

Conspectus
Although graphene’s physical structure is a single atom thick, two-dimensional, hexagonal crystal of sp2 bonded carbon, this simple description belies the myriad interesting and complex physical properties attributed to this fascinating material. Because of its unusual electronic structure and superlative properties, graphene serves as a leading candidate for many next generation technologies including high frequency electronics, broadband photodetectors, biological and gas sensors, and transparent conductive coatings. Despite this promise, researchers could apply graphene more routinely in real-world technologies if they could chemically adjust graphene’s electronic properties. For example, the covalent modification of graphene to create a band gap comparable to silicon (~1 eV) would enable its use in digital electronics, and larger band gaps would provide new opportunities for graphene-based photonics. Towards this end, researchers have focused considerable effort on the chemical functionalization of graphene. Due to its high thermodynamic stability and chemical inertness, new methods and techniques are required to create covalent bonds without promoting undesirable side reactions or irreversible damage to the underlying carbon lattice.
In this Account, we review and discuss recent theoretical and experimental work studying covalent modifications to graphene using gas phase atomic radicals. Atomic radicals have sufficient energy to overcome the kinetic and thermodynamic barriers associated with covalent reactions on the basal plane of graphene but lack the energy required to break the C-C sigma bonds that would destroy the carbon lattice. Furthermore, because they are atomic species, radicals substantially reduce the likelihood of unwanted side reactions that confound other covalent chemistries. Overall, these methods based on atomic radicals show promise for the homogeneous functionalization of graphene and the production of new classes of two-dimensional materials with fundamentally different electronic and physical properties.
Specifically, we focus on recent studies of the addition of atomic hydrogen, fluorine, and oxygen to the basal plane of graphene. In each of these reactions a high energy, activating step initiates the process, breaking the local π structure and distorting the surrounding lattice. Scanning tunneling microscopy experiments reveal that substrate mediated interactions often dominate when the initial binding event occurs. We then compare these substrate effects with the results of theoretical studies that typically assume a vacuum environment. As the surface coverage increases, clusters often form around the initial distortion, and the stoichiometric composition of the saturated end product depends strongly on both the substrate and reactant species. In addition to these chemical and structural observations, we review how covalent modification can extend the range of physical properties that are achievable in two-dimensional materials.
Keywords: Graphene, Scanning Tunneling Microscopy, Surface Science, Density Functional Theory, Electronic Materials
1. Introduction
Graphene, a single layer of sp2-bonded carbon atoms arranged in a honeycomb lattice, is the first two-dimensional material shown to exist in a suspended form, defying previously held notions that suspended 2D materials were thermodynamically unstable.1 Since its initial isolation, graphene has demonstrated a plethora of remarkable physical properties, many of which stem from its unusual electronic structure near the Fermi level.2 In particular, electrons and holes in graphene respond rapidly to applied electric fields due to exceptionally high charge carrier mobilities in excess of 200,000 cm2/Vs.3 This superlative mobility has recently been exploited in high frequency transistors with operating frequencies in excess of 100 GHz,4 suggesting that graphene has the potential to become the foundation for next generation electronics.
Despite its many outstanding electronic attributes, several issues must be overcome before the full promise of graphene can be realized. For example, in contrast to conventional semiconductors, graphene does not possess a band gap, which implies that it cannot be directly employed in digital electronics where high transistor on/off ratios are imperative.5 Furthermore, imperfect methods for integrating graphene with other materials, including dielectrics, metals, and dopants, compromises performance in fully fabricated devices and circuits.6 Underlying these challenges is the chemical inertness of the basal plane of graphene, which confounds efforts to tailor its properties and limits the formation of well-defined interfaces with other materials.
The lack of chemical reactivity in graphene can be traced to its homogeneity and highly delocalized electronic structure. Typically, chemical reactions occur at locations that have weak or labile bonds, highly localized orbitals, dangling bonds, or localized charges, none of which are present in graphene. On the other hand, in the honeycomb lattice of graphene, each carbon atom possesses a 3-fold symmetric sp2 electronic hybridization in which the π orbitals extend out of the atomic plane, forming a self-passivating, highly delocalized network. Disrupting this chemical structure is not only thermodynamically unfavorable, but also requires the formation of high energy radicals localized on adjacent carbons that are difficult to support.
Covalent chemistry on the basal plane of graphene is further hindered by a high kinetic barrier associated with rearrangements of the carbon lattice. The equilibrium geometry for aromatic carbons is planar, which is a direct result of the symmetry of the sp2 hybridization. Covalent modification of graphene, however, converts sp2 carbons to sp3, which prefer to be in a tetrahedral geometry with longer bonds as shown in Figure 1. This transformation affects not only the sp3 carbon directly affected by the reaction, but also creates a geometric distortion that extends over multiple lattice positions. The resulting energetic penalty for this lattice deformation significantly increases the transition state energy associated with covalent chemistry on graphene.
Figure 1.

Typical lattice distortion resulting from a single covalent adsorbate on graphene. Atoms are colored according to their out of plane height to better visualize the surrounding distortion.
As a consequence of the absence of reactive sites and a substantial kinetic barrier to reaction, covalent modification of the basal plane of graphene requires the presence of high energy reactants such as strong acids or radicals. However, high energy reactants often induce unintended side reactions or a lack of homogeneity in the resulting chemical functionalization. For instance, reduction of aryl diazonium cations lead to organic radicals that covalently react with graphene,7 but also are susceptible to self-polymerization that results in spatially inhomogeneous attachment and a disordered energy landscape.8 Similarly, acidic oxidation of graphene results in a complicated chemical structure including hydroxides, epoxides, topological defects, and nanometer-sized holes.9 While inhomogeneously functionalized graphene may be acceptable in some applications, it remains suboptimal for high performance electronics that are invariably based on well-defined, crystalline materials.
In this Account, we highlight recent work concerning the chemistry and emergent properties of covalent atomic adsorbates on the basal plane of graphene. Compared to organic free radicals, atomic species (e.g., H, F, and O) minimize the possibility of side reactions and thus hold promise for covalently functionalizing graphene in a chemically uniform and homogeneous manner. Furthermore, the relative simplicity of atomic adsorbates allows them to be well described by theoretical calculations, thereby facilitating further elucidation of the stable chemical compositions and structures that result from these reactions. In addition to providing a snapshot of the current understanding and achievements for covalently modified graphene, this Account also provides perspective on the remaining challenges and future research directions for this field.
2. Atomic Hydrogen on Graphene
2.1. Theoretical Studies of Hydrogenated Graphene
Hydrogen is the most thoroughly studied atomic adsorbate on graphene. From a theoretical standpoint, covalent addition of hydrogen to graphene has been studied at a variety of coverages including single atomic adsorbates,10–12 dimers,11, 13 clusters,11 and saturation coverage.14–15 In the low coverage limit, hydrogen binds preferentially as a dimer. Specifically, the reaction of graphene with hydrogen has a substantial barrier that results from the reorganization of the carbon lattice to accommodate an sp3 defect and the creation of a radical. Following the first atomic hydrogen adsorption event, the deformed lattice can more easily accommodate a second carbon-hydrogen bond, resulting in a dimer structure. Additional hydrogen atoms are expected to cluster, and the most stable conformation, known as graphane, has been theoretically established as the chair conformation shown in Figure 2a, in which each carbon atom is covalently bound to hydrogen with alternating C-H bonds pointing in the same direction.
Figure 2.

(A) Structure of graphane (i.e., the high coverage limit of hydrogenated graphene). White atoms are hydrogen, and gray atoms are carbon. Reproduced from Sofo et al.14 Copyright 2007 American Physical Society. (B) Electronic band structure for graphane. Solid lines are with the GGA approximation, while red dots represent the more accurate GW band structure. Reproduced from Lebegue et al.15 Copyright 2009 American Physical Society. (C) Evolution of the band gap as a function of hydrogen coverage. Reproduced from Gao et al.11 Copyright 2011 American Chemical Society.
The addition of hydrogen to graphene has been predicted to radically change its electronic, elastic, optical, and even magnetic properties. Fully hydrogenated graphane is expected to be a wide band gap material, with a direct gap at the center of the bouillon zone. In particular, high-level GW approximation electronic structure calculations predict a band gap of 5.4 eV.15 If hydrogen adsorption at lower coverage occurs in a spatially periodic fashion, then a controlled smaller band gap has been predicted. However, spatially random hydrogen dimers preclude the opening of a band gap until the coverage has reached ~66%, thus highlighting the advantages of chemically homogeneous and spatially periodic hydrogen adsorption.11 The expected direct gap of hydrogenated graphene also suggests the possibility for realizing novel optical properties. For example, Samarakoon et al. have calculated the optical absorption spectrum of graphane, and shown that despite the presence of strong excitonic peaks in the dielectric function, the absorption onset occurs in the ultraviolet, thus making graphane completely transparent.16
2.2. Experimental Studies of Hydrogenated Graphene
Hydrogenated graphene has been produced primarily using atomic hydrogen beams, in which molecular hydrogen is cracked on a hot filament,17–19 or via exposure to hydrogen-based plasmas.20–21 Typically, atomic hydrogen beams have been employed for low coverage studies with scanning tunneling microscopy (STM), while hydrogen plasmas provide sufficient hydrogen flux to approach the 1:1 carbon to hydrogen ratio of graphane. The detailed conditions for hydrogenation determine the level of irreversible damage to the graphene lattice. In particular, hot, high-power plasmas result in hydrogenated graphene with significant irreversible defect density, while low temperature plasmas and atomic hydrogen beams add hydrogen in a manner that is thermally reversible to pristine graphene as measured by Raman spectroscopy, electrical transport measurements, and STM.18–19
The structure of atomic hydrogen bound to a single side of graphene has been resolved by STM for epitaxial graphene (EG) on silicon carbide17–18, 22 and graphene on single crystal transition metals grown by chemical vapor deposition (CVD).23 In agreement with theoretical predictions, hydrogen atoms adsorb on graphene primarily as dimer species, although monomers have been reported as a minority component on EG in contrast to graphite.24 The adsorption of atomic hydrogen on EG and CVD graphene is shown in Figure 3. On both EG and CVD graphene, lattice mismatch between graphene and the substrate produce long range corrugations in the physical and electronic structure known as a Moiré pattern. This graphene-substrate interaction templates the hydrogen adsorption in registry with the Moiré pattern as shown in Figures 3b and 3e. On CVD graphene, the attachment of hydrogen has been predicted to displace the carbon atoms down towards the metallic substrate, resulting in the formation of covalent metal carbide bonds. Consequently, hydrogen adsorption is more favorable in areas in which the graphene ring is situated directly atop a metal substrate atom. The relatively strong metal carbide interaction results in the Moiré pattern being preserved even at high hydrogen coverage on CVD graphene on Ir(111). The importance of the metal carbide bond has been further confirmed by studying the hydrogenation of CVD graphene grown on multiple metals with varying graphene substrate interaction strengths.25 In contrast, the Moiré pattern is not observed on EG at high coverage; instead, the hydrogen adsorption appears randomly clustered.
Figure 3.

(A) 100 nm × 100 nm STM image of clean epitaxial graphene on SiC. (B) STM image of epitaxial graphene after a low exposure to atomic hydrogen, showing two types of hydrogen dimers. (C) Atomic resolution of a single hydrogen dimer showing two adatoms. (D) 100 nm × 100 nm STM image of epitaxial graphene exposed to a saturation coverage of atomic hydrogen. (E,F) STM images of graphene on Ir(111) after exposure to low (E) and saturation (F) doses of atomic hydrogen. (A) & (D) are reproduced from Guisinger et al.17 Copyright 2009 American Chemical Society. (B) and (C) are reproduced from Sessi et al.18 Copyright 2009 American Chemical Society. (E) and (F) are reproduced from Balog et al.23 Copyright 2010 Nature Publishing Group.
Electronically, hydrogenation reduces the local conductivity, with differential tunneling conductance STM imaging revealing a ring of depressed local density of states with a diameter of up to 5 nm surrounding a single hydrogen dimer pair on EG.18 At saturated hydrogen coverage on EG, scanning tunneling spectroscopy reveals the loss of the Dirac point associated with graphene and the presence of a small band gap. Hydrogenated CVD graphene on Ir(111) also possesses a band gap of 400 meV as measured by angle-resolved photoemission.23
For suspended graphene, hydrogen adsorption can occur on both sides of the graphene membrane. By exposing exfoliated graphene to a low temperature plasma of hydrogen, Elias et al. created large areas of hydrogenated graphene, reaching a fully saturated graphane structure in the suspended regions.19 Because these graphene samples were not mounted on conductive substrates, they were not imaged with STM. However, the structure was inferred by transmission electron microscopy (TEM) diffraction, which shows sharp diffraction spots and a characteristic contraction of the lattice from graphene to graphane. The degree of hydrogenation has also been monitored by the evolution of the Raman D band, which is associated with symmetry breaking defects in the sp2 lattice such as sp3 carbons.19, 21 For hydrogenated graphene on SiO2, Raman measurements show that aromatic carbons remain even after extensive exposure, indicating that hydrogen adsorption is clustered and saturates well below a 1:1 ratio. This observation was confirmed by charge transport measurements, which reveal a decrease in mobility and p-type doping, but no significant band gap for hydrogenated graphene on SiO2.19
3. Fluorographene
3.1. Theoretical Studies of Fluorinated Graphene
The reaction of fluorine with graphene has been modeled as addition reactions for both its acidic and radical components.26–34 Fluorination of graphene is similar to hydrogenation in that fluorine forms a single bond to carbon but with a reversed dipole and increased binding strength. Furthermore, unlike hydrogenation, the binding of a single fluorine atom is exothermic, and nucleation is not required for film growth.32 Since fluorination does not require nucleation, it should be easier to produce saturated coverages of fluorographene compared to graphane. When fluorine covalently reacts with graphene, the carbon lattice is distorted upward around the resulting sp3 carbon, and the binding energy of additional fluorine atoms is increased. For graphene in which only a single side is exposed to fluorine, fluorination is expected to saturate at 25% coverage (C4F).30 This structure proposed by Robinson et al. is shown in Figure 4a. For double-sided fluorine exposure, the binding energy decreases monotonically until a fully saturated coverage is reached.32 The most stable calculated structure of saturated fluorographene is an alternating conformation analogous to graphane as shown in Figure 4c.29, 32, 35
Figure 4.

(A) Geometric structure of stable single-sided fluorinated graphene, C4F. (B) Calculated electronic band structure for C4F. Gray atoms are carbon, and cyan atoms are fluorine. Blue lines represent the LDA band structure, and red dots are the more accurate GW0 corrected valence bands. The yellow bar highlights the LDA band gap along the Brillouin zone. (C) The most stable geometric structure of fully fluorinated graphene, CF. (D) Electronic band structure of CF within the LDA and GW0 approximations. Images are reproduced from \ahin et al.36 Copyright 2011 American Physical Society.
Once graphene has been fully fluorinated, its electronic properties are predicted to change substantially. Fluorographene is expected to be electrically insulating with a band gap of 3.1 eV, as calculated within the local density approximation (LDA).16 When dynamic screening is taken into account with the GW approximation, the calculated band gap further increases to 7.4 eV.29, 36 The calculated LDA and GW band structure for C4F and fluorographene are shown in Figures 4b and 4d. Similar to graphane, fluorographene is a direct band gap insulator at the Γ point, suggesting the possibility for strong excitonic effects in the optical absorption spectrum. For example, Samarakoon et al. calculated the absorption spectrum for isolated fluorographene and graphane, and found that the higher polarity of the C-F bond significantly increases the electron-hole interactions.16 This enhanced Coulombic interaction results in an absorption onset that is significantly red shifted for fluorographene, and increases the potential utility of fluorographene in electro-optical and excitonic devices.
3.2. Experimental Studies of Fluorinated Graphene
Although bulk graphite fluoride has been used as a lubricant for nearly one hundred years, graphene fluoride was not experimentally realized until 2010. Three prominent methods for creating fluorographene exist: (1) Exposure to the compound XeF2;30, 37 (2) Plasma etching using a fluorinated compound;38 (3) Liquid phase exfoliation of bulk graphite fluoride.34 Among these methods, plasma etching and XeF2 exposure yield similar results as will be outlined below. In contrast, graphite fluoride is often a non-stoichiometric compound39 with a fluorine to carbon ratio between 0.5 and 1.3, which indicates the presence of CF2 and CF3 functional groups that are incompatible with pristine graphene surfaces.
Robinson et al. first reported single-sided fluorination of CVD graphene on copper foil in which they concluded that the fluorine coverage saturated at a composition of C4F based on X-ray photoemission spectroscopy (XPS) characterization.30 On the other hand, for graphene on SiO2 or silicon-on-insulator substrates, they found that fluorination produces a carbon to fluorine ratio approaching 1:1. The resulting XPS data are shown in Figure 5a. In an independent study, Nair et al. fluorinated exfoliated graphene flakes and epitaxial graphene on SiC with XeF2.37 Interestingly, while saturated fluorination of CVD graphene and graphene on SiO2 can be achieved within minutes or hours, fluorination of epitaxial graphene proceeds slowly, and ultimately requires approximately two months of exposure at elevated temperatures to reach saturation. TEM diffraction measurements of suspended samples confirm that fluorinated graphene is a hexagonal crystal with an expanded unit cell due to the longer sp3 bonds as shown in Figure 5.37
Figure 5.

(A) XPS spectra of the carbon peak for CVD graphene as it is exposed to XeF2, confirming the binding of fluorine to the carbon lattice. (B) Optical image of a patterned fluorinated graphene film, showing that fluorination of graphene induces transparency. (C) Raman spectra of fully fluorinated graphene and graphite fluoride. (D) TEM diffraction image of fluorographene. (E) An expansion of the in-plane lattice constant verifies that the fluorine is creating elongated sp3 bonds. (A) and (B) are reproduced from Robinson et al.30 Copyright 2010 American Chemical Society. (C), (D), and (E) are reproduced from Nair et al.37 Copyright 2010 Wiley Publishers.
Fluorographene is highly insulating at room temperature, which indicates the opening of a band gap. Furthermore, fluorographene is transparent in the visible, providing further evidence of a band gap (Figure 5b).30, 37 During the early stages of fluorination, Raman measurements show rises in the D peak, which indicates the creation of defects and sp3 carbons. As more fluorine is added, the band gap extends into the ultraviolet, and without resonance enhancement, Nair et al. report no detectable Raman signal from fully fluorinated graphene under visible excitation.37 In addition, Jeon et al. were able to detect fluorescence from both excitonic and direct emission states from dispersed layers of graphite fluoride.40 An optical band gap of 3.8 eV was measured, which is close to that predicted by Samarakoon et al.16 The observation of fluorescence, combined with the change in the optical absorption spectrum and high electrical resistivity, confirms the direct band gap nature of fluorographene.
4. Atomic Oxygen on Graphene
The addition of atomic oxygen to graphene is a more complex and rich reaction than fluorine or hydrogen since oxygen is expected to form two covalent bonds rather than one. The potential complexity of oxygen chemistry on graphene is most clearly seen for the case of Hummers method graphite oxide (GO), which is a solution-based oxidation method for graphite using nitric acid and other strong oxidizing reagents.41 While this is the most common method for covalently functionalizing graphene, the resulting GO material is highly inhomogeneous. In particular, GO contains a plethora of functional groups including epoxides and alcohols on the basal plane, and ketones, carboxylic acid, ether, and enol functional groups on the edges. For a systematic review of GO and its chemistry, we recommend the work of Dreyer et al. and the references therein.42 Here, we focus on recent theoretical and experimental studies for graphene functionalized by atomic oxygen that result in homogeneous epoxidation.
4.1. Theoretical Studies of Graphene Epoxide
The complexity of the structure of GO has led to a large body of computational and theoretical work attempting to model and understand the interaction between oxygen and graphene. Attachment of atomic oxygen to defect-free regions of the honeycomb lattice is expected to occur as an epoxide, in which one oxygen atom bridges two adjacent carbon atoms with a binding energy of 1.9 eV.43–44 The structure of an isolated epoxide on graphene is shown in Figure 6a. Solís-Fernández et al. calculated that the Mulliken charge associated with each epoxide is −0.29 e, which suggests that epoxide groups should p-type dope graphene in the dilute limit. Because the epoxide always binds to two adjacent carbons, no radicals are created, and the ground state is always nonmagnetic, regardless of coverage.43
Figure 6.
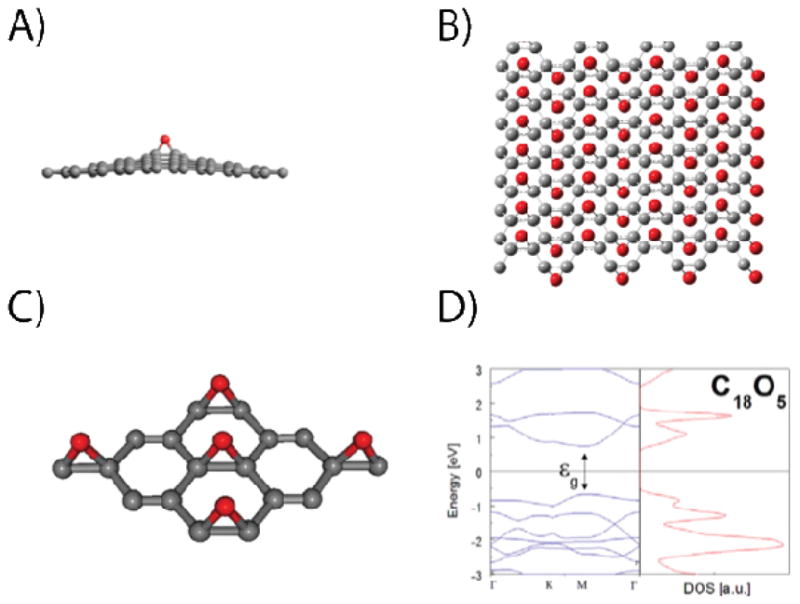
(A) Structure of a graphene epoxide bond. (B) Structure proposed by Boukhvalov et al. for double-sided graphene epoxide. (C) Structure of single-sided graphene epoxide. (D) Electronic band structure of (C) within the LDA. (A) is reproduced from Hossain et al.49 Copyright 2012 Nature Publishing Group. (C) and (D) are reproduced from Nourbakhsh et al.46 Copyright 2011 Institute of Physics Publishing.
As the carbon lattice becomes distorted due to the presence of sp3carbons, the binding energy for additional oxygen increases.45 If atomic oxygen has access to both sides of suspended graphene, then the most stable coverage occurs at a stoichiometry of C2O, with epoxides forming stripes on alternating sides of the carbon membrane as shown in Figure 6b.45 Stable high coverage structures have also been predicted for oxygen bound to only a single side of graphene such as alternating stripes of carbon rings and epoxides.46 As the coverage in both single-sided46 and double-sided exposure45 approaches saturation, a band gap of greater than 3 eV is expected to open. Interestingly, when the top layer of bilayer graphene is decorated with epoxide groups, the semi-metallic electronic structure of monolayer graphene is expected to be recovered.47
4.2. Experimental Studies of Graphene Epoxide
Experimentally, the formation of epoxide groups on graphene has been achieved by exposure to oxygen plasmas48 and atomic oxygen beams created by cracking molecular oxygen on a hot filament.49 As in the case of hydrogen plasmas, the processing conditions for exposure of graphene to an oxygen plasma must be carefully controlled to avoid damage to the carbon lattice. In fact, attempts to measure the oxidation of graphite by an oxygen plasma have found that plasmas can etch graphite even at low exposure levels, creating single-atom vacancies and nanometer-sized pits,50–51 while additional exposure induces the formation of micro-pores.52 This potential for irreversible lattice damage in plasma-based oxidation is enhanced by the presence of higher energy ions such as O2+ that can sputter carbonaceous material. In addition to the plasma conditions, the substrate temperature during oxidation must also be carefully controlled as high temperatures promote etching and loss of carbon.
Recent work has shown that etching can be suppressed in the dilute limit by performing oxidation in ultra-high vacuum (UHV) conditions with thermally cracked atomic oxygen instead of plasmas.49, 53 Hossain et al. exposed epitaxial graphene on SiC(0001) to atomic oxygen produced with a hot tungsten filament in UHV, and imaged the resulting surface with STM as shown in Figure 7a. Unlike the adsorption of hydrogen on epitaxial graphene,17 only a single shape is observed for the oxygen protrusions, which suggests a highly homogeneous chemical functionalization. Raman spectra show an increase in the D band of the graphene, indicating that the oxygen adsorbates are creating sp3 carbon through covalent attachment. This result is consistent with changes to the Raman spectrum observed in the wet chemical oxidation of graphene and epitaxial graphene.54–55 No depressions as a result of pitting or etching are observed. Furthermore, core level XPS confirms that the oxygen atoms are bound only as epoxide groups.
Figure 7.

(A,B) STM images of epitaxial graphene on SiC after exposure to atomic oxygen before (A) and after (B) thermal desorption. (C) Core level XPS spectra of epitaxial graphene before and after exposure to atomic oxygen. (D,E) STM images showing low (D) and saturated (E) exposure of CVD graphene on Ir(111) to atomic oxygen. The insets are the autocorrelation patterns from each image and show that hexagonal symmetry is preserved. (A), (B), and (C) are reproduced from Hossain et al.49 Copyright 2012 Nature Publishing Group. (D) and (E) are reproduced from Vinogradov et al. 53 Copyright 2011 American Chemical Society.
The dilute epoxidation of epitaxial graphene is completely reversible by thermal annealing as shown in Figure 7b. Following annealing at 260 °C, the epoxide features in the STM image disappear, and pristine graphene is recovered with no evidence of substrate damage or defects. The thermal reversibility of this UHV expoxidation process is further confirmed via Raman spectroscopy, ultraviolet photoelectron spectroscopy, and core level XPS. Overall, this study shows that chemically homogeneous and thermally reversible epoxidation of graphene can be achieved via UHV atomic oxygen exposure.
Vinogradov et al. found similar results for the addition of atomic oxygen on graphene on transition metals.53 STM images reveal that oxygen atoms preferentially bind to locations on the graphene/Ir(111) Moiré pattern, which correspond to carbon atoms closer to the metal substrate. After extended oxidation, the Moiré pattern becomes disordered and is no longer observed in the STM images, although the autocorrelation of the STM data shows that the symmetry underlying the Moiré pattern remains. This result suggests that the epoxide is not etching the graphene even at saturated coverages. In addition, a low energy shoulder in the oxygen peak appears as well as multiple peaks for the transition metals in the XPS measurements. The presence of the epoxide bond pushes the carbon down towards the metal surface, similar to the effect of atomic hydrogen on graphene on Ir(111).23 Even at saturated coverage, the linear structure predicted by DFT (and observed in STM images of exfoliated GO flakes)56 is not observed for graphene epoxide prepared in UHV. Unlike the epoxide functionality on epitaxial graphene on SiC, the epoxide groups on graphene/Ir(111) suffer from thermal irreversibility with strong evidence for carbon removal following annealing above 500 °C in both STM and XPS measurements.
5. Conclusion
In this Account, we have reviewed recent work focused on the covalent functionalization of graphene. In particular, reactions with atomic species (e.g., H, F, and O) represent a promising path to creating new classes of chemically tunable two-dimensional materials. While theoretical calculations have predicted a suite of distinct structures with desirable electronic and optical properties, experimental realization of fully saturated graphene-based materials remains a challenge in most cases. Consequently, additional research is needed to better characterize and understand these covalent modification strategies, especially atomically resolved structural studies that reveal the detailed nature of these chemical reactions. In addition, the chemistry of covalently modified graphene should be explored. Specifically, since all of the aforementioned reactions alter the delocalized π network of graphene into highly localized and directional bonds, covalently functionalized graphene is likely to be amenable to subsequent chemical attack. In this manner, the atomically modified graphene structures described here can serve as chemical intermediates for additional classes of chemically active two-dimensional materials.
Acknowledgments
This work was supported by the Office of Naval Research (N00014-11-1-0463) and a W.M. Keck Foundation Science and Engineering Grant. J.E.J. gratefully acknowledges support from an IIN Postdoctoral Fellowship and the Northwestern University International Institute for Nanotechnology as well as the National Institutes of Health and National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH/NIAMS T32 AR007611).
Biographies
James E. Johns received his B.S. in chemistry from the University of Virginia in 2004, and his Ph.D. from the University of California, Berkeley in 2010. He is currently a postdoctoral fellow of the International Institute of Nanotechnology, and is working in the laboratory of Mark Hersam at Northwestern University on the development of new methods to chemically modify graphene with atomic level precision.
Mark C. Hersam is a Professor of Materials Science and Engineering, Chemistry, and Medicine at Northwestern University. He earned a B.S. in Electrical Engineering from the University of Illinois at Urbana-Champaign (UIUC) in 1996, M.Phil. in Physics from the University of Cambridge in 1997, and a Ph.D. in Electrical Engineering from UIUC in 2000. His research interests include nanofabrication, scanning probe microscopy, semiconductor surfaces, and carbon nanomaterials.
References
- 1.Novoselov KS, Jiang D, Schedin F, Booth TJ, Khotkevich VV, Morozov SV, Geim AK. Two-Dimensional Atomic Crystals. Proc Natl Acad Sci U S A. 2005;102:10451–10453. doi: 10.1073/pnas.0502848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avouris P. Graphene: Electronic and Photonic Properties and Devices. Nano Lett. 2010;10:4285–4294. doi: 10.1021/nl102824h. [DOI] [PubMed] [Google Scholar]
- 3.Morozov SV, Novoselov KS, Katsnelson MI, Schedin F, Elias DC, Jaszczak JA, Geim AK. Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer. Phys Rev Lett. 2008;100:016602. doi: 10.1103/PhysRevLett.100.016602. [DOI] [PubMed] [Google Scholar]
- 4.Lin YM, Dimitrakopoulos C, Jenkins KA, Farmer DB, Chiu HY, Grill A, Avouris P. 100-GHz Transistors from Wafer-Scale Epitaxial Graphene. Science. 2010;327:662. doi: 10.1126/science.1184289. [DOI] [PubMed] [Google Scholar]
- 5.Xia FN, Farmer DB, Lin YM, Avouris P. Graphene Field-Effect Transistors with High On/Off Current Ratio and Large Transport Band Gap at Room Temperature. Nano Lett. 2010;10:715–718. doi: 10.1021/nl9039636. [DOI] [PubMed] [Google Scholar]
- 6.Alaboson JMP, Wang QH, Emery JD, Lipson AL, Bedzyk MJ, Elam JW, Pellin MJ, Hersam MC. Seeding Atomic Layer Deposition of High-k Dielectrics on Epitaxial Graphene with Organic Self-Assembled Monolayers. ACS Nano. 2011;5:5223–5232. doi: 10.1021/nn201414d. [DOI] [PubMed] [Google Scholar]
- 7.Bekyarova E, Itkis ME, Ramesh P, Berger C, Sprinkle M, de Heer WA, Haddon RC. Chemical Modification of Epitaxial Graphene: Spontaneous Grafting of Aryl Groups. J Am Chem Soc. 2009;131:1336–1337. doi: 10.1021/ja8057327. [DOI] [PubMed] [Google Scholar]
- 8.Hossain MZ, Walsh MA, Hersam MC. Scanning Tunneling Microscopy, Spectroscopy, and Nanolithography of Epitaxial Graphene Chemically Modified with Aryl Moieties. J Am Chem Soc. 2010;132:15399–15403. doi: 10.1021/ja107085n. [DOI] [PubMed] [Google Scholar]
- 9.Gómez-Navarro C, Meyer JC, Sundaram RS, Chuvilin A, Kurasch S, Burghard M, Kern K, Kaiser U. Atomic Structure of Reduced Graphene Oxide. Nano Lett. 2010;10:1144–1148. doi: 10.1021/nl9031617. [DOI] [PubMed] [Google Scholar]
- 10.Ivanovskaya VV, Zobelli A, Teillet-Billy D, Rougeau N, Sidis V, Briddon PR. Hydrogen Adsorption on Graphene: A First Principles Study. Eur Phys J B. 2010;76:481–486. [Google Scholar]
- 11.Gao HL, Wang L, Zhao JJ, Ding F, Lu JP. Band Gap Tuning of Hydrogenated Graphene: H Coverage and Configuration Dependence. J Phys Chem C. 2011;115:3236–3242. [Google Scholar]
- 12.Jeloaica L, Sidis V. DFT Investigation of the Adsorption of Atomic Hydrogen on a Cluster-Model Graphite Surface. Chem Phys Lett. 1999;300:157–162. [Google Scholar]
- 13.Boukhvalov DW, Katsnelson MI, Lichtenstein AI. Hydrogen on Graphene: Electronic Structure, Total Energy, Structural Distortions and Magnetism from First-Principles Calculations. Phys Rev B. 2008;77:035427. [Google Scholar]
- 14.Sofo JO, Chaudhari AS, Barber GD. Graphane: A Two-Dimensional Hydrocarbon. Phys Rev B. 2007;75:153401. [Google Scholar]
- 15.Lebegue S, Klintenberg M, Eriksson O, Katsnelson MI. Accurate Electronic Band Gap of Pure and Functionalized Graphane from GW Calculations. Phys Rev B. 2009;79:245117. [Google Scholar]
- 16.Samarakoon DK, Chen Z, Nicolas C, Wang XQ. Structural and Electronic Properties of Fluorographene. Small. 2011;7:965–969. doi: 10.1002/smll.201002058. [DOI] [PubMed] [Google Scholar]
- 17.Guisinger NP, Rutter GM, Crain JN, First PN, Stroscio JA. Exposure of Epitaxial Graphene on SiC(0001) to Atomic Hydrogen. Nano Lett. 2009;9:1462–1466. doi: 10.1021/nl803331q. [DOI] [PubMed] [Google Scholar]
- 18.Sessi P, Guest JR, Bode M, Guisinger NP. Patterning Graphene at the Nanometer Scale via Hydrogen Desorption. Nano Lett. 2009;9:4343–4347. doi: 10.1021/nl902605t. [DOI] [PubMed] [Google Scholar]
- 19.Elias DC, Nair RR, Mohiuddin TMG, Morozov SV, Blake P, Halsall MP, Ferrari AC, Boukhvalov DW, Katsnelson MI, Geim AK, Novoselov KS. Control of Graphene’s Properties by Reversible Hydrogenation: Evidence for Graphane. Science. 2009;323:610–613. doi: 10.1126/science.1167130. [DOI] [PubMed] [Google Scholar]
- 20.Wojtaszek M, Tombros N, Caretta A, van Loosdrecht PHM, van Wees BJ. A Road to Hydrogenating Graphene by a Reactive Ion Etching Plasma. J Appl Phys. 2011;110:063715. [Google Scholar]
- 21.Luo ZQ, Yu T, Kim KJ, Ni ZH, You YM, Lim S, Shen ZX, Wang SZ, Lin JY. Thickness-Dependent Reversible Hydrogenation of Graphene Layers. ACS Nano. 2009;3:1781–1788. doi: 10.1021/nn900371t. [DOI] [PubMed] [Google Scholar]
- 22.Balog R, Jørgensen B, Wells J, Lægsgaard E, Hofmann P, Besenbacher F, Hornekær L. Atomic Hydrogen Adsorbate Structures on Graphene. J Am Chem Soc. 2009;131:8744–8745. doi: 10.1021/ja902714h. [DOI] [PubMed] [Google Scholar]
- 23.Balog R, Jorgensen B, Nilsson L, Andersen M, Rienks E, Bianchi M, Fanetti M, Laegsgaard E, Baraldi A, Lizzit S, Sljivancanin Z, Besenbacher F, Hammer B, Pedersen TG, Hofmann P, Hornekaer L. Bandgap Opening in Graphene Induced by Patterned Hydrogen Adsorption. Nat Mater. 2010;9:315–319. doi: 10.1038/nmat2710. [DOI] [PubMed] [Google Scholar]
- 24.Hornekær L, Šljivančanin Ž, Xu W, Otero R, Rauls E, Stensgaard I, Lægsgaard E, Hammer B, Besenbacher F. Metastable Structures Recombination Pathways for Atomic Hydrogen on the Graphite (0001) Surface. Phys Rev Lett. 2006;96:156104. doi: 10.1103/PhysRevLett.96.156104. [DOI] [PubMed] [Google Scholar]
- 25.Ng ML, Balog R, Hornekær L, Preobrajenski AB, Vinogradov NA, Mårtensson N, Schulte K. Controlling Hydrogenation of Graphene on Transition Metals. J Phys Chem C. 2010;114:18559–18565. [Google Scholar]
- 26.Medeiros PVC, Mascarenhas AJS, de Brito Mota F, de Castilho CMC. A DFT Study of Halogen Atoms Adsorbed on Graphene Layers. Nanotechnology. 2010;21:485701. doi: 10.1088/0957-4484/21/48/485701. [DOI] [PubMed] [Google Scholar]
- 27.Artyukhov VI, Chernozatonskii LA. Structure and Layer Interaction in Carbon Monofluoride and Graphane: A Comparative Computational Study. J Phys Chem A. 2010;114:5389–5396. doi: 10.1021/jp1003566. [DOI] [PubMed] [Google Scholar]
- 28.Zhou J, Liang QF, Dong JM. Enhanced Spin-Orbit Coupling in Hydrogenated and Fluorinated Graphene. Carbon. 2010;48:1405–1409. [Google Scholar]
- 29.Leenaerts O, Peelaers H, Hernandez-Nieves AD, Partoens B, Peeters FM. First-Principles Investigation of Graphene Fluoride and Graphane. Phys Rev B. 2010;82:195436. [Google Scholar]
- 30.Robinson JT, Burgess JS, Junkermeier CE, Badescu SC, Reinecke TL, Perkins FK, Zalalutdniov MK, Baldwin JW, Culbertson JC, Sheehan PE, Snow ES. Properties of Fluorinated Graphene Films. Nano Lett. 2010;10:3001–3005. doi: 10.1021/nl101437p. [DOI] [PubMed] [Google Scholar]
- 31.Peelaers H, Hernandez-Nieves AD, Leenaerts O, Partoens B, Peeters FM. Vibrational Properties of Graphene Fluoride and Graphane. Appl Phys Lett. 2011;98:051914. [Google Scholar]
- 32.Ribas M, Singh A, Sorokin P, Yakobson B. Patterning Nanoroads and Quantum Dots on Fluorinated Graphene. Nano Research. 2011;4:143–152. [Google Scholar]
- 33.Boukhvalov DW, Katsnelson MI. Chemical Functionalization of Graphene. J Phys -Condes Matter. 2009;21:344205. doi: 10.1088/0953-8984/21/34/344205. [DOI] [PubMed] [Google Scholar]
- 34.Zbořil R, Karlický F, Bourlinos AB, Steriotis TA, Stubos AK, Georgakilas V, Šafářová K, Jančík D, Trapalis C, Otyepka M. Graphene Fluoride: A Stable Stoichiometric Graphene Derivative and its Chemical Conversion to Graphene. Small. 2010;6:2885–2891. doi: 10.1002/smll.201001401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boukhvalov DW. Modeling of Epitaxial Graphene Functionalization. Nanotechnology. 2011;22:055708. doi: 10.1088/0957-4484/22/5/055708. [DOI] [PubMed] [Google Scholar]
- 36.Şahin H, Topsakal M, Ciraci S. Structures of Fluorinated Graphene and Their Signatures. Phys Rev B. 2011;83:115432. [Google Scholar]
- 37.Nair RR, Ren WC, Jalil R, Riaz I, Kravets VG, Britnell L, Blake P, Schedin F, Mayorov AS, Yuan SJ, Katsnelson MI, Cheng HM, Strupinski W, Bulusheva LG, Okotrub AV, Grigorieva IV, Grigorenko AN, Novoselov KS, Geim AK. Fluorographene: A Two-Dimensional Counterpart of Teflon. Small. 2010;6:2877–2884. doi: 10.1002/smll.201001555. [DOI] [PubMed] [Google Scholar]
- 38.Baraket M, Walton SG, Lock EH, Robinson JT, Perkins FK. The Functionalization of Graphene Using Electron-Beam Generated Plasmas. Appl Phys Lett. 2010;96:231501. [Google Scholar]
- 39.Touhara H, Okino F. Property Control of Carbon Materials by Fluorination. Carbon. 2000;38:241–267. [Google Scholar]
- 40.Jeon KJ, Lee Z, Pollak E, Moreschini L, Bostwick A, Park CM, Mendelsberg R, Radmilovic V, Kostecki R, Richardson TJ, Rotenberg E. Fluorographene: A Wide Bandgap Semiconductor with Ultraviolet Luminescence. ACS Nano. 2011;5:1042–1046. doi: 10.1021/nn1025274. [DOI] [PubMed] [Google Scholar]
- 41.Hummers WS, Offeman RE. Preparation of Graphite Oxide. J Am Chem Soc. 1958;80:1339. [Google Scholar]
- 42.Dreyer DR, Park S, Bielawski CW, Ruoff RS. The Chemistry of Graphene Oxide. Chem Soc Rev. 2010;39:228–240. doi: 10.1039/b917103g. [DOI] [PubMed] [Google Scholar]
- 43.Barinov A, Malcioglu OB, Fabris S, Sun T, Gregoratti L, Dalmiglio M, Kiskinova M. Initial Stages of Oxidation on Graphitic Surfaces: Photoemission Study and Density Functional Theory Calculations. J Phys Chem C. 2009;113:9009–9013. [Google Scholar]
- 44.Sorescu DC, Jordan KD, Avouris P. Theoretical Study of Oxygen Adsorption on Graphite and the (8,0) Single-walled Carbon Nanotube. J Phys Chem B. 2001;105:11227–11232. [Google Scholar]
- 45.Boukhvalov DW, Katsnelson MI. Modeling of Graphite Oxide. J Am Chem Soc. 2008;130:10697–10701. doi: 10.1021/ja8021686. [DOI] [PubMed] [Google Scholar]
- 46.Nourbakhsh A, Cantoro M, Vosch T, Pourtois G, Clemente F, Van der Veen MH, Hofkens J, Heyns MM, Gendt SD, Sels BF. Bandgap Opening in Oxygen Plasma-Treated Graphene. Nanotechnology. 2010;21:435203. doi: 10.1088/0957-4484/21/43/435203. [DOI] [PubMed] [Google Scholar]
- 47.Nourbakhsh A, Cantoro M, Klekachev AV, Pourtois G, Vosch T, Hofkens J, van der Veen MH, Heyns MM, De Gendt S, Sels BF. Single Layer vs Bilayer Graphene: A Comparative Study of the Effects of Oxygen Plasma Treatment on Their Electronic and Optical Properties. J Phys Chem C. 2011;115:16619–16624. [Google Scholar]
- 48.Barinov A, Malcioglu OB, Fabris S, Sun T, Gregoratti L, Dalmiglio M, Kiskinova M. Initial Stages of Oxidation on Graphitic Surfaces: Photoemission Study and Density Functional Theory Calculations. J Phys Chem C. 2009;113:9009–9013. [Google Scholar]
- 49.Hossain MZ, Johns JE, Bevan KH, Karmel HJ, Liang YT, Yoshimoto S, Mukai K, Koitaya T, Yoshinobu J, Kawai M, Lear AM, Kesmodel LL, Tait SL, Hersam MC. Chemically Homogeneous and Thermally Reversible Oxidation of Epitaxial Graphene. Nature Chemistry. 2012;4:305–309. doi: 10.1038/nchem.1269. [DOI] [PubMed] [Google Scholar]
- 50.Paredes JI, Martínez-Alonso A, Tascón JMD. Early Stages of Plasma Oxidation of Graphite: Nanoscale Physicochemical Changes As Detected by Scanning Probe Microscopies. Langmuir. 2002;18:4314–4323. [Google Scholar]
- 51.Solís-Fernández P, Paredes JI, Cosío A, Martínez-Alonso A, Tascón JMD. A Comparison between Physically and Chemically Driven Etching in the Oxidation of Graphite Surfaces. J Colloid Interface Sci. 2010;344:451–459. doi: 10.1016/j.jcis.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 52.Paredes JI, Martínez-Alonso A, Tascón JMD. Characterization of Microporosity and Mesoporosity in Carbonaceous Materials by Scanning Tunneling Microscopy. Langmuir. 2000;17:474–480. [Google Scholar]
- 53.Vinogradov NA, Schulte K, Ng ML, Mikkelsen A, Lundgren E, Mårtensson N, Preobrajenski AB. Impact of Atomic Oxygen on the Structure of Graphene Formed on Ir(111) and Pt(111) J Phys Chem C. 2011;115:9568–9577. [Google Scholar]
- 54.Yang D, Velamakanni A, Bozoklu G, Park S, Stoller M, Piner RD, Stankovich S, Jung I, Field DA, Ventrice CA, Jr, Ruoff RS. Chemical Analysis of Graphene Oxide Films after Heat and Chemical Treatments by X-ray Photoelectron and Micro-Raman Spectroscopy. Carbon. 2009;47:145–152. [Google Scholar]
- 55.Kim S, Zhou S, Hu Y, Acik M, Chabal YJ, Berger C, de Heer W, Bongiorno A, Riedo E. Room-Temperature Metastability of Multilayer Graphene Oxide Films. Nature Materials. 2012;11:544–549. doi: 10.1038/nmat3316. [DOI] [PubMed] [Google Scholar]
- 56.Pandey D, Reifenberger R, Piner R. Scanning Probe Microscopy Study of Exfoliated Oxidized Graphene Sheets. Surf Sci. 2008;602:1607–1613. [Google Scholar]