

Abstract
Menopausal hormone treatment (MHT) may limit progression of cardiovascular disease (CVD) but poses a thrombosis risk. To test targeted candidate gene variation for association with subclinical CVD defined by carotid artery intima-media thickness (CIMT) and coronary artery calcification (CAC), 610 women participating in the Kronos Early Estrogen Prevention Study (KEEPS), a clinical trial of MHT to prevent progression of CVD, were genotyped for 13,229 single nucleotide polymorphisms (SNPs) within 764 genes from anticoagulant, procoagulant, fibrinolytic, or innate immunity pathways. According to linear regression, proportion of European ancestry correlated negatively, but age at enrollment and pulse pressure correlated positively with CIMT. Adjusting for these variables, two SNPs, one on chromosome 2 for MAP4K4 gene (rs2236935, β = 0.037, P value = 2.36 × 10−06) and one on chromosome 5 for IL5 gene (rs739318, β = 0.051, P value = 5.02 × 10−05), associated positively with CIMT; two SNPs on chromosome 17 for CCL5 (rs4796119, β = −0.043, P value = 3.59 × 10−05; rs2291299, β = −0.032, P value = 5.59 × 10−05) correlated negatively with CIMT; only rs2236935 remained significant after correcting for multiple testing. Using logistic regression, when we adjusted for waist circumference, two SNPs (rs11465886, IRAK2, chromosome 3, OR = 3.91, P value = 1.10 × 10−04; and rs17751769, SERPINA1, chromosome 14, OR = 1.96, P value = 2.42 × 10−04) associated positively with a CAC score of >0 Agatston unit; one SNP (rs630014, ABO, OR = 0.51, P value = 2.51 × 10−04) associated negatively; none remained significant after correcting for multiple testing. Whether these SNPs associate with CIMT and CAC in women randomized to MHT remains to be determined.
Keywords: ABO, IRAK2, MAP4K4, menopause, serpina1
menopausal hormone treatment (MHT) may limit progression of cardiovascular disease (33) but poses a risk for thrombosis (5, 7, 13, 20, 75a). Genetic factors contributing to a benefit-risk profile for an individual woman remain underexplored. Most studies approached analysis of genetic factors for single traits rather than assessment of cardiovascular disease/thrombotic risk as a complex genetic trait (26, 48). For example, polymorphisms in genes for the estrogen receptors are associated with accelerated atherosclerosis (1, 17, 44, 49, 65, 90) and high-density lipoproteins, insulin sensitivity, and apolipoprotein A-1 and other cardiovascular disease risk factors (88). Genotypic markers of procoagulant activity of the blood such as factor V Leiden and prothrombin G20210A have been evaluated almost exclusively in the context of defining risk for venous thrombosis and not cardiovascular disease in general, even though both arterial and venous thromboses have been linked to estrogen therapy. Epidemiology of coronary disease and stroke also has established familial risk factors for occurrence of events (30). Thus, linking genetic variation in estrogen (or other sex-steroid) receptors to genetic variation in proteins of the coagulation cascade could help identify an at-risk thrombotic genotype. For instance, polymorphisms in platelet surface receptors glycoprotein IIIa for the fibrinogen receptor (9) may interact with estrogen receptor activation of platelets perhaps through modulation of cyclooxygenase metabolism. Alternatively, polymorphisms in estrogen receptors could affect endothelial production of vasoactive factors such as nitric oxide, which inhibits platelet aggregation (58, 68, 98), or affect synthase of proteins of the coagulation cascade (52, 53, 86, 89). As platelet aggregation and secretion contribute to formation of vascular lesions at sites of endothelial dysfunction (75), estrogenic effects on endothelium, platelets, and proteins of the coagulation cascade potentially provide a link between development of subclinical cardiovascular disease and thrombosis.
Variation in plasma concentrations of procoagulant factors and activation markers of coagulation and fibrinolysis exhibit a high degree of heritability among apparently healthy individuals (4, 18, 59, 71, 83, 84). Mutations may occur in several pathways: 1) components of the anticoagulant pathway, 2) mutations that down- or upregulate the procoagulant pathway, 3) mutations that downregulate fibrinolysis, and 4) mutations of the immunological pathway (69). However, isolated impairment of one of these pathways is insufficient to cause thrombosis (10, 28, 59). Thus, in the setting of changes in hormonal status (estrogen-deplete or -replete), interactions among pathways linking hemostasis to cardiovascular risk represent complex genetic phenomena. Indeed, penetrance for thrombotic risk is compounded among carriers that are exposed to clinical risk factors such as estrogen therapy (37, 40, 55, 73, 74, 80, 93, 94, 100). These findings suggest that identification of genetic variation, single nucleotide polymorphisms (SNPs), or haplotypes within these pathways for coagulation and immunity with those related to hormonal status and diagnostic genotyping (or haplotyping), could better stratify patients exposed to clinical risk factors into high- and low-thrombotic risk, identify unexposed patients at risk for development of cardiovascular disease, and better target prophylaxis.
Although the genetic variant, factor V Leiden, is associated with increased relative risk for thrombosis with estrogen treatment (31, 47, 72), estrogenic-associated thrombosis occurs in individuals without this genetic variant (6, 32, 75a). Evidence linking genetic variation in other hemostatic factors such as fibrinogen, factor VII, and prothrombin with thrombosis is equivocal (11). However, as treatment with selective estrogen receptor modulators tamoxifen and raloxifene also increases risk of venous thrombosis, ligand-bound estrogen receptors may be causal to this increased risk (6). To date, other trials/studies that have evaluated effects of hormone treatment on progression/outcomes of cardiovascular disease have not evaluated estrogen receptor polymorphism with the genetic component of proteins of the coagulation cascade with procoagulant activity of the blood. Therefore, this study was undertaken to evaluate genetic polymorphisms in estrogen receptors and pathways of genes within the anticoagulant, procoagulant, fibrinolytic, or innate immunity pathways with subclinical cardiovascular disease in women enrolled in the Kronos Early Estrogen Prevention Study (KEEPS). KEEPS is a multicentered randomized, placebo-controlled, double-blinded, prospective trial of effects of low-dose hormone therapy (oral or transdermal) in recently menopausal women on progression of subclinical atherosclerosis defined by changes in carotid intima-media thickening and coronary arterial calcification over 4 years (27).
Focusing on genotyping for estrogen receptor polymorphisms and establishing risk for hormone therapy carry immediate and practical implications for decisions as to whether or not to use estrogen treatments but also for decisions regarding what type of estrogen treatment is most appropriate to reduce risk of adverse events (46, 75a, 77, 78, 95).
METHODS
Participants.
Women meeting inclusion criteria for the KEEPS (NCT00154180) and who gave informed consent to have their DNA used for research purposes were included in this study. There were nine centers participating in KEEPS: Brigham and Women's Hospital; Columbia University College of Physicians and Surgeons; the Kronos Longevity Research Institute; Mayo Clinic, Rochester, MN; Montefiore Medical Center; University of California at San Francisco; University of Utah; University of Washington; and Yale University. KEEPS inclusion criteria were age 42–58 yr, having at least 6 mo but no more than 36 mo of amenorrhea from last spontaneous menses occurring after age 40 yr, follicle stimulating hormone values ≥35 ng/ml and/or estradiol levels <35 pg/ml, and good general health. Women were excluded from KEEPS if they had a history of clinically defined cardiovascular disease; were current smokers [>10 cigarettes (half pack)/day by self-report]; had a coronary artery calcification (CAC) score ≥50 Agatston units (AU), body mass index (BMI) >35 kg/m2; or if they had dyslipidemia (low-density lipoprotein cholesterol >190 mg/dl), hypertriglyceridemia (triglycerides, >400 mg/dl), 17β-estradiol >40 mg/dl, uncontrolled hypertension (systolic blood pressure >150 mmHg and/or diastolic blood pressure >95 mmHg), or fasting blood glucose >126 mg/dl (27, 60).
Enrollment began in July 2005 and was completed in June 2008 with 728 women meeting inclusion criteria and randomized to treatment: oral conjugated equine estrogen (Premarin, 0.45 mg/day), transdermal 17β-estradiol (via skin patch, Climara, 50 μg/day) both with progesterone (oral Prometrium, 200 mg/day) for the first 12 days of the month, or placebo group (inactive pill/patch) (60). Of women randomized to treatment, 684 consented to allow analysis of their DNA. Clinical and lifestyle parameters used in this study were collected at screening and baseline visits prior to randomization to treatment.
The institutional review boards of all participating institutions approved this study, and all women signed informed consent.
Blood collection and preparation of DNA.
All blood specimens were collected following an overnight fast and then frozen at −70°C on site until they were either processed locally or sent to the Kronos Science Laboratory (Phoenix, AZ) for storage or assays. Complete blood count and chemistry panel were performed at the clinical laboratories at each recruiting center. Genomic DNA was extracted from whole blood using the QIAamp DNA Blood Midi Kit (Qiagen), and the DNA concentration was measured by the PicoGreen technique (Invitrogen). SNPs for the custom 16,720 bead Illumina Infinium [13,229 SNPs including 492 ancestry informative markers (AIMs)] included candidate genes selected from electronic databases that annotate the anticoagulant, procoagulant, fibrinolytic, and/or innate immunity pathways with focus on platelet, monocyte, neutrophil and endothelial cell agonists, receptors, ligands, signal transduction and adhesion molecules, granule content and effectors; plasma proteases and inhibitors; matrix metalloproteases; inflammatory cytokines and receptors; estrogen, progesterone and androgen receptors, co-regulators and enzymes related to metabolism of estrogen; and enzymes associated metabolism of catechols, homocysteine, thromboxane A2, prostacyclin and 3-hydroxy-3-methylglutaryl coenzyme A reductase (30). Furthermore, the Estrogen Response Element Database (ERE DB) and Hormone Receptor Target Binding Loci Data Base (HRTDLDB) were checked for the genes of interest using the official gene symbol as well as official gene name aliases on the respective chromosomes.
Genotyping and quality control.
Leukocyte genomic DNA was extracted, quantified, and diluted to the appropriate concentration for Illumina Infinium iSelect genotyping on all samples collected. Controls included 2% sample replicates and a trio from the Centre d'Etude du Polymorphism Humain (CEPH) for quality control. In addition, DNA sample addresses were randomly assigned across both the 96-well plate as well as the 12-address iSelect BeadChip to avoid potential plate and chip effects, respectively. Genotyping results from high-quality control DNA (SNP call rate ≥95%) were used to generate a cluster algorithm.
Anthropometric and lifestyle exposures.
Baseline smoking status (current use yes/no) and race/ethnicity were obtained through self-reported questionnaire data. The height (cm) and weight (kg) of participants, wearing light clothing and without shoes, were measured with stadiometers and calibrated balance-beam scales, respectively. BMI was calculated as weight in kilograms divided by the square of height in meters. Waist circumference (cm) was measured with a nonstretchable tape after a normal expiration at the smallest horizontal circumference between the ribs and iliac crest. Blood pressure was measured in the right arm in the seated position, after at least a 5 min rest, and averaged across two readings. Women ranked their menopausal symptoms (hot flashes, night sweats, vaginal dryness, dyspareunia, palpitations, insomnia, depression, mood swings, and irritability) on a score of 1–3 as either none (0), mild (1), moderate (2), or severe (3). A weighted sum of the scores for menopausal symptoms was calculated based on the following formula: 3(hot flashes) + 2(dyspareunia + vaginal dryness + night sweats) + mood swings + insomnia + 0.5(palpitations + depression + irritability).
Measures of atherosclerosis.
Carotid artery intima-media thickness (CIMT) was measured via B-mode ultrasound at each study center by certified ultrasound technicians trained at the Core Imaging and Reading Center (CIRC; PI: Dr. Howard Hodis, USC) to perform a standard acquisition sequence (patents 2005, 2006, 2011). Image acquisition procedures were optimized for minimal measurement variability (33). The ultrasound power, echo detector gain, and dynamic range were recorded to establish identical conditions for serial examinations. All instruments were high-resolution imagers with a multiarray 7.5 MHz probe. Electrocardiogram and ultrasound images were simultaneously recorded. All images were evaluated at the CIRC by an experienced investigator with in-house developed automated computerized edge-detection software (patents 2005, 2006, 2011) (33, 81).
CAC was measured on nonenhanced cardiac computed tomography scans. CAC was defined as a plaque of at least 3 contiguous pixels (area 1.02 mm2) with a density of >130 HU. A total CAC score was determined by summing the individual Agatston scores from each of four anatomic sites (left main, left anterior descending, circumflex, and right coronary). A single experienced investigator, blinded to the subject identity, interpreted all the scans using commercially available software (TeraRecon, Foster City, CA) (12, 36, 62, 76).
Statistical analysis.
The analysis consists of two primary outcomes, CIMT as a continuous measure and CAC as a binary measure. Descriptive statistics were used to summarize the cohort variables including means with standard deviations (SD) and medians with interquartile ranges (Q1, Q3) for all continuous parameters. Since the distribution of CIMT is skewed, this variable was log-transformed. We used three HapMap 2 populations (CEU: Utah residents from the CEPH; YRI: Yoruba from Ibudan, Nigeria; and CHB/JPT: Han Chinese from Beijing, China, and Japanese from Tokyo, Japan) to characterize the set of allele frequencies at each locus of the 492 AIMs (79). To identify the population structure of KEEPS we used these 492 AIMs and the program STRUCTURE (66). Within the KEEPS sample most were Caucasian, and we used percent CEU from the STRUCTURE program to determine the proportion of European ancestry within each individual. STRUCTURE allows for population admixture and assigns individuals in the sample of interest (the KEEPS sample) population probabilities determined by individual allele frequencies. The technique assumes the loci are unlinked, and it assumes Hardy-Weinberg equilibrium within the populations.
Prior to the single SNP analysis, we identified for each outcome the following nongenetic risk factors (demographic and clinical variables). The proportion of European ancestry as determined by STRUCTURE, self-reported “white,” age in years at study enrollment, low-density lipoprotein cholesterol level (mg/dl), high-density lipoprotein cholesterol level (mg/dl), triglyceride level (mg/dl), fasting glucose (mg/dl), hypertension diagnosis (by history on phone screen), weighted algorithm of menopausal scores, waist circumference (cm), pulse pressure [mean of systolic blood pressure at pre-enrollment, baseline visit (visit 0) and enrollment, randomization visit (visit 1) minus mean of diastolic blood pressure at visits 0 and 1 in mmHg], and current smoking status (yes/no). The clinical variables were regressed against the natural log of CIMT using linear regression. Two analyses were performed. First, all 12 variables were jointly regressed against log CIMT. Second, a stepwise linear regression was performed using alpha equal 0.10 to enter or leave. Model residuals were examined to see that modeling assumptions were met. We considered only variables with P ≤ 0.0001 significant for use as adjusting variables when examining the association with the genotyped SNPs. Proportion of European ancestry, age at enrollment, and pulse pressure were significant in the stepwise linear regression and were used as adjusting variables in subsequent analysis of log-transformed CIMT.
Similarly, the relationship between demographic and clinical variables and CAC was examined in an unconditional logistic regression. The same demographic and clinical variables were included in models of CAC as listed above. As above, all 12 variables were jointly examined, and then, subsequently, a stepwise logistic regression was performed. Model assumptions were checked; only model variables with P ≤ 0.01 were used as adjusting variables. Only waist circumference was significantly associated with CAC.
The association between each SNP and log-transformed CIMT was tested by a linear regression model with additive genetic effect adjusting for age, proportion of European ancestry, and pulse pressure and for CAC by unconditional logistic regression with additive genetic effect adjusting for waist circumference. All above analyses were done with SAS software version 9.1.
These analyses were corrected for multiple comparisons with an extension of false discovery rates (8). The false discovery rate is an analog measure of the P value that takes into account the number of statistical tests and estimates the expected proportion of false positive tests incurred when a particular SNP is significant. All analyses were performed using PLINK v 1.07 (67).
RESULTS
From the 684 DNA samples, 74 were removed for the following reasons: HapMap CEPH genotype controls (18), misidentified (16), call rates <0.95 (8), relatedness to another sample (5), duplicate samples (26), ineligible sample (1). Due to incomplete phenotypic data, 11 samples were removed from the CIMT analysis and 10 from the CAC analysis.
From the 13,229 SNPs, only 11,955 were used for association analysis. A total of 1,274 SNPs were excluded from analysis due to call rate <0.95 (714), monomorphic (no minor alleles, 68), and ancestry informative markers (492).
Population stratification from the individual DNA samples using three HapMap Phase 11 populations identified the majority of participants to be of Central European ancestry (Fig. 1 and Table 1). Individuals reporting themselves as Hispanic, Black, or Asian were of mixed genetic ancestry (Table 1).
Fig. 1.

Kronos Early Estrogen Prevention Study (KEEPS) population stratification using the program STRUCTURE and 492 ancestry informative markers. Cluster 1 represents 89 HapMap CHB and JPT (Asian) samples, cluster 2 represents 60 HapMap CEU (Caucasian) samples, and cluster 3 represents 60 HapMap YRI (African) samples. KEEPS samples are represented by circles.
Table 1.
Proportion of ancestry for 4 main races (collapsing all the Asians under 1 category)
| Proportion of Ancestry |
||||
|---|---|---|---|---|
| Reported Race | n | CHB/JPT | CEU | YRI |
| No answer/other | 40 | 0.019 | 0.938 | 0.043 |
| Asian | 17 | 0.723 | 0.256 | 0.021 |
| Black | 43 | 0.013 | 0.181 | 0.807 |
| White | 466 | 0.008 | 0.986 | 0.007 |
| Hispanic | 44 | 0.164 | 0.669 | 0.167 |
CEU, Utah residents from the Centre d'Etude du Polymorphism Humain; YRI, Yoruba from Ibudan, Nigeria; CHB/JPT, Han Chinese from Beijing, China, and Japanese from Tokyo, Japan.
Collective phenotypic characteristics of the 610 women for whom SNP analyses were performed are provided in Table 2. The mean and standard deviation for CIMT for these genotyped individuals were 0.72 and 0.09 mm, respectively (median = 0.70 mm, range 0.53–1.17 mm). Covariates that significantly associated positively with the log-transformed CIMT were age at study enrollment and pulse pressure, whereas proportion of European ancestry (CEU) per subject associated negatively with CIMT (Table 3). Each unit of CEU proportion decreased the log-transformed CIMT, and each unit of age at study enrollment and pulse pressure increased the log-transformed CIMT. The adjusted R-square of these three variables or the contribution to overall variance of log CIMT is low (8.54%). To avoid potential false positive SNPs we adjusted our analysis for these covariates. With these adjustments, four SNPs showed significant association with log-transformed CIMT with two SNPs, one on chromosome 2 (rs2236935 on mitogen-activated protein kinase, MAP4K4) and one on chromosome 5 (rs739718 on interleukin 5, IL5/IRF1), associated significantly with increases in log-transformed CIMT, and two SNPS on chromosome 17 [rs4796119 and 532291299, chemokine (C-C motif) ligand 5 (CCL5) also known as regulated upon activation, normal T-cell expressed, and secreted (RANTES)] negatively associated with CIMT (Fig. 2, Table 4); however, after multiple testing correction only one SNP on chromosome 2 remained significant (rs2236935, Bonferroni P-corrected = 0.02818), which was similar to Benjamini's correction (8).
Table 2.
Characteristics of genotyped participants in KEEPS
| Covariate | n | Mean ± SD | Range |
|---|---|---|---|
| Age at menopause, yr | 609 | 51.3 ± 2.5 | 42.2–57.5 |
| Time past menopause, yr | 610 | 1.43 ± 0.72 | 0.5–3.0 |
| Weighted menopausal symptom score | 610 | 12.7 ± 6.8 | 0.00–33.0 |
| Smoking status (current cigarette smoker) | 610 | 39 (6.4%) | |
| Waist circumference, cm | 602 | 84.9 ± 11.8 | 57.2–120.0 |
| Body mass index, kg/mm2 | 610 | 26.3 ± 4.3 | 16.9–40.0 |
| Systolic blood pressure, mmHg | 610 | 119.0 ± 15.1 | 82.0–189.0 |
| Diastolic blood pressure, mmHg | 610 | 74.9 ± 9.3 | 50.0–113.0 |
| Fasting blood glucose, mg/dl | 610 | 89.4 ± 9.8 | 55.0–126.0 |
| HDL cholesterol, mg/dl | 610 | 65.4 ± 17.4 | 24.00–129.0 |
| LDL cholesterol, mg/dl | 610 | 128.7 ± 30.1 | 11.0–194.0 |
| Triglycerides, mg/dl | 610 | 89.7 ± 50.2 | 7.0–374.0 |
Data are shown as means ± SD, n, number of genotyped individuals; KEEPS, Kronos Early Estrogen Prevention Study.
Table 3.
Significant independent covariates for CIMT and CAC used in the regression analyses
| β-Coefficient (SE) | P | |
|---|---|---|
| CIMT | ||
| Proportion of European ancestry | −0.088 (0.018) | <0.001 |
| Age at enrollment | 0.0076 (0.0019) | <0.001 |
| Pulse pressure, mmHg | 0.0017 (0.0004) | <0.001 |
| CAC | β-coefficient (SE)* | |
| Waist circumference | 0.028 (0.0098) | 0.0044 |
CIMT, carotid artery intima-media thickness; CAC, coronary artery calcification.
Odds ratio = 1.03.
Fig. 2.
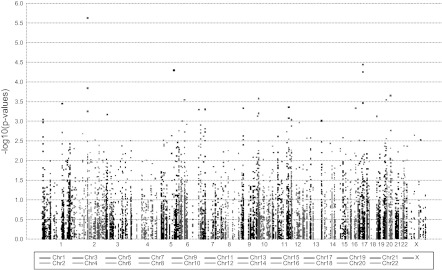
Manhattan plot for log-transformed carotid artery intima-media thickness adjusted for proportion of European ancestry, age at enrollment, and pulse pressure. The most significant single nucleotide polymorphism (SNP) is located on chromosome 2 (rs2236935, MAP4K4 gene).
Table 4.
Significant SNPs associated with CIMT
| SNP | Gene Name | Chr. | Position, base pair | Risk Allele | n | Beta | P* |
|---|---|---|---|---|---|---|---|
| rs2236935 | MAP4K4 | 2 | 101810474 | G | 606 | 0.03697 | 2.36E-06 |
| rs4796119 | CCL5 | 17 | 31217201 | G | 607 | −0.0427 | 3.59E-05 |
| rs739718 | IL5 | 5 | 131900972 | G | 607 | 0.05122 | 5.02E-05 |
| rs2291299 | CCL5 | 17 | 31215519 | G | 607 | −0.03179 | 5.59E-05 |
SNP, single nucleotide polymorphism.
Uncorrected P.
Of the 600 women for whom complete phenotypic data were available for CAC measurements, 80 (13%) had a positive Agatston score. The only covariant significantly associated with a positive CAC score was waist circumference (Table 3). When we adjusted for this covariate, three SNPS associated with CAC scores (Fig. 3 and Table 5). Of these, two SNPs (rs11465886) of the interleukin-1 receptor-associated kinase-like 2 (IRAK2) gene and (rs17751769) of the SERPINA 1 (alpha 1-antitrypsin) gene were associated positively with the presence of coronary arterial calcification, whereas SNP (rs630014) of the ABO gene associated negatively with CAC. However, none of the SNPs were significant after correcting for multiple testing.
Fig. 3.
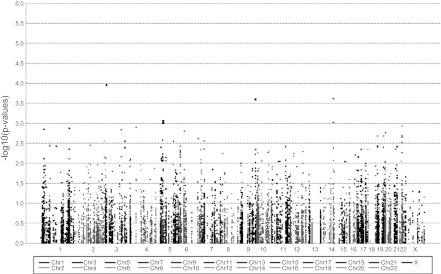
Manhattan plot for coronary artery calcification adjusted for waist circumference. The most significant SNP is located on chromosome 3 (rs11465886, IRAK2 gene).
Table 5.
Description of significant SNPs associated with CAC
| SNP | Gene Name | Chr. | Position, base pair | Risk Allele | n | Odds Ratio | P* |
|---|---|---|---|---|---|---|---|
| rs11465886 | IRAK2 | 3 | 10225783 | G | 599 | 3.909 | 0.000110 |
| rs17751769 | SERPINA1 | 14 | 93926410 | A | 575 | 1.955 | 0.000242 |
| rs630014 | ABO | 9 | 135139543 | A | 599 | 0.508 | 0.000251 |
| rs702689 | MAP3K1 | 5 | 56213200 | G | 595 | 1.784 | 0.000869 |
| rs1610317 | SERPINA1 | 14 | 93928109 | G | 599 | 1.834 | 0.000949 |
| rs832573 | MAP3K1 | 5 | 56195335 | A | 599 | 1.766 | 0.000995 |
Uncorrected P.
Searches of both the ERE DB and the HRTDL DB indicated only one of the six genes of interest, the SERPINA 1 gene, was registered with an estrogen receptor response element.
DISCUSSION
In this targeted candidate genetic analysis to evaluate potential effects of MHT on progression of cardiovascular disease and thrombotic risk as complex genetic traits, genetic variation only within genes of the innate and humoral immunity pathways and not of the anticoagulant, procoagulant, or fibrinolytic pathways associated with subclinical cardiovascular disease defined by CIMT and CAC. Although statistical significance of some associations was lost after correcting for multiple testing, the contribution of the identified genes will be discussed relative to current investigations of single trait analyses.
Contrary to our hypothesis, neither CIMT nor CAC showed a relationship to polymorphisms of either estrogen receptor alpha or beta. However, women were estrogen deplete for a mean of 1.5 yr at the time of testing. It remains to be determined whether or not polymorphisms in estrogen receptors or in enzymes involved in estrogen metabolism will associate with changes in either CIMT or CAC after 4 yr of MHT. Genetic variance in both estrogen receptors and enzymes involved in steroid metabolism associated with cardiovascular risk factors in women of four different ethnic groups in the Study of Women Across the Nation (SWAN) (85, 87, 88). Although some cardiovascular risk factors segregated by race/ethnicity in the SWAN, no analyses were performed for AIMs, and differences among race/ethnic groups were not consistently observed and may have reflected life style, cultural, and/or environmental differences (57, 87). In the present study, CIMT was negatively associated with proportion of European ancestry.
By design, women eligible for randomization to KEEPS had low cardiovascular risk profiles (BMI, fasting serum blood glucose, triglycerides, HDL-C, LDL-C, and blood pressure within normative ranges). However, as it has been reported in studies of other populations, CIMT correlated with age and blood pressure (pulse pressure in the current study) (39, 56, 70, 82).
Activation of inflammatory processes contributes to progression of cardiovascular disease (50). Therefore, it might be expected for variants in genes encoding proteins within the inflammatory pathway to associate with subclinical cardiovascular disease measured as CIMT and CAC. However, the differential association of SNPs with CIMT and CAC suggests heterogeneous mechanisms for development of arterial wall thickening within carotid arteries and calcification of the coronary arteries. The concept of distinct risk factors and processes for development of disease based on the anatomical origin of the blood vessel is supported by other studies (34, 35, 38, 97).
In the present study, an SNP in the MAP4K4 gene, a serine/threonine kinase mediating TNF-α signaling, and the IL5 gene, encoding a cytokine that stimulates growth and differentiation of B cells (humoral immunity) and eosinophils, associated positively with CIMT. Both of these pathways have been implicated in development of vascular lesions in experimental animals and are regulated by estrogen (2, 19, 45, 54). MAP kinase is one of the signaling pathways activated by binding of pathogen-associated molecular patterns to the toll-like receptor 4 (TLR4)-CD14 dimer on the surface of macrophages and considered a component of innate immunity. Estrogenic regulation of these genes would alter expression of modified gene product.
Alternatively, two SNPS within the CCL5 (RANTES) gene associated negatively with CIMT. Activation of CCL5 and its receptor accelerates development of atherosclerosis in mice models of the disease (41, 43). RANTES is also considered a component of the humoral inflammatory pathway (51), indicating that direct and indirect interactions among components of the hemostasis and inflammatory pathways are complex and components of each cannot be considered in isolation. Whether these SNPs result in a defect in RANTES signaling remains to be tested in isolated human tissues. Furthermore, the product of this SNP may be interacting with another gene that has other direct or indirect effects on development of CIMT. Previously, CAPN10-haplotype on chromosome 2 was also found through targeted gene studies, as well as through genome-wide association study (GWAS) to have strong association with CIMT (22, 23, 96).
Consistent with other studies, waist circumference associated with a positive CAC score in KEEPS participants (21, 42). When we adjusted for waist circumference, two SNPs associated positively with CAC. One SNP was found on the gene for IRAK2, which encodes a protein affecting transcriptional regulation, mRNA stability, and the IL-1-induced upregulation of NF-κB, thus linking innate immunity with development of CAC.
The other SNP positively associated with CAC was on the SERPINA1 gene (14q32.13), which encodes the protease inhibitor alpha 1-antitrypsin that breaks down elastase. This SNP most frequently associates with lung fibrosis. Although this defect could affect elastase in arterial tissue leading to arterial stiffening, the relationship of this SNP to development calcific processes remains to be explored. This gene was found to have an estrogen response element in the promoter region. In the Framingham Heart Study, the African Americans in the Hypertension Genetic Epidemiology Network study, and a study in Dominican Republican families, quantitative trait loci for CIMT associated with arterial stiffness also were mapped to chromosome 14 (97). Of interest is that SNP (rs7152362) resides on chromosome 14q32.12. The relationship of that SNP to calcification processes also remains to be explored.
An SNP of the ABO gene that negatively correlated with CAC encodes proteins affecting posttranslational modification of proteins, for example, von Willebrand factor, a protein carrier for coagulation factor VIII. In a case-controlled study including mostly pre- or postmenopausal women, a case-controlled study including both men and women, and a GWAS, risk of venous thrombosis was associated with a non-O blood type (29, 30, 99). Estrogen is known to modulate both mRNA stability and posttranslational modification of proteins (61). Therefore, it will be interesting to determine whether these SNPs for IRAK2 and ABO remain associated with CAC in women treated with estrogen. After we corrected for multiple comparisons, none of these SNPs remained significantly associated with CAC, reflecting perhaps the low level of calcification (<50 AU in KEEPS), which is considered nonclinically relevant or inadequate power to detect an effect as only 80 women eligible for the study had a positive CAC score.
Other genome-wide and targeted studies have identified associations of several other genetic variants with CIMT and CAC including genes encoding proteins for lipid metabolism, angiotensin-converting enzyme, TLR4 (innate immunity), and bone formation (3, 16, 25, 63, 64, 91). The value of these analyses in cardiovascular risk prediction in humans is inconsistent and does not necessarily provide value beyond those of more conventional risk phenotypic characteristics (92, 97). These inconsistencies may result from associations with events reflecting advanced disease rather than determinates of subclinical disease, analysis adjusted for sex rather than dichotomized by sex, not accounting for hormonal status, and influences of ancestry-related genotypes (14, 15, 24, 92, 97). Strengths of the current investigation lie in a targeted genetic approach in healthy individuals in which it will be possible to determine longitudinal assessment of these genetic associations with disease progression following a single intervention (MHT) in women completing the KEEPS trial.
In summary, in a targeted gene analysis of healthy, recently menopausal women, distinct genetic variants associated with measures of subclinical cardiovascular disease. Estrogen is known to modulate some of the processes regulated by the identified genes. Therefore, it will be possible to evaluate how MHTs affect the phenotypic expression of these modulators of disease progression in KEEPS participants randomized to MHT completing the study.
GRANTS
KEEPS is funded by grants from the Aurora Foundation to the Kronos Longevity Research Institute, the Mayo Foundation, the NIH HL-90639 to V. M. Miller, Mayo CTSA1 UL1 RR-024150, Brigham and Women's Hospital/Harvard Medical School CTSA UL1 RR-024139, and UCSF CTSA UL1 RR-024131 from the National Center for Advancing Translational Sciences (NCATS), a component of the NIH and NIH Roadmap for Medical Research. The manuscript's contents are solely the responsibility of the authors and do not necessarily represent the official view of NCATS or NIH. Information on the National Center for Research Resources is available at http://www.ncrr.nih.gov. Study medications were supplied in part by Bayer Health Care and by Abbott Pharmaceuticals.
Role of the Sponsors: The Aurora Foundation, Bayer Health Care, and Abbott Pharmaceuticals did not have input into the design or conduct of the study or the review or approval of this article.
DISCLOSURES
Matthew J Budoff, research grants from General Electric Company; Nanette Santoro, investigator initiated grant from Bayer Pharmaceuticals.
AUTHOR CONTRIBUTIONS
Author contributions: V.M.M., J.A.H., H.N.H., S.M.H., and M.d.A. conception and design of research; V.M.M., T.M.P., E.N.J., A.S.L., D.N.R., J.A.H., J.M.C., M.J.B., N.S., and M.d.A. analyzed data; V.M.M., T.M.P., A.S.L., D.N.R., J.A.H., J.M.C., G.S.H., H.N.H., N.S., P.N.H., and M.d.A. interpreted results of experiments; V.M.M., E.N.J., and M.d.A. prepared figures; V.M.M., T.M.P., and M.d.A. drafted manuscript; V.M.M., T.M.P., E.N.J., J.A.H., J.M.C., G.S.H., H.N.H., M.J.B., N.S., P.N.H., R.A.L., J.E.M., F.N., H.S.T., S.M.H., and M.d.A. edited and revised manuscript; V.M.M., T.M.P., E.N.J., A.S.L., D.N.R., J.A.H., J.M.C., G.S.H., H.N.H., M.J.B., N.S., P.N.H., R.A.L., J.E.M., F.N., H.S.T., S.M.H., and M.d.A. approved final version of manuscript; J.M.C. and G.S.H. performed experiments.
ACKNOWLEDGMENTS
KEEPS would not be possible without the dedicated volunteers participating in this study and collaborators and coworkers at each study center who include: at Albert Einstein College of Medicine: Genevieve Neal-Perry, Ruth Freeman, Hussein Amin, Barbara Isaac, Maureen Magnani, Rachel Wildman; at Brigham and Women's Hospital/Harvard Medical School: JoAnn Manson, Maria Bueche, Marie Gerhard-Herman, Kate Kalan, Jan Lieson, Kathryn M. Rexrode, Barbara Richmond, Frank Rybicki, Brian Walsh; at Columbia College of Physicians and Surgeons: Rogerio Lobo, Luz Sanabria, Jolene Lalas, Michelle Warren; at Kronos Longevity Research Institute: S. Mitchell Harman, Mary Dunn, Panayiotis D. Tsitouras, Viola Zepeda; at Mayo Clinic: Virginia M. Miller, Philip A. Araoz, Rebecca Beck, Dalene Bott-Kitslaar, Sharon L. Mulvagh, Lynne T. Shuster, Teresa G. Zais; at University of California, Los Angeles, CAC Reading Center: Matthew Budoff, Chris Dailing, Yanlin Gao, Angel Solano; at University of California, San Francisco Medical Center: Marcelle I. Cedars, Nancy Jancar, Jean Perry, Rebecca S. Wong, Robyn Pearl, Judy Yee, Brett Elicker, Gretchen A. W. Gooding; UCSF Statistical Reading Center: Dennis Black, Lisa Palermo; at University of Southern California, Los Angeles Atherosclerosis Research Unit, Core Imaging and Reading Center: Howard N. Hodis, Yanjie Li, Mingzhu Yan; at University of Utah School of Medicine: Eliot Brinton, Paul N. Hopkins, M. Nazeem Nanjee, Kirtly Jones, Timothy Beals, Stacey Larrinaga-Shum; at VA Puget Sound Health Care System and University of Washington School of Medicine: George Merriam, Pamela Asberry, SueAnn Brickle, Colleen Carney, Molly Carr, Monica Kletke, Lynna C. Smith; at Yale University, School of Medicine: Hugh Taylor, Kathryn Czarkowski, Lubna Pal, Linda McDonald, Mary Jane Minkin, Diane Wall, Erin Wolff [now at National Institutes of Health (NIH)/National Institute of Child Health and Human Development]. Others: Frederick Naftolin (New York University), Nanette Santoro (University of Colorado).
Institutional Review numbers for KEEPS Centers: The central KEEPS and Phoenix KEEPS (IRB protocol by the Western IRB) STUDY NUM 1058663, and WIRB PRO NUM 20040792. Brigham and Women's Hospital (Partners), #2004-P-002144 BWH; Mayo Clinic, 2241-04; Columbia, AAAA-8062; Yale, 0409027022; University of Utah, 13257; Einstein/Montefiore, 04-08-213; University of Wisconsin, H-2005-0059; UCSF, KEEPS (main study & cognitive substudy) #10-02980; University of Washington IRB #26702; VAPSHCS IRB #01048.
REFERENCES
- 1. Aavik E, du Toit D, Myburgh E, Frosen J, Hayry P. Estrogen receptor beta dominates in baboon carotid after endothelial denudation injury. Mol Cell Endocrinol 182: 91–98, 2001 [DOI] [PubMed] [Google Scholar]
- 2. Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, Taleb S, Van Vre E, Esposito B, Vilar J, Sirvent J, Van Snick J, Tedgui A, Tedder TF, Mallat Z. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med 207: 1579–1587, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ameziane N, Beillat T, Verpillat P, Chollet-Martin S, Aumont MC, Seknadji P, Lamotte M, Lebret D, Ollivier V, de Prost D. Association of the Toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscler Thromb Vasc Biol 23: 61–64, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Ariens RA, de Lange M, Snieder H, Boothby M, Spector T, Grant P. Activation markers of coagulation and fibrinolysis in twins: heritability of the prethrombotic state. Lancet 359: 667–671, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Barrett-Connor E, Bush TL. Estrogen and coronary heart disease in women. J Am Med Assoc 265: 1861–1867, 1991 [PubMed] [Google Scholar]
- 6. Barrett-Connor E, Grady D, Sashegyi A, Anderson PW, Cox DA, Hoszowski K, Rautaharju P, Harper KD. Raloxifene and cardiovascular events in osteoporotic postmenopausal women. Four-year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. JAMA 287: 847–857, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Bass KM, Bush TL. Estrogen therapy and cardiovascular risk in women. J La State Med Soc 143: 33–39, 1991 [PubMed] [Google Scholar]
- 8. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125: 279–284, 2001 [DOI] [PubMed] [Google Scholar]
- 9. Boudoulas KD, Cooke GE, Roos CM, Bray PF, Goldschmidt-Clermont PJ. The PIA polymorphism of glycoprotein IIIa functions as a modifier for the effect of estrogen on platelet aggregation. Arch Pathol Lab Med 125: 112–115, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Bovill E, Bauer K, Dickerman J, Callas P, West B. The clinical spectrum of heterozygous protein C deficiency in a large New England kindred. Blood 73: 712–717, 1989 [PubMed] [Google Scholar]
- 11. Braunstein JB, Warner Kershner D, Bray P, Gerstenblith G, Schulman SP, Post WS, Blumenthal RS. Interaction of hemostatic genetics with hormone therapy. New insights to explain arterial thrombosis in postmenopausal women. Chest 121: 906–920, 2002 [DOI] [PubMed] [Google Scholar]
- 12. Budoff MJ, Chen GPW, Hunter CJ, Takasu J, Agrawal N, Sorochinsky B, Mao S. Effects of hormone replacement on progression of coronary calcium as measured by electron beam tomography. J Womens Health 14: 410–417, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Bush TL, Cowan LD, Barrett-Connor E, Criqui MH, Karon JM, Wallace RB, Tyroler HA, Rifkind BM. Estrogen use and all-cause mortality. Preliminary results from the Lipid Research Clinics Program Follow-Up Study. JAMA 249: 903–906, 1983 [DOI] [PubMed] [Google Scholar]
- 14. Charchar FJ, Bloomer LDS, Barnes TA, Cowley MJ, Nelson CP, Wang Y, Denniff M, Radoslaw D, Christofidou P, Nankervis S, Dominiczak AF, Bani-Mustafa A, Balmforth AJ, Hall AS, Erdmann J, Cambien F, Deloukas P, Hengstenberg C, Packard C, Schunkert H, Ouwehand WH, Ford I, Goodall AH, Jobling MA, Samani NJ, Tomaszewski M. New insights into inheritance of coronary artery disease in men - the role of the Y sex chromosome. Lancet 379: 915–922, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Charchar FJ, Tomaszewski M, Lacka B, Zakrzewski J, Zukowska-Szczechowska E, Grzeszczak W, Dominiczak AF. Association of the human Y chromosome with cholesterol levels in the general population. Arterioscler Thromb Vasc Biol 24: 308–312, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Chen Q, Reis SE, Kammerer C, Craig W, McNamara DM, Holubkov R, Sharaf BL, Sopko G, Pauly DF, Merz CN, Kamboh MI. Association of anti-oxidized LDL and candidate genes with severity of coronary stenosis in the Women's Ischemia Syndrome Evaluation study. J Lipid Res 52: 801–807, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christian RC, Harrington S, Edwards WD, Oberg AL, Fitzpatrick LA. Estrogen status correlates with the calcium content of coronary atherosclerotic plaques in women. J Clin Endocrinol Metab 87: 1062–1067, 2002 [DOI] [PubMed] [Google Scholar]
- 18. de Lange M, Snieder H, Ariens RA, Spector TD, Grant PJ. The genetics of haemostasis: a twin study. Lancet 357: 101–105, 2001 [DOI] [PubMed] [Google Scholar]
- 19. Douin-Echinard V, Calippe B, Billon-Gales A, Fontaine C, Lenfant F, Tremollieres F, Bayard F, Guery JC, Arnal JF, Gourdy P. Estradiol administration controls eosinophilia through estrogen receptor-alpha activation during acute peritoneal inflammation. J Leukoc Biol 90: 145–154, 2011 [DOI] [PubMed] [Google Scholar]
- 20. Ernster VL, Bush TL, Huggins GR, Hullka BS, Kelsey JL, Schottenfeld D. Benefits and risks of menopausal estrogen and/or progestin hormone use. Prev Med 17: 201–223, 1988 [DOI] [PubMed] [Google Scholar]
- 21. Fox CS, Hwang S, Massaro J, Lie K, Vasan R, O'Donnell C, H U. Relation of subcutaneous and visceral adipose tissue to coroanry and abdominal aortic calcium (from the Framingham Study). Am J Cardiol 104: 543–547, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goodarzi MO, Taylor KD, Guo X, Quinones MJ, Cui J, Li Y, Saad MF, Yang H, Hsueh WA, Hodis HN, Rotter JI. Association of the diabetes gene calpain-10 with subclinical atherosclerosis: the Mexican-American Coronary Artery Disease Study. Diabetes 54: 1228–1232, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Goodarzi MO, Taylor KD, Jones MR, Fang B, Guo X, Xiang AH, Buchanan TA, Hodis HN, Raffel LJ, Rotter JI. Replication of calpain-10 genetic association with carotid intima-media thickness. Atherosclerosis 205: 503–505, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guthery SL, Salisbury BA, Pungliya MS, Stephens JC, Bamshad M. The structure of common genetic variation in United States populations. Am J Hum Genet 81: 1221–1231, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hallman DM, Brown SA, Ballantyne CM, Sharrett AR, Boerwinkle E. Relationship between low-density lipoprotein subclasses and asymptomatic atherosclerosis in subjects from the Atherosclerosis Risk in Communities (ARIC) Study. Biomarkers 9: 190–202, 2004 [DOI] [PubMed] [Google Scholar]
- 26. Hancock D, Scott W. Population-based case-control associations studies. Curr Protoc Hum Genet 1: Unit 1.17, 2012 [DOI] [PubMed] [Google Scholar]
- 27. Harman SM, Brinton EA, Cedars M, Lobo R, Manson JE, Merriam GR, Miller VM, Naftolin F, Santoro N. KEEPS: the Kronos Early Estrogen Prevention Study. Climacteric 8: 3–12, 2005 [DOI] [PubMed] [Google Scholar]
- 28. Hasstedt SJ, Bovill EG, Callas PW, Long GL. An unknown genetic defect increases venous thrombosis risk, through interaction with protein C deficiency. Am J Hum Genet 63: 569–576, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heit JA, Armasu SM, Asmann YW, Cunningham JM, Matusumoto ME, Petterson TM, De Andrade M. A genome-wide association study of venous thromboembolism identifies risk variants in chromosomes 1q24.2 and 9q. J Thromb Haemost 10: 1521–1531, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heit JA, Cunningham JM, Petterson TM, Armasu SM, Rider DN, De Andrade M. Genetic variation within the anticoagulant, procoagulant, fibrinolytic and innate immunity pathways as risk factors for venous thromboembolism. J Thromb Haemost 9: 113–1142, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Herrington DM, Klein KP. Genome and Hormones: Gender Differences in Physiology Invited Review: Pharmacogenetics of estrogen replacement therapy. J Appl Physiol 91: 2776–2784, 2001 [DOI] [PubMed] [Google Scholar]
- 32. Herrington DM, Vittinghoff E, Howard TD, Major DA, Owen J, Reboussin DM, Bowden D, Vittner V, Simon JA, Grady D, Hulley SB. Factor V Leiden, hormone replacement therapy, and risk of venous thromboembolic events in women with coronary disease. Arterioscler Thromb Vasc Biol 22: 1012–1017, 2002 [DOI] [PubMed] [Google Scholar]
- 33. Hodis HN, Mack WJ, Lobo RA, Shoupe D, Sevanian A, Mahrer PR, Selzer RH, Liu C, Liu C, Azen SP; Estrogen in the Prevention of Atherosclerosis Trial Research Group Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 135: 939–953, 2001 [DOI] [PubMed] [Google Scholar]
- 34. Hsue PY, Ordovas K, Lee T, Reddy G, Gotway M, Schnell A, Ho JE, Selby V, Madden E, Martin JN, Deeks SG, Ganz P, Waters DD. Carotid intima-media thickness among human immunodeficiency virus-infected patients without coronary calcium. Am J Cardiol 109: 742–747, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jayachandran M, Litwiller RD, Lahr BD, Bailey KR, Owen WG, Mulvagh SL, Heit JA, Hodis HN, Harman SM, Miller VM. Alterations in platelet function and cell-derived microvesicles in recently menopausal women: relationship to metabolic syndrome and atherogenic risk. J Cardiovasc Transl Res 4: 811–822, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jayachandran M, Litwiller RD, Owen WG, Heit JA, Behrenbeck TR, Mulvagh SL, Araoz PA, Budoff MJ, Harman SM, Miller VM. Characterization of blood borne microparticles as markers of premature coronary calcification in newly menopausal women. Am J Physiol Heart Circ Physiol 295: H931–H938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Juul K, Tybjaerg-Hansen A, Schnnohr P, Nordestgaard BG. Factor V Leiden and the risk of venous thromboembolism in the adult Danish population. Ann Intern Med 116: 851–854, 2004 [DOI] [PubMed] [Google Scholar]
- 38. Karim R, Hodis HN, Stanczyk FZ, Lobo RA, Mack WJ. Relationship between serum levels of sex hormones and progression of subclinical atherosclerosis in postmenopausal women. J Clin Endocrinol Metab 93: 131–138, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karim R, Mack WJ, Lobo RA, Hwang J, Liu CR, Liu CH, Sevanian A, Hodis HN. Determinants of the effect of estrogen on the progression of subclinical atherosclerosis: Estrogen in the Prevention of Atherosclerosis Trial. Menopause 12: 366–373, 2005 [DOI] [PubMed] [Google Scholar]
- 40. Koeleman B, Reitsma P, Allaart C, Bertina R. Activated protein C as an additional risk factor for thrombosis in protein C-deficient families. Blood 84: 1031–1035, 1994 [PubMed] [Google Scholar]
- 41. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med 15: 97–103, 2009 [DOI] [PubMed] [Google Scholar]
- 42. Kramer C, von Muhlen D, Gross J, Barrett-Connor E. A prospective study of abdominal obesity and coronary artery calcium progression in older adults. J Clin Endocrinol Metab 94: 5093–5044, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krohn R, Raffetseder U, Bot I, Zernecke A, Shagdarsuren E, Liehn EA, van Santbrink PJ, Nelson PJ, Biessen EA, Mertens PR, Weber C. Y-box binding protein-1 controls CC chemokine ligand-5 (CCL5) expression in smooth muscle cells and contributes to neointima formation in atherosclerosis-prone mice. Circulation 116: 1812–1820, 2007 [DOI] [PubMed] [Google Scholar]
- 44. Kunnas TA, Laippala P, Penttila A, Lehtimaki T, Karhunen PJ. Association of polymorphism of human alpha oestrogen receptor gene with coronary artery disease in men: a necropsy study. BMJ 321: 273–274, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, Kehry M, Dunn R, Agrotis A, Tipping P, Bobik A, Toh BH. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol 185: 4410–4419, 2010 [DOI] [PubMed] [Google Scholar]
- 46. Lacut K, Oger E, Le Gal G, Blouch MT, Abgrall JF, Kerlan V, Scarabin PV, Mottier D, SARAH Investigators Differential effects of oral and transdermal postmenopausal estrogen replacement therapies on C-reactive protein. Thromb Haemost 90: 124–131, 2003 [PubMed] [Google Scholar]
- 47. Lane DA, Grant PJ. Role of hemostatic gene polymorphisms in venous and arterial thrombotic disease. Blood 95: 1517–1532, 2000 [PubMed] [Google Scholar]
- 48. Lewis C. Genetic associations studies: Design, analysis and interpretation. Brief Bioinform 3: 146–153, 2002 [DOI] [PubMed] [Google Scholar]
- 49. Liang M, Ekblad E, Gustafsson JA, Nilsson BO. Stimulation of vascular protein synthesis by activation of oestrogen receptor beta. J Endocrinol 171: 417–423, 2001 [DOI] [PubMed] [Google Scholar]
- 50. Libby P. Inflammation in atherosclerosis. Nature 420: 868–874, 2002 [DOI] [PubMed] [Google Scholar]
- 51. Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 54: 2129–2138, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lobo RA, Bush T, Carr BR, Pickar JH. Effects of lower doses of conjugated equine estrogens and medroxyprogesterone acetate on plasma lipids and lipoproteins, coagulation factors, and carbohydrate metabolism. Fertil Steril 76: 13–24, 2001 [DOI] [PubMed] [Google Scholar]
- 53. Luyer MDP, Khosla S, Owen WG, Miller VM. Prospective randomized study of effects of unopposed estrogen replacement therapy on markers of coagulation and inflammation in postmenopausal women. J Clin Endocrinol Metab 86: 3629–3634, 2000 [DOI] [PubMed] [Google Scholar]
- 54. Mahmoodzadeh S, Dworatzek E, Fritschka S, Pham TH, Regitz-Zagrosek V. 17Beta-estradiol inhibits matrix metalloproteinase-2 transcription via MAP kinase in fibroblasts. Cardiovasc Res 85: 719–728, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marinelli I, Sacchi E, Landi G, Taioli E, Duca F, Mannuci PM. High risk of cerebral-vein thrombosis in carriers of a prothrombin-gene mutation and in users of oral contraceptives. N Engl J Med 338: 1793–1797, 1998 [DOI] [PubMed] [Google Scholar]
- 56. Matthews KA, Owens JF, Kuller LH, Sutton-Tyrrell K, Lassila HC, Wolfson SK. Stress-induced pulse pressure change predicts women's carotid atherosclerosis. Stroke 29: 1525–1530, 1998 [DOI] [PubMed] [Google Scholar]
- 57. Matthews KA, Sowers MF, Derby CA, Stein E, Miracle-McMahill H, Crawford SL, Pasternak RC. Ethnic differences in cardiovascular risk factor burden among middle-aged women: Study of Women's Health Across the Nation (SWAN). Am Heart J 149: 1066–1073, 2005 [DOI] [PubMed] [Google Scholar]
- 58. Mehta JL, Chen LY, Kone BC, Mehta P, Turner P. Identification of constitutive and inducible forms of nitric oxide synthase in human platelets. J Lab Clin Med 125: 370–377, 1995 [PubMed] [Google Scholar]
- 59. Miletich J, Sherman L, Broze G., Jr Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med 317: 991–996, 1987 [DOI] [PubMed] [Google Scholar]
- 60. Miller VM, Black DM, Brinton EA, Budoff MJ, Cedars MI, Hodis HN, Lobo RA, Manson JE, Merriam GR, Naftolin F, Santoro N, Taylor HS, Harman SM. Using basic science to design a clinical trial: baseline characteristics of women enrolled in the Kronos Early Estrogen Prevention Study (KEEPS). J Cardiovasc Transl Res 2: 228–239, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev 60: 210–241, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mulvagh SL, Behrenbeck T, Lahr BA, Bailey KR, Zais TG, Araoz PA, Miller VM. Endothelial function and cardiovascular risk stratification in menopausal women. Climacteric 13: 45–54, 2010 [DOI] [PubMed] [Google Scholar]
- 63. Oei HH, Sayed-Tabatabaei FA, Hofman A, Oudkerk M, van Duijn CM, Witteman JC. The association between angiotensin-converting enzyme gene polymorphism and coronary calcification. The Rotterdam Coronary Calcification Study. Atherosclerosis 182: 169–173, 2005 [DOI] [PubMed] [Google Scholar]
- 64. Paramsothy P, Knopp RH, Bertoni AG, Blumenthal RS, Wasserman BA, Tsai MY, Rue T, Wong ND, Heckbert SR. Association of combinations of lipid parameters with carotid intima-media thickness and coronary artery calcium in the MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol 56: 1034–1041, 2010 [DOI] [PubMed] [Google Scholar]
- 65. Poelzl G, Kasai Y, Mochizuki N, Shaul PW, Brown M, Mendelsohn ME. Specific association of estrogen receptor beta with the cell cycle spindle assembly checkpoint protein, MAD2. Proc Natl Acad Sci USA 97: 2836–2839, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics 155: 945–959, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Radomski MW, Zakar T, Salas E. Nitric oxide in platelets. Methods Enzymol 269: 88–107, 1996 [DOI] [PubMed] [Google Scholar]
- 69. Reitsma P, Rosendaal F. Activation of Innate immunity in patients with venous thrombosis: the Leiden Thrombophilia Study. J Thromb Haemost 2: 619–622, 2004 [DOI] [PubMed] [Google Scholar]
- 70. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation 125: e2–e220, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rosendaal FR, Bovill EG. Heritability of clotting factors and the revival of the prothrombotic state. Lancet 359: 638–639, 2002 [DOI] [PubMed] [Google Scholar]
- 72. Rosendaal FR, Helmerhorst FM, Vandenbroucke JP. Female hormones and thrombosis. Arterioscler Thromb Vasc Biol 22: 201–210, 2002 [DOI] [PubMed] [Google Scholar]
- 73. Rosendaal FR, Koster T, Vandebroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 85: 1504–1508, 1995 [PubMed] [Google Scholar]
- 74. Rosendaal FR, Vessey MP, Rumley A. Hormonal replacement therapy, prothrombotic mutations and the risk of venous thrombosis. Br J Haematol 116: 851–854, 2002 [DOI] [PubMed] [Google Scholar]
- 75. Ross R. Cell biology of atherosclerosis. Annu Rev Physiol 57: 791–804, 1995 [DOI] [PubMed] [Google Scholar]
- 75a. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J; Writing Group for the Women's Health Initiative Investigators Risks and benefits of estrogen plus progestin in healthy postmenopausal women. Principal results from the Women's Health Initiative randomized controlled trial. JAMA 288: 321–333, 2002 [DOI] [PubMed] [Google Scholar]
- 76. Rumberger JA, Schwartz RS, Sheedy PF, III, Edwards WD, Fitzpatrick LA. Coronary calcification and pathologic stenosis: an ROC analysis to predict atherosclerotic severity and the influence of gender using ultrafast computed tomography. Am J Cardiol 74: 1169–1173, 1994 [DOI] [PubMed] [Google Scholar]
- 77. Scarabin PY, Alhenc-Gelas M, Plu-Bureau G, Taisne P, Agher R, Aiach M. Effects of oral and transdermal estrogen/progesterone regimens on blood coagulation and fibrinolysis in postmenopausal women: a randomized controlled trial. Arterioscler Thromb Vasc Biol 17: 3071–3078, 1997 [DOI] [PubMed] [Google Scholar]
- 78. Scarabin PY, Oger E, Plu-Bureau G. Differential association of oral and transdermal oestrogen-replacement therapy with venous thromboembolism risk. Lancet 362: 428–432, 2003 [DOI] [PubMed] [Google Scholar]
- 79. Seldin MF, Shigeta R, Villoslada P, Selmi C, Tuomilehto J, Silva G, Belmont JW, Klareskog L, Gregersen PK. European population substructure: clustering of northern and southern populations. PLoS Genet 2: e143, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Seligsohn U, Berger A, Abend M, Rubin L, Attias D, Zivelin A, Rapaport SI. Homozygous protein C deficiency manifested by massive venous thrombosis in the newborn. N Engl J Med 310: 559–562, 1984 [DOI] [PubMed] [Google Scholar]
- 81. Selzer RH, Hodis HN, Kwong-Fu H, Mack WJ, Lee PL, Liu CR, Liu CH. Evaluation of computerized edge tracking for quantifying intima-media thickness of the common carotid artery from B-mode ultrasound images. Atherosclerosis 111: 1–11, 1994 [DOI] [PubMed] [Google Scholar]
- 82. Sherva R, Miller MB, Lynch AI, Devereux RB, Rao DC, Oberman A, Hopkins PN, Kitzman DW, Atwood LD, Arnett DK. A whole genome scan for pulse pressure/stroke volume ration in African Americans: the HyperGEN study. Am J Hypertens 20: 398–402, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Souto J, Almasy L, Borrell M, Blanco-Vaca F, Mateo J, Soria J, Coll I, Felices R, Stone W, Fontcuberta J, Blangero J. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: the GAIT study. Genetic analysis of idiopathic thrombophilia. Am J Hum Genet 67: 1452–1459, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Souto J, Almasy L, Borrell M, Gari M, Martinez E, Mateo J, Stone W, Blangero J, Fontcuberta J. Genetic determinants of hemostasis phenotypes in Spanish families. Circulation 101: 1546–1551, 2000 [DOI] [PubMed] [Google Scholar]
- 85. Sowers MR, Jannausch ML, McConnell DS, Kardia SR, Randolph JF., Jr Endogenous estradiol and its association with estrogen receptor gene polymorphisms. Am J Med 119: S16–S22, 2006 [DOI] [PubMed] [Google Scholar]
- 86. Sowers MR, Matthews KA, Jannausch M, Randolph JF, McConnell D, Sutton-Tyrrell K, Little R, Lasley B, Pasternak R. Hemostatic factors and estrogen during the menopausal transition. J Clin Endocrinol Metab 90: 5942–5948, 2005 [DOI] [PubMed] [Google Scholar]
- 87. Sowers MR, Symons JP, Jannausch ML, Chu J, Kardia SR. Sex steroid hormone polymorphisms, high-density lipoprotein cholesterol, and apolipoprotein A-1 from the Study of Women's Health Across the Nation (SWAN). Am J Med 119: S61–S68, 2006 [DOI] [PubMed] [Google Scholar]
- 88. Sowers MR, Wilson AL, Karvonen-Gutierrez CA, Kardia SR. Sex steroid hormone pathway genes and health-related measures in women of 4 races/ethnicities: the Study of Women's Health Across the Nation (SWAN). Am J Med 119: S103–S110, 2006 [DOI] [PubMed] [Google Scholar]
- 89. Stefanick ML, Legault C, Tracy RP, Howard G, Kessler CM, Lucas DL, Bush TL. Distribution and correlates of plasma fibrinogen in middle-aged women: Initial findings of the postmenopausal estrogen/progestin interventions (PEPI) study. Arterioscler Thromb Vasc Biol 15: 2085–2093, 1995 [DOI] [PubMed] [Google Scholar]
- 90. Sudhir K, Chou TM, Chatterjee K, Smith EP, Williams TC, Kane JP, Malloy MJ, Korach KS, Rubanyi GM. Premature coronary artery disease associated with a disruptive mutation in the estrogen receptor gene in a man. Circulation 96: 3774–3777, 1997 [DOI] [PubMed] [Google Scholar]
- 91. Taylor BC, Schreiner PJ, Doherty TM, Fornage M, Carr JJ, Sidney S. Matrix Gla protein and osteopontin genetic associations with coronary artery calcification and bone density: the CARDIA study. Hum Genet 116: 525–528, 2005 [DOI] [PubMed] [Google Scholar]
- 92. Thanassoulis G, Peloso GM, Pencina MJ, Hoffmann U, Fox CS, Cupples LA, Levy D, D'Agostino RB, Hwang SJ, O'Donnell CJ. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet 5: 113–121, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. van Boven HA, Reitsma PH, Rosendaal FR, Bayston TA, Chowdhury V, Bauer KA, Scharrer I, Conrad J, Lane DA. Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb Haemost 75: 417–421, 1996 [PubMed] [Google Scholar]
- 94. Vandenbroucke JP, Koster T, Briet E, Reitsma PH, Bertina RM, Rosendaal FR. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 344: 1453–1457, 1994 [DOI] [PubMed] [Google Scholar]
- 95. Vongpatanasin W, Tuncel M, Wang Z, Arbique D, Mehrad B, Jialal I. Differential effects of oral versus transdermal estrogen replacement therapy on C-reactive protein in postmenopausal women. J Am Coll Cardiol 41: 1358–1363, 2003 [DOI] [PubMed] [Google Scholar]
- 96. Wang D, Yang H, Quinones MJ, Bulnes-Enriquez I, Jimenez X, De La Rosa R, Modilevsky T, Yu K, Li Y, Taylor KD, Hsueh WA, Hodis HN, Rotter JI. A genome-wide scan for carotid artery intima-media thickness: the Mexican-American Coronary Artery Disease family study. Stroke 36: 540–545, 2005 [DOI] [PubMed] [Google Scholar]
- 97. Wang L, Beecham A, Zhuo D, Dong C, Blanton SH, Rundek T, Sacco RL. Fine mapping study reveals novel candidate genes for carotid intima-media thickness in Dominican Republican families. Circ Cardiovasc Genet 5: 234–241, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wennmalm A. Nitric oxide (NO) in the cardiovascular system: Role in atherosclerosis and hypercholesterolemia. Blood Press 3: 279–282, 1994 [DOI] [PubMed] [Google Scholar]
- 99. Wiggins KL, Smith NL, Glazer NL, Rosendaal FR, Heckbert SR, Psaty BM, Rice KM, Lumley T. ABO genotype and risk of thrombotic events and hemorrhagic stroke. J Thromb Haemost 7: 263–269, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zoller B, Berntsdotter A, Carcia de Frutos P, Dahlback B. Resistance to activated protein C as an additional genetic risk factor in hereditary deficiency of protein S. Blood 85: 3518–3523, 1995 [PubMed] [Google Scholar]