

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia, affecting 35 million people today. The search for new treatments is made ever more urgent by prospects for increasing prevalence due to population aging. Mouse models are one of the most important research tools for finding new treatments for AD. Here, we review those models. We begin by briefly reviewing the AD genetics on which mouse models are based and then consider the most common mouse models of AD, including mice transgenic for human amyloid precursor protein (hAPP) and beta-amyloid (Aβ), mice expressing mutant presenilin genes, mice modeling tau’s role in AD, and apolipoprotein E models. The discussion highlights key features and important differences between these mouse models. We conclude with a discussion about the role of AD mouse models in the translational pipeline.
Keywords: Alzheimer’s disease, Amyloid-beta, Presenilin, Tau, Apolipoprotein E (ApoE), Behavior, Transgenic
1. Introduction
Alzheimer’s disease (AD) is the most common cause of dementia. AD affects 35 million people today and its worldwide prevalence is expected to reach 115 million by 2050 due to aging of the population [130]. AD is now recognized to progress through three stages: preclinical, mild cognitive impairment (MCI), and dementia [1,107,157]. In preclinical AD, AD biomarkers are present but symptoms have not yet appeared [157]. In MCI, patients have cognitive deficits but no functional impairments [1]. And in AD dementia, a decline in two or more cognitive domains has gradually progressed to the point that functioning at work or daily activities is impaired [107]. Pathologically, AD remains diagnosed on the basis of protein aggregates in the brain including amyloid plaques composed of amyloid-beta (Aβ) peptides and neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau. Although cholinesterase inhibitors and the NMDA receptor antagonist memantine provide modest symptomatic benefit, currently there is no disease-modifying treatment for AD [138].
Mouse models are one of the most important research tools for finding new treatments for AD. Yet many of the potential disease-modifying treatments that have failed in clinical trials in recent years claimed to show some promise in mouse models [104]. Thus, it is an opportune time to evaluate the existing mouse models of AD and their role in the translational pipeline. Since the most important AD mouse models are based on disease-causing mutations, we begin by reviewing the genetics of AD. We then consider commonly used AD mouse models, focusing on potentially important differences between the models. We conclude by evaluating these models as tools for target identification and validation and as potential tools for preclinical testing of lead compounds.
2. Genetics of AD
Genetics plays an important role in AD, and the discovery of AD-associated genes has provided the foundation for development of mouse models. AD-associated genes can be divided into those in which mutations cause autosomal dominant AD and those in which polymorphisms serve as risk factors for AD.
2.1. Autosomal dominant AD genes
Mutations in three genes cause autosomal dominant AD: amyloid precursor protein (APP) and the presenilin genes (PSEN1 and PSEN2) [55,98,142,154]. Although autosomal dominant AD mutations make up a small fraction of AD cases [62], its symptoms and pathology have pronounced similarities with sporadic AD. Thus, expressing these disease-associated genes in mice has served as the basis for most AD mouse models.
All of the identified mutations that cause autosomal dominant AD directly alter the production of Aβ through APP processing [26,31,129,162]. APP is a type I transmembrane protein with a large amino-terminal extracellular domain (Fig. 1). To produce Aβ, this extracellular domain is first cleaved by β-secretase (also known as beta-site APP cleaving enzyme, BACE). The remaining carboxy-terminal fragment is then cleaved within the membrane by γ-secretase, which is composed of presenilin and other components [36,45]. γ-secretase can cleave APP at different sites, leading to Aβ peptides of 40 or 42 amino acids. Aβ42 is more prone to oligomerization and is more toxic than Aβ40 [106].
Fig. 1.
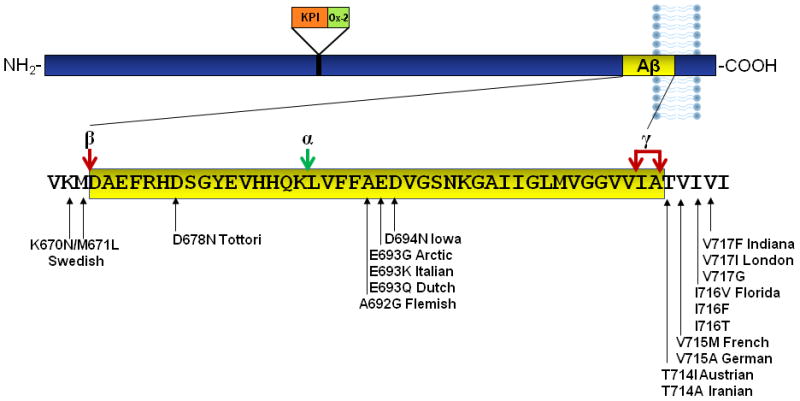
APP processing and APP mutations. Aβ42 is encoded by amino acids 672–713 of APP (numbered according to the longest isoform, APP770). Aβ is produced through sequential cleavage by β-secretase, then γ-secretase. γ-secretase can cleave at alternate sites to produce Aβ40 or Aβ42. Alternative APP processing by α-secretase prevents Aβ production. Common APP mutations include the Swedish mutation at the β-secretase cleavage site and multiple named and unnamed mutations at the γ-secretase cleavage site. Intra-Aβ mutations are also shown.
APP mutations
The first gene mutation identified as a cause of autosomal dominant AD is in the APP gene [55]. AD-causing mutations in APP occur predominantly at the two cleavage sites that lead to Aβ production (Fig. 1). APP mutations are named according to the geographic location in which the affected family originated. The K670N/M671L double mutation at the β-secretase cleavage site, originally found in a Swedish family, results in increased BACE cleavage and thus increased Aβ production, both Aβ40 and Aβ42 [31,162]. The London (V717I), Indiana (V717F), and other mutations at the γ-secretase cleavage site favor production of the more toxic Aβ42, relative to Aβ40 [26,129,162]. These mutations are commonly used in mouse models of AD.
APP mutations within Aβ, such as the Dutch (E693Q) and Arctic (E693G) mutations, increase fibrillogenesis or resistance to proteolysis [105,117]. The Arctic mutation has been used in AD models [28]. The Dutch mutation results in a vascular disorder called hereditary cerebral hemorrhage with amyloidosis and has been used to model that disease [99,173].
In addition to point mutations, increases in APP gene copy number can cause AD. Duplications in the APP gene result in early-onset AD in multiple families [19]. Because the APP gene is on chromosome 21, patients with Down’s Syndrome have three copies of the APP gene and develop AD, usually in their 40s [63]. Thus, mice overexpressing wild-type APP, even without mutations, may serve as useful models for AD.
Presenilin mutations
Mutations in the presenilin genes are another cause of autosomal dominant Alzheimer’s disease [98,142,154]. The two presenilin genes encode proteins with similar function, although PSEN1 mutations are more severe and much more common than PSEN2 mutations [11], and thus have been the focus for presenilin mouse models. The presenilin genes encode the catalytic subunit of γ-secretase [36,45]. AD-associated presenilin mutations increase the Aβ42/Aβ40 ratio [42,125,153,154]. However, presenilins have several other functions, including cleavage of other γ-secretase substrates, cell adhesion, calcium homeostasis, transport, trafficking/ localization, and apoptosis [168,174]. Some of these functions are disrupted by presenilin mutations [9,180,182]. Thus, there is debate over whether presenilin mutations cause AD due to a toxic gain of function that increases the Aβ42/Aβ40 ratio or to a detrimental loss of one of presenilin’s other functions [152].
2.2. AD risk factor genes
Autosomal dominant forms of AD are quite rare. Most cases of AD are sporadic and late onset, but several genes modulate the risk of this more common form of the disease. The strongest risk factor gene is APOE, encoding apolipoprotein E (apoE). There are 3 alleles of APOE; ε3 is the most common, ε4 is associated with increased risk and earlier of age of AD onset, and ε2 is protective [33,46]. Although present in only about 15% of the general population, the ε4 allele occurs in 40–65% of AD cases [144]. A single copy of ε4 increases AD risk 4-fold over noncarriers, and homozygosity for ε4 increases the risk 9- to 15-fold [46,159]. Each copy lowers the age of onset about 10 years [33]. Therefore, apoE4-related models are highly relevant to late-onset AD.
ApoE4 has both Aβ-dependent and Aβ-independent roles in AD. Patients with the ε4 allele have elevated brain Aβ [148], decreased Aβ in the CSF [161] and increased plaque deposition [148,169]. Independent of Aβ, apoE4 increases tau phosphorylation [60] and induces mitochondrial dysfunction [53]. Thus, APOE models both with and without Aβ may yield important clues about AD pathogenesis.
Genes including BIN1, CLU, ABCA7, CR1, PICALM, and others have also recently been associated with late-onset, sporadic AD through genome-wide association studies [10,69,86,115]. All of these genes have fairly small effects, with odds ratios around 1.15, compared with almost 4 for APOE [http://www.Alzgene.org]. Thus, these genes are less likely to be the basis of robust new AD mouse models. However, knockout and transgenic mice will likely be key tools for learning the function of these genes, which will advance our overall understanding of AD mechanisms and pathology.
3. Genetic mouse models of AD
There are many genetic models to choose from when studying AD. Space limitations prevent us from considering them all. We will focus here on important prototypes in each class, the models that are most commonly used, and those that are available from repositories such as the Jackson Laboratories and are thus most easily accessible (Table 1).
Table 1. Representative mouse models of AD.
This list is not comprehensive, but includes important prototypes, some commonly used models, and those that are available from repositories and thus are most easily available.
| Tg Line | Gene/Isoform | Mutation | Promoter | Plaques (mo) | Cognitive deficits (mo) | Vendor Number | Reference | |
|---|---|---|---|---|---|---|---|---|
| hAPP models | ||||||||
| PDAPP | hAPP695<751,7701 | Ind | PDGF-B | 6–9 | 6 | n.a. | [27, 41, 49, 73, 110] | |
| J20 | hAPP695<751,7701 | Swe, Ind | PDGF-B | 6 | 4 | JAX 006293 | [114, 126] | |
| Tg2576 | hAPP695 | Swe | HamPrP | 9 | 10 | Taconic 1349 | [5, 163, 177] | |
| APP23 | hAPP751 | Swe | Thy1 | 6 CAA: 12 |
3 | n.a. | [21, 91] | |
| TgCRND8 | hAPP695 | Swe, Ind | HamPrP | 3 CAA: 11 |
3 | n.a. | [29, 79] | |
| TASD-41 | hAPP751 | Swe, Lon | Thy1 | 3 CAA: 7 |
6 | n.a. | [140] | |
| R1.40 | hAPP YAC2 | Swe | hAPP | 14–15 | 16–17 | JAX 005300 | [66, 93, 94] | |
| Aβ Models | ||||||||
| BRI-Aβ42A | BRI-Aβ42 | n.a. | MoPrP | 3 | ? | JAX 007182 | [106] | |
| hAPP/PS1 models | ||||||||
| PSAPP (Tg2576 × PS1) | hAPP695 PSEN1 |
Swe M146L |
HamPrP PDGF-B |
6 | 4 | n.a. | [4, 40, 67] | |
| APPswe/PS1ΔE9 | m/hAPP6953 PSEN1 |
Swe ΔE9 |
MoPrP MoPrP |
6 | 6 | JAX 004462 | [82, 83, 146] | |
| 5XFAD | hAPP695 PSEN1 |
Swe, Lon, Flo M146L, L28V |
Thy1 Thy1 |
2 | 6 | JAX 008730 | [89, 118, 122, 123] | |
| 2xKI | m/hAPP3 PSEN1 |
Swe P264L |
mAPP mPS1 |
6 | 9–12 | n.a. | [25, 184] | |
| Models with hTau | ||||||||
| TAPP (Tg2576 × JNPL3) | hAPP695 hTau-4R0N |
Swe P301L |
HamPrP MoPrP |
8–15 | motor deficits | Taconic 2469 | [100] | |
| 3xTg | hAPP695 hTau-4R0N PSEN1 |
Swe P301L M146V |
Thy1 Thy1 mPS1 |
6 | 4.5 | JAX 004807 | [12, 120, 121] | |
| htau | hTau PAC4 | Wild-type | hTau | – | 12 | JAX 005491 | [3, 44, 128] | |
APP minigene expressing all three isoforms, mostly KPI-positive APPs.
Yeast artificial chromosome, expressing entire human APP gene, including all isoforms.
Humanized mouse APP, in which three amino acids in Aβ are changed to the human sequence.
P1 artificial chromosome, expressing entire human tau gene, including all isoforms (H1 haplotype).
Our discussion will center on issues important for evaluating the models and their relevance to various forms of AD, interpreting data derived from the model, and – for those who may be entering the field choosing a model for new AD studies. This includes information on how the models were constructed and their primary AD-related phenotypes. Because different models may be most appropriate for addressing different questions, no one model should be considered the best.
It is important to emphasize that no existing mouse model exhibits all features of AD. The ideal model of AD would develop the full range of clinical and pathological features of AD, including cognitive and behavioral deficits, amyloid plaques, neurofibrillary tangles, gliosis, synapse loss, axonopathy, neuron loss and neurodegeneration. Different mouse lines develop these phenotypes to varying degrees and in different combinations. For example, cognitive deficits and amyloid plaques are observed in almost all of the models, neurofibrillary tangles are generally seen only when human tau is also expressed, and neuronal loss is seen in only a few models. This is an issue for the use of mouse models for preclinical drug testing, where one would desire a model that incorporates most features of the disease. In general, however, this is less of a problem for studies aimed at dissecting mechanisms, when it can helpful to isolate some phenotypes from others. In this regard, AD mouse models are best viewed as reductionist tools for understanding the effects on brain function of genes/proteins that have been implicated in AD, and for identifying strategies to block them.
3.1. hAPP transgenic models
The first and most widely used mouse models of AD are based on transgenic expression of human APP (hAPP). Although autosomal dominant AD accounts for relatively few AD cases [62], these mutations serve as the basis for most AD models. Although this is a caveat to working with these models, many clinicians find the similarities between autosomal dominant and sporadic forms of AD to be more noteworthy than their differences.
Many hAPP transgenic lines exist. In general, these lines develop robust amyloid pathology and have memory deficits. They model the synaptotoxicity of AD but do not typically exhibit significant neuron loss. Among the differences between hAPP lines are the promoters driving hAPP expression, the hAPP isoform(s) and mutation(s) expressed, and the background strain. Next we consider each of these features in more detail.
Promoters
hAPP has been expressed from numerous promoters, most commonly under the promoters of the platelet-derived growth factor B-chain (PDGF), thymocyte differentiation antigen 1 (Thy-1), and prion protein (PrP) genes. These promoters drive different levels and spatial patterns of expression (Table 2). The PDGF promoter drives expression mainly in the brain, across widespread regions and selectively in neurons [143]. The Thy-1 promoter drives somewhat higher levels of expression and is also neuron-specific [23]. Thy-1 is unique in that expression does not turn on until around postnatal day 7, avoiding possible developmental effects. The Prp promoter drives the strongest expression, up to 15-fold above endogenous levels, but is less selective, expressing in both neurons and glia and also in extraneural tissues (Table 2).
Table 2.
Characteristics of promoters commonly used in AD mouse models.
| Promoter | Brain Expression | Extraneural Expression | Timing | |
|---|---|---|---|---|
| Regions | Cell Types | |||
| PDGF-B | Widespread [143] | Neurons [143] | Heart, Lung (low) [143] | Begins by E15 [143] |
| Thy-1 | Widespread [23,56] | Neurons [23] | None, with ΔIVS3 modification of promoter [175] | Begins P7 [23] |
| Prp | Widespread [16,170] | Neurons, Astrocytes, Oligodendroyctes, Microglia [16] | Liver > Kidney, Spleen, and other organs [7] | Begins E12.5 [7] |
APP isoforms
The APP mRNA undergoes alternative splicing of exons 7 and 8, resulting in three isoforms named by the number of amino acids in the final product: APP695, APP751, and APP770. The two longer isoforms include a Kunitz protease inhibitor (KPI) domain, and APP770 also has an Ox-2 antigen domain of unknown function (Fig. 2). KPI domains inhibit serine proteases, primarily trypsin [71,88]. The KPI domain mediates certain protein-protein interactions in KPI-positive APP isoforms, including with tumor necrosis factor-alpha-converting enzyme, low-density lipoprotein receptor-related protein, and Notch1 [108]. KPI-positive APP isoforms appear to predominate in axons during the establishment of neural connections [113].
Fig. 2.
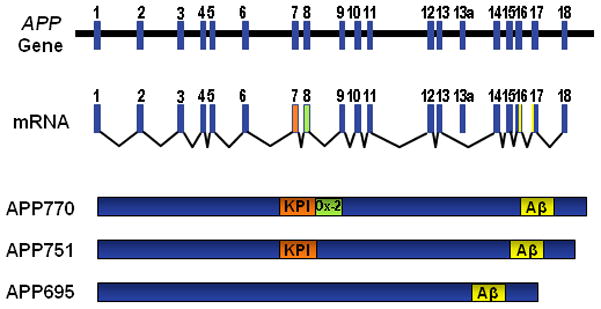
Alternatively spliced isoforms of APP. The APP gene contains 19 exons. Exon 13a is not included in brain isoforms. Exons 7 and 8 can be alternatively spliced to produce APP695, APP751, and APP770. APP751 and APP770 include exon 7 (orange), which encodes a Kunitz protease inhibitor (KPI) domain. APP770 also includes exon 8 (green), which encodes an OX-2 domain.
Evidence is accumulating that the KPI-positive isoforms may be involved in AD pathology. In the normal brain, the ratio of APP695:APP751:APP770 is 20:10:1 [165], but with age, the proportion of KPI-positive APP isoforms increases [164]. KPI-positive APP isoforms are also more prevalent in AD brain cortex [109]. Furthermore, overexpressing wild-type human APP751 causes more AD-like pathology and cognitive deficits than overexpressing wild-type human APP695 [64]. This may be due to the fact that, compared to APP695, KPI-positive APP isoforms are more likely to undergo amyloidogenic β-secretase cleavage, relative to nonamyloidogenic α-secretase cleavage [65].
Given these isoform differences, it is important to consider the APP isoforms expressed in an AD model. Some lines have a cDNA transgene and express a single isoform, most commonly APP695 (Table 1). Others express multiple isoforms, either from the entire APP gene (knock-in and YAC transgenic models) or a hybrid minigene containing the introns around exons 7 and 8 to allow alternative splicing (PDAPP and J20). Interestingly, in contrast to the expression pattern of endogenous APP isoforms in the normal human and mouse brain, where APP695 is most prevalent, the PDAPP line has higher expression of the KPI-positive isoforms that are associated with aging and AD [141].
Mutation
Different hAPP transgenic lines express different AD-associated mutations. Some of the earliest lines have a mutation only at the γ-secretase cleavage site (e.g., PDAPP). However, most currently used mouse lines express the K670N/M671L Swedish double mutation at the β-secretase cleavage site. (Recent litigation over a patent on the Swedish mutation has created controversy regarding these lines [95].) Some lines express only the Swedish mutation (e.g., Tg2576), while others combine the Swedish mutation with a γ-secretase cleavage site mutation (e.g., TgCRND8 and J20). Others lines add a mutation within the Aβ sequence, such as the E693G Arctic mutation [28].
Lines that do not contain FAD mutations, but model the effect of overexpressing wild-type hAPP, have also been produced. The I63 line is related to the J20 line, but expresses wild-type hAPP at high levels [114]. Ts65Dn mice, a model of Down’s syndrome with a partial translocation of mouse chromosome 16 (homologous to human chromosome 21) show age-related learning and memory deficits and neurodegeneration of basal forebrain cholinergic neurons [34,70,136].
Background strain
Background strain differences can strongly modulate phenotypes in mouse models. Background refers to the genetic makeup, apart from the transgene or gene of interest, which varies between the different inbred strains that are commonly used. A line’s original background strain is determined by the strain of the fertilized egg (for transgenic lines generated by pronuclear injection) or by the source of the ES cells (for lines generated by homologous recombination). A mouse line can be moved onto a different background strain by backcrossing with mice of the new strain. After 10 generations, the line is considered congenic on the new background. Common background strains are C57BL/6, 129, FVB/N, DBA, and C3H.
Background strain can impact the phenotype of AD models at several levels. First, strains vary in their levels of anxiety and activity, and some are prone to vision and hearing issues, all of which can affect performance on behavioral tests used to study AD. For example, FVB/N mice have visual impairments that affect their performance on most tasks [131]. Second, different strains have differing susceptibility to excitotoxicity, inflammation, and neurodegeneration, and differing learning/memory abilities. For example, C57BL/6 mice perform better on the Morris water maze than 129/Sv and DBA/2 strains [51,124,172]. Finally, some strains are more susceptible to hAPP/Aβ effects than others [22]. For example, crossing to a 129 strain masks cognitive deficits that were apparent on a 129–C57BL/6 mixed background [179]. Similarly, the TgCRND8 line has a more severe phenotype in the Morris water maze and significantly higher plaque deposition on a 129–C57BL/6 mixed background than on a C3H–C57BL/6 mixed background [54]. Finally, the same hAPP transgene produces a more severe phenotype on the FVB/N background than on a mixed C57BL/6–SJL background [74].
These background strain effects do not diminish the relevance or reproducibility of findings in mouse models, but rather reflect the fact that, as in humans, many genetic variations can affect susceptibility to AD-related dysfunction. Part of the power of mouse models using inbred genetic backgrounds is that one can isolate the effects of the intended manipulation from those of unknown genetic background differences. It is noteworthy when findings are observed on multiple genetic backgrounds, which speaks for their robustness, but time and cost prevent routinely performing mouse experiments on more than one strain background.
3.2 Aβ transgenic models
APP transgenic mice express not only Aβ, but also several other APP fragments that can be biologically active. To isolate the effects of Aβ, lines directly expressing Aβ have been generated. These mice express a fusion protein between Aβ and the BRI protein involved in familial British dementia (FBD). BRI is a transmembrane protein that is cleaved by furin and related proteases to release a peptide called ABri that forms amyloid in FBD. Replacing the ABri sequence with Aβ produces a fusion protein that results in Aβ secretion when expressed in cells [102]. Transgenic mice expressing a BRI-Aβ42 fusion protein develop amyloid pathology [106]. However, no data about cognitive deficits have yet been published, so it is not clear how useful these mice will be as AD models.
3.3. Presenilin & hAPP/presenilin double transgenic models
Presenilin mutations have also been used to make mouse models for AD research. Most models are based on PSEN1 (encoding presenilin 1 or PS1), the most common autosomal dominant AD-associated gene. Transgenic lines are available expressing one [14,30,32,38,43,78,97,125,176] or multiple [97] presenilin mutations. Additionally, knock-in mice utilizing the endogenous promoter have also been produced [24,47,59,116].
Singly transgenic presenilin mutant mice express increased Aβ42 levels with no effect on Aβ40 [43]. However, they do not develop AD pathology or cognitive deficits [37,68,75,85,92]. This lack of phenotype is likely due to sequence differences between mouse and human APP/Aβ [151]. Mouse APP differs from human APP by 17 amino acids, 3 of which are within the Aβ region (Fig. 3). These amino-terminal differences have at least two effects on Aβ. First, the amino acid differences in mouse Aβ cause less efficient aggregation [17]. Second, while mouse BACE cleaves hAPP to produce Aβ1-x, it is more likely to cleave mouse APP (mAPP) to form Aβ11-x [20,76,103]. Thus, while there are examples of mouse Aβ causing cognitive deficits when overproduced for long periods [87], for the most part human Aβ appears necessary for development of AD phenotypes in mice.
Fig. 3.
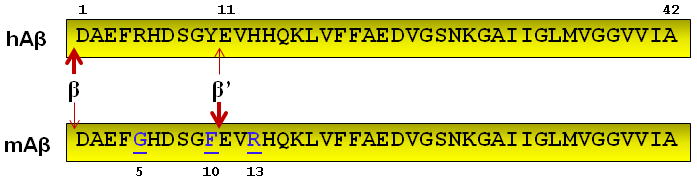
Comparison of human and mouse Aβ. Mouse Aβ (mAβ) differs from human Aβ (hAβ) at positions 5, 10, and 13, which affects the aggregation properties of Aβ. β-secretease also has a greater preference for cleavage at the β’ site in mouse Aβ, to produce Aβ11-x instead of Aβ1-x.
This hypothesis was tested by crossing presenilin mutant mice with mice transgenic for either mAPP or hAPP. Presenilin mutant mice crossed with a mouse line overexpressing mAPP have no AD pathology or cognitive deficits [84]. On the other hand, the same presenilin mutant mice crossed with hAPP transgenic mice have extensive plaque deposition and behavioral deficits [145]. A prototype is the PSAPP line, derived from a cross between transgenic mice expressing the M146L presenilin mutation and the Tg2576 line of hAPP transgenic mice. This line develops Aβ plaque deposition earlier than Tg2576 and has increased Aβ42 [40,43,82]. PSAPP mice have deficits in the Y-maze even before plaque deposition, a finding that separates cognitive deficits from plaque burden [67].
Today’s more commonly used hAPP/PS1 lines were created by co-injecting the presenilin and hAPP transgenes so they breed as a single transgene. One such line created in the laboratory of David Borchelt combines hAPP containing the Swedish mutation and PS1 containing the ΔE9 mutation (APPswe/PS1ΔE9 mice). These mice develop amyloid plaques and behavioral deficits around 6–7 months of age [82]. An even more rapidly progressing line, 5XFAD, was created by combining five AD-related mutations, including the Swedish, Florida (I716V), and London (V717I) mutations in hAPP and the M146L and L286V mutations in PS1 [118]. 5XFAD mice express high levels of Aβ42 and develop amyloid pathology and cognitive deficits by around 4 months [118]. In addition, the 5XFAD line develops neuron loss, unlike most other hAPP and hAPP/PS1 models [118].
APP and presenilin mutations have also been co-expressed in double knock-in (2xKI) mice. Here, the mouse APP gene was engineered to express the Swedish mutation plus a humanized Aβ sequence [135], and the presenilin gene was engineered to express the P264L mutation associated with familial AD [156]. Neither the APP knock-in nor PS1 knock-in lines alone develops amyloid pathology, but 2xKI mice do [184]. 2xKI mice are not impaired on the standard version of the Morris water maze, but have deficits on a more challenging version of the task in which the platform is moved once they learn its location, requiring the mice to learn its new location [25]. One advantage of the 2xKI model is that APP is not overexpressed; deficits arise due to increasing Aβ levels, particularly Aβ42. A disadvantage is that the breeding strategy required to produce mice with homozygous mutations does not produce littermate controls; separately bred, age-matched, wild-type mice are used instead.
3.4. Modeling the role of tau
The mouse models discussed thus far reliably recapitulate the Aβ pathology of AD, but not the tau pathology. Efforts to model neurofibrillary pathology in AD mouse models have mostly relied on expressing transgenic human tau with mutations that cause frontotemporal dementia (FTD) [100,120]. It is important to note, however, that tau mutations do not cause AD, and thus it is unclear that the mechanisms induced by tau mutations are involved in AD pathophysiology. Therefore, we have not included singly transgenic mice with FTD-associated tau mutations in this discussion of AD mouse models, as they are better considered to be mouse models of FTD. Instead, we will consider lines expressing mutant human tau in conjunction with hAPP [100,120] and then examine lines expressing wild-type human tau [3].
The original hAPP/tau double transgenic mice, called TAPP mice, were produced by crossing the Tg2576 line of hAPP transgenic mice and the JNPL3 line expressing P301L human tau [100]. P301L is the most common FTD-associated tau mutation and favors tau aggregation [77,150,158]. The JNPL3 line expresses human tau under control of the mouse PrP promoter at levels similar to endogenous tau and develops motor and behavioral deficits [101]. Aβ deposition in TAPP mice is similar to the Tg2576 line, but tau pathology is more severe than the JNPL3 line, indicating that Aβ can accelerate the tau pathology [13,57,100]. The utility of the TAPP line is limited by early and significant motor deficits in the parent JNPL3 mice that confound most learning and memory tests [101].
Combining mutant APP, PS1, and tau transgenes, the 3xTg line takes a similar approach. hAPP with the Swedish mutation and human tau with the P301L mutation were co-injected into embryonic cells homozygous for a PS1 M146V mutation, generating a strain in which the hAPP and tau breed as a single transgene on the mutant PS1 background. 3xTg mice develop extracellular Aβ plaques before tangle pathology, as in human AD [120]. However, the Aβ and tau pathologies in 3xTg mice appear to develop independently, without a causal link, since tau pathology was unaffected by crossing with BACE-deficient mice to eliminate Aβ production [178].
While the TAPP and 3xTg lines bear the caveat that they carry a tau mutation not associated with AD, these lines provide a combination of plaque and tangle pathology that is not seen in the other lines we have discussed. Although these lines expressing mutant tau are widely used, lines expressing wild-type tau also exist and may be more appropriate for studying AD mechanisms. In fact, two different comparisons between wild-type tau transgenic and mutant tau transgenic lines have shown that while the mutant tau line shows more aggregation, the wild-type tau line shows earlier neuronal dysfunction [90,111,167]. Combined with recent studies suggesting that neurofibrillary pathology may not be critical to tau-related neurodegeneration [35], these data suggest that studying the role of wild-type tau on neuronal dysfunction in AD models may be very fruitful, even in the absence of significant tau aggregation. The htau line, which expresses the entire human tau gene (including all 6 isoforms driven by the tau promoter) on a mouse tau knockout background, is one example [3]. These mice develop tau phosphorylation and aggregation, dendritic spine loss and impaired synaptic plasticity, cognitive deficits, and cell death [2,3,39,128].
In addition to these human tau transgenic models, it is also becoming clear that the role of tau can be studied in hAPP transgenic mice, because endogenous tau plays an essential role in multiple lines of hAPP mice [81,137,139]. Reducing or eliminating tau prevents deficits in learning and memory [80,137,139] and synaptic plasticity [137,155], and blocks increased susceptibility to early mortality and neuronal overactivity in hAPP mice [80,137,139]. Thus, even though tau does not aggregate or form neurofibrillary tangles in hAPP mice, it is critically involved in mediating or enabling their hAPP/Aβ-induced deficits. Tau acts downstream of Aβ, because the protective effects of reducing tau occur without changes in Aβ levels or aggregation [80,139]. Aβ also does not induce degeneration in primary neuronal cultures from tau knockout mice [134], suggesting that a fundamental feature of Aβ-induced neuronal dysfunction may depend on tau.
3.5. ApoE models
As discussed above, the ε4 allele of APOE (encoding apoE4) is the strongest risk factor for AD. ApoE4 differs from the more common apoE3 by a single amino acid at position 112, with arginine in apoE4 and cysteine in apoE3. The endogenous mouse apoE has arginine at the equivalent position, but is not structurally or functionally apoE4-like because of other sequence differences [133]. Thus, the primary apoE models involve human isoforms. With transgenic models, the choice of promoter is important, since apoE is primarily produced in astrocytes but can be produced in neurons under stress [181]. ApoE4 causes cognitive deficits whether expressed from astrocyte [61] or neuronal [132] promoters, but in direct comparisons, lines expressing apoE4 in neurons have more adverse effects than lines expressing apoE4 in astrocytes [18].
Given these differences, knock-in mice in which apoE is expressed under control of the endogenous promoter and regulatory elements may be the most appropriate models. Both apoE3 and apoE4 knock-in mice are available. ApoE4 knock-in mice have cognitive and synaptic plasticity impairments that are not seen in apoE3 knock-in mice [15,58,171]. When crossed with hAPP mice, apoE4 knock-in mice have higher plaque deposition than ApoE3 knock-in mice [8,48].
4. The role of mouse models in AD research
By and large, mouse models fill a unique niche in AD research. Mice have a high degree of phylogenetic conservation with humans in the architecture and function of the hippocampal and entorhinal cortex circuits that mediate episodic memory and are vulnerable in AD. They also have a similar number of genes and considerable chromosomal synteny with humans [112]. At the same time, mouse models provide a system that is reductionist enough to facilitate experimental manipulation. These facts notwithstanding, mouse models of AD are sometimes criticized because they incompletely recapitulate AD pathology or because some compounds that appeared promising in preclinical mouse studies went on to unsuccessful clinical trials. One must be aware of the limitations of any research tool, and there are many important issues to keep in mind in evaluating data from mouse models of AD:
There may be limitations in applying data from AD mouse models to sporadic AD in humans, since the mice most closely model autosomal dominant AD. There are certain pathophysiological differences between autosomal dominant and sporadic AD, chiefly the fact that one is driven primarily by Aβ overproduction and the other is not. In the extreme, a treatment that acts by neutralizing the preference of β-secretase for the Swedish mutation in hAPP might have huge benefit in a mouse model with that mutation, but none in sporadic AD. On the other hand, both conditions seem to be characterized by high Aβ levels, so treatments that reduce Aβ or block its detrimental effects may be equally effective in the two conditions. The degree to which treatments for sporadic and autosomal dominant AD will overlap is an issue that the field in general, including clinical researchers, will have to face as interest grows in studying autosomal dominant AD patients as a pool of subjects for testing AD treatments in the presymptomatic phase of the disease [160].
AD mice might model earlier stages of the disease than the mild–moderate AD that has been the focus of many clinical trials. AD mice may be a better model of the early preclinical stages of AD than the later dementia stages [183]. This predicts that treatments effective in mouse models might have clinical benefit if administered presymptomatically, which is increasingly a focus of AD clinical trial design.
AD mouse models have been highly useful as a tool for target identification and validation… The conventional pathway for rational drug discovery begins with target identification and validation, followed by high-throughput screening, lead compound optimization, preclinical testing in animal models, then clinical trials (Fig. 4). Mouse models are well suited to the early stages of this process, when an arsenal of approaches including genetic manipulation can be applied to identify and validate new targets.
… but there are challenges in using mouse models for preclinical testing of lead compounds. The other phase of the drug discovery process in which mouse models are useful is preclinical testing (Fig. 4). However, there are limitations in this regard, including dramatic differences in drug metabolism, pharmacokinetics, and routes of administration that make it difficult to compare the effectiveness of specific compounds across species [50].
The choice of outcome measures is critical. A variety of outcome measures is available in mouse models, including both neuropathological (e.g., plaque deposition) and functional (e.g., deficits in learning and memory). Different outcome measures are likely to have different predictive validity. Initial studies of AD mouse models focused on pathological outcome measures, which in general are robust and consistent. However, pathology, particularly plaque pathology, does not correlate with cognitive impairment in either mouse models or AD patients [6,177]. Outcome measures such as behavioral and electrophysiological studies that reflect function may be more relevant and predictive of efficacy in AD. Although more variable than pathology, functional outcomes reflect the deficits that are most important to correct in AD. Compounds that have proceeded to clinical trials based on effectiveness in clearing plaques, without functional data, have not fared well in humans with AD [52].
There is no substitute for good experimental design. Like any experiment, preclinical mouse studies can suffer from poor reproducibility due to experimental design issues, such as low numbers of mice. These issues have haunted the ALS field, which has witnessed several preclinical candidates that showed efficacy in a mouse model fail when moved quickly into clinical trials without first being independently replicated. Retesting the failed compounds in mice after properly controlling for potential experimental confounds showed no significant effects, and the positive results in the original trials were attributed to measurement noise [149]. There is a related issue of publication bias, such that even very carefully performed studies with negative results may never be published and so are not available to counterbalance a weak positive result. Better avenues for publishing negative data could help address the problem of preclinical false positive results.
Fig. 4.
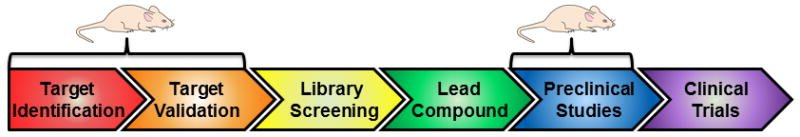
Roles for mouse models in the drug discovery pipeline. Mouse models have been very useful in the “basic science” stages of target identification and validation. Mouse models are also used for preclinical testing of lead compounds.
5. Conclusions
Tremendous progress in understanding AD pathophysiology has been made in the last twenty years. While human clinical research has contributed greatly, human subjects research is generally constrained by the inability to experimentally manipulate variables, so that most conclusions are descriptive or correlational. Mouse models, on the other hand, enable studies of causal relationships because of the power to manipulate the system. For example, human studies suggested that the synapse was the primary target in AD and that synapse loss correlated most closely with cognitive deficits [166]. The synaptic hypothesis was then directly tested in mouse models, demonstrating that Aβ alters synaptic transmission and impairs synaptic plasticity, and even more importantly, enabling detailed dissection of the molecular pathways involved to identify new therapeutic targets [127].
There are many other examples of the power of mouse models. Our understanding that small soluble aggregates of Aβ and tau play a greater role in neuronal dysfunction than the obvious neuropathological hallmarks of AD, amyloid plaques and neurofibrillary tangles, is due in large part to experiments that could not have been performed in humans, including most recently the ability to directly visualize aggregation through in vivo microscopy [35,72,96]. Mouse models were also central in winding down battles between “baptists” and “tauists” about whether Aβ or tau was more important, by providing experimental evidence that both are important, with tau downstream of Aβ [119,139]. And one of the most promising therapies now in clinical trials, Aβ immunotherapy, was initially developed in mouse models [147]. Especially with the wide variety of lines available and continuing technological advances, there is every reason to believe that mouse models will continue to be invaluable tools in the drug discovery pathway for AD treatment.
Acknowledgments
We thank Tony Filiano, Dheepa Sekar, Robert Steele, and Brian Warmus for helpful comments on earlier drafts of the manuscript. This work was supported by grants to E.D.R from the NIH (NS054811 and NS075487), the Stephen Bechtel Fund, and American Health Assistance Foundation. A.M.H. is supported by NIH grant GM008111.
References
- 1.Albert MS, Dekosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. Alzheimer’s Dement. 2011 doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde Y-A, Duff K, Davies P. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 4.Arendash GW, Gordon MN, Diamond DM, Austin LA, Hatcher JM, Jantzen P, Dicarlo G, Wilcock D, Morgan D. DNA Cell Biol. 2001;20:737–744. doi: 10.1089/10445490152717604. [DOI] [PubMed] [Google Scholar]
- 5.Arendash GW, King DL. Physiol Behav. 2002;75:643–652. doi: 10.1016/s0031-9384(02)00640-6. [DOI] [PubMed] [Google Scholar]
- 6.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 7.Asante EA, Gowland I, Linehan JM, Mahal SP, Collinge J. Neurobiol Dis. 2002;10:1–7. doi: 10.1006/nbdi.2002.0486. [DOI] [PubMed] [Google Scholar]
- 8.Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, Hansen JC, Sullivan PM, Paul SM. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berezovska O, Jack C, McLean P, Aster JC, Hicks C, Xia W, Wolfe MS, Kimberly WT, Weinmaster G, Selkoe DJ, Hyman BT. J Neurochem. 2000;75:583–593. doi: 10.1046/j.1471-4159.2000.0750583.x. [DOI] [PubMed] [Google Scholar]
- 10.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 11.Bertram L, Tanzi RE. Pharmacol Res. 2004;50:385–396. doi: 10.1016/j.phrs.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 12.Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 13.Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M. Am J Pathol. 2007;171:2012–2020. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 15.Bour A, Grootendorst J, Vogel E, Kelche C, Dodart JC, Bales K, Moreau PH, Sullivan PM, Mathis C. Behav Brain Res. 2008;193:174–182. doi: 10.1016/j.bbr.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 16.Boy J, Leergaard TB, Schmidt T, Odeh F, Bichelmeier U, Nuber S, Holzmann C, Wree A, Prusiner SB, Bujard H, Riess O, Bjaalie JG. NeuroImage. 2006;33:449–462. doi: 10.1016/j.neuroimage.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 17.Bush AI, Pettingell WH, Multhaup G, Paradis MD, Vonsattel J-P, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 18.Buttini M, Masliah E, Yu GQ, Palop JJ, Chang S, Bernardo A, Lin C, Wyss-Coray T, Huang Y, Mucke L. Am J Pathol. 2010;177:563–569. doi: 10.2353/ajpath.2010.090973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cabrejo L, Guyant-Marechal L, Laquerriere A, Vercelletto M, De La Fourniere F, Thomas-Anterion C, Verny C, Letournel F, Pasquier F, Vital A, Checler F, Frebourg T, Campion D, Hannequin D. Brain. 2006;129:2966–2976. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- 20.Cai HB, Wang YS, McCarthy D, Wen HJ, Borchelt DR, Price DL, Wong PC. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 21.Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 22.Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, Eckman C, Meiners J, Nilsen SP, Younkin SG, Hsiao KK. Hum Mol Genet. 1997;6:1951–1959. doi: 10.1093/hmg/6.11.1951. [DOI] [PubMed] [Google Scholar]
- 23.Caroni P. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- 24.Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. Proc Natl Acad Sci USA. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charier-Harlin M-C. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 27.Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- 28.Cheng I, Palop J, Esposito L, Bien-Ly N, Yan F, Mucke L. Nat Med. 2004;10:1190–1192. doi: 10.1038/nm1123. [DOI] [PubMed] [Google Scholar]
- 29.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 30.Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 31.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 32.Citron M, Westaway D, Xia WM, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, Hyslop PS, Selkoe DJ. Nature Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 33.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 34.Davisson MT, Schmidt C, Akeson EC. Prog Clin Biol Res. 1990;360:263–280. [PubMed] [Google Scholar]
- 35.de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 37.Dewachter I, Ris L, Croes S, Borghgraef P, Devijver H, Voets T, Nilius B, Godaux E, Van Leuven F. Neurobiol Aging. 2008;29:639–652. doi: 10.1016/j.neurobiolaging.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Dewachter I, Van Dorpe J, Smeijers L, Gilis M, Kuiperi C, Laenen I, Caluwaerts N, Moechars D, Checler DR, Vanderstichele H, Van Leuven F. J Neurosci. 2000;20:6452–6458. doi: 10.1523/JNEUROSCI.20-17-06452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dickstein DL, Brautigam H, Stockton SD, Jr, Schmeidler J, Hof PR. Brain Struct Funct. 2010;214:161–179. doi: 10.1007/s00429-010-0245-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dineley KT, Xia XF, Bui D, Sweatt JD, Zheng H. JBiolChem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- 41.Dodart JC, Meziane H, Mathis C, Bales KR, Paul SM, Ungerer A. Behav Neurosci. 1999;113:982–990. doi: 10.1037//0735-7044.113.5.982. [DOI] [PubMed] [Google Scholar]
- 42.Duff K. Trends Neurosci. 1997;20:279–280. doi: 10.1016/s0166-2236(97)01093-x. [DOI] [PubMed] [Google Scholar]
- 43.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-Tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 44.Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- 45.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 46.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. J Am Med Assoc. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 47.Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. Neurobiol Aging. 2002;23:335–348. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- 48.Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. J Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 50.Geerts H. CNS Drugs. 2009;23:915–926. doi: 10.2165/11310890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 51.Gerlai R. Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 52.Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, Lacombe D, Kong X, Aman A, Laurin J, Szarek WA, Tremblay P. Neurobiol Aging. 2007;28:537–547. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 53.Gibson GE, Haroutunian V, Zhang H, Park LCH, Shi Q, Lesser M, Mohs RC, Sheu RKF, Blass JP. Ann Neurol. 2000;48:297–303. [PubMed] [Google Scholar]
- 54.Glazner KA, Odero GL, Anema E, Motnenko A, Schapansky J, Grossman D, Oliver DR, Glazner GW, Albensi BC. Life Sci. 2010;86:942–950. doi: 10.1016/j.lfs.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 55.Goate A, Chartier-Harlin M-C, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 56.Gordon JW, Chesa PG, Nishimura H, Rettig WJ, Maccari T, Endo T, Seravalli E, Seki T, Silver J. Cell. 1987;50:445–452. doi: 10.1016/0092-8674(87)90498-3. [DOI] [PubMed] [Google Scholar]
- 57.Gotz J, Chen F, van Dorpe J, Nitsch RM. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 58.Grootendorst J, Bour A, Vogel E, Kelche C, Sullivan PM, Dodart JC, Bales K, Mathis C. Behav Brain Res. 2005;159:1–14. doi: 10.1016/j.bbr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 59.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Nat Med. 1999;5:101–116. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 60.Harris F, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley R, Huang Y. Proc Natl Acad Sci USA. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hartman RE. Exp Neurol. 2001;170:326–344. doi: 10.1006/exnr.2001.7715. [DOI] [PubMed] [Google Scholar]
- 62.Harvey RJ, Skelton-Robinson M, Rossor MN. J Neurol Neurosurg Psychiatry. 2003;74:1206–1209. doi: 10.1136/jnnp.74.9.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heston LL. Psychiatr Dev. 1984;2:287–294. [PubMed] [Google Scholar]
- 64.Higgins LS, Catalano R, Quon D, Cordell B. Ann NY Acad Sci. 1993;695:224–227. doi: 10.1111/j.1749-6632.1993.tb23056.x. [DOI] [PubMed] [Google Scholar]
- 65.Ho L, Fukuchi K, Younkin SG. J Biol Chem. 1996;271:30929–30934. doi: 10.1074/jbc.271.48.30929. [DOI] [PubMed] [Google Scholar]
- 66.Hock BJ, Lattal KM, Kulnane LS, Abel T, Lamb BT. Curr Aging Sci. 2009;2:205–213. doi: 10.2174/1874609810902030205. [DOI] [PubMed] [Google Scholar]
- 67.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 68.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 69.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, Destefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J I the Alzheimer’s Disease Neuroimaging; C consortium, E consortium. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holtzman DM, Santucci D, Kilbridge J, Chua-Couzens J, Fontana DJ, Daniels SE, Johnson RM, Chen K, Sun YL, Carlson E, Alleva E, Epstein CJ, Mobley WC. Proc Natl Acad Sci USA. 1996;93:13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hook VY, Sei C, Yasothornsrikul S, Toneff T, Kang YH, Efthimiopoulos S, Robakis NK, Van Nostrand W. J Biol Chem. 1999;274:3165–3172. doi: 10.1074/jbc.274.5.3165. [DOI] [PubMed] [Google Scholar]
- 72.Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang FS, Cole G. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 74.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Clark HB, Carlson G. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 75.Huang XG, Yee BK, Nag S, Chan ST, Tang F. Exp Neurol. 2003;183:673–681. doi: 10.1016/s0014-4886(03)00242-5. [DOI] [PubMed] [Google Scholar]
- 76.Huse JT, Liu K, Pijak DS, Carlin D, Lee VM, Doms RW. J Biol Chem. 2002;277:16278–16284. doi: 10.1074/jbc.M111141200. [DOI] [PubMed] [Google Scholar]
- 77.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 78.Hwang DY, Chae KR, Kang TS, Hwang JH, Lim CH, Kang HK, Goo JS, Lee MR, Lim HJ, Min SH, Cho JY, Hong JT, Song CW, Paik SG, Cho JS, Kim YK. FASEB J. 2002;16:805–813. doi: 10.1096/fj.01-0732com. [DOI] [PubMed] [Google Scholar]
- 79.Hyde LA, Kazdoba TM, Grilli M, Lozza G, Brusa R, Zhang Q, Wong GT, McCool MF, Zhang L, Parker EM, Higgins GA. Behav Brain Res. 2005;160:344–355. doi: 10.1016/j.bbr.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 80.Ittner LM, Gotz J. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 81.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 82.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 83.Jankowsky JL, Slunt HH, Gonzales V, Jenkins NA, Copeland NG, Borchelt DR. Neurobiol Aging. 2004;25:885–892. doi: 10.1016/j.neurobiolaging.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 84.Jankowsky JL, Younkin LH, Gonzales V, Fadale DJ, Slunt HH, Lester HA, Younkin SG, Borchelt DR. J Biol Chem. 2007;282:22707–22720. doi: 10.1074/jbc.M611050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Janus C, D’Amelio S, Amitay O, Chishti MA, Strome R, Fraser P, Carlson GA, Roder JC, St George-Hyslop P, Westaway D. Neurobiol Aging. 2000;21:541–549. doi: 10.1016/s0197-4580(00)00107-x. [DOI] [PubMed] [Google Scholar]
- 86.Jun G, Naj AC, Beecham GW, Wang LS, Buros J, Gallins PJ, Buxbaum JD, Ertekin-Taner N, Fallin MD, Friedland R, Inzelberg R, Kramer P, Rogaeva E, St George-Hyslop P, Cantwell LB, Dombroski BA, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Lunetta KL, Martin ER, Montine TJ, Goate AM, Blacker D, Tsuang DW, Beekly D, Cupples LA, Hakonarson H, Kukull W, Foroud TM, Haines J, Mayeux R, Farrer LA, Pericak-Vance MA, Schellenberg GD Alzheimer’s Disease Genetics Consortium. Arch Neurol. 2010;67:1473–1484. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kawasumi M, Chiba T, Yamada M, Miyamae-Kaneko M, Matsuoka M, Nakahara J, Tomita T, Iwatsubo T, Kato S, Aiso S, Nishimoto I, Kouyama K. The European journal of neuroscience. 2004;19:2826–2838. doi: 10.1111/j.0953-816X.2004.03397.x. [DOI] [PubMed] [Google Scholar]
- 88.Kido H, Fukutomi A, Schilling J, Wang Y, Cordell B, Katunuma N. Biochem Biophys Res Commun. 1990;167:716–721. doi: 10.1016/0006-291x(90)92084-d. [DOI] [PubMed] [Google Scholar]
- 89.Kimura R, Ohno M. Neurobiol Dis. 2009;33:229–235. doi: 10.1016/j.nbd.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kimura T, Fukuda T, Sahara N, Yamashita S, Murayama M, Mizoroki T, Yoshiike Y, Lee B, Sotiropoulos I, Maeda S, Takashima A. J Biol Chem. 2010;285:38692–38699. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lalonde R, Dumont M, Staufenbiel M, Sturchler-Pierrat C, Strazielle C. Brain Res. 2002;956:36–44. doi: 10.1016/s0006-8993(02)03476-5. [DOI] [PubMed] [Google Scholar]
- 92.Lalonde R, Qian S, Strazielle C. Behav Brain Res. 2003;138:71–79. doi: 10.1016/s0166-4328(02)00230-9. [DOI] [PubMed] [Google Scholar]
- 93.Lamb BT, Bardel KA, Kulnane LS, Anderson JJ, Holtz G, Wagner SL, Sisodia SS, Hoeger EJ. Nat Neurosci. 1999;2:695–697. doi: 10.1038/11154. [DOI] [PubMed] [Google Scholar]
- 94.Lamb BT, Call LM, Slunt HH, Bardel KA, Lawler AM, Eckman CB, Younkin SG, Holtz G, Wagner SL, Price DL, Sisodia SS, Gearhart JD. Hum Mol Genet. 1997;6:1535–1541. doi: 10.1093/hmg/6.9.1535. [DOI] [PubMed] [Google Scholar]
- 95.Landhuis E, Strobel G. Law and Disorder APPswe Patent Suits Raise Ruckus Again. Alzheimer Research Forum. 2010 http://www.alzforum.org/new/detail.asp?id=2472.
- 96.Lesne S, KMT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 97.Leutner S, Czech C, Schindowski K, Touchet N, Eckert A, Muller WE. Neurosci Lett. 2000;292:87–90. doi: 10.1016/s0304-3940(00)01449-x. [DOI] [PubMed] [Google Scholar]
- 98.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 99.Levy E, Carman MD, Fernandez Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 100.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 101.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 102.Lewis PA, Piper S, Baker M, Onstead L, Murphy MP, Hardy J, Wang R, McGowan E, Golde TE. Biochim Biophys Acta. 2001;1537:58–62. doi: 10.1016/s0925-4439(01)00054-0. [DOI] [PubMed] [Google Scholar]
- 103.Liu K, Doms RW, Lee VM. Biochemistry. 2002;41:3128–3136. doi: 10.1021/bi015800g. [DOI] [PubMed] [Google Scholar]
- 104.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Lancet Neurol. 2010;9:702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 105.Massi F, Klimov D, Thirumalai D, Straub JE. Protein Sci. 2002;11:1639–1647. doi: 10.1110/ps.3150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carillo MC, Thies B, Weintraub S, Phelps CH. Alzheimers Dement. 2011 doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Menendez-Gonzalez M, Perez-Pinera P, Martinez-Rivera M, Calatayud MT, Blazquez Menes B. Neurodegener Dis. 2005;2:277–283. doi: 10.1159/000092315. [DOI] [PubMed] [Google Scholar]
- 109.Moir RD, Lynch T, Bush AI, Whyte S, Henry A, Portbury S, Multhaup G, Small DH, Tanzi RE, Beyreuther K, Masters CL. J Biol Chem. 1998;273:5013–5019. doi: 10.1074/jbc.273.9.5013. [DOI] [PubMed] [Google Scholar]
- 110.Morgan D. Neurochem Res. 2003;28:1029–1034. doi: 10.1023/a:1023255106106. [DOI] [PubMed] [Google Scholar]
- 111.Morris M, Maeda S, Vossel K, Mucke L. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O’Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES Mouse Genome Sequencing Consortium. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 113.Moya KL, Confaloni A, Allinquant B. JNeurochem. 1994;63:1971–1974. doi: 10.1046/j.1471-4159.1994.63051971.x. [DOI] [PubMed] [Google Scholar]
- 114.Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, George-Hyslop PS, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, Decarli C, Dekosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nakano Y, Kondoh G, Kudo T, Imaizumi K, Kato M, Miyazaki JI, Tohyama M, Takeda J, Takeda M. The European journal of neuroscience. 1999;11:2577–2581. doi: 10.1046/j.1460-9568.1999.00698.x. [DOI] [PubMed] [Google Scholar]
- 117.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 118.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 120.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 121.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 122.Ohno M. Neurobiol Learn Mem. 2009;92:455–459. doi: 10.1016/j.nlm.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Owen EH, Logue SF, Rasmussen DL, Wehner JM. Neuroscience. 1997;80:1087–1099. doi: 10.1016/s0306-4522(97)00165-6. [DOI] [PubMed] [Google Scholar]
- 125.Oyama F, Sawamura N, Kobayashi K, Morishima-Kawashima M, Kuramochi T, Ito M, Tomita T, Maruyama K, Saido TC, Iwatsubo T, Capell A, Walter J, Grunberg J, Ueyama Y, Haass C, Ihara Y. J Neurochem. 1998;71:313–322. doi: 10.1046/j.1471-4159.1998.71010313.x. [DOI] [PubMed] [Google Scholar]
- 126.Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, Mucke L. Proc Natl Acad Sci USA. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Palop JJ, Mucke L. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. J Neurosci. 2009;29:10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Price DL, Sisodia SS. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 130.Prince MJ. J, Alzheimer’s Disease International. 2009 [Google Scholar]
- 131.Pugh PL, Ahmed SF, Smith MI, Upton N, Hunter AJ. Behav Brain Res. 2004;155:283–289. doi: 10.1016/j.bbr.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 132.Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L. Proc Natl Acad Sci USA. 1998;95:10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Raffai RL, Dong LM, Farese RV, Jr, Weisgraber KH. Proc Natl Acad Sci USA. 2001;98:11587–11591. doi: 10.1073/pnas.201279298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Proc Natl Acad Sci USA. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- 136.Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. Nat Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 137.Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Roberson ED, Mucke L. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, Mucke L. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 140.Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 141.Rockenstein EM, McConlogue L, Tan H, Gordon M, Power M, Masliah E, Mucke L. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 142.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Piacentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, St George-Hyslop PH. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 143.Sasahara M, Fries JWU, Raines EW, Gown AM, Westrum LE, Frosch MP, Bonthron DT, Ross R, Collins T. Cell. 1991;64:217–227. doi: 10.1016/0092-8674(91)90223-l. [DOI] [PubMed] [Google Scholar]
- 144.Saunders AM, Strittmatter WJ, Schmechel D, StGeorge-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 145.Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, Price DL, Tang F, Markowska AL, Borchelt DR. Neurobiol Dis. 2005;18:602–617. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 146.Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, Price DL, Tang F, Markowska AL, Borchelt DR. Neurobiol Dis. 2005;18:602–617. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 147.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 148.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, Bostrom A, Theodoss J, Al-Nakhala BM, Vieira FG, Ramasubbu J, Heywood JA. Amyotroph Lateral Scler. 2008;9:4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- 150.Seelaar H, Kamphorst W, Rosso SM, Azmani A, Masdjedi R, de Koning I, Maat-Kievit JA, Anar B, Kaat LD, Breedveld GJ, Dooijes D, Rozemuller JM, Bronner IF, Rizzu P, van Swieten JC. Neurology. 2008;71:1220–1226. doi: 10.1212/01.wnl.0000319702.37497.72. [DOI] [PubMed] [Google Scholar]
- 151.Selkoe DJ. Annu Rev Neurosci. 1989;12:463–490. doi: 10.1146/annurev.ne.12.030189.002335. [DOI] [PubMed] [Google Scholar]
- 152.Shen J, Kelleher RJ., 3rd Proc Natl Acad Sci USA. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeva EA, Levesque G, Rogaev EI, Lin C, Liang Y, Ikeda M, Mar L, Brice A, Agid Y, Percy ME, Clerget-Darpoux F, Piacentini S, Marcon G, Nacmias B, Amaducci L, Frebourg T, Lannfelt L, Rommens JM, St George-Hyslop PH. Hum Mol Genet. 1996;5:985–988. doi: 10.1093/hmg/5.7.985. [DOI] [PubMed] [Google Scholar]
- 154.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Nature. 1995;375:754–760. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 155.Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Alzheimers Dement. 2011 doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Engchild J, Salvesen GS, Roses AD. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Strobel G. Scientists and regulators discuss preclinical AD trials. Alzheimer Research Forum. 2011 http://www.alzforum.org/new/detail.asp?id=2727.
- 161.Sunderland T, Mirza N, Putnam KT, Linker G, Bhupali D, Durham R, Soares H, Kimmel L, Friedman D, Bergeson J, Csako G, Levy JA, Bartko JJ, Cohen RM. Biol Psychiatry. 2004;56:670–676. doi: 10.1016/j.biopsych.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 162.Suzuki N, Cheung TT, Cai X-D, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 163.Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Am J Pathol. 2000;157:331–339. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tanaka S, Liu L, Kimura J, Shiojiri S, Takahashi Y, Kitaguchi N, Nakamura S, Ueda K. Mol Brain Res. 1992;15:303–310. doi: 10.1016/0169-328x(92)90122-r. [DOI] [PubMed] [Google Scholar]
- 165.Tanaka S, Shiojiri S, Takahashi Y, Kitaguchi N, Ito H, Kameyama M, Kimura J, Nakamura S, Ueda K. Biochem Biophys Res Commun. 1989;165:1406–1414. doi: 10.1016/0006-291x(89)92760-5. [DOI] [PubMed] [Google Scholar]
- 166.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 167.Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. J Biol Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- 168.Thinakaran G, Parent AT. Pharmacol Res. 2004;50:411–418. doi: 10.1016/j.phrs.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 169.Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Neurology. 2004;62:1977–1983. doi: 10.1212/01.wnl.0000128091.92139.0f. [DOI] [PubMed] [Google Scholar]
- 170.Tremblay P, Meiner Z, Galou M, Heinrich C, Petromilli C, Lisse T, Cayetano J, Torchia V, Mobley W, Bujard H, DeArmond SJ, Prusiner SB. Proc Natl Acad Sci USA. 1998;95:12580–12585. doi: 10.1073/pnas.95.21.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Ye GL, Sotak M, Sullivan PM, Pasternak JF, LaDu MJ. Neuroreport. 2004;15:2655–2658. doi: 10.1097/00001756-200412030-00020. [DOI] [PubMed] [Google Scholar]
- 172.Upchurch M, Wehner JM. Behav Genet. 1988;18:55–68. doi: 10.1007/BF01067075. [DOI] [PubMed] [Google Scholar]
- 173.Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos RA. Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 174.Vetrivel KS, Zhang YW, Xu H, Thinakaran G. Mol Neurodegener. 2006;1:4. doi: 10.1186/1750-1326-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vidal M, Morris R, Grosveld F, Spanopoulou E. EMBO J. 1990;9:833–840. doi: 10.1002/j.1460-2075.1990.tb08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. Exp Neurol. 2004;188:224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 177.Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 179.Wolfer DP, Muller U, Stagliar M, Lipp H-P. Brain Res. 1997;771:1–13. doi: 10.1016/s0006-8993(97)00673-2. [DOI] [PubMed] [Google Scholar]
- 180.Xia XF, Wang P, Sun XY, Soriano S, Shum WK, Yamaguchi H, Trumbauer ME, Takashima A, Koo EH, Zheng H. Proc Natl Acad Sci USA. 2002;99:8760–8765. doi: 10.1073/pnas.132045399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yuasa S, Nakajima M, Aizawa H, Sahara N, Koizumi KI, Sakai T, Usami M, Kobayashi SI, Kuroyanagi H, Mori H, Koseki H, Shirasawa T. J Neurosci Res. 2002;70:501–513. doi: 10.1002/jnr.10430. [DOI] [PubMed] [Google Scholar]
- 183.Zahs KR, Ashe KH. Trends Neurosci. 2010;33:381–389. doi: 10.1016/j.tins.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 184.Zhang C, McNeil E, Dressler L, Siman R. Exp Neurol. 2007;204:77–87. doi: 10.1016/j.expneurol.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]