolution X-ray structure are indicated as diamonds. The rmsd (σ) and correlation coefficient (r) values are indicated. Residue identification for the highest resolution data has been previously reported [27].
There is a reported unligated structure of chymotrypsin inhibitor CI2 at 2.00 Å resolution (PDB code 2CI2 [31]) and two structures of the subtilisin-bound state at 1.50 Å resolution (pdb code 1LW6 [33]) and 2.10 Å resolution (pdb code 2SNI [61]). As noted in the X-ray analysis [61], the structure of the inhibitor is very closely preserved when the complex with subtilisin is formed. To minimize ambiguities that might arise due to the differing ligation states, the electrostatic analysis of hydrogen exchange was limited to the 14 backbone amide hydrogens that are exposed to solvent in both ligated and unligated crystal structures. The two subtilisin-complexed CI2 crystal structures yield similar quality log kOH− predictions (Fig. 4B). The larger rmsd value obtained for the log kOH− predictions derived from unligated structure 2CI2 is entirely accounted for by the inaccuracy of the prediction for Lys 53. The sidechain carboxylate of the preceding residue Asp 52 is oriented more strongly toward the amide of Lys 53 in the 2CI2 structure than in either of the other two crystal forms, resulting in a stronger suppression in the predicted acidity for that peptide.
The log kOH− values for the static solvent-exposed amides in the X-ray structure of the K29Q variant of ubiquitin (1.00 Å resolution) predicted the experimental values to an rmsd of 0.92 [27]. Analogous calculations on the ubiquitin X-ray structures 1YIW (1.39 Å resolution) and 1UBQ (1.80 Å resolution) yielded log kOH− predictions with rmsd values of 1.03 and 1.17, respectively (Fig. 4C).
Although the rmsd values of the errors in the log exchange rate constants predicted for these four proteins span a fairly narrow range, the correlation coefficients for these predictions indicate that the robustness of these predictions is significantly system dependent. The comparatively high level of correlation observed for the rubredoxin predictions reflects the large range in experimental rates exhibited by the static solvent exposed amide hydrogens of this protein (σ for log kOH− of 2.12). The average dispersion for the experimental log rate constants for the analogous amides of FKBP12 and CI2 are somewhat smaller (1.53 and 1.50, respectively), while the dispersion in the experimental values for ubiquitin is only 40% as large as that of rubredoxin.
As has long been discussed in the field of protein sidechain pK predictions by continuum dielectric methods, a limited range of experimental pK values for a particular ionizable sidechain type offers a weak test bed for evaluating model predictions. When compared to the present peptide pK predictions, the much lower statistical significance typically obtained from continuum dielectric predictions of sidechain pK values have prompted the use of only residues exhibiting highly shifted pK values for testing predictions [62]. As illustration, despite the wide range of theoretical analyses that have been applied to ubiquitin, it has not been a popular target for general sidechain pK predictions. This likely reflects the limited range in pK values exhibited by its aspartate and glutamate sidechains (rmsd of 0.24 and 0.27, respectively [63]).
An analogous limitation applies to staphylococcal nuclease for which early magnetization transfer-based measurements of the rapidly exchanging amides by Mori et al. [64] yielded an rmsd for the log rate constants of only 0.64 for the experimentally monitored static solvent-exposed backbone amides. Indeed, a recent continuum dielectric-based prediction of hydrogen exchange for a mutationally stabilized variant of staphylococcal nuclease failed to obtain a statistically significant correlation for the static solvent-exposed amides as predicted from a modeled structure [65]. This recent study noted the very limited range of exchange rates for this subset of staphylococcal nuclease amides. However, in rejecting an electrostatics-based analysis, these authors invoked their steric blocking paradigm [19–21] to argue that the billion-fold range of experimental exchange rates exhibited by the static solvent-exposed amide hydrogens in the proteins considered in our previous [26,27] and present studies arises from amide-bound crystallographic waters blocking the encounters with the hydroxide exchange catalyst [65]. Crystallographically ordered water molecules are hydrogen bonded to nearly all surface exposed backbone amides in the ~1 Å resolution cryo-cooled crystal structures considered herein. A steric blocking mechanism for this wide range of exchange rates implies that there must be at least a billion-fold (>12 kcal/mol) variation in the binding constants for these individual hydrogen bonded water molecules. At the same time these individual blocking water molecules must remain impervious to being transformed into an hydroxide equivalent as predicted by the classic Grotthuss model for proton transfer in water [66]. This steric blocking analysis does not address why protein exchange rates can be robustly predicted by continuum dielectric calculations of the free energy barrier to forming the hydrated peptide anion complex anticipated by the Grotthuss model.
In the context of ensemble-based hydrogen exchange rate predictions, the staphylococcal nuclease system offers considerable potential for bringing together joint predictions of both sidechain and peptide ionization behavior to probe transient protein conformational states. Garcia-Moreno and collaborators have combined pK titration measurements on a wide range of mutational variants with molecular dynamics simulations to gain insight into the conformational transitions involved in the dielectric shielding of structurally buried ionizable sidechains [67–69]. It can be anticipated that at least some of the conformational transitions that provide transient exposure to solvent for these buried sidechains will prove to be effective in facilitating peptide hydrogen exchange as well.
Although the errors in kOH− predictions will reflect the inaccuracies inherent in the dominant acidic conformer analysis and the approximations underlying the continuum dielectric prediction of conformer reactivity, by comparing the rate prediction errors for a given protein obtained using a range of experimentally derived structures, systematic variations should reflect how reliably each structural model provides a representation of the dominant conformation in solution. Even considering the high resolution X-ray structures for each of these four proteins, there appears to be a modest sensitivity to the quality of the hydrogen exchange predictions as a function of crystallographic resolution. As noted above, the one apparent exception to this trend arises from the orientation of a single aspartate carboxylate on the preceding residue in one CI2 X-ray structure. As discussed below, a considerably wider range of predictive performance is obtained when protein structures derived from homology modeling and NMR solution structure determination are considered.
3.4. Predicting hydrogen exchange from protein homology models
The Modeller 9v6 program [34] was used to generate homology structures for each of the four proteins using X-ray structures from proteins exhibiting ungapped alignment of approximately 60% sequence identity, excepting CI2 for which the sole homologous deposited structure (bitter gourd trypsin inhibitor) is only 36% identical. The 2.50 Å resolution X-ray structure of yeast FKBP12 from Saccharomyces cerevisiae (pdb code 1YAT, 57% sequence identity [70]) yields hydrogen exchange rate predictions that are only modestly worse than those of the lowest resolution structure of the human protein 1FKK (Fig. 5A). The 0.93 Å resolution X-ray structure of the bitter gourd trypsin inhibitor (pdb code 1VBW) predicts the log kOH− rate constants with an rmsd of 1.38 and a correlation coefficient of 0.76 (Fig. 5B), appreciably below the performance derived from the barley CI2 crystal structures. The 1.70 Å resolution structure (pdb code 1BT0 [71]) of the 63% identical Arabidopsis thaliana Rub-1 protein yields a similarly modest quality prediction for the hydrogen exchange rates of ubiquitin (Fig. 5C).
Fig. 5.

Correlation of hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann-derived pK values predicted from X-ray structure-based homology models. In panel A are given the predictions for the homology modeled human FKBP12 derived from the X-ray structure of yeast FKBP12 (PDB code 1YAT [70]). In panel B are given the predictions for the homology modeled barley CI2 derived from the X-ray structure of bitter gourd trypsin inhibitor (PDB code 1VBW). In panel C are given the predictions for the homology modeled human ubiquitin derived from the X-ray structure of the Rub-1 protein from Arabidopsis thaliana (PDB code 1BT0 [71]). Amides exposed to solvent > 0.5 Å2 that are solvent inaccessible in the highest resolution cognate X-ray structure are indicated as diamonds. The rmsd (σ) and correlation coefficient (r) values are indicated.
Strikingly worse statistics are obtained when the hydrogen exchange rate constants for Pf rubredoxin are predicted using an homology model derived from the 59% identical Clostridium pasteurianum (Cp) protein (pdb code 1IRN [72]). The rmsd for the predicted log kOH− values is nearly twice as large as those obtained using the three Pf rubredoxin crystal structures (Fig. 6), while the correlation coefficient of 0.69 is markedly below the value of 0.93 obtained with the three Pf structures.
Fig. 6.

Correlation of Pf rubredoxin hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann-derived pK values predicted from the Clostridium pasteurianum rubredoxin-based homology model (PDB code 1IRN [72]). Amides exposed to solvent > 0.5 Å2 that are solvent inaccessible in the highest resolution cognate X-ray structure are indicated as diamonds. The rmsd (σ) and correlation coefficient (r) values are indicated.
The poor statistics for the Cp rubredoxin-based predictions largely arise from the highly inaccurate prediction for the exchange behavior of Phe 30. The amide hydrogen of this residue is predicted to be well exposed to solvent in the Cp rubredoxin-based homology model of Pf rubredoxin, although it is completely inaccessible in each of the Pf rubredoxin X-ray structures. In the Pf rubredoxin X-ray structures, the amide hydrogen of Phe 30 is hydrogen bonded to the structurally buried carboxylate sidechain of Glu 15. When the Pro 15 of the Cp rubredoxin was converted to a glutamate, the automated application of the Modeler program was unable to predict the local conformational rearrangement that leads to the burial of the glutamate sidechain and instead the carboxylate was directed out into the solvent phase. Along with the substitution of Trp for Tyr at residue 4, the Pro to Glu substitution at residue 15 is considered to provide a characteristic signature of the hyperthermophile archaeal rubredoxins as compared to the mesophile eubacterial rubredoxins and is believed to account for a significant proportion of the difference in thermal stability exhibited by the Pyrococcus furiosus rubredoxin relative to the Cp protein [56,73]. Removal of the exchange rate prediction for Phe 30 reduces the rmsd for the log kOH− values to 1.47 with a correlation coefficient of 0.82, considerably more similar to the values derived from the homology modeling predictions for the other three proteins.
Although the rmsd for the log kOH− predictions derived from the yeast FKBP12 is only 17% higher than the average of the values obtained from the three human FKBP12 structures, the homology models for the other three proteins yield rmsd values close to 50% higher (excluding the aberrant prediction for Phe 30 of Pf rubredoxin). The correlation coefficients for the homology models are on average 0.10 less than the predictions derived from the X-ray structures of the proteins under study. As is well known, homology modeling is markedly less robust when mutational variations induce substantial changes in local structure as illustrated by the burial of the Glu 15 carboxylate in the hyperthermophile rubredoxins. In this example, the hydrogen exchange analysis cleanly identifies the site of error in the homology modeling.
3.5. Predicting hydrogen exchange in ubiquitin from the models of an NMR solution structure
NMR structure determinations were reported for each of these four proteins in the early days of solution structure technique development. However, NOE-based structures utilizing current analysis protocols have only been deposited for ubiquitin and CI2. Although ubiquitin continues to be the most widely used protein for demonstrating the utility of residual dipolar couplings in the determination of protein structure and conformational dynamics, the most recently deposited solution structure of the free ubiquitin monomer utilizing a conventional NOE-based protocol is that of Akasaka and colleagues (PDB code 1V80 [74]). By many criteria, the 1V80 study is a typical solution structure analysis. The CYANA structure analysis program [75] was used to generate 100 views of a single structure solution to the experimental constraints and the 10 models yielding the lowest DYANA target function values were then deposited. The mean rmsd value of 1.1 Å obtained for these 10 models with respect to the backbone of the 1UBQ X-ray structure compares quite favorably to the rmsd values obtained for other pairs of analogous NMR and X-ray structures. Garbuzynskiy and colleagues [8] compared structures for 60 proteins determined by both X-ray and NMR methods in which no large scale structural differences were observed. For only 11 of these 60 pairs of protein structure determinations do the backbones of the X-ray and mean NMR structures differ by 1.0 Å or less, and for only one of these 11 proteins was the NMR structure deposited in the Protein Data Bank before the deposition of the corresponding crystal structure.
Akasaka and colleagues identified model 3 as the most representative from the set of 10 models in the 1V80 structure determination. Using this model, essentially no correlation was obtained between the predicted and observed hydrogen exchange rates (Fig. 7). Analogous calculations for the other nine models in the 1V80 structure determination indicate a general inability to yield statistically significant predictions (Table 1, Fig. S1 - Supplementary Materials). The average rmsd error in the log kOH− predictions is 80% larger than the average dispersion for the experimental log rate constants. Since averaging the reactivity of highly exposed amides over the entire 2NR2 ensemble yields an rmsd of only 0.48 with a correlation coefficient of 0.95 [5], the approximations underlying the continuum dielectric prediction of conformer reactivity are not a limiting aspect of this comparison.
Fig. 7.

Correlation of ubiquitin hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann-derived pK values predicted from the third model from the 1V80 NMR structure [74], identified by those authors as the most representative model in the pdb file. Consistent with the analysis of the X-ray and homology model-derived predictions for ubiquitin hydrogen exchange, Asp 52 is not included. The rmsd (σ) and correlation coefficient (r) values are indicated.
Table 1.
Hydrogen exchange predictions derived from the 1V80 solution structure of ubiquitin
| Model | Δlog kOH− | correlation | access in X-raya |
|---|---|---|---|
| 1 | 1.57 | −0.36 | 9 |
| 2 | 1.47 | 0.21 | 10 |
| 3 | 1.01 | 0.26 | 10 |
| 4 | 1.61 | 0.23 | 9 |
| 5 | 2.02 | −0.18 | 10 |
| 6 | 1.02 | 0.39 | 10 |
| 7 | 1.86 | 0.23 | 10 |
| 8 | 1.81 | 0.19 | 10 |
| 9 | 1.66 | −0.27 | 9 |
| 10 | 1.22 | 0.46 | 9 |
| ave | 1.52 | 0.12 | 9.6 |
Number of amide hydrogens exposed to solvent > 0.5 Å that are similarly exposed in the 1.00 Å ubiquitin X-ray structure (16 total).
Another relevant measure of similarity between two protein structures is the number of amide hydrogens that are solvent-exposed in both models. In the earlier figures of this manuscript, static solvent-exposed amides that are not similarly exposed in the highest resolution X-ray structure have been symbolized as diamonds. Among each of the 10 models in the 1V80 NMR structure, the amide hydrogens for between 5 and 8 residues are accessible by > 0.5 Å2 in either the NMR model or the highest resolution X-ray structure, but not in both. In contrast, the set of static solvent-exposed amides for 1UBQ and 1YIW X-ray structures differ from the 1.00 Å resolution ubiquitin structure at 1 and 3 residues, respectively (Fig. 4C). The large discrepancy for the 1V80 NMR models primarily reflects the fact that significantly fewer amides are solvent-exposed than for the 16 residues in the highest resolution X-ray structure (Table 1).
A related question is whether a given model structure is within the energetically accessible neighborhood of conformations that provide accurate predictions of the hydrogen exchange reactivity throughout the protein-aqueous interface. Kolmogorov-Smirnov analysis [76] for the distribution of intra- and inter-ensemble pairwise structural rmsd values provides a robust means of assessing the degree to which two sets of protein conformations are mutually consistent with a single underlying distribution or are either partially or fully disjoint [6]. In the simplest application, analysis of the structural rmsd values provide insight into whether the three ubiquitin X-ray structures, the 1BT0-derived homology model or any of the 1V80 NMR models can be viewed as lying within the conformational distribution sampled by the 144 models of the 2NR2 ensemble. Each of the three X-ray structures yield smaller mean backbone heavy atom rmsd values to these 144 conformations than do the majority of the 2NR2 ensemble members with respect to the other 143 conformations (Fig. 8), indicative of the fact that the X-ray structures lie near the center of the 2NR2 conformational distribution. In contrast, the 1BT0-derived homology model lies near the edge of the 2NR2 distribution while each of the 1V80 NMR structures lies significantly farther from the 2NR2 distribution than does any member of that distribution with respect to the rest of the model ensemble. Analysis of the mean rmsd values for all heavy atoms of the structural models yields a similar conclusion (Fig. S2 – Supplementary Materials).
Fig. 8.

Mean rmsd values among conformers of ubiquitin. Indicated in gray is the distribution of rmsd values for each model of the NMR relaxation-restrained 2NR2 ensemble [52] with respect to the other 143 ensemble models for the backbone C, Cα, N and O atoms. The mean rmsd values for the 1UBQ [84], 1YIW [29] and K29Q variant X-ray structures with respect to the 144 2NR2 ensemble models are denoted by (●), while that of the 1BT0-derived [71] homology model is indicated by (■). The mean rmsd values for each of the 10 models of the 1V80 NMR solution structure analysis [74] with respect to the 2NR2 ensemble models are denoted by (◆).
The practical significance of the disjointedness between the 2NR2 ensemble and the models of 1V80 NMR structure can be illustrated by a molecular docking protocol in which a large number of initial protein conformations are utilized in parallel. To the degree that the 2NR2 ensemble accurately represents 144 random samplings of the Boltzmann distribution, a protocol that is based on selecting protein conformations in an energetically balanced fashion would very rarely utilize the conformational space represented by the models in 1V80 NMR structure.
3.6. Predicting hydrogen exchange in CI2 from the models of an NMR solution structure
The solution structure of CI2 was reported by Poulsen and colleagues in 1991 (PDB code 3CI2 [77]). Fig. 9A illustrates the hydrogen exchange predictions obtained using their first model. The statistics for model 1 are modestly better than the average values obtained from all 20 models (Table 2). Analogous to the considerations for Asp 52 of ubiquitin, the gauche χ1 rotamer states for Asp 45 and Asp 55 in the 3CI2 models led to their being removed from the hydrogen exchange analysis.
Fig. 9.

Correlation of CI2 hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann-derived pK values predicted from the first (A) and twelfth (B) models of the original NMR solution structural analysis [77]. Amides exposed to solvent > 0.5 Å2 that are solvent inaccessible in the highest resolution cognate X-ray structure are indicated as diamonds. The rmsd (σ) and correlation coefficient (r) values are indicated.
Table 2.
Hydrogen exchange predictions derived from the 3CI2 solution structure analysis
| Model | Δlog kOH− | correlation | access in X-raya |
|---|---|---|---|
| 1 | 2.32 | 0.58 | 8 |
| 2 | 2.57 | −0.94 | 4 |
| 3 | 2.52 | 0.72 | 9 |
| 4 | 2.08 | 0.62 | 6 |
| 5 | 2.36 | 0.57 | 6 |
| 6 | 1.89 | 0.84 | 7 |
| 7 | 2.14 | 0.77 | 9 |
| 8 | 1.87 | 0.81 | 9 |
| 9 | 1.68 | 0.35 | 7 |
| 10 | 3.47 | 0.45 | 8 |
| 11 | 3.92 | 0.06 | 5 |
| 12 | 1.24 | 0.85 | 6 |
| 13 | 3.43 | 0.13 | 9 |
| 14 | 2.74 | 0.03 | 6 |
| 15 | 1.71 | 0.72 | 9 |
| 16 | 3.18 | −0.20 | 7 |
| 17 | 1.63 | 0.69 | 6 |
| 18 | 2.74 | 0.72 | 8 |
| 19 | 3.60 | 0.38 | 9 |
| 20 | 3.87 | 0.22 | 6 |
| ave | 2.55 | 0.42 | 7.2 |
Number of amide hydrogens exposed to solvent > 0.5 Å2 that are similarly exposed in the CI2 X-ray structures (14 total).
Similar to the case of the 1V80 NMR structure of ubiquitin, the average rmsd error in the log kOH− predictions from the 3CI2 models is 70% larger than the average dispersion for the experimental log rate constants. Only 1 of the 20 models from the original 3CI2 study (model 12 – Fig. 9B) yielded hydrogen exchange predictions that were comparable to those from the X-ray and homology models. However, it should be noted that only 6 of the 14 amides that are exposed to solvent in all three CI2 X-ray structures are also accessible in this model (Table 2). Indeed, on average, only 7.2 of these 14 residues are exposed to solvent in the 20 models of the original NMR structure. Although several of these 20 models yielded reasonably large correlation coefficients (Table 2), as illustrated by the −0.94 correlation coefficient of model 2, the optimized slope of the correlation between the predicted and observed values often markedly deviates from the expected unit slope.
More recently, the NOE distance bounds and other NMR restraints for the 3CI2 set were reanalyzed in the RECOORD study [78] which used CYANA [79] and CNS [80]-based structure refinement with explicit solvation applied to 545 deposited NMR structures. The central result of the RECOORD analysis was that a consistent protocol with current versions of CYANA [79] and CNS [80] using explicitly solvated protein models produced an increase in the average rmsd values of 0.4 Å for the well-ordered regions of the individual families of structures without compromising the distance and dihedral angle restraints, while markedly improving the local geometry as assessed by PROCHECK [81] and WHATCHECK [82]. The RECOORD study offered compelling evidence that a large fraction of the deposited NMR protein structure families are overly precise.
The hydrogen exchange predictions from both the CYANA- and CNS-based refinements of CI2 are markedly superior to those obtained from the original 3CI2 NMR structure (Table 3). The mean rmsd for the log kOH− predictions from all 25 models in the CYANA-based refinement was 1.22 with a mean correlation coefficient of 0.70, comparable to the values obtained from the homology model based on the 36% sequence identity 1VBW crystal structure. While model 10 yielded hydrogen exchange prediction statistics that were similar to the average performance (Fig. 10B), several models (e.g., model 1 – Fig. 10A) yielded predictions comparable to those from the CI2 X-ray structures. The RECOORD re-refinement also led to sets of static solvent-exposed amides that are markedly more similar to the X-ray structures (Table 3) than those obtained in the original 3CI2 analysis. Analysis of the hydrated CNS-based refinement structures provided similarly robust predictions.
Table 3.
Hydrogen exchange predictions derived from the CYANA-based RECOORD re-refinement of CI2
| Model | Δlog kOH− | correlation | access in X-raya |
|---|---|---|---|
| 1 | 1.06 | 0.89 | 10 |
| 2 | 1.41 | 0.78 | 10 |
| 3 | 1.49 | 0.68 | 10 |
| 4 | 1.07 | 0.59 | 11 |
| 5 | 1.33 | 0.60 | 10 |
| 6 | 1.00 | 0.83 | 9 |
| 7 | 1.08 | 0.59 | 10 |
| 8 | 1.06 | 0.80 | 9 |
| 9 | 1.28 | 0.70 | 10 |
| 10 | 1.19 | 0.73 | 10 |
| 11 | 1.60 | 0.52 | 10 |
| 12 | 0.89 | 0.92 | 9 |
| 13 | 0.92 | 0.90 | 8 |
| 14 | 1.61 | 0.36 | 10 |
| 15 | 1.23 | 0.84 | 11 |
| 16 | 0.98 | 0.65 | 12 |
| 17 | 1.44 | 0.52 | 11 |
| 18 | 1.21 | 0.79 | 9 |
| 19 | 1.60 | 0.69 | 11 |
| 20 | 1.30 | 0.56 | 10 |
| 21 | 1.29 | 0.68 | 9 |
| 22 | 1.14 | 0.73 | 11 |
| 23 | 1.18 | 0.66 | 10 |
| 24 | 1.15 | 0.65 | 11 |
| 25 | 0.90 | 0.79 | 9 |
| ave | 1.22 | 0.70 | 10.0 |
Number of amide hydrogens exposed to solvent > 0.5 Å that are similarly exposed in the CI2 X-ray structures (14 total).
Fig. 10.

Correlation of CI2 hydroxide-catalyzed hydrogen exchange log rate constants with Poisson-Boltzmann-derived pK values predicted from the first (A) and tenth (B) models of the RECOORD NMR solution structural analysis. Amides exposed to solvent > 0.5 Å2 that are solvent inaccessible in the highest resolution cognate X-ray structure are indicated as diamonds. The rmsd (σ) and correlation coefficient (r) values are indicated.
The Asp 55 amide hydrogen is exposed to solvent by more than 0.5 Å2 in 20 of the 25 RECOORD re-refined 3CI2 models. Nevertheless, this residue was excluded from the hydrogen exchange analysis, in part, due to its gauche χ1 rotamer conformation in these re-refined structures. More importantly, a cross-hydrogen bond 15N-13C′ scalar coupling has been reported between the Asp 55 amide and the Asp 52 carbonyl [83], inconsistent with the local structure reported in the RECOORD models. All three X-ray structures indicate this same hydrogen bond.
Since the RECOORD re-refinement of CI2 markedly improved the peptide acidity predictions, it is warranted to consider how this re-analysis affects the overall differences in structure vis-a-vis the highest resolution X-ray structure. For the 20 models in the original 3CI2 structure study, the mean rmsd value with respect to the 1LW6 X-ray structure is 1.55 Å for backbone atoms and 2.29 Å for all heavy atoms. These values are reduced to 1.10 Å and 1.81 Å, respectively, for the 25 models from the RECOORD re-refinement. All but 1 of the 25 RECOORD re-refined CI2 models are closer to the highest resolution X-ray coordinates than is any model of the original NMR structure. The substantial increased consistency between the sets of static solvent-exposed amides for the RECOORD re-refinement and X-ray structures likely implies an appreciable improvement in the modeling of local mainchain geometry due to the inclusion of a more realistic explicitly hydrated force field during the structural refinement protocol.
The RECOORD re-refinement yielded a systematic shift in the NMR models toward the CI2 X-ray coordinates, while simultaneously improving the peptide acidity predictions. The quality of hydrogen exchange rate predictions provided by the CI2 X-ray structures is consistent with a close preservation of protein conformation between crystal and solution phase, as has previously been argued on the basis of chemical shifts and cross-hydrogen bond coupling interactions [83].
4. Conclusion
The experimental rates at which hydroxide-catalyzed exchange of amide hydrogens occur at sites along the protein backbone are determined by the reactivity of all of the energetically accessible conformations in the Boltzmann distribution. Nevertheless, for each of the four proteins considered in this study, individual structural models can prove sufficient to provide statistically significant predictions of these exchange rates. Since for many practical applications it is desirable to identify a most representative structure for the conformation of a protein in solution, chemical reactivity-based hydrogen exchange analysis offers a powerful experimental approach to assessing the degree to which a given model fulfills that most representative ideal.
The high resolution X-ray structures of rubredoxin, FKBP12, CI2 and ubiquitin provide more robust predictions of the exchange rates for backbone amide hydrogens that are exposed to solvent in the static structures than do either modeled structures based on sequence identities near 60% or NMR solution structure analyses (Table 4). The rmsd between the backbone atoms of each model and the highest resolution structure of that protein generally follows the correlation coefficient for the hydrogen exchange predictions. The RECOORD analysis of the NMR restraints of CI2 yielded hydrogen exchange predictions that were, on average, comparable to those derived from the 1BT0-based homology model. In contrast, the models of the 1V80 solution structure were unable to provide statistically significant predictions of ubiquitin hydrogen exchange, presumably due to a combination of conformations that are more divergent from the high resolution X-ray structures and the relatively limited range of experimental exchange rates for the static solvent exposed amides of this protein.
Table 4.
Summary of hydrogen exchange predictions derived from X-ray, homology modeling and NMR structures
| protein | method | PDB code | resolution | Δlog kOH− | correlation | rmsdc |
|---|---|---|---|---|---|---|
| ubiquitin | X-ray | a | 1.00 Å | 0.92 | 0.79 | --- |
| X-ray | 1YIW | 1.39 Å | 1.03 | 0.61 | 0.66 Å | |
| X-ray | 1UBQ | 1.80 Å | 1.17 | 0.82 | 0.43 Å | |
| homology | 1BT0 | 1.70 Å | 1.44 | 0.61 | 0.61 Å | |
| NMR | 1V80 | 1.52 | 0.12 | 1.10 Å | ||
| CI2 | X-ray | 1LW6 | 1.50 Å | 0.83 | 0.83 | --- |
| X-ray | 2CI2 | 2.00 Å | 1.04 | 0.77 | 0.46 Å | |
| X-ray | 2SNI | 2.10 Å | 0.84 | 0.83 | 0.38 Å | |
| homology | 1VBW | 0.93 Å | 1.38 | 0.76 | 1.15 Å | |
| NMR | 3CI2 | 2.55 | 0.42 | 1.55 Å | ||
| NMR | b | 1.22 | 0.70 | 1.10 Å | ||
| FKBP12 | X-ray | 2PPN | 0.92 Å | 0.82 | 0.85 | --- |
| X-ray | 1D6O | 1.85 Å | 1.07 | 0.82 | 0.32 Å | |
| X-ray | 1FKK | 2.20 Å | 1.14 | 0.85 | 0.40 Å | |
| homology | 1YAT | 2.50 Å | 1.18 | 0.74 | 0.84 Å | |
| rubredoxin | X-ray | 1BQ8 | 1.10 Å | 0.96 | 0.93 | --- |
| X-ray | 1VCX | 1.50 Å | 0.97 | 0.94 | 0.22 Å | |
| X-ray | 1CAA | 1.80 Å | 1.03 | 0.93 | 0.15 Å | |
| homology | 1IRN | 1.20 Å | 1.85 | 0.69 | 0.54 Å |
Hydrogen exchange analysis appears capable of distinguishing the relative quality of protein X-ray structures as representations of the conformation in solution. Although resolution limit is not a perfect measure of structure quality, the ability to observe modest correlation between resolution limit and the accuracy of hydrogen exchange predictions supports the interpretation that the increased structural accuracy which is achieved by pursuing X-ray analysis to the maximum diffraction limit may often be reflected in the increased accuracy with which the structure of the protein in solution is represented. In the absence of any apparent evolutionary pressure, the differences in electrostatic environments yield up to billion-fold variations in the rate at which sterically accessible amides undergo reactions at the protein surface. Although hydroxide-catalyzed peptide exchange is arguably the simplest reaction of proteins, its exquisite sensitivity to protein conformation strikingly demonstrates the rigorous demands placed upon establishing chemical accuracy in protein modeling.
Supplementary Material
Highlights.
The acidity of protein amides is exquisitely sensitive to the surrounding charge and dielectric volume distribution.
Poisson-Boltzmann (PB) predictions appear sensitive to the resolution of independent crystal structure determinations.
X-ray structures of four proteins yield amide exchange predictions superior to homology model and NMR structures.
The previously reported RECOORD re-refinement of chymotrypsin inhibitor 2 markedly improves the peptide acidity predictions.
Acknowledgments
This work was supported in part by National Institutes of Health grant GM 088214.
Abbreviations
- PDB
protein data bank
- CI2
chymotrypsin inhibitor 2
- FKPB
FK506 binding protein
- rmsd
root mean square deviation
- NOE
nuclear Overhauser enhancement
- PB
Poisson-Boltzmann
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometrical approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 2.Cozzini P, Kellog GE, Spyrakis F, Abraham DJ, Constantino G, Emerson A, Fanelli F, Gohlke H, Kuhn LA, Morris GA, Orozco M, Pertinhez TA, Rizzi M, Sotriffer CA. Target Flexibility: An Emerging Consideration in Drug Discovery and Design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meiler J, Baker D. ROSETTALIGAND: Protein-Small Molecule Docking with Full Side-Chain Flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 4.Davis IW, Baker D. ROSETTALIGAND docking with full ligand and receptor flexibility. J Mol Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 5.Hernández G, Anderson JS, LeMaster DM. Experimentally Assessing Molecular Dynamics Sampling of the Protein Native State Conformational Distribution. Biophys Chem. 2012;163–164:21–34. doi: 10.1016/j.bpc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernández G, Anderson JS, LeMaster DM. Assessing the native state conformational distribution of ubiquitin by peptide acidity. Biophys Chem. 2010;153:70–82. doi: 10.1016/j.bpc.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garbuzynskiy SO, Melnik BS, Lobanov MY, Finkelstein AV, Galzitskaya OV. Comparison of X-ray and NMR structures: Is there a systematic difference in residues contacts between X-ray and NMR-resolved protein structures? Proteins. 2005;60:139–147. doi: 10.1002/prot.20491. [DOI] [PubMed] [Google Scholar]
- 9.Andrec M, Snyder DA, Zhou ZY, Young J, Montelione GT, Levy RM. A large data set comparision of protein structures determined by crystallography and NMR: Statistical test for structural differences and the effect of crystal packing. Proteins. 2007;69:449–465. doi: 10.1002/prot.21507. [DOI] [PubMed] [Google Scholar]
- 10.Brünger AT, Campbell RL, Clore GM, Gronenborn AM, Karplus M, Petsko GA, Teeter MM. Solution of a protein crystal structure with a model obtained from NMR interproton distance restraints. Science. 1987;235:1049–1053. doi: 10.1126/science.235.4792.1049. [DOI] [PubMed] [Google Scholar]
- 11.Chen YW, Dodson EJ, Kleywegt GJ. Does NMR mean “Not for molecular replacement”? Using NMR-based search models to solve protein crystal structures. Structure. 2000;8:R213–R220. doi: 10.1016/s0969-2126(00)00524-4. [DOI] [PubMed] [Google Scholar]
- 12.Qian B, Raman S, Das R, Bradley P, McCoy AJ, Read RJ, Baker D. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–264. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramelot TA, Raman S, Kuzin AP, Xiao R, Ma LC, Acton TB, Hunt JF, Montelione GT, Baker D, Kennedy MA. Improving NMR protein structure quality by ROSETTA refinement: A molecular replacement study. Proteins. 2009;75:147–167. doi: 10.1002/prot.22229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diamond R. On the use of normal modes in thermal parameter refinement: Theory and application to the bovine pancreatic trypsin inhibitor. Acta Crystallogr Sect A. 1990;46:425–435. doi: 10.1107/s0108767390002082. [DOI] [PubMed] [Google Scholar]
- 15.Kidera A, Go N. Refinement of protein dynamic structure: Normal mode refinement. Proc Natl Acad Sci U S A. 1990;87:3718–3722. doi: 10.1073/pnas.87.10.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schomaker V, Trueblood KN. On the rigid-body motion of molecules in crystals. Acta Cryst. 1968;24:63–76. [Google Scholar]
- 17.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst D. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 18.Moore PB. On the relationship between diffraction patterns and motions in macromolecular crystals. Structure. 2009;17:1307–1315. doi: 10.1016/j.str.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Bai YW, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen-exchange. Proteins: Struct, Funct, Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai Y, Englander SW. Hydrogen-bond strength and beta-sheet propensities - the role of a side-chain blocking effect. Proteins: Struct, Funct, Genet. 1994;18:262–266. doi: 10.1002/prot.340180307. [DOI] [PubMed] [Google Scholar]
- 21.Bai YW, Milne JS, Mayne L, Englander SW. Protein stability parameters measured by hydrogen exchange. Proteins: Struct, Funct, Genet. 1994;20:4–14. doi: 10.1002/prot.340200103. [DOI] [PubMed] [Google Scholar]
- 22.Fogolari F, Esposito G, Viglino P, Briggs JM, McCammon JA. pKa shift effects on backbone amide base-catalyzed hydrogen exchange rates in peptides. J Am Chem Soc. 1998;120:3735–3738. [Google Scholar]
- 23.Avbelj F, Baldwin RL. Origin of the neighboring residue effect on peptide backbone conformation. Proc Natl Acad Sci USA. 2004;101:10967–10972. doi: 10.1073/pnas.0404050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson JS, Hernández G, LeMaster DM. Backbone conformational dependence of peptide acidity. Biophys Chem. 2009;141:124–130. doi: 10.1016/j.bpc.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 25.LeMaster DM, Anderson JS, Hernández G. Role of native-state structure in rubredoxin native-state hydrogen exchange. Biochemistry. 2006;45:9956–9963. doi: 10.1021/bi0605540. [DOI] [PubMed] [Google Scholar]
- 26.Anderson JS, Hernández G, LeMaster DM. A billion-fold range in acidity for the solvent-exposed amides of Pyrococcus furiosus rubredoxin. Biochemistry. 2008;47:6178–6188. doi: 10.1021/bi800284y. [DOI] [PubMed] [Google Scholar]
- 27.Hernández G, Anderson JS, LeMaster DM. Polarization and polarizability assessed by protein amide acidity. Biochemistry. 2009;48:6482–6494. doi: 10.1021/bi900526z. [DOI] [PubMed] [Google Scholar]
- 28.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285:1733–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 29.Bang D, Makhatadze GI, Tereshko V, Kossiakoff AA, Kent SB. Total chemical synthesis and X-ray crystal structure of a protein diastereomer: [D-Gln 35]ubiquitin. Angew Chem Int Ed Engl. 2005;44:3852–3856. doi: 10.1002/anie.200463040. [DOI] [PubMed] [Google Scholar]
- 30.Parker LL, Houk AR, Jensen JH. Cooperative hydrogen bonding effects are key determinants of backbone amide proton chemical shifts in proteins. J Am Chem Soc. 2006;128:9863–9872. doi: 10.1021/ja0617901. [DOI] [PubMed] [Google Scholar]
- 31.McPhalen CA, James MNG. Crystal and molecular structure of the serine proteinase inhibitor CI-2 from barley seeds. Biochemistry. 1987;26:261–269. doi: 10.1021/bi00375a036. [DOI] [PubMed] [Google Scholar]
- 32.Jackson SE, Moracci M, elMasry N, Johnson CM, Fersht AR. Effect of cavity-creating mutations in the hydrophobic core of chymotrypsin inhibitor 2. Biochemistry. 1993;32:11259–11269. doi: 10.1021/bi00093a001. [DOI] [PubMed] [Google Scholar]
- 33.Radisky ES, Koshland JDE. A clogged gutter mechanism for protease inhibitors. Proc Natl Acad Sci USA. 2002;99:10316–10321. doi: 10.1073/pnas.112332899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial constraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 35.Rocchia W, Sridharan S, Nicholls A, Alexov E, Chiabrera A, Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: Applications to the molecular systems and geometric objects. J Comput Chem. 2002;23:128–137. doi: 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- 36.LeMaster DM, Anderson JS, Hernández G. Peptide conformer acidity analysis of protein flexibility monitored by hydrogen exchange. Biochemistry. 2009;48:9256–9265. doi: 10.1021/bi901219x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson JS, Hernandez G, LeMaster DM. Sidechain conformational dependence of hydrogen exchange in model peptides. Biophys Chem. 2010;151:61–70. doi: 10.1016/j.bpc.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 38.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 39.Ibarra-Molero B, Loladze VV, Makhatadze GI, Sanchez-Ruiz JM. Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry. 1999;38:8138–8149. doi: 10.1021/bi9905819. [DOI] [PubMed] [Google Scholar]
- 40.Yu L, Fesik SW. pH titration of the histidine residues of cyclophilin and FK506 binding protein in the absence and presence of immunosuppressant ligands. Biochim Biophys Acta. 1994;1209:24–32. doi: 10.1016/0167-4838(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 41.Sridharan S, Nicholls A, Honig B. A new vertex algorithm to calculate solvent accessible surface-areas. FASEB J. 1992;61:A174. [Google Scholar]
- 42.Rashin AA. Buried surface area, conformational entropy, and protein stability. Biopolymers. 1984;23:1605–1620. doi: 10.1002/bip.360230813. [DOI] [PubMed] [Google Scholar]
- 43.Molday RS, Kallen RG. Substituent effects on amide hydrogen exchange rates in aqueous solution. J Am Chem Soc. 1972;94:6739–6745. [Google Scholar]
- 44.Wang WH, Cheng CC. General base catalyzed proton exchange in amides. Bull Chem Soc Jpn. 1994;67:1054–1057. [Google Scholar]
- 45.Eigen M. Proton transfer, acid-base catalysis, and enzymatic hydrolysis. (I) Elementary processes. Angew Chem Int Ed. 1964;3:1–19. [Google Scholar]
- 46.Luz Z, Meiboom S. The activation energies of proton transfer reactions in water. J Am Chem Soc. 1964;86:4768–4769. [Google Scholar]
- 47.Tolbert LM, Solntsev KM. Excited-state proton transfer: From constrained systems to “Super” photoacids to superfast proton transfer. Acc Chem Res. 2002;35:19–27. doi: 10.1021/ar990109f. [DOI] [PubMed] [Google Scholar]
- 48.Leiderman P, Genosar L, Huppert D. Excited-state proton transfer: Indication of three steps in the dissociation and recombination process. J Phys Chem A. 2005;109:5965–5977. doi: 10.1021/jp050037b. [DOI] [PubMed] [Google Scholar]
- 49.LeMaster DM, Anderson JS, Hernández G. Spatial distribution of dielectric shielding in the interior of Pyrococcus furiosus rubredoxin as sampled in the subnanosecond timeframe by hydrogen exchange. Biophys Chem. 2007;129:43–48. doi: 10.1016/j.bpc.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seeman JI. Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin-Hammett/Winstein-Holness Kinetics. Chem Rev. 1983;83:83–134. [Google Scholar]
- 51.Bau R, Rees DC, Kurtz DM, Jr, Scott RA, Huang HS, Adams MWW, Eidsness MK. Crystal-structure of rubredoxin from Pyrococcus furiosus at 0.95 Angstrom resolution, and the structures of N-terminal methionine and formylmethionine variants of Pf Rd. Contributions of N-terminal interactions to thermostability. J Biol Inorg Chem. 1998;3:484–493. [Google Scholar]
- 52.Richter B, Gsponer J, Varnai P, Salvatella X, Vendruscolo M. The MUMO (minimal under-restraining minimal over-restraining) method for the determination of native state ensembles of proteins. J Biomol NMR. 2007;37:117–135. doi: 10.1007/s10858-006-9117-7. [DOI] [PubMed] [Google Scholar]
- 53.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, Becker S, Meiler J, Grubmuller H, Griesinger C, deGroot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 54.Fenwick RB, Esteban-Martin S, Richter B, Lee D, Walter KFA, Milovanovic D, Becker S, Lakomek NA, Griesinger C, Salvatella X. Weak Long-Range Correlated Motions in a Surface Patch of Ubiquitin Involved in Molecular Recognition. J Am Chem Soc. 2011;133:10336–10339. doi: 10.1021/ja200461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kurihara K, Tanaka I, Chatake T, Adams MWW, Jenney JFE, Moiseeva N, Bau R, Niimura N. Neutron crystallographic study on rubredoxin from Pyrococcus furiosus by BIX-3, a single-crystal diffractometer for biomacromolecules. Proc Natl Acad Sci USA. 2004;101:11215–11220. doi: 10.1073/pnas.0403807101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Day MW, Hsu BT, Joshua-Tor L, Park JB, Zhou ZH, Adams MWW, Rees DC. X-ray crystal structures of the oxidized and reduced forms of the rubredoxin from the marine hyperthermophilic archaebacterium Pyrococcus furiosus. Prot Sci. 1992;1:1494–1507. doi: 10.1002/pro.5560011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardberg AS, delCastillo AR, Weiss KL, Meilleur F, Blakeley MP, Myles DA. Unambiguous determination of H-atom positions: Results from neutron and high-resolution X-ray crystallography. Acta Cryst D. 2010;66:558–567. doi: 10.1107/S0907444910005494. [DOI] [PubMed] [Google Scholar]
- 58.Szep S, Park S, Boder ET, VanDuyne GD, Saven JG. Structural coupling between FKBP12 and buried water. Proteins. 2008;74:603–611. doi: 10.1002/prot.22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burkhard P, Taylor P, Walkinshaw MD. X-ray structures of small ligand-FKBP complexes provide an estimate for the hydrophobic interaction energies. J Mol Biol. 2000;295:953–962. doi: 10.1006/jmbi.1999.3411. [DOI] [PubMed] [Google Scholar]
- 60.Wilson KP, Yamashita MM, Sintchak MD, Rotstein SH, Murcko MA, Boger J, Thomson JA, Fitzgibbon MJ, Black JR, Navia MA. Comparative X-ray structures of the major binding protein for the immunosupressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Cryst D. 1995;51:511–521. doi: 10.1107/S0907444994014514. [DOI] [PubMed] [Google Scholar]
- 61.McPhalen CA, James MN. Structural comparison of two serine proteinase-protein inhibitior complexes: Eglin c-subtilisin Carlsberg and CI-2-subtilisin. Biochemistry. 1988;27:6582–6598. [PubMed] [Google Scholar]
- 62.Garcia-Moreno B, Dwyer JJ, Gittis AG, Lattman EE, Spencer DS, Stites WE. Experimental Measurement of the Effective Dielectric in the Hydrophobic Core of a Protein. Biophys Chem. 1997;64:211–224. doi: 10.1016/s0301-4622(96)02238-7. [DOI] [PubMed] [Google Scholar]
- 63.Sundd M, Iverson N, Ibarra-Molero B, Sanchez-Ruiz JM, Robertson AD. Electrostatic interactions in ubiquitin: Stabilization of carboxylates by lysine amino groups. Biochemistry. 2002;41:7586–7596. doi: 10.1021/bi025571d. [DOI] [PubMed] [Google Scholar]
- 64.Mori S, Abeygunawardana C, Berg JM, vanZijl PCM. NMR study of rapidly exchanging backbone amide protons in staphylococcal nuclease and the correlation with structural and dynamic properties. J Am Chem Soc. 1997;119:6844–6852. [Google Scholar]
- 65.Skinner JJ, Lim WK, Bedard S, Black BE, Englander SW. Protein hydrogen exchange: Testing current models. Prot Sci. 2012;21:987–995. doi: 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grotthuss CJT. Sur la décomposition de l’eau et des corps qu’elle tient en dissolution à l’aide de l’électricité galvanique. Ann Chim LVIII. 1806:54–74. [Google Scholar]
- 67.Karp DA, Gittis AG, Stahley MR, Fitch CA, Stites WE, García-Moreno BE. High apparent dielectric constant inside a protein reflects structural reorganization coupled to the ionization of an internal Asp. Biophys J. 2007;92:2041–2053. doi: 10.1529/biophysj.106.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Damjanovic A, Brooks BR, Garcia-Moreno B. Conformational relaxation and water penetration coupled to ionization of internal groups in proteins. J Phys Chem A. 2011;115:4042–4053. doi: 10.1021/jp110373f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karp DA, Stanley MR, Garcia-Moreno B. Conformational consequences of ionization of Lys, Asp, and Glu buried at position 66 in staphylococcal nuclease. Biochemistry. 2010;49:4138–4146. doi: 10.1021/bi902114m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rotonda J, Burbaum JJ, Chan HK, Marcy AI, Becker JW. Improved calcineurin inhibition by yeast FKBP12-drug complexes. Crystallographic and functional analysis. J Biol Chem. 1993;268:7607–7609. doi: 10.2210/pdb1yat/pdb. [DOI] [PubMed] [Google Scholar]
- 71.Rao-naik C, delaCruz W, Laplaza JM, Tan S, Callis J, Fisher AJ. The Rub-1 family of ubiquitin-like proteins. Crystal structure of Arabidopsis Rub-1 and expression of multiples rubs in Arabidopsis. J Biol Chem. 1998;273:34976–34982. doi: 10.1074/jbc.273.52.34976. [DOI] [PubMed] [Google Scholar]
- 72.Dauter Z, Wilson KS, Sieker LC, Moulis JM, Meyer J. Zinc- and iron-rubredoxins from Clostridium pasteurianum at atomic resolution: A high precision model of a ZnS4 coordination unit in a protein. Proc Natl Acad Sci USA. 1996;93:8836–8840. doi: 10.1073/pnas.93.17.8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eidsness MK, Richie KA, Burden AE, Kurtz DM, Scott RA. Dissecting contributions to the thermostability of Pyrococcus furiosus rubredoxin: Beta-sheet chimeras. Biochemistry. 1997;36:10406–10413. doi: 10.1021/bi970110r. [DOI] [PubMed] [Google Scholar]
- 74.Kitahara R, Yokoyama S, Akasaka K. NMR snapshots of a fluctuating protein structure: Ubiquitin at 30 bar-3 kbar. J Mol Biol. 2005;347:277–285. doi: 10.1016/j.jmb.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 75.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 76.Eadie WT, Drijard D, James FE, Roos M, Sadoulet B. Statistical Methods in Experimental Physics. North-Holland, Amsterdam: 1971. [Google Scholar]
- 77.Ludvigsen S, Shen HY, Kjaer M, Madsen JC, Poulsen FM. Refinement of the three-dimensional solution structure of barley serine proteinase inhibitor 2 and comparison with the structures in crystals. J Mol Biol. 1991;222:621–635. doi: 10.1016/0022-2836(91)90500-6. [DOI] [PubMed] [Google Scholar]
- 78.Nederveen AJ, Doreleijers JF, Vranken W, Miller Z, Spronk CAEM, Nabuurs SB, Güntert P, Livny M, Markley JL, Nilges M, Ulrich EL, Kaptein R, Bonvin AMJJ. RECOORD: A recalculated coordinate database of 500+ proteins form the PDB using restraints from the BioMagResBank. Proteins. 2005;59:662–672. doi: 10.1002/prot.20408. [DOI] [PubMed] [Google Scholar]
- 79.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 80.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 81.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 82.Hooft RWW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 83.Bonvin AMJJ, Houben K, Guenneugues M, Kaptein R, Boelens R. Rapid protein fold determination using secondary chemical shifts and cross-hydrogen bond 15N-13C′ scalar couplings (3hbJNC′) J Biomolec NMR. 2001;21:221–233. doi: 10.1023/a:1012935005256. [DOI] [PubMed] [Google Scholar]
- 84.Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 Å resolution. J Mol Biol. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.