

Abstract
Lung transplant recipients experience poor long-term survival, largely due to chronic rejection. The pathogenesis of chronic rejection is incompletely understood, but bacterial colonization of the lung is associated with chronic rejection, while antibiotic use slows its progression. The lung harbors a bacterial community, termed the microbiome, which is present both in health and disease. We hypothesize that the lung microbiome will change following transplantation, and these changes may correspond to the development of rejection. Twelve bronchoalveolar lavage fluid (BALF) samples were obtained from four patients at three time points after transplantation and two BALF samples were obtained from healthy, non-transplant controls. The microbiome of each sample was determined by pyrosequencing the 16S rDNA hypervariable 3 region. The data were analyzed using mothur, Ribosomal Database Project Classifier, Fast UniFrac, and Metastats. Transplanted lungs contained more bacterial sequences and demonstrated more microbial diversity than did control lungs. Bacteria in the phyla Proteobacteria (class Betaproteobacteria) predominated in the transplant samples. In contrast, the microbiome of the healthy lung consisted of the phyla Proteobacteria (class Gammaproteobacteria) and Firmicutes. The microbiome of the transplanted lung is vastly different from that of healthy lungs, mainly due to the presence of the family Burkholderiaceae in transplant samples.
Keywords: Lung Transplantation, Lung Microbiome, 16S rRNA Pyrosequencing, Lung Transplant Rejection
Introduction
Despite the remarkable success of organ transplantation, lung transplant recipients experience poor long-term survival compared to recipients of other solid organ transplants. Poor survival is primarily due to the development of bronchiolitis obliterans syndrome (BOS), a clinical marker of chronic lung transplant rejection (Jaramillo et al., 2005). BOS is characterized by shortness of breath and cough, with progressive airflow obstruction, airway inflammation, and fibrosis demonstrated on histopathology. This chronic inflammatory and fibrotic state leads to decreased lung function, the hallmark of chronic rejection.
Several recent studies have identified Pseudomonas colonization of the lung allograft as a risk factor for BOS (Botha et al., 2008; Nakajima et al., 2011; Vos et al., 2008). Lung transplant recipients are at risk for bacterial colonization or infection due to poor clearance of airway secretions, impaired blood flow to the transplanted lung, impaired cough reflex, airway stricture, and use of immunosuppressant medications. These bacteria may persist to form a community of bacteria in the lung, termed the microbiome (Kiley, 2011). Although the pathogenesis of BOS remains unclear, risk factors for the disorder include allograft infection, primary allograft dysfunction, gastroesophageal reflux, acute rejection, and lymphocytic bronchitis. These risk factors all augment the host response to the allograft in a manner similar to the pulmonary response to microbes, suggesting a link between infection and chronic rejection (Jaramillo et al., 2005; Nakajima et al., 2011).
Furthermore, a link between the lung microbiome and BOS is suggested by several studies that show chronic treatment of BOS patients with the antibiotic azithromycin results in decreased airway inflammation and stabilization or improvement in lung function in approximately 50% of affected patients (Gerhardt et al., 2003; Vanaudenaerde et al., 2008). The mechanism by which azithromycin alters the course of BOS is unknown and alteration in the microbiome remains a possibility. Therefore, we hypothesize that bacterial infection or colonization of the lung plays a role in the development of BOS.
The microbiome of healthy and diseased lungs has been studied. Charlson et al. demonstrated the presence of 16S rDNA sequences in the bronchoalveolar lavage fluid (BALF) of healthy volunteers. The authors concluded that the normal lung microbiome consists of organisms that gain access to the lower respiratory tract through microaspiration (Charlson et al., 2011). The lung microbiomes of healthy smokers (Erb-Downward et al., 2011) as well as patients with asthma (Hilty et al., 2010), chronic obstructive pulmonary disease (COPD) (Erb-Downward et al., 2011; Huang et al., 2010; Pragman et al., 2012; Sze et al., 2012), and cystic fibrosis (CF) (van der Gast et al., 2011) have been described as well. These three diseases are characterized by periods of relatively stable lung function interrupted by disease exacerbations. Disease exacerbations are often accompanied by bacterial infection, and shifts in the microbiome may pre-date infection and exacerbation. As has been described by the “hygiene hypothesis” for asthma pathogenesis, exposure to a normal commensal microbiome may promote immune tolerance, which is necessary for the normal immune system maturation and control of inflammation (Couzin-Frankel, 2010). We suggest that alterations in the transplanted lung microbiome and/or its interactions with a suppressed host immune system may lead to disordered immune tolerance and the development of BOS.
Although infections have been implicated in BOS, no systematic characterization of the transplanted lung microbiome has yet been performed. We hypothesize that alterations in the lung microbiome following transplantation play a role in the development of BOS. We undertook this pilot study of the microbiome in BALF from 4 lung transplant patients over 3 successive time points in order to describe the transplanted lung microbiome.
Materials and Methods
Sample Selection
The University of Minnesota O’Brien Lung Transplant Database and Biobank collects BALF samples longitudinally from consented lung transplant recipients at the University of Minnesota since 2001. Patients undergo bronchoscopy of the transplanted lung at the University of Minnesota every 2-3 months or when clinically indicated to detect rejection, for an average of 18 months after transplantation. BALF samples from four patients were selected for this study: two patients with BOS grade 3, one with BOS grade 2, and one without BOS (grade 0). For each patient with BOS, the samples consisted of two collected prior to the clinical diagnosis of BOS and one collected after the diagnosis of BOS, all taken at approximately 6-month intervals. These ranged in time from 5-22 months prior to the diagnosis of BOS and 0-26 months after the diagnosis of BOS. Three of the four patients received a single lung transplant, while the fourth patient received a double lung transplant. All patients had COPD as their underlying disease and two of these patients had alpha-1 anti-trypsin deficiency. The patients with alpha-1 anti-trypsin deficiency were not on replacement therapy after transplantation. All BALF used in this study was taken from the transplanted lung. The four transplant recipients resided in the Minneapolis area for a minimum of 2-3 months following transplantation and then returned to their homes. One lived in a suburb of Minneapolis and the other three lived in different small towns in Iowa or Minnesota. We also obtained control BALF samples from two healthy, non-transplant residents of the Minneapolis area. These bronchoscopies were performed at the Minneapolis Veterans Affairs Medical Center. Standard clinical protocols were followed to prevent upper airway contamination of all the BALF samples. All patients provided informed consent and their identities were not provided to the research team. The institutional review board for human studies approved the protocols (IRB Study #0601E80869 and IRB study #0202M17621).
DNA isolation, PCR amplification, and sequencing
BALF samples were thawed and 0.75 ml of fluid used for DNA isolation. We used a previously described protocol (Yu & Morrison, 2004) for DNA isolation that included bead beating to lyse bacterial cells, followed by precipitation with isopropanol, and digestion with RNase. Purified DNA was subjected to Multiple Displacement Amplification with REPLI-g (Qiagen, Valencia, CA), which provided highly uniform DNA amplification with minimal amplification bias (Hosono et al., 2003). PCR amplification using 16S rDNA primers specific to the constant regions flanking the V3 region (Muyzer et al., 1993; Kim et al., 2011) was performed using 25 cycles. The primer sequences were: GCCTCCCTCGCGCCATCAG - 10 base barcode - CCTACGGGAGGCAGCAG 3′ (forward) and 5′ GCCTTGCCAGCCCGCTCAG - ATTACCGCGGCTGCTGG 3′ (reverse). For each sample, a 10 base bar code was included to distinguish patient number and sampling time. Amplicons were gel purified and sequenced at the University of Minnesota Biomedical Genomic Center on a Roche 454 FLX DNA sequencer using titanium chemistry. To minimize effects of random sequencing errors, we used RDP Pipeline (Cole et al., 2009) to eliminate (a) sequences that did not appropriately match the PCR primer and the barcode at the beginning of a read, (b) sequence reads with <50 bases after the proximal PCR primer if they terminated before reaching the distal primer, and (c) sequences that contained more than one undetermined nucleotide (N). Trim.seqs and chimera.uchime implemented in mothur (Schloss et al., 2009) were used to truncate low-quality sequences and remove chimeras, respectively. Both primers were trimmed from high-quality reads before sequences were submitted to RDP Classifier for taxonomic identification using a bootstrap cutoff of 50%. Operational Taxonomic Units (OTUs) were defined at a similarity cutoff of 97% using mothur. The 16S rDNA sequences are available at https://main.g2.bx.psu.edu/u/alexapragman/h/transplant-microbiome-sequences.
Data visualization
The heat map was created using mothur v.1.21.0 and Java TreeView (Saldanha, 2004; Schloss et al., 2009). The DNA sequences were obtained from mothur and then sorted from the most to least abundant OTUs. Sequence abundance values within each OTU were normalized for comparisons of abundance between samples. The sequence abundance values were transformed to log10, and the heat map generated in TreeView. For PCoA analysis, sequences were dereplicated and ClustalW was used to align the dereplicated sequences (Chenna et al., 2003). The aligned sequences were used to generate a phylogenetic tree using Phylip (University of Washington) with a weighted UniFrac distance algorithm (Hamady et al., 2010). Metastats (White et al., 2009) was used to detect differentially abundant taxa using taxonomic data from RDP Classifier at p-value threshold of 0.05.
Results
The total number of sequences obtained per sample and the number of unique operational taxonomic units (OTUs) in each sample are shown in Table 1. The number of unique OTUs per transplant sample ranged from 81 to 214, while control samples demonstrated far fewer OTUs. The number of sequences obtained increased at later time points, and the number of OTUs observed increased with the number of sequences obtained. Shannon-Weaver and Simpson diversity indices (1-D) were calculated to evaluate the microbial diversity in the various samples. Values of 3.5 in the Shannon-Weaver algorithm indicate a highly species-rich community. Thus, with values that range from 2.94-3.94, several of the transplant patient samples exhibited rich microbiomes. The control samples demonstrated much lower Shannon-Weaver indices. The Simpson index values range from 0-1, with 1 indicating a highly diverse community. Simpson indices ranged from 0.77-0.9 for the transplant samples. One of the control samples exhibited similar values, but the second sample demonstrated a value of 0.51, indicating poor OTU diversity. There was no consistent pattern of change in the diversity index values as transplant patients developed BOS.
Table 1.
Overview of DNA sequencing data
| Patient number | Time point/BOS Scorea |
Number of Sequences |
OTU’s | Shannon | Simpson (1-D) |
|---|---|---|---|---|---|
| 480 | 1 | 541 | 81 | 3.17 | 0.82 |
| 2 | 1796 | 181 | 3.71 | 0.89 | |
| 3 | 2910 | 214 | 3.47 | 0.87 | |
| 360 | 1 | 546 | 84 | 3.45 | 0.89 |
| 2 | 1899 | 180 | 3.34 | 0.85 | |
| 3* | 1005 | 118 | 3.28 | 0.86 | |
| 8 | 1 | 979 | 123 | 3.65 | 0.90 |
| 2 | 1627 | 169 | 3.30 | 0.83 | |
| 3* | 1551 | 182 | 3.47 | 0.84 | |
| 171 | 1 | 2166 | 170 | 3.23 | 0.83 |
| 2 | 1118 | 119 | 2.94 | 0.77 | |
| 3* | 1755 | 214 | 3.94 | 0.85 | |
| Control 5 | NAb | 449 | 26 | 2.22 | 0.86 |
| Control 6 | NAb | 375 | 16 | 1.11 | 0.51 |
Samples obtained after the diagnosis of BOS are depicted with a *.
Not Applicable
Rarefaction curves were prepared for each of the samples (Figure 1). The results indicated that deeper sequencing among the transplant samples would likely identify additional OTUs. Control sample curves, especially Control 5, appear closer to reaching an asymptote. This suggests that deeper sequencing of the controls would be less likely to identify additional OTUs.
Figure 1.

Rarefaction curves. Each curve represents one of the 12 BALF samples from lung transplant patients or 2 BALF samples from controls. Each curve has an associated designation of patient and time point number or control number, and curves for the same patient are shown as shades of the same color.
To compare the taxonomies identified in each sample, graphs were prepared using data from RDP Classifier (Figure 2). The phylum-level classification is depicted on the top, the class-level classifications are depicted in the middle, and the genus-level classifications are depicted at bottom of the figure. The taxa are ranked in abundance from the top to the bottom in the figure legend. At the phylum level, Proteobacteria make up the majority of the bacteria, including >90% of the sequences in each transplant sample. The remaining 10% of the transplant sequences are in the phyla Actinobacteria and Firmicutes. At the class level, the transplant samples mostly contain Betaproteobacteria, with a smaller number of Alphaproteobacteria. The control samples consist of Gammaproteobacteria and Bacilli (a class in the phylum Firmicutes). At the genus level, the taxa were color-coded such that all genera within the same class are shades of the class color. The transplant samples consist mostly of Cupriavidus, unclassified Burkholderiaceae, Ralstonia, and Wautersia (all class Betaproteobacteria) and Ochrobactrum (class Alphaproteobacteria). The control samples consist of Haemophilus (class Gammaproteobacteria) and Streptococcus, Lactobacillus, and Dolosigranulum (class Bacilli).
Figure 2.
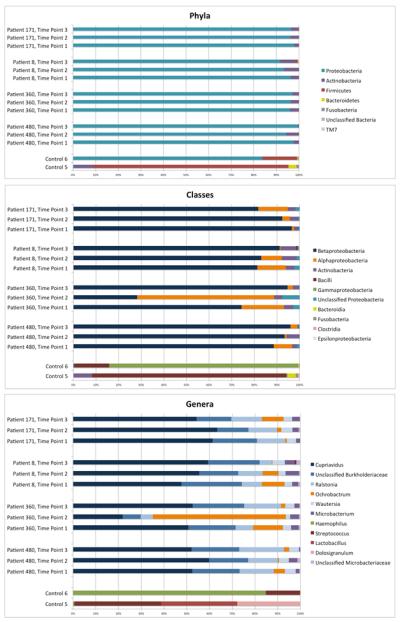
Taxonomic Classification at the Phylum, Class and Genus Level. The taxonomic classification of all sequences in each sample is represented at the phylum level (top), class level (middle) and genus level (bottom). The legends (at right) are organized with the most frequent taxons (overall) at the top and least frequent at the bottom. In order maintain the readability of the genus-level histogram, the results only depict genera with >50 sequences noted when totaled among all samples. This simplification omits only 1.7% of all sequences. Each class is represented by a unique color, and each genus within a class is represented by a different shade of the class color.
Principal coordinate analysis (PCoA) was used to establish relationships among all samples (Figure 3). For this analysis, the OTUs found in the 12 lung transplant patient samples were compared to each other and to the control lung samples. The PCoA graph demonstrates that the OTUs detected in the lungs of control patients were well separated from transplanted lung samples along principal coordinate 1, which accounted for ~30% of the observed differences. This indicates that the microbial populations found in the normal lungs were different and distinct from those in the lungs of transplant patients, and thus confirms the impression obtained from the taxonomic classification. We did not observe any significant separation of the pre- and post-BOS samples using PCoA.
Figure 3.
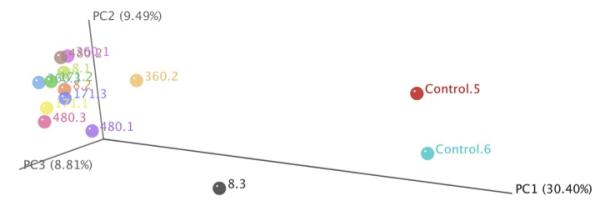
Principal Coordinate Analysis of Transplant Lung and Control Samples. Principal coordinate 1 is represented along the horizontal axis, while principal coordinate 2 is represented along the vertical axis. Control samples separate from the transplant samples along principal coordinate 1.
To determine the similarity of the microbiomes between patients and within the same patient over time, Venn diagrams were constructed (not shown). Within each of the four transplant patients, the number of OTUs present at all three time points represented a small fraction of the total OTUs in that patient, ranging from 8.1% to 13.5%. This suggests that there was considerable variability in the microbial populations over the study time period, with approximately 10% of each patient’s OTUs being present at all three time points. We also used Venn diagrams to determine which OTUs were present in all four patients (not shown). Among all four patients, 631 distinct OTUs were identified. Ninety-four were found in all four patients (14.8%). In contrast, only 2 OTUs were found in common between control and transplant samples (data not shown).
To better visualize the transplanted lung microbiomes between patients and over time following transplantation, we created a heat map of all observed OTUs (Figure 4). To perform this analysis, the sequences were first de-replicated and then sorted by the observed OTU frequency. The top of the heat map represents the most abundant OTUs. As expected based on the Venn diagram comparisons, none of the samples appeared similar, especially when the less abundant samples at the bottom of the diagram are considered. These less abundant OTU’s are most apparent in the BALF samples collected in the second and third time points, perhaps because these samples included more sequences overall. The control samples appeared markedly different than the transplant samples. No clear pattern emerged over time, or with the development of BOS.
Figure 4.
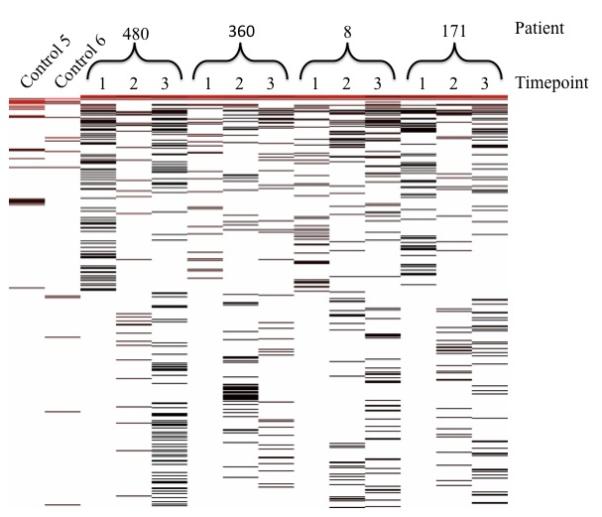
Heat map. The OTUs are arrayed by relative abundance with the most frequently identified OTUs at the top. The patient or control number is indicated along the top of the figure, while the time points are indicated by 1, 2, or 3.
At the top of the heat map there were OTUs that appeared to be present in all 12 lung transplant samples. Consequently, we sorted the OTUs to identify the genera present in all transplant samples. The DNA sequences in this group were retrieved from the data set and classified using RDP classifier and BLAST (when RDP Classifier was unable to classify the sequence at the genus level). Five unique genera were found in all 12 transplant samples: Ensifer, Cupriavidus, Microbacterium, Ochrobactrum, and Ralstonia. Ralstonia (Jhung et al., 2007; Ryan et al., 2006), Ochrobactrum (Higgins et al., 2001), Microbacterium (Funke et al., 1995; Giammanco et al., 2006), and Cupriavidus (Higgins et al., 2001) have each been associated with respiratory infections, bacteremia, and nosocomial infections. Their presence in the lungs of the four transplant patients is consistent with their having a potential role in disease or the lack of effective lung clearance mechanisms allowing them to persist. Furthermore, both Ralstonia and Cupriavidus are members of the family Burkholderiaceae which also includes Burkholderia, a known lung pathogen (Lyczak et al., 2002; Mahenthiralingam et al., 2002). On the other hand, Ensifer (Germida & Casida, 1983) is a soil bacterium that has not previously been described as a lung pathogen.
We sought to statistically evaluate the differential abundance of the various genera across our samples. We used Metastats to compare the abundance of each genus in control versus transplant samples, and pre-BOS transplant samples to post-BOS transplant samples. When we compared control and transplant samples, we discovered 6 genera that were more abundant in the transplant samples and 5 genera that were less abundant in the transplant samples, when compared to control samples (Table 2). The 6 genera more abundant in the transplant samples included 4 of the 5 genera we identified in all transplant samples. The remaining 2 genera identified in our Metastats analysis were Wautersia, a member of Betaproteobacteria closely related to Cupriavidus (Martinez-Aguilar et al., 2012), and Burkholderia, a known lung pathogen. The 5 genera that were less abundant in transplanted lung samples consisted of 4 members of the phylum Firmicutes, of which 3 (Catonella, Gamella, and Streptococcus) are members of the oral flora. The list also includes Pelomonas, a member of the class Betaproteobacteria. Metastats also was used to compare the 3 post-BOS transplant samples to the other 9 transplant samples. Although no genera reached statistical significance, both Actinomyces (a known oral and lung pathogen in the phylum Actinobacteria) and unclassified Xanthomonadaceae (a family in the class Gammaproteobacteria) were more common in the post-BOS samples with a p-value of 0.06.
Table 2.
Metastats Analysis
| Less Likely in Transplant Samples a | p value |
|---|---|
| Catonella | 0.046 |
| Gamella | 0.046 |
| Pelomonas | 0.046 |
| Staphylococcus | 0.002 |
| Unclassified Lactobacillaceaec | 0.046 |
| Streptococcus | 0.032 |
|
| |
| More Likely in Transplant Samples b | |
|
| |
| Unclassified Micrococcineaec | 0.003 |
| Burkholderia | 0.004 |
| Ensifer | 0.042 |
| Unclassified Microbacteriaceaec | 0.006 |
| Microbacterium | 0.003 |
| Wautersia | 0.001 |
| Ralstonia | 0.002 |
| Unclassified Burkholderiaceaec | 0.001 |
| Cupriavidus | 0.000 |
Genera less likely to be found in transplant patient samples, compared to control samples.
Genera more likely to be found in transplant patient samples, compared to control samples.
Sequences that RDP Classifier was unable to classify at the genus level, but could be assigned to the families noted above.
Discussion
Culture-independent detection and identification of bacteria has resulted in the ability to intensively study the microbiome of almost any habitat. Here we describe our pilot study of the transplanted lung microbiome in a longitudinal fashion, using BAL fluid acquired from 4 patients at 3 successive time points. We compared the transplanted lung microbiome to healthy control patients and found that the identified taxa are quite distinct. The transplanted lung microbiome was divided among several different phyla, but >90% of sequences were from Proteobacteria. The majority of these fell into the class Betaproteobacteria, and the genera Cupriavidus, Ralstonia, Wautersia, or unclassified Burkholderiaceae.
In healthy control patients the predominant microbes were in the taxa Firmicutes (genera Streptococcus, Lactobacillus, and Dolosigranulum) and Proteobacteria (genus Haemophilus). Three previous publications on the lung microbiome have also described the most common taxa in healthy lungs. The first publication identified the phyla Proteobacteria, Firmicutes, Fusobacteria, and Bacteroidetes (genera Pseudomonas, Streptococcus, Veillonella, Fusobacterium, Prevotella, and Porphyromonas) most frequently in healthy lungs (Erb-Downward et al., 2011). The second publication identified the phyla Firmicutes and Bacteroidetes (families Veillonellaceae, Streptococcaceae and Prevotellaceae) most frequently (Charlson et al., 2011). The third publication identified Proteobacteria and Bacteroidetes most frequently among healthy controls (Sze et al., 2012). We also identified Firmicutes and Proteobacteria in our control samples, but did not identify any members of the anaerobic phyla Fusobacteria or Bacteroidetes. We recently published the sequencing results of 10 additional control BAL samples from this same cohort and neither Bacteroidetes nor Fusobacteria were well represented in these additional samples (Pragman et al., 2012).
The most striking difference between transplant and control samples was the family Burkholderiaceae, which was the most abundant family found in all transplant samples but was not found in either control sample. Although our diversity indices indicated that transplant samples exhibited greater microbial diversity than control samples, the transplant samples were dominated by several genera that belonged to the family Burkholderiaceae. This family contains Burkholderia, which is a significant cause of morbidity and mortality among cystic fibrosis (CF) sufferers. All patients in our study were transplanted because of COPD, rather than other lung diseases such as CF. Our patients did not have bronchoscopy performed prior to transplantation and only one patient had a sputum culture done prior to transplantation. This culture did not grow any lung pathogens, including Burkholderia. Post-transplantation BAL cultures did not grow Burkholderia even through it was present in all BAL fluid by pyrosequencing.
The central question of our study involved whether or not the transplant lung microbiome was distinct from the healthy lung microbiome. Our data demonstrated that the transplanted lung microbiome is significantly different from the healthy lung microbiome. However, only ~10% of OTUs were found at multiple time points or in all patients. Therefore only a small minority of microbes were common or persisted over time. The remaining 90% of microbes may represent transient microbes and those present in the environments of each of the patients. These microbes could represent true residents of the transplanted lung, but there may be turnover and replacement over time. Although the majority of each transplant sample was made up of the same 4 genera, additional sequencing of more patient samples will be needed in order to definitively establish a core lung transplant microbiome.
A second, related question involved the extent to which the transplanted lung microbiome changed with the development of BOS. Our PCoA analysis did not establish clustering of post-BOS samples, nor did our Metastats analysis detect differentially abundant taxa among the post-BOS samples. Additional sampling of the transplanted lung microbiome will be required to address whether or not microbiome changes pre-date the development of BOS.
This small study is the first to describe the transplanted lung microbiome and compare it to the normal lung microbiome. We have examined this microbiome longitudinally, both before and after the development of BOS. The transplanted lung microbiome is distinct from the healthy lung microbiome and includes organisms known to cause lung disease. Our study included 12 patient samples (4 patients at 3 time points) from individuals transplanted due to COPD. Additional patient samples, including samples from patients with different indications for transplantation, will be needed to fully characterize how the microbiome changes with time and its relationship with the development of BOS. We do not know whether the unique set of microbes found in the BALF of the lung transplant recipients are the result of anatomic and physiologic deficits associated with transplantation, the use of immunosuppressive drugs, or the microbes’ interaction with the host immune system. Nevertheless, this study demonstrated that the transplanted lung microbiome is vastly different from the healthy lung microbiome, mainly due to the presence of the family Burkholderiaceae in transplant samples. This study will provide the foundation for further work on the role of the transplant lung microbiome in health and disease, including BOS.
Acknowledgements
We thank the University of Minnesota BioMedical Genomics Center for assistance with sequencing, Minnesota Supercomputing Institute for their technical support, and Bonnie Holmes and Cheryl Stibbe of the O’Brien Biobank for database management and sample acquisition. This work was supported by NIH grant T32-AI55433 from the National Institutes of Allergy and Infectious Diseases. All authors report no conflicts of interest.
References
- Botha P, Archer L, Anderson RL, Lordan J, Dark JH, Corris PA, Gould K, Fisher AJ. Pseudomonas aeruginosa colonization of the allograft after lung transplantation and the risk of bronchiolitis obliterans syndrome. Transplantation. 2008;85:771–774. doi: 10.1097/TP.0b013e31816651de. [DOI] [PubMed] [Google Scholar]
- Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–5. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzin-Frankel J. Bacteria and asthma: untangling the links. Science. 2010;330:1168–1169. doi: 10.1126/science.330.6008.1168. [DOI] [PubMed] [Google Scholar]
- Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke G, Falsen E, Barreau C. Primary identification of Microbacterium spp. encountered in clinical specimens as CDC coryneform group A-4 and A-5 bacteria. J Clin Microbiol. 1995;33:188–192. doi: 10.1128/jcm.33.1.188-192.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt SG, McDyer JF, Girgis RE, Conte JV, Yang SC, Orens JB. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med. 2003;168:121–125. doi: 10.1164/rccm.200212-1424BC. [DOI] [PubMed] [Google Scholar]
- Germida JJ, Casida LE. Ensifer adhaerens Predatory Activity Against Other Bacteria in Soil, as Monitored by Indirect Phage Analysis. Appl Environ Microbiol. 1983;45:1380–1388. doi: 10.1128/aem.45.4.1380-1388.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giammanco GM, Pignato S, Grimont PA, Grimont F, Santangelo C, Leonardi G, Giuffrida A, Legname V, Giammanco G. Interstitial pulmonary inflammation due to Microbacterium sp. after heart transplantation. J Med Microbiol. 2006;55:335–339. doi: 10.1099/jmm.0.46219-0. [DOI] [PubMed] [Google Scholar]
- Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CS, Avison MB, Jamieson L, Simm AM, Bennett PM, Walsh TR. Characterization, cloning and sequence analysis of the inducible Ochrobactrum anthropi AmpC beta-lactamase. J Antimicrob Chemother. 2001;47:745–754. doi: 10.1093/jac/47.6.745. [DOI] [PubMed] [Google Scholar]
- Hilty M, Burke C, Pedro H, et al. Disordered Microbial Communities in Asthmatic Airways. Plos One. 2010;5:372–381.e1-3. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono S, Faruqi AF, Dean FB, Du Y, Sun Z, Wu X, Du J, Kingsmore SF, Egholm M, Lasken RS. Unbiased whole-genome amplification directly from clinical samples. Genome Res. 2003;13:954–964. doi: 10.1101/gr.816903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Kim E, Cox MJ, Brodie EL, Brown R, Wiener-Kronish JP, Lynch SV. A Persistent and Diverse Airway Microbiota Present during Chronic Obstructive Pulmonary Disease Exacerbations. Omics-a Journal of Integrative Biology. 2010;14:9–59. doi: 10.1089/omi.2009.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo A, Fernandez FG, Kuo EY, Trulock EP, Patterson GA, Mohanakumar T. Immune mechanisms in the pathogenesis of bronchiolitis obliterans syndrome after lung transplantation. Pediatr Transplant. 2005;9:84–93. doi: 10.1111/j.1399-3046.2004.00270.x. [DOI] [PubMed] [Google Scholar]
- Jhung MA, Sunenshine RH, Noble-Wang J, et al. A national outbreak of Ralstonia mannitolilytica associated with use of a contaminated oxygen-delivery device among pediatric patients. Pediatrics. 2007;119:1061–1068. doi: 10.1542/peds.2006-3739. [DOI] [PubMed] [Google Scholar]
- Kiley JP. Advancing respiratory research. Chest. 2011;140:497–501. doi: 10.1378/chest.11-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HB, Borewicz K, White BA, Singer RS, Sreevatsan S, Tu ZJ, Isaacson RE. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet Microbiol. 2011;153:124–133. doi: 10.1016/j.vetmic.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahenthiralingam E, Baldwin A, Vandamme P. Burkholderia cepacia complex infection in patients with cystic fibrosis. J Med Microbiol. 2002;51:533–538. doi: 10.1099/0022-1317-51-7-533. [DOI] [PubMed] [Google Scholar]
- Martinez-Aguilar L, Caballero-Mellado J, Estrada-de Los, Santos P. Transfer of Wautersia numazuensis to Cupriavidus genus as Cupriavidus numazuensis comb. nov. Int J Syst Evol Microbiol. 2012 doi: 10.1099/ijs.0.038729-0. [DOI] [PubMed] [Google Scholar]
- Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Palchevsky V, Perkins DL, Belperio JA, Finn PW. Lung transplantation: infection, inflammation, and the microbiome. Semin Immunopathol. 2011;33:135–156. doi: 10.1007/s00281-011-0249-9. [DOI] [PubMed] [Google Scholar]
- Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One. 2012;7:e47305. doi: 10.1371/journal.pone.0047305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MP, Pembroke JT, Adley CC. Ralstonia pickettii: a persistent gram-negative nosocomial infectious organism. J Hosp Infect. 2006;62:278–284. doi: 10.1016/j.jhin.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, Cooper J, Sin DD, Mohn WW, Hogg JC. The Lung Tissue Microbiome in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2012 doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Gast CJ, Walker AW, Stressmann FA, Rogers GB, Scott P, Daniels TW, Carroll MP, Parkhill J, Bruce KD. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011;5:780–791. doi: 10.1038/ismej.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaudenaerde BM, Meyts I, Vos R, Geudens N, De Wever W, Verbeken EK, Van Raemdonck DE, Dupont LJ, Verleden GM. A dichotomy in bronchiolitis obliterans syndrome after lung transplantation revealed by azithromycin therapy. Eur Respir J. 2008;32:832–843. doi: 10.1183/09031936.00134307. [DOI] [PubMed] [Google Scholar]
- Vos R, Vanaudenaerde BM, Geudens N, Dupont LJ, Van Raemdonck DE, Verleden GM. Pseudomonal airway colonisation: risk factor for bronchiolitis obliterans syndrome after lung transplantation? Eur Respir J. 2008;31:1037–1045. doi: 10.1183/09031936.00128607. [DOI] [PubMed] [Google Scholar]
- White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36:808–812. doi: 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]