

Abstract
C57BL/6 mice develop dermatitis and scarring alopecia resembling human cicatricial alopecias (CA), particularly the central centrifugal cicatricial alopecia (CCCA) type. To evaluate the role of retinoids in CA, expression of retinoid metabolism components were examined in these mice with mild, moderate, or severe CA compared to hair cycle matched mice with no disease. Two feeding studies were performed with dams fed either NIH 31 diet (study 1) or AIN93G diet (study 2). Adult mice were fed AIN93M diet with 4 (recommended), 28, or 56 IU vitamin A/g diet. Feeding the AIN93M diet to adults increased CA frequency over NIH 31 fed mice. Increased follicular dystrophy was seen in study 1 and increased dermal scars in study 2 in mice fed the 28 IU diet. These results indicate that retinoid metabolism is altered in CA in C57BL/6J mice that require precise levels of dietary vitamin A. Human patients with CCCA, pseudopelade (end stage scarring), and controls with no alopecia were also studied. Many retinoid metabolism proteins were increased in mild CCCA, but were undetectable in pseudopelade. Studies to determine if these dietary alterations in retinoid metabolism seen in C57BL/6J mice are also involved in different types of human CA are needed.
Introduction
Vitamin A and its derivatives (retinoids) are important for development and maintenance of multiple epithelial tissues including skin, hair, and sebaceous glands, as severe, adverse effects are seen in vitamin A deficiency or excess (Everts, 2012; Smith and Thiboutot, 2008; Zouboulis, 2006). Vitamin A is stored in the liver and extra-hepatic tissues as retinyl esters (O’Byrne et al., 2005). Retinol bound to retinol binding protein (RBP4) is the main circulating form of vitamin A (Blaner et al., 2002) and maintained at a constant blood level (Blomhoff et al., 1991). After a meal some retinyl esters enter nonhepatic tissues from circulating chylomicron remnants (Blaner et al., 2002). Retinoic acid (RA) synthesis occurs locally in or near cells that utilize it. Precise spatial and temporal levels of RA in skin are achieved by regulating key steps in cellular vitamin A metabolism: storage as retinyl esters, RA synthesis, and RA degradation (Everts, 2012). Retinol is transported into the cell via the protein called Stimulated by RA6 (STRA6) and binds cellular retinol binding protein 1 (RBP1, aka CRBP). This bound retinol is either esterified by lecithin:retinol acyltransferase (LRAT) for storage or reversibly oxidized to retinal via retinol dehydrogenases (DHRS9, RDH10). Retinal is further oxidized to RA by retinal dehydrogenases 1-3 (ALDH1A1-3). RA then moves to the nucleus bound to cellular RA binding protein 2 (CRABP2), binds to RA receptors alpha, beta, and gamma (RARA, B, C) where it activates transcription of over 500 genes (Balmer and Blomhoff, 2002; Dong et al., 1999). Retinyl ester hydrolases (REH) convert stored retinyl esters back to retinol when needed (Blomhoff et al., 1991; Gao and Simon, 2005).
When RBP1/CRBP is saturated or absent, retinol interacts with different enzymes. Acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) esterifies retinol for storage. Alopecia with accelerated telogen to anagen transition was seen in basal epidermis and outer root sheath specific (Krt14 cre) Dgat1tm2Far null mice (Shih et al., 2009). This increased anagen induction and alopecia was reduced in Dgat1tm2Far null mice by severely reducing dietary vitamin A intake indicating alopecia was due to excess retinol and RA. Free retinol can also be oxidized by medium chain alcohol dehydrogenases (ADH1, 3, and 4; current gene names Adh1, 5, and 7, Duester, 2001). It has been argued by some (Kumar et al., 2012) and refuted by others (Napoli, 2012) that excess retinol is oxidized to retinal by ADH1, and then to RA by ALDH1A1. When given a toxic retinol dose, mice that lack Adh7 tm1Gdu (class IV, previously Adh4) have decreased retinol oxidation in the kidney (Deltour et al., 1999a, b), but not in the liver (Molotkov et al., 2002). These Adh7tm1Gdu null mice are also more sensitive to vitamin A deficiency. ADH1 and ADH7 (class IV) are present in skin (unpublished observation, (Haselbeck et al., 1997), but their roles in skin have not been studied.
RA is further metabolized by cytochrome P450 26 family members (CYP26A1, B1, C1) with the assistance of cellular RA binding protein 1 (CRABP1). Both Cyp26a1 and Cyp26b1 message levels are RA inducible in both organotypic cultures and human epidermis (Lorie et al., 2009a; Lorie et al., 2009b).
Primary cicatricial alopecias (CA) are scarring hair loss diseases with permanent hair loss due to destruction of follicles and their stem cells by lymphocytic and neutrophilic mediated inflammation (Bergfeld and Elston, 2003). Central Centrifugal Cicatrial Alopecia (CCCA) is a progressive permanent alopecia seen primarily in African American females usually beginning in their mid-thirties in an anterior vertex and mid-crown pattern. C57BL/6J (B6) mice develop CA with similar characteristics as patients with CCCA, including premature desquamation of the inner root sheath (follicular dystrophy, (Sperling, 2001; Sperling and Cowper, 2006; Sperling and Sau, 1992; Sperling et al., 1994; Sperling et al., 2000; Sundberg et al., 2011). In B6 mice, as the hair shafts twist they penetrate through the outer root sheath and induce an inflammatory response. Studies in asebia mutant mice (ABJ/Le-Scd1ab-J/Scd1ab-J and DBA/1lacJ-Scd1ab-2J/Scd1ab-2J) also suggest that the inflammatory response in CA follows hair shaft penetration through the follicle (Sundberg et al., 2000). Other theories for the cause of the various CAs (McElwee, 2008) include: altered sebaceous gland function with reduced sebum (Al-Zaid et al., 2011; Stenn et al., 1999; Sundberg et al., 2000) and sebaceous gland duct inflammation (Al-Zaid et al., 2011); reduced PPARG and lipid metabolism (Karnik et al., 2009); and hair follicle loss of immune privilege (Harries et al., 2009).
Few studies have examined the role of diet in CA. Retinoid supplementation improved CA in four patients with low serum retinol levels (Kalz, 1958). Because of this study and the unique localization pattern of RA synthesis within the hair follicle and sebaceous gland during the hair cycle (Everts et al., 2007), the expression of retinoid metabolism proteins were examined in the B6 mouse model of CA and human patients with CA. The initial study reported all substrains of B6 mice had a polymorphism in Adh7, which reduced its function (Dolney et al., 2001a; Dolney et al., 2001b), and only B6 substrains with a higher frequency of disease had higher levels of DHRS9 in telogen follicles (Sundberg et al., 2011). Since CA in B6 mice spared telogen follicles and only late anagen and catagen follicles were affected, studies examined retinoid metabolism in anagen hair follicles. The current report identifies RA synthesis and signaling protein changes in both mouse and human CA and that modifying vitamin A intake modifies CA severity in B6 mice.
Results
RA synthesis, signaling, and degradation proteins were increased in early/mild CA, but became undetectable in severe disease
Expression of retinoid synthesis components was previously measured in telogen follicles of C57BL/6 mouse substrains (Sundberg et al., 2011). The current study evaluated anagen follicles, as it is the stage when B6 CA is most severe and RA synthesis components peaked (Everts et al., 2007; Sundberg et al., 2011). Biopsies of B6 mice that spontaneously developed CA in The Jackson Laboratory production facility were evaluated by immunohistochemistry (IHC) using antibodies against RBP1, DHRS9, ALDH1A1, ALDH1A2, ALDH1A3, CRABP2, CYP26A1, RARA, and RARB. These mice were grouped by severity of disease into no disease, mild, moderate, or severe disease with hair follicles in mid-late anagen using standard staging criteria (Muller-Rover et al., 2001). Biopsies from human patients with CCCA, end stage CA (Pseudopelade), and controls with no alopecia (tinea capitis and follicles adjacent to pilar cysts) were also examined. DHRS9 was increased in mild disease in mice and humans with CCCA yet undetectable in severe disease (Fig. 1, Fig. S1, Fig. S2). A similar pattern of expression was also seen for ALDH1A1, ALDH1A2, and RARB in mice and humans (Fig. 3, Fig. S3, data not shown). CYP26A1 showed the same pattern as well, but was only tested in mice (Fig. S3). In moderate mouse CA levels of these proteins were similar to controls (data not shown). In areas of dermal scars in mice, DHRS9 and ALDH1A2 were high, but ALDH1A1 localized to only the outer root sheath of telogen follicles and ALDH1A3 was not altered (data not shown). CRABP2 IR also increased in mild disease, but remained high within dystrophic hair follicles in all disease states (Fig. 2). RARA localization changed with more RARA positive cells seen in the dermis; especially surrounding the bulge in mild, moderate, and severe mouse disease (Fig. 2).
Figure 1. Immunoreactivity of DHRS9 increased in C57BL/6J mice with mild dermatitis and biopsies from human patients with CCCA, but was reduced in mice with severe disease and biopsies from human patients with pseudopelade.

Immunohistochemistry was performed with an antibody against DHRS9 in dorsal skin from chow fed B6 mice with no disease (a, n=18), mild disease (c, n=6), severe disease (e, n=38), biopsies from patients with no alopecia (b, normal Caucasian skin adjacent to Pilar Cyst removed by excision n=2, Tinea capitus from African American n=1), CCCA (d, n=14), or pseudopelade (f, n=3). B and f are from Caucasian patients, while d is from an African American patient. Bar = 101 μM for main picture and 10.1 μM for insets.
Figure 3. Overview of retinoid metabolism and how these proteins changed during the progression of cicatricial alopecia.

The left shows a schematic of RA synthesis, degradation, and signalling pathways in B6 mice. The right shows a summary of IHC results in chow fed B6 mice with no (control), mild, moderate, or severe disease. Immunoreactivity intensity ranges from dark to light; very strong (maroon), strong (red), moderate (hot pink), mild (pink), or undetectable (white). CL= companion layer, PM= premedulla.
Figure 2. CRABP2 and RARA were increased in C57BL/6J mice with cicatricial alopecia.

Immunohistochemistry was performed with antibodies against CRABP2 (a,c,e), and RARA (b,d,f), in mice with no disease (a,b), mild disease (c,d), or severe disease (e,f). Bar = 101 μM for main picture and 10.1 μM for insets.
Dietary vitamin A altered the frequency and severity of CA in B6 mice
To test the hypothesis that abnormal vitamin A metabolism is one of the primary mechanisms causing CA in B6 mice, 12-week-old female B6 mice (n= 9–10) were fed purified diets containing one of three levels of Vitamin A (Research Diets, Inc, New Brunswick, NJ). Diets contained either the American Institute of Nutrition (AIN; (Reeves et al., 1993) recommended level (4 IU/g diet), the highest level found in a commercial diet used at The Jackson Laboratory (high; 28 IU/g diet), or twice the highest level (excess; 56 IU/g). Mice fed excess vitamin A had less alopecia than either of the other groups and all of these mice had significantly fewer hair follicles in anagen indicating follicles were in their normal telogen phase (Fig. 4a,b). Histological analysis showed more hair follicle dystrophy occurred when hair follicles were in anagen or catagen with an inverse correlation between percent of hair follicle dystrophy and telogen follicles (Fig. 4c). Mice fed high vitamin A had more follicular dystrophy and granulomas (Fig. 4d,e).
Figure 4. Cicatrical alopecia was altered by dietary vitamin A and wax stripping.
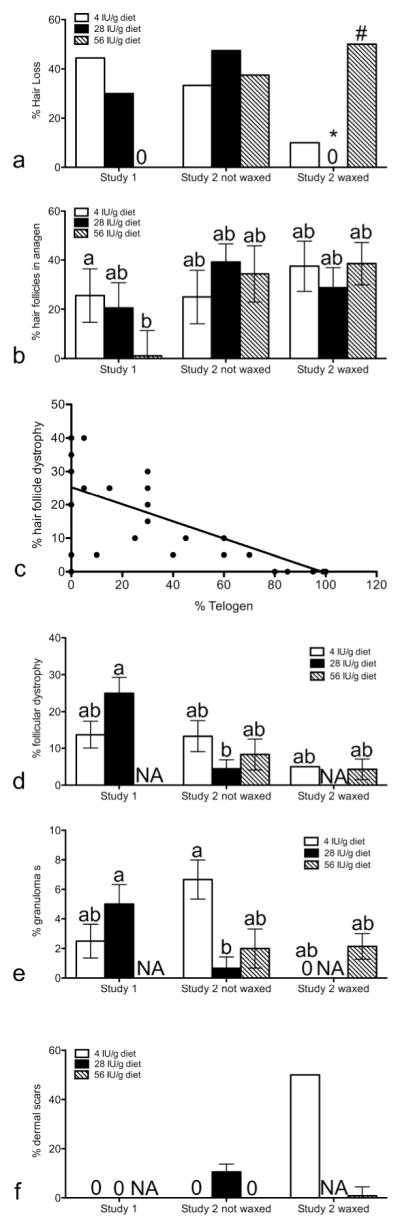
The percent of mice with hair loss (a) was determined by visual inspection in B6 mice fed 4 (open bars), 28 (closed bars), or 56 (hatched bars) IU vitamin A/g diet. H&E slides were then scored for % of hair follicles in anagen (b) and telogen (c), % of follicular dystrophy (d), % granulomas (e), and % of dermal scars (f). * Significant effect of wax stripping as analyzed by chi square (p<0.05), # significant diet effect in the wax stripped mice (p<0.05). Results with different letters are significantly different from each other in mice with hair loss (p<0.05). e) r = −0.86, p<0.001. n=9–10 study 1, n=8–19 study 2.
A second feeding study was done to test the hypotheses that significantly lowering dietary vitamin A would reduce the frequency and severity of CA. Typical mouse commercial diets (chow) contain up to 7 times the AIN recommended level of dietary vitamin A (4 IU/g diet, Reeves et al., 1993). The chow diet fed to mice at The Jackson Laboratory was analyzed and contained 6-28 IU vitamin A/g depending on the lot. This variability is important since vitamin A is stored in the liver and dams fed chow during breeding produce pups with large amounts of vitamin A in their liver. Feeding dams a vitamin A deficient diet during breeding reduced liver stores of vitamin A in their pups (Smith et al., 1987) and reduced alopecia in Dgat1tm2Far null mice (Shih et al., 2009). To significantly lower vitamin A levels in this study dams were fed AIN93G diet for two generations. This study also evaluated if inducing anagen by wax stripping would lead to CA but these hypotheses were incorrect. Alopecia was consistently high in all mice not wax stripped. However, wax stripping reduced the frequency of alopecia in mice fed recommended and high levels of vitamin A (Fig. 4a). The one wax stripped mouse with alopecia fed the recommended diet developed significantly more ulcers, dermal scars, and epidermal hyperplasia than any other mouse in the study. The only other mice with epidermal hyperplasia and dermal scars were the mice fed high vitamin A in study 2 (Fig. 4f). They also had significantly less follicular dystrophy and granulomas than mice fed the same diet in study 1 (Fig. 4d,e, p<0.05). Within study 2, mice fed high vitamin A had significantly lower number of granulomas than mice fed recommended levels of vitamin A (Fig. 4e, p<0.05). This suggests that these mice fed high vitamin A in study 2 had a previous bout of severe disease.
Liver retinoid analysis revealed dams fed the 4 IU diet did not lower liver retinyl palmitate levels in their pups as predicted (Fig. 5b, Smith et al., 1987). Both liver retinol and retinyl palmitate increased with dietary vitamin A as expected (Fig. 5a,b, p<0.001). Liver retinol was significantly greater in the second study (Fig. 5a, p<0.01). In addition, wax stripping significantly reduced liver retinyl palmitate levels (Fig. 5b, p<0.01). Skin retinol increased with dietary vitamin A in both studies (Fig 5c, p<0.05). In mice fed high vitamin A, skin retinol was greater in study 1 than study 2. This trend was seen when mice wax stripped and not wax stripped were compared separately, but was significant when all mice in study 2 fed high vitamin A were combined (Fig 5c*, p<0.05). There were no significant effects of pup or dam diets on skin retinyl ester levels (Fig 5d). Skin retinyl ester levels were significantly reduced in wax stripped mice. (Fig 5c,d, p<0.05).
Figure 5. Dietary vitamin A altered liver and skin retinoids differently in the various studies.

Liver retinol (a), liver retinyl palmitate (b), skin retinol (c), and skin retinyl esters (d) were analyzed by HPLC in study 1 (open bars, n=9–10), study 2 with no wax stripping (closed bars, n=8–19), and study 2 with wax stripping (hatched bars, n=10–16). *Significant study effect (p< 0.001 in a, and p< 0.05 in c), # significant effect of wax stripping (p < 0.005), results with different letters are significantly different from each other (p<0.05).
IHC analysis was performed to compare results from these diet studies to those performed in NIH31 fed mice. RARB was significantly reduced as the percent of follicular dystrophy increased (Fig. S4b, d, Fig. S5). RARB was also low in dermal scars (Fig. S4f, Fig. S5). In contrast, DHRS9 remained high in dystrophic hair follicles, but dropped slightly in dermal scars (Fig. S4a, c, e). None of the mice in these diet controlled studies had severe follicular dystrophy as seen in NIH31 fed mice.
Discussion
This study emphasizes that the effects of an essential nutrient, vitamin A, must be tightly regulated to prevent adverse effects due to excess or deficiency. In this study, both low and high levels of dietary vitamin A given to B6 mice affected the development and degree of follicular scarring indicating precise control of retinoid metabolism is required for normal function of hair follicles. Initial studies localized RA synthesis and signaling proteins in normal mouse skin (Everts et al., 2004; Everts et al., 2007). A subsequent study found that only B6 substrains with a higher frequency of inflamed and follicular scarring had higher levels of DHRS9 in telogen follicles (Sundberg et al., 2011), suggesting a possible basis for their tendency to develop CA. Since CA occurs primarily during mid-late anagen, the current study analyzed mid-late anagen hair follicles and found that RA synthesis, degradation, and binding proteins were altered during progression of CA in B6 mice. These proteins were increased in mild disease and reduced in severe disease. A similar pattern of expression was also seen in biopsies from patients with CCCA, although the full progression of disease was not obtained.
This study characterized changes in retinoid metabolism throughout the entire progression of CA in a mouse model of primary human CA. Others (Flowers et al., 2011) found at only one time point Rbp1 (Crbp) and Crabp2 increased in skin specific (Krt14-cre recombinase) Scd1tm2Ntam null male B6 mice with mild CA. They also reported that several other retinoid metabolism genes were increased in their microarray analyses, but changes in expression of these genes were not further validated at the message or protein level. They also found that retinol, retinyl esters, and RA levels were increased in the skin of these null mice. Feeding a vitamin A deficient diet did not reduce skin RA levels or change the phenotype, arguing that the lack of Scd1 led to the increased skin RA levels. In the current study RA metabolism was altered in the absence of a Scd1 mutation and significantly reducing dietary vitamin A made the disease worse. These current studies expand on the observations of Flowers et al. (2011) by showing that as follicular dystrophy increases, expression of proteins responsible for RA synthesis and degradation begin to decrease and reach undetectable levels in severe CA. This observation was further confirmed by the reduction of RARB, a classic RA target gene, as follicular dystrophy increased in the two diet studies preceding reduction of DHRS9. In addition, these alterations in RA synthesis proteins were also seen in biopsies from patients with CA. It is unknown at this time whether altered RA synthesis and signaling caused the alopecia or this alteration in retinoid metabolism is secondary to the disease.
CYP26A1 also localized within the hair follicle and was altered during CA. CYP26A1 was previously reported only in basal keratinocytes, eccrine sweat glands, and sebaceous glands (Heise et al., 2006). CYP26A1 plays a key role in maintaining RA levels through a feedback inhibitory loop (Reijntjes et al., 2005). High levels of RA directly induce CYP26A1 expression (Loudig et al., 2000). Then CYP26A1 degrades this excess RA. In addition, when RA levels are low CYP26A1 levels are also low. In retinol dehydrogenase 1 (Rdh1tm1.1Jln) null mice reduction in CYP26A1 expression compensated for the reduced RA synthesis and maintained normal RA levels (Zhang et al., 2007). These observations are consistent with the concurrent alterations of CYP26A1 with DHRS9 in this study.
Epigenetic silencing may explain the expression results seen. Mutations in either CRABP2 or RARA resulted in hypermethylation and epigenetic silencing of RARA target genes such as RARB, CRBP, and CYP26A1 (Corlazzoli et al., 2009; Laursen et al., 2012; Ren et al., 2005). Since CRABP2 functions to deliver RA to RARA (Dong et al., 1999) and RARA represses transcription in the absence of RA (Chen and Evans, 1995; Horlein et al., 1995), the above studies suggest that any defect resulting in reduced RA binding to its receptor could cause hypermethylation and gene silencing of RARA target genes. Such defects would include: reduced levels of RA precursors retinol or retinyl esters, reduced uptake of retinol or retinyl esters into the skin, reduced RA synthesis, reduced delivery of RA to RARA via CRABP2, increased RA degradation, or any defect in RARA itself. It is possible that the reduction in RA synthesis enzymes, RARB and CYP26A1 in moderate and severe CA, was due to this mechanism of aberrant RA signaling induced hypermethylation. Alternatively, a primary effect of increased dietary vitamin A could be due to toxic effects during mild disease on specific hair follicle cells leading to their death or decreased biochemical function such that the levels of RA synthesis enzymes and receptors decrease as CA disease progresses. The current studies indicate the balance between RA synthesis and retinol degradation is altered in a mouse model of human CA and imply altered retinoid metabolism is involved in the pathogenesis of CA. Future studies will determine if CA is due to altered retinoid metabolism or is due to loss of retinoid related enzymes present in skin due to scarring induced by another cause.
The current study implies that precise levels of dietary vitamin A are required to compensate for the defects in retinoid metabolism found in substrains of C57BL/6 mice. A polymorphism in Adh7 (Class IV, previously reported as Adh4) was previously found in C57BL/6 mice that reduced its function (Dolney et al., 2001a; Dolney et al., 2001b; Sundberg et al., 2011). C57BL/6 mice have arginine120, while C3H/HeJ and other strains of mice have cysteine120. Human ADH7 (class IV) also contains arginine120, similar to C57BL/6 mice (Kedishvili et al., 1995). Additional polymorphisms in ADH7 (class IV) were also found in humans linked to alcohol dependence, heroin addiction, and reduced risk of head and neck squamous cell carcinoma (Edenberg et al., 2006; Levran et al., 2009; Wei et al., 2010). Polymorphisms in ADH7 (class IV) may predispose B6 mice and humans to CA. Studies in transgenic mice suggest two roles for Adh7 (class IV). Adh7 tm1Gdu null mice have reduced fetal survival when dams are deficient in vitamin A (Deltour et al., 1999a). When these Adh7 tm1Gdu null mice are stressed with excess retinol there was reduced retinol clearance in the kidney (Deltour et al., 1999a, b), but not the liver (Molotkov et al., 2002). These data imply reduced Adh7 (class IV) activity increase susceptibility to both vitamin A deficiency and toxicity in B6 mice and some humans. This phenomenon may explain why more severe disease was seen in the second study when dams were fed lower vitamin A. The mice fed the AIN93G diet were also slow to breed; the number of litters, and the number of pups per litter were decreased compared to chow fed mice. These pups appeared runted at first, but appeared to catch up their growth after several days. In addition, many of the dam’s first litters were stillborn. It is possible that C57BL/6 mice require a narrower window of vitamin A for optimal function than other strains of mice. In the current study this optimal window is above 4 and below 28 IU vitamin A/g of diet. Variations in vitamin A levels in commercial unpurified diets range from 6.2–28.7 IU in 7 tests from 2 laboratories, 11.8–24.3 IU within the same lot (Sundberg unpublished data) implying mixing is not uniform. In susceptible strains this variation in vitamin A concentration in commercial diets can have a profound impact on the frequency of alopecia and dermatitis, and explain the apparent randomness of CA incidence in B6 mice in The Jackson Laboratory facility. Humans with similar polymorphisms may also require more precise levels of dietary vitamin A. Anecdotal reports suggest reduced alopecia with retinyl palmitate supplementation at roughly 3.5 times the recommended daily allowance (RDA, 8,000 IU/day) in patients with low serum vitamin A (Wilma Bergfeld, personal communication). Future studies are needed to determine the optimal dose of vitamin A in humans and mice susceptible to CA.
Retinoids are not the first treatment choice in CA patients, but they do help some patients and are listed as a second or third line of treatment in some forms of CA (Harries et al., 2008). This variable benefit of retinoid therapy is similar to the initial clinical trials in cancer (reviewed in, Freemantle et al., 2003). It is now well established that retinoid resistance occurs in cancer due to epigenetic hypermethylation of RARB and other RA synthesis enzymes (Tang and Gudas, 2011). Current studies combining low dose retinoid treatment with methyl transferase inhibitors in mouse models of cancer were more successful (Tang et al., 2009). A similar situation of retinoid resistance may occur in CA. If this is the case then this dual treatment strategy may be worth testing for the treatment of CA. Since high vitamin A worsened CA in this mouse study, this implies that pharmacological doses of retinoids may have been toxic. High vitamin A intake (12 IU/g diet) also accelerated alopecia areata in graft induced C3H/HeJ mice (Duncan et al., in press). Telogen effluvium was also seen in patients treated with retinoids (Ruzicka et al., 1992). Thus, a better treatment option may be to restore retinoid signalling with methyl transferase inhibitors while maintaining adequate dietary vitamin A levels. The exact “adequate” level of dietary vitamin A likely is very different in mice and humans with different genetic backgrounds.
In summary, this study suggests retinoid metabolism is altered in CA and high dietary vitamin A may worsen the disease. Future studies are needed to determine if the changes observed occur in all primary lymphocytic and neutrophilic forms of CA and the optimal level of dietary vitamin A to prevent the occurrence and severity of human CA. In B6 mice studies the diet should start with 1.5 to 3 times the recommended levels of vitamin A (6–12 IU/g diet in B6 mice). This optimal level maybe different in different genotypes so future studies should analyze linkage between Adh7 (class IV) SNPs with CA. Since all substrains of B6 mice have Adh7 polymorphisms, but differ in disease frequency, future studies should also identify other genetic mutations in RA synthesis or signaling genes. Epigenetic changes in retinoid metabolism in CA should also be explored. In addition, RARB expression levels may be a useful biomarker for severity of CA in specific substrains of B6 mice and types of human CA. This study also found that the frequency of this rare disease could be increased by diet so that future studies can be done in B6 mice to define the pathogenesis of CA and develop specific treatments of human CA.
Materials & Methods
Mice
The Jackson Laboratory IACUC approved all procedures and hair loss was observed weekly. H&E slides of biopsies were scored for specific follicle stage, follicular dystrophy, granulomas, epidermal hyperplasia, ulcers and dermal scars by a board certified pathologist (JPS, Sundberg et al., 2008).
Initial expression study mice
Mice were fed the NIH 31 diet with 6% fat; (LabDiet 5K52, Purina Mills, St. Louis, MO) throughout the study. There were 51 female controls. Cases were divided into mild (n=6), moderate (n=15), severe (n=38), and dermal scars (26). Hair follicles were staged as anagen II–IIIc verses anagen IV-catagen I, and hair follicles in these precise stages were compared between the groups (Muller-Rover et al., 2001).
Diet study 1 mice
Dams and female pups were fed the NIH 31 diet until 12 weeks of age until switched to the AIN93 Maintenance (M) diet containing 4 (n=9), 28 (n=10), or 56 (n=10) IU vitamin A/g diet (Research Diets, Inc., New Brunswick, NJ) for 4 months.
Diet study 2 mice
Dams were fed the AIN93 growth (G) diet throughout gestation. Female pups from these dams were maintained on this AIN93G diet during their growth phase and when they became pregnant themselves. Mice from this second generation of dams fed the AIN93G diet were used in this study. At 6 weeks of age these mice were fed the AIN93M diet containing 4 (n=19), 28 (n=35), or 56 (n=22) IU vitamin A/g diet (Research Diets, Inc., New Brunswick, NJ). At 16 weeks of age mice (n=10, 4 IU; n=16, 28 IU; n=14, 56 IU) were depilated then euthanized 2 weeks later.
Human samples
Paraffin blocks of biopsies from fourteen human patients with central centrifugal cicatricial alopecia (CCCA), three human patients with pseudopelade, and three non-alopecic dermatoses (tinea capitus and skin adjacent to pilar cysts) were obtained with Institutional Review Board approval from archives at Vanderbilt University (Nashville, TN; Table S1).
Procedures
IHC was performed as described previously (Everts et al., 2007). Antibodies were purchased for CYP26A1 (Alpha Diagnostic Intl. Inc., San Antonio, TX.), RARA, and RARB (Santa Cruz Biotech, Santa Cruz, CA.). Other antibodies used were produced and validated in Dr. Ong’s laboratory (Everts et al., 2007). A blinded IHC score of RARB Immunoreactivity was measured using a scale of 0–4 multiplied by percent of cells/mid-late anagen hair follicles. Two blinded researchers scored human samples for % of cells and strength on a 0 to 4 scale with 0 = no IR; 1 = 1–25% cells, weak IR; 2 = 26–50% cells, moderate IR; 3 = 51–75% cells, intense IR; 4 = 76–100% cells, very intense IR. The IHC-Level was calculated as the sum of intensity plus % cells as previously validated (Shamonki et al., 2006).
Retinoids were extracted (Pappas et al., 1993) and analyzed by HPLC with a modification of (Fleshman et al., 2012) using a C18 4.6 × 100mm, 3.5 μM column. The solvents were (A) 80:20 MeOH/0.1% formic acid and (B) MtBE. 10–20 μl of sample in 100%A was injected and retinoids separated with initial conditions of 100%A, a linear gradient to 100%B over 13 minutes, held at 100%B for 3 minutes, then equilibrated back to 100%A over 2 minutes. Spectra were analyzed using a photodiode array (Agilent Technologies, Santa Clara, CA) at 325 nm. Standards were purchased from Sigma (St. Louis, MO).
Chi squared analysis was performed to determine differences in hair loss using Prism 5 (Graph Pad; La Jolla, CA). All other data were analyzed by analysis of variance, followed by Tukey post hoc tests when appropriate using SPSS, v19 (IBM; Armonk, New York).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (AR052009 to HBE, AR052710 to JPS), the North American Hair Research Society (HBE), the Cicatricial Alopecia Research Foundation (CARF to JPS), and the Wauford Foundation (HBE).
Abbreviations
- B6
C57BL/6J inbred mouse strain
- RA
retinoic acid
- STRA6
Stimulated by retinoic acid 6
- RBP1
(formerly CRBP) Cellular retinol binding protein 1
- DHRS9
Dehydrogenase reductase (SDR family) member 9
- ALDH1A1, 2, and 3
Retinal dehydrogenase 1, 2, and 3
- CRABP1
Cellular retinoic acid binding protein I
- CRABP2
Cellular retinoic acid binding protein II
- RARA, B, and G
Retinoic acid receptor alpha, beta, and gamma
- ROLDHs
Retinol dehydrogenases
- RALDHs
Retinal dehydrogenases
- IR
-
Immunoreactivity.
Gene symbols and RNA message expression is italicized while proteins are not. Protein symbols from mouse, rat, and human are all in capitalized and not italicized
Footnotes
See Supplemental Data for more detailed methods.
References
- Al-Zaid T, Vanderweil S, Zembowicz A, Lyle S. Sebaceous gland loss and inflammation in scarring alopecia: A potential role in pathogenesis. J Am Acad Dermatol. 2011;65:597–603. doi: 10.1016/j.jaad.2010.09.774. [DOI] [PubMed] [Google Scholar]
- Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- Bergfeld WF, Elston DM. Cicatricial alopecia. In: Olsen EA, editor. Disorders of Hair Growth: Diagnosis and Treatment. 2. New York: McGraw-Hill Medical Publishing Division; 2003. pp. 363–398. [Google Scholar]
- Blaner WS, Gamble MV, Vogel S, Piantedosi R, Paik J, Gottesman ME. Retinol-binding protein (RBP): Essential physiologic functions. J Nutr. 2002;132:2979S–2979S. [Google Scholar]
- Blomhoff R, Green MH, Green JB, Berg T, Norum KR. Vitamin-A metabolism - New perspectives on absorption, transport, and storage. Physiol Rev. 1991;71:951–990. doi: 10.1152/physrev.1991.71.4.951. [DOI] [PubMed] [Google Scholar]
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- Corlazzoli F, Rossetti S, Bistulfi G, Ren MQ, Sacchi N. Derangement of a Factor Upstream of RAR alpha Triggers the Repression of a Pleiotropic Epigenetic Network. Plos One. 2009;4:e4305. doi: 10.1371/journal.pone.0004305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltour L, Foglio MH, Duester G. Impaired retinol utilization in Adh4 alcohol dehydrogenase mutant mice. Dev Genet. 1999a;25:1–10. doi: 10.1002/(SICI)1520-6408(1999)25:1<1::AID-DVG1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Deltour L, Foglio MH, Duester G. Metabolic deficiencies in alcohol dehydrogenase Adh1, Adh3, and Adh4 null mutant mice. Overlapping roles of Adh1 and Adh4 in ethanol clearance and metabolism of retinol to retinoic acid. J Biol Chem. 1999b;274:16796–16801. doi: 10.1074/jbc.274.24.16796. [DOI] [PubMed] [Google Scholar]
- Dolney DE, Szalai G, Duester G, Felder MR. Molecular analysis of genetic differences among inbred mouse strains controlling tissue expression pattern of alcohol dehydrogenase 4. Gene. 2001a;267:145–156. doi: 10.1016/s0378-1119(01)00409-7. [DOI] [PubMed] [Google Scholar]
- Dolney DE, Szalai G, Felder MR. Differences in charge and kinetic properties of alcohol dehydrogenase 4 from C57BL/6 mice compared to other inbred strains are associated with a cysteine120 to arginine120 substitution. Biochem Genet. 2001b;39:239–250. doi: 10.1023/a:1010278631535. [DOI] [PubMed] [Google Scholar]
- Dong D, Ruuska SE, Levinthal DJ, Noy N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem. 1999;274:23695–23698. doi: 10.1074/jbc.274.34.23695. [DOI] [PubMed] [Google Scholar]
- Duester G. Genetic dissection of retinoid dehydrogenases. Chem-Biol Interact. 2001;130:469–480. doi: 10.1016/s0009-2797(00)00292-1. [DOI] [PubMed] [Google Scholar]
- Duncan FJ, Silva KA, Johnson C, King B, Szatkiewicz JP, Kamdar S, et al. Endogenous retinoids in the pathogenesis of alopecia areata. J Invest Dermatol. doi: 10.1038/jid.2012.344. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Xuei XL, Chen HJ, Tian HJ, Wetherill LF, Dick DM, et al. Association of alcohol dehydrogenase genes with alcohol dependence: a comprehensive analysis. Human Molecular Genetics. 2006;15:1539–1549. doi: 10.1093/hmg/ddl073. [DOI] [PubMed] [Google Scholar]
- Everts HB. Endogenous retinoids in the hair follicle and sebaceous gland. Biochim Biophys Acta. 2012;1821:222–229. doi: 10.1016/j.bbalip.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts HB, King LE, Jr, Sundberg JP, Ong DE. Hair cycle-specific immunolocalization of retinoic acid synthesizing enzymes Aldh1a2 and Aldh1a3 indicate complex regulation. J Invest Dermatol. 2004;123:258–263. doi: 10.1111/j.0022-202X.2004.23223.x. [DOI] [PubMed] [Google Scholar]
- Everts HB, Sundberg JP, King LE, Jr, Ong DE. Immunolocalization of enzymes, binding proteins, and receptors sufficient for retinoic acid synthesis and signaling during the hair cycle. J Invest Dermatol. 2007;127:1593–1604. doi: 10.1038/sj.jid.5700753. [DOI] [PubMed] [Google Scholar]
- Fleshman MK, Riedl KM, Novotny JA, Schwartz SJ, Harrison EH. An LC/MS method for d8-beta-carotene and d4-retinyl esters: beta-carotene absorption and its conversion to vitamin A in humans. J Lipid Res. 2012;53:820–827. doi: 10.1194/jlr.D021139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers MT, Paton CM, O’Byrne SM, Schiesser K, Dawson JA, Blaner WS, et al. Metabolic Changes in Skin Caused by Scd1 Deficiency: A Focus on Retinol Metabolism. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22:7305–7315. doi: 10.1038/sj.onc.1206936. [DOI] [PubMed] [Google Scholar]
- Gao J, Simon M. Identification of a novel keratinocyte retinyl ester hydrolase as a transacylase and lipase. J Invest Dermatol. 2005;124:1259–1266. doi: 10.1111/j.0022-202X.2005.23761.x. [DOI] [PubMed] [Google Scholar]
- Harries MJ, Meyer KC, Paus R. Hair loss as a result of cutaneous autoimmunity: Frontiers in the immunopathogenesis of primary cicatricial alopecia. Autoimmun Rev. 2009;8:478–483. doi: 10.1016/j.autrev.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Harries MJ, Sinclair RD, MacDonald-Hull S, Whiting DA, Griffiths CEM, Paus R. Management of primary cicatricial alopecias: options for treatment. Br J Dermatol. 2008;159:1–22. doi: 10.1111/j.1365-2133.2008.08591.x. [DOI] [PubMed] [Google Scholar]
- Haselbeck RJ, Ang HL, Duester G. Class IV alcohol/retinol dehydrogenase localization in epidermal basal layer: Potential site of retinoic acid synthesis during skin development. Dev Dyn. 1997;208:447–453. doi: 10.1002/(SICI)1097-0177(199704)208:4<447::AID-AJA1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Heise R, Mey J, Neis MM, Marquardt Y, Joussen S, Ott H, et al. Skin retinoid concentrations are modulated by CYP26AI expression restricted to basal keratinocytes in normal human skin and differentiated 3D skin models. J Invest Dermatol. 2006;126:2473–2480. doi: 10.1038/sj.jid.5700432. [DOI] [PubMed] [Google Scholar]
- Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, et al. Ligand-independent repression by the thyroid-hormone receptor-mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- Kalz F. Cicatricial alopecia and vitamin A. AMA Arch Derm. 1958;78:740–743. doi: 10.1001/archderm.1958.01560120060009. [DOI] [PubMed] [Google Scholar]
- Karnik P, Tekeste Z, McCormick TS, Gilliam AC, Price VH, Cooper KD, et al. Hair follicle stem cell-specific PPARg deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243–1257. doi: 10.1038/jid.2008.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedishvili NY, Bosron WF, Stone CL, Hurley TD, Peggs CF, Thomasson HR, et al. Expression and kinetic charaterization of recombinant human stocach alcohol-dehydrogenase-active-site amino-acid-sequece explains substrate specificity compared with liver isozymes. J Biol Chem. 1995;270:3625–3630. doi: 10.1074/jbc.270.8.3625. [DOI] [PubMed] [Google Scholar]
- Kumar S, Sandell LL, Trainer PA, Koentgen F, Duester G. Alcohol and aldehyde dehydrogenases: Retinoid metabolic effects in mouse knockout models. Biochim Biophys Acta, Mol Cell Biol Lipids. 2012;1821:198–205. doi: 10.1016/j.bbalip.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen KB, Wong PM, Gudas LJ. Epigenetic regulation by RAR alpha maintains ligand-independent transcriptional activity. Nucleic Acids Research. 2012;40:102–115. doi: 10.1093/nar/gkr637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Londono D, O’Hara K, Randesi M, Rotrosen J, Casadonte P, et al. Heroin addiction in African Americans: a hypothesis-driven association study. Genes Brain and Behavior. 2009;8:531–540. doi: 10.1111/j.1601-183X.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorie EP, Chamcheu JC, Vahlquist A, Torma H. Both all-trans retinoic acid and cytochrome P450 (CYP26) inhibitors affect the expression of vitamin A metabolizing enzymes and retinoid biomarkers in organotypic epidermis. Arch Dermatol Res. 2009a;301:475–485. doi: 10.1007/s00403-009-0937-7. [DOI] [PubMed] [Google Scholar]
- Lorie EP, Cools M, Borgers M, Wouters L, Shroot B, Hagforsen E, et al. Topical treatment with CYP26 inhibitor talarozole (R115866) dose dependently alters the expression of retinoid-regulated genes in normal human epidermis. Br J Dermatol. 2009b;160:26–36. doi: 10.1111/j.1365-2133.2008.08895.x. [DOI] [PubMed] [Google Scholar]
- Loudig O, Babichuk C, White J, Abu-Abed S, Mueller C, Petkovich M. Cytochrome P450RAI(CYP26) promoter: a distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol Endocrinol. 2000;14:1483–1497. doi: 10.1210/mend.14.9.0518. [DOI] [PubMed] [Google Scholar]
- McElwee KJ. Etiology of cicatricial alopecias: a basic science point of view. Dermatologic Therapy. 2008;21:212–220. doi: 10.1111/j.1529-8019.2008.00202.x. [DOI] [PubMed] [Google Scholar]
- Molotkov A, Fan X, Duester G. Excessive vitamin A toxicity in mice genetically deficient in either alcohol dehydrogenase Adh1 or Adh3. Eur J Biochem. 2002;269:2607–2612. doi: 10.1046/j.1432-1033.2002.02935.x. [DOI] [PubMed] [Google Scholar]
- Muller-Rover S, Handjiski B, van der Veen C, Eichmuller S, Foitzik K, McKay IA, et al. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Invest Dermatol. 2001;117:3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- Napoli JL. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim Biophys Acta, Mol Cell Biol Lipids. 2012;1821:152–167. doi: 10.1016/j.bbalip.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, et al. Retinoid absorption and storage is impaired in mice lacking lecithin : retinol acyltransferase (LRAT) J Biol Chem. 2005;280:35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas RS, Newcomer ME, Ong DE. Endogenous retinoids in rat epididymal tissue and rat and human spermatozoa. Biol Reprod. 1993;48:235–247. doi: 10.1095/biolreprod48.2.235. [DOI] [PubMed] [Google Scholar]
- Reeves PG, Nielsen FH, Fahey GC., Jr AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- Reijntjes S, Blentic A, Gale E, Maden M. The control of morphogen signalling: Regulation of the synthesis and catabolism of retinoic acid in the developing embryo. Dev Biol. 2005;285:224–237. doi: 10.1016/j.ydbio.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Ren MQ, Pozzi S, Bistulfi G, Somenzi G, Rossetti S, Sacchi N. Impaired retinoic acid (RA) signal leads to RAR beta 2 epigenetic silencing and RA resistance. Mol Cell Biol. 2005;25:10591–10603. doi: 10.1128/MCB.25.23.10591-10603.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka T, Sommerburg C, Goerz G, Kind P, Mensing H. Treatment of cutaneous lupus-erythematosus with acitretin and hydroxychloroquine. Br J Dermatol. 1992;127:513–518. doi: 10.1111/j.1365-2133.1992.tb14851.x. [DOI] [PubMed] [Google Scholar]
- Shamonki MI, Kligman I, Shamonki JM, Schattman GL, Hyjek E, Spandorfer SD, et al. Immunohistochemical expression of endometrial L-selectin ligand is higher in donor egg recipients with embryonic implantation. Fertil Steril. 2006;86:1365–1375. doi: 10.1016/j.fertnstert.2006.04.035. [DOI] [PubMed] [Google Scholar]
- Shih MYS, Kane MA, Zhou P, Yen CLE, Streeper RS, Napoli JL, et al. Retinol Esterification by DGAT1 Is Essential for Retinoid Homeostasis in Murine Skin. J Biol Chem. 2009;284:4292–4299. doi: 10.1074/jbc.M807503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Thiboutot DM. Sebaceous gland lipids: friend or foe? J Lipid Res. 2008;49:271–281. doi: 10.1194/jlr.R700015-JLR200. [DOI] [PubMed] [Google Scholar]
- Smith SM, Levy NS, Hayes CE. Impaired immunity in vitamin A deficient mice. J Nutr. 1987;117:857–865. doi: 10.1093/jn/117.5.857. [DOI] [PubMed] [Google Scholar]
- Sperling LC. Scarring alopecia and the dermatopathologist. J Cutan Pathol. 2001;28:333–342. doi: 10.1034/j.1600-0560.2001.280701.x. [DOI] [PubMed] [Google Scholar]
- Sperling LC, Cowper SE. The histoplathology of primary cicatricial alopecia. Semin Cutan Med Surg. 2006;25:41–50. doi: 10.1016/j.sder.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Sperling LC, Sau P. The follicular degeneration syndrome in black patients- ‘hot comb alopecia’ revisited and revised. Arch Dermatol. 1992;128:68–74. [PubMed] [Google Scholar]
- Sperling LC, Skelton HG, Smith KJ, Sau P, Friedman K. Follicular degeneration syndrome in men. Arch Dermatol. 1994;130:763–769. [PubMed] [Google Scholar]
- Sperling LC, Solomon AR, Whiting DA. A new look at scarring alopecia. Arch Dermatol. 2000;136:235–242. doi: 10.1001/archderm.136.2.235. [DOI] [PubMed] [Google Scholar]
- Stenn KS, Sundberg JP, Sperling LC. Hair follicle biology, the sebaceous gland, and scarring alopecias. Arch Dermatol. 1999;135:973–974. doi: 10.1001/archderm.135.8.973. [DOI] [PubMed] [Google Scholar]
- Sundberg JP, Boggess D, Sundberg BA, Eilertsen K, Parimoo S, Filippi M, et al. Asebia-2J (Scd1ab2J): A new allele and a model for scarring alopecia. Am J Pathol. 2000;156:2067–2075. doi: 10.1016/S0002-9440(10)65078-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg JP, Sundberg BA, Schofield P. Integrating mouse anatomy and pathology ontologies into a phenotyping database: Tools for data capture and training. Mammalian Genome. 2008;19:413–419. doi: 10.1007/s00335-008-9123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg JP, Taylor D, Lorch G, Miller J, Silva KA, Sundberg BA, et al. Primary Follicular Dystrophy With Scarring Dermatitis in C57BL/6 Mouse Substrains Resembles Central Centrifugal Cicatricial Alopecia in Humans. Vet Pathol. 2011;48:513–524. doi: 10.1177/0300985810379431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, Albert M, Scognamiglio T, Gudas LJ. A DNA Methyltransferase Inhibitor and All-trans Retinoic Acid Reduce Oral Cavity Carcinogenesis Induced by the Carcinogen 4-Nitroquinoline 1-Oxide. Cancer Prevention Research. 2009;2:1100–1110. doi: 10.1158/1940-6207.CAPR-09-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, Gudas LJ. Retinoids, Retinoic Acid Receptors, and Cancer. Annual Review of Pathology: Mechanisms of Disease. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- Wei S, Liu ZS, Zhao H, Niu JG, Wang LE, El-Naggar AK, et al. A Single Nucleotide Polymorphism in the Alcohol Dehydrogenase 7 Gene (Alanine to Glycine Substitution at Amino Acid 92) Is Associated With the Risk of Squamous Cell Carcinoma of the Head and Neck. Cancer. 2010;116:2984–2992. doi: 10.1002/cncr.25058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Hu PR, Krois CR, Kane MA, Napoli JL. Altered vitamin A homeostasis and increased size and adiposity in the rdh1-null mouse. Faseb J. 2007;21:2886–2896. doi: 10.1096/fj.06-7964com. [DOI] [PubMed] [Google Scholar]
- Zouboulis CC. Isotretinoin revisited: Pluripotent effects on human sebaceous gland cells. J Invest Dermatol. 2006;126:2154–2156. doi: 10.1038/sj.jid.5700418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.