

Abstract
Alginate lyase (AlgL) catalyzes the cleavage of the polysaccharide alginate through a β-elimination reaction. In Pseudomonas aeruginosa algL is part of the alginate biosynthetic operon, and although it is required for alginate biosynthesis, it is not clear why. Steady-state kinetic studies were performed to characterize its substrate specificity, and revealed that AlgL operates preferentially on non-acetylated alginate or its precursor mannuronan. Mature alginate is secreted as a partially acetylated polysaccharide, so this observation is consistent with suggestions that AlgL serves to degrade mislocalized alginate that is trapped in the periplasmic space. The kcat/Km for the reaction increased linearly with the number of residues in the substrate, from 2.1×105 M−1s−1 for substrate containing 16 residues to 7.9×106 M−1s−1 for substrate with 280 residues. Over the same substrate size range, kcat varied between 10 s−1 and 30 s−1. The variation in kcat/Km with substrate length suggests that AlgL operates in a processive manner. AlgL displayed a surprising lack of stereospecificity, in that it was able to catalyze cleavage adjacent to either mannuronate or guluronate residues in alginate. Thus, the enzyme is able to remove the C5 proton from both mannuronate and guluronate, which are C5 epimers. Exhaustive digestion of alginate by AlgL generated dimeric and trimeric products, which were characterized by 1H NMR spectroscopy and mass spectrometry. Rapid-mixing chemical quench studies revealed that there was no lag in dimer or trimer production, indicating that AlgL operates as an exopolysaccharide lyase.
Alginate is a linear polysaccharide that is secreted by Pseudomonas aeruginosa in response to various environmental stimuli, and is responsible for the mucoid phenotype exhibited by the bacteria when they infect the lungs of cystic fibrosis patients. The onset of mucoidy in the lungs correlates with decreased prognosis for survival for those patients (1), and alginate production has been shown to promote bacterial persistence (2).
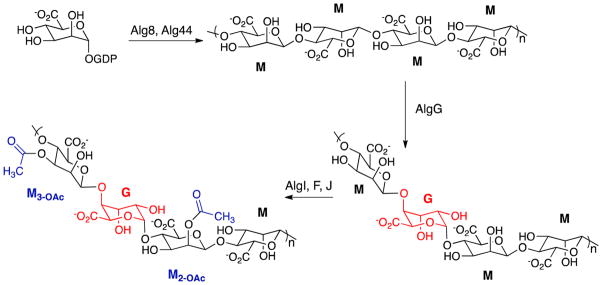
The pathway for alginate synthesis from fructose 6-phosphate has been described and the enzymes required for synthesis of the sugar-nucleotide precursor have been characterized biochemically (3–5). Many questions remain about the final stages of synthesis and secretion, although structural studies of the proteins involved and extensive microbiological studies have provided a wealth of information (reviewed in (6)). The chemical steps that occur in the latter stages of alginate biosynthesis are shown in Scheme 1. The first polymeric species in the pathway is mannuronan, a homopolymer of β-(1→4)-D-mannuronic acid, which is formed from GDP-mannuronic acid that is present in the cytoplasm. Mannuronan formation requires Alg8 and Alg44, which are associated with the inner membrane. The newly synthesized mannuronan is found in the periplasmic space where the final steps in synthesis occur. AlgG catalyzes epimerization of some residues to form α-L-guluronic acid and AlgF, J and I are required for acetylation of some mannuronic acid hydroxyl groups at C2 and C3. Somewhat paradoxically, functional alginate biosynthesis requires AlgL, which is a periplasmic alginate lyase. Deletion of algL is lethal, and microscopic examination of the cells reveals that alginate or a precursor accumulates in the periplasmic space until the cells burst (7).
Scheme 1.
Some features of the AlgL reaction have been reported (8), but the mechanism has not been examined in detail. Interest in alginate lyase stems not only from its involvement in the synthesis of alginate, a virulence factor that is important for the establishment of chronic lung infections, but also from the compelling nature of its catalytic reaction. The reaction is a β-elimination requiring abstraction of the C5 proton, which is adjacent to a carboxylate, and therefore is extremely nonacidic. The specificity of AlgL has not been examined before, and is interesting because the epimers mannuronate and guluronate adopt different conformations, so that the C5 proton in the two sugars is unlikely to occupy the same position with respect to catalytic residues at the active site. In the present study we have determined the substrate specificity and product distribution of the AlgL reaction.
Materials and Methods
Laboratory reagents were purchased from Sigma-Aldrich unless otherwise indicated and used without further purification. MES was obtained from Research Organics.
Purification of AlgL
Recombinant AlgL with a (His)6-tag at the C-terminus was purified from Escherichia coli cells harboring an expression plasmid that was constructed by inserting the algL coding sequence into pET-26b using the NcoI and XhoI restriction sites (9). The construct included a periplasmic-localizing pelB leader sequence, which was cleaved during translation. Transformed E. coli C41(DE3) cells were selected by growth on plates with kanamycin. For production of AlgL, cells were grown in LB medium supplemented with kanamycin (50 μg/mL) at 37° C with rotary shaking until the OD600 reached 0.6–0.8. Subsequently, 1 mM IPTG was added to induce expression of AlgL, and growth was continued at 25°C for 16 h. Cells were harvested by centrifugation at 6000g for 15 min, yielding 3–4 g cell paste per liter of media.
The purification procedure was based on a modification of the published protocol (9). In a typical purification, 6 g cell paste was suspended in 20 ml buffer composed of 50 mM Tris pH 7, 500 mM NaCl, 2 mM DTT, 2% glycerol and 5 mM imidazole. Before lysis 250 U benzonase (Novagen) and 1 mM PMSF was added. Cells were lysed by passage through a French press four times. Insoluble cell debris was removed by centrifugation at 12,200g for 45 min. The cell-free extract was loaded onto a 5 mL His Trap HP (GE) affinity column charged with Co2+. The column was washed with 30 mL resuspension buffer, and 30 mL resuspension buffer containing 20 mM imidazole. AlgL was eluted from the column with 30 mL resuspension buffer containing 200 mM imidazole. AlgL-containing fractions were combined and dialyzed against fresh resuspension buffer, then chromatographed on the same column a second time under the same conditions. Purity of the final product was checked by SDS-PAGE and the sample was concentrated to greater than 2 mg/mL using spin concentrators (Pierce). Enzyme concentration was determined by absorbance measurements using an extinction coefficient at 280 nm of 67380 M−1 cm−1, which was calculated from the sequence of the mature protein by ProtParam.(10) Glycerol was added to the protein to a final concentration of 10% (v/v), and aliquots were stored at −80°C.
Substrate Isolation and Characterization
PolyM was isolated from Pseudomonas aeruginosa strain FRD462, which contains a point mutation in algG, and so does not have mannuronan epimerase activity (11). Alginate was isolated from the overproducing P. aeruginosa strains PDO486, which was the generous gift of Professor Dennis Ohman, (Virginia Commonwealth University) and PDO300. In each case, the appropriate strain was grown overnight in liquid LB media at 37° C. Aliquots were then spread on solid media that contained 22.5 g Pseudomonas Isolation Agar (Remel), 12.5 g glycerol, 12.5 g LB broth, 37.5 g sucrose and 7.5 g agar, per liter. Inoculated plates were incubated at 28°C for 3 days. The viscous mass was scraped from the surface of the plates and suspended in 0.85% NaCl and stirred at room temperature until a homogeneous suspension was obtained. The cells were removed by centrifugation at 1700g for 15 min. An equal volume of isopropanol was added to the supernatant to precipitate the polysaccharide. The polysaccharide was dissolved and precipitated with isopropanol two more times, first from 0.85% (w/v) NaCl, then from 1 M NaCl. The collected polysaccharide was redissolved in 0.85% NaCl and incubated with 5–10 mg Pronase for one hour and reprecipitated with isopropanol. Deacetylated alginate and polyM were obtained by incubating dissolved samples in 0.1 M NaOH for 30 min at room temperature, followed by reprecipitation with isopropanol. To obtain oligosaccharides of various lengths, samples were subjected to acid hydrolysis. Samples were refluxed at pH 5.6 for 1 hour, then at pH 3.5 for a variable length of time (0–60 min) to generate samples containing ~10 to 250 monomeric units. Uronic acid content was quantified by the carbazole assay as described (12). Reducing end concentration was quantified with 3-methyl-2-benzothiazolinonehydrazone as described(13) and the polymer length (dp) was calculated as the ratio of monomeric units to reducing ends.
The composition of the isolated material was determined by 1H NMR as described (14). Samples were subjected to partial acid hydrolysis in order to decrease the polymer size so that well-resolved signals could be obtained. Lyophilized samples were dissolved in D2O and spectra were obtained at 80°C using solvent suppression with a Bruker DRX 300 MHz spectrometer.
AlgL Assay
Product formation was detected by the increase in absorbance at 240 nm arising from formation of unsaturated uronates. Assays were conducted at 25° C in a Carey 50 Bio UV-Vis spectrophotometer. An extinction coefficient of 6150 M−1 cm−1 (15) was used to convert absorbance to product concentration. The reaction buffer contained 100 mM sodium phosphate, 100 mM MES, 100 mM Tris and 200 mM NaCl, pH 7.1 unless otherwise indicated. Initial velocity kinetics data were fitted to the Michaelis-Menten equation.
Purification of dimers & trimers
Dimers and trimers obtained from complete AlgL digestion of polyM or alginate were purified using a DEAE column prior to HPLC analysis. Reaction mixtures containing up to 20 mg digested polysaccharides were loaded onto a DEAE-Sephadex A-25 column (2.8 cm × 19 cm, which was equilibrated with 25 mM ammonium acetate pH 6.8. A gradient from the equilibration buffer to 1 M NH4OAc over 1 L with a flow rate of 5 mL/min was used to elute the oligosaccharides. The separation was carried out at room temperature. Fractions containing AlgL digestion products were identified from their absorbance at 240 nm.
Dimers and trimers for mass spectral analysis were purified by size exclusion chromatography. Reaction mixtures containing up to 10 mg digested polysaccharides were loaded onto a Bio-Gel® P-2 column (1.5 cm × 28 cm, Bio-Rad) equilibrated with 1 mM phosphate pH 6.8. The column was developed at 0.5 mL/min and fractions containing AlgL digestion products were pooled and concentrated by rotary evaporation. Because MES from the AlgL stock solution was found to co-elute with reaction products and interfere with MS analysis, AlgL digestions of samples for MS analysis were conducted without MES.
HPLC characterization
Samples were dissolved in 0.1 mL of 100 mM NaOH containing 1 mM saccharate as an internal standard, and injected onto a CarboPac PA-100 column (4 × 250 mm) using a Dionex DX-500 HPLC system equipped with an ED40 electrochemical detector. The solvent flow rate was 1 mL/min, and products were eluted using the following gradient: 0–10 min 0% B, 10–24 min gradient to 60% B, 24–40 min gradient to 80% B, 40–50 min gradient to 100% B, where solvent A was 0.1 M NaOH and solvent B was 1 M Na acetate in 0.1 M NaOH. Under these conditions saccharate eluted at 22 minutes, dimeric product at 25 min and trimer at 28 min.
Chemical characterization of products
ESI-MS was performed on a Thermo-Finnigan TSQ7000 triple-quadrupole MS with the API2 source and Performance Pack (ThermoFinnigan, San Jose, CA). Oligosaccharides were desalted by size exclusion chromatography with a P-2 column as described above, and delivered to the electrospray source using a syringe pump at a flow rate of 10 μl/min. The MS was run in negative mode, with the mass scan range from 100 to 1500 Da. The capillary temperature was kept at 250°C, and the electrospray needle voltage was 4.5 kV. Nitrogen sheath gas was provided to the source from a nearby dewar of liquid nitrogen at 40–80 psi. Oligosaccharides identified by ESI-MS were further confirmed by MS/MS on the same instrument under the same conditions except that argon was used as the CID gas. Purified products from the AlgL reaction were characterized by 1H NMR with a Bruker DRX 300MHz spectrometer. Samples containing 0.5 – 1 mg product were dissolved in D2O; spectra were obtained at 60°C and 90°C using a pulse sequence that suppressed the HOD peak.
Transient-State Kinetic Studies
Rapid-mixing chemical quench experiments were conducted using a Kintek RQF-3 instrument. PolyM was dissolved in 25 mM Tris, 25 mM sodium phosphate, 25 mM MES, 50 mM NaCl pH 7 at a final concentration of 2.5 mg/mL. The average dp was 71. AlgL was used at a concentration of 8.4 mg/mL. Equal volumes of polyM and AlgL were mixed in the chemical-quench apparatus thermostated at 25°C, and the reactions were terminated at fixed times ranging from 20 to 250 ms by the addition of 100 mM HCl. For each time point, 12 individual samples were pooled; the pooled samples were neutralized by addition of NaOH, lyophilized and dissolved in 100 μl 100 mM NaOH with 1 mM internal standard (saccharate). Two to three replicate runs of each sample (25 El) were analyzed by HPLC as described above.
Results
AlgL expression and purification
Robust overexpression of AlgL was observed, and purification using metal ion affinity chromatography yielded protein that was greater than 95% pure, based on Coomassie-stained SDS-PAGE. Higher yields of AlgL were obtained using Co2+-NTA columns instead of Ni2+-NTA, which was used in previous work (9) (data not shown). Samples supplemented with 10% (v/v) glycerol and stored at −80 °C were stable for several months. Although purified AlgL was stable and active, with no observable activity decrease with even 2 weeks’ storage at 4°C, its activity was observed to vary dramatically with protein concentration (Figure 1). Initiating the reaction by addition of substrate after allowing the enzyme to incubate in the assay solution for up to 30 minutes had little effect on the measured velocity. These results suggest that AlgL aggregates in a nonreversible or slowly reversible manner, but this has not been investigated further.
Figure 1.
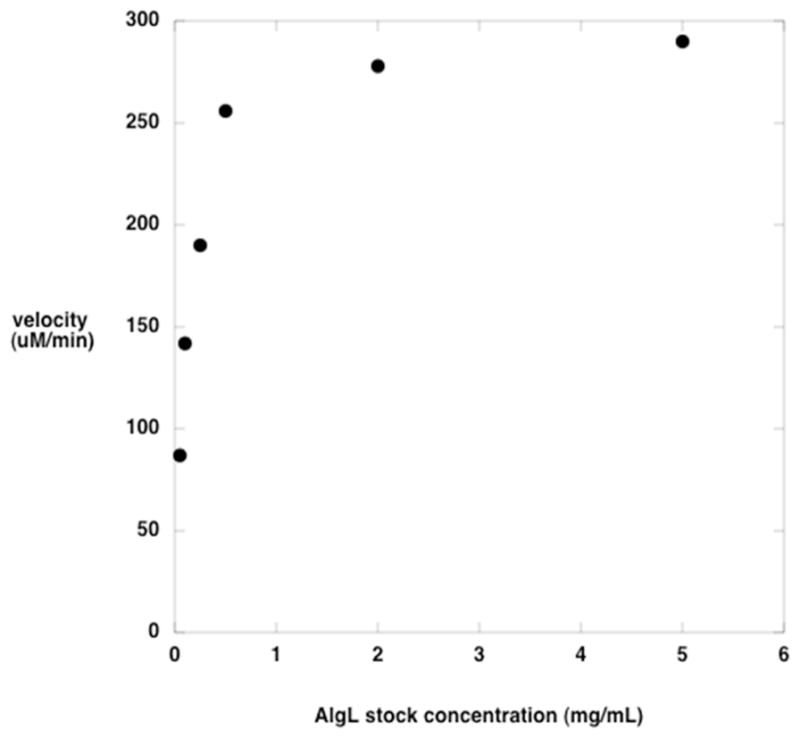
Activity of AlgL as a function of protein stock concentration. The polyM concentration was 20 μM, and had an average dp of 34. Each reaction was initiated by the addition of 30 μg of AlgL from a stock solution at the concentration indicated.
In characterizing the AlgL reaction, we sought to determine characteristics that would speak to its functional role in the bacterium. Thus, we examined the dependence of the reaction on the length of the substrate, and whether the enzyme exhibited specificity for acetylated or deacetylated substrates, since exogenous alginate encountered by the bacteria would be long, acetylated material. To gain insight into the catalytic mechanism of AlgL we examined its specificity for cleavage at mannuronate versus guluronate residues and whether it acted as an exo or endo lyase.
Acetylated versus deacetylated substrates
Four potential substrates for AlgL were tested: alginate1, polyM, acetylated alginate and acetylated polyM. The kinetic parameters were determined by monitoring product formation with the spectrophotometric assay described above (Table 1). Because the kinetic parameters vary with the number of sugar residues in the polymer, comparisons between acetylated and deacetylated substrates were made using substates containing the same number of residues. Since the kcat/Km values for deacetylated substrates are 10-fold higher than for the corresponding acetylated substrates, it appears that AlgL has been optimized for acting on unacetylated alginate or polyM.
Table 1.
Steady-state kinetic parameters for the AlgL reaction.
| substratea | kcat/Km (s−1M−1) | kcat (s−1) | Km (μM) |
|---|---|---|---|
| polyM | 2.5±0.6 × 106 | 32±3 | 13±3 |
| polyM-OAc | 0.30±0.06 × 106 | 1.5±0.1 | 5±1 |
| alginate | 3.1±0.9 × 106 | 32±4 | 11±3 |
| alginate-OAc | 0.5±0.1 × 106 | 1.2±0.1 | 2.6±0.7 |
PolyM and polyM-OAc used in the experiment were dp 133; alginate and alginate-OAc were dp 263.
Size specificity
The steady-state kinetic parameters for cleavage of various sizes of polyM were determined (Figure 2). There was only modest variation in kcat across the size range examined, but Km decreased with increasing substrate length, and kcat/Km increased. Oligomers containing fewer than 9–10 residues were not substrates for AlgL, or reacted too slowly to permit characterization at the concentrations used.
Figure 2.



Steady state kinetic parameters for AlgL-catalyzed degradation of polyM as a function of substrate length. A) kcat; B) Km; C) kcat/Km.
Cleavage at mannuronate versus guluronate
Exhaustive incubation of AlgL with alginate or polyM resulted in formation of dimeric and trimeric products. The products were characterized by 1H NMR and mass spectrometry. Mass spectra revealed peaks consistent with trimers and dimers containing an unsaturated residue (m/z 527 and 351, respectively) as well as the mono-sodiated and di-sodiated trimers. MS/MS of each of these species revealed fragmentation consistent with mannuronate or oligomannuronan. The 1H NMR spectra matched published spectra for dimers and trimers of mannuronate and guluronate containing the 4,5-unsaturated residues at the nonreducing end (16). The distribution of dimers and trimers was characterized by HPLC analysis, and was found to vary with the guluronate content of the sample (Table 2).
Table 2.
Composition of AlgL digestion products.
| strain | % guluronate content (starting material) | % dimer (product 1) | % trimer (product 2) |
|---|---|---|---|
| PDO486 | 40 | 93 | 7 |
| PDO300 | 20 | 58 | 42 |
| FRD462 | 0 | 39 | 61 |
The site of cleavage was identified by 1H NMR spectroscopic characterization of isolated dimeric and trimeric products. Although the same 4,5-unsaturated residue is formed at the nonreducing end of the polymer by lyase action at mannuronate or guluronate, the site of action of AlgL can be inferred by comparing the mannuronate and guluronate content of the final product mixture with that of the substrate. Alginate composition was determined by 1H NMR as described (17). The anomeric proton from guluronate residues appears in an uncrowded region of the spectrum, at 5.07 ppm. Overlapping peaks centered at 4.72 ppm arise from H5 on guluronate residues and H1 on mannuronate residues. Multiple peaks for each proton are observed because the chemical shift is dependent on the chemical identity of neighboring residues. H5 on guluronate residues that are the central residue in GGM, GGG and MGG triplets appear away from the 4.72 ppm signal, but GG diads are not found in significant concentrations in alginate isolated from P. aeruginosa cultures. Therefore, the peak at 4.72 ppm in the present samples is composed of one proton from guluronate and one proton from mannuronate, so the ratio of the areas of the peaks at 5.07 ppm and 4.72 ppm gives the fractional guluronate content of the alginate. As shown in Figure 3, the guluronate content of the alginate from P. aeruginosa strain PDO486 was 40%. The chemical shift of H4 in the unsaturated residue at the nonreducing end of the product is sensitive to the identity of its neighboring residue, so that the peak at 5.95 ppm arises from terminal residues adjacent to guluronate and the peak at 5.86 ppm arises from terminal residues adjacent to mannuronate. Based on the areas of these signals, 23% of the residues adjacent to the terminal residue are guluronates; since the guluronate content of the product decreased relative to that of the starting material, AlgL must be converting some guluronate residues into the 4,5-unsaturated residue at the nonreducing end of the product, i.e., AlgL acts on guluronate residues as well as mannuronate residues.
Figure 3.

1H NMR spectra of PDO486 alginate before (A) and after (B) digestion with AlgL. G and M refer to guluronate and mannuronate, respectively; the number is the proton from which the signal arises. Δ-G and Δ-M refer to unsaturated residues at the nonreducing end of the products adjacent to guluronate and mannuronate, respectively. The area under each labeled peak is given below the chemical shift axis.
Site of action
To determine whether AlgL acted at internal sites in its polysaccharide substrate or at the end, product formation from a long substrate was monitored in the pre-steady state time regime (Figure 4). If AlgL catalyzed cleavage at internal sites of the substrate, then several rounds of catalysis would be required before dimeric and trimeric product would be formed. Conversely, if AlgL acts at the ends of the substrate, each catalytic cycle would release dimeric or trimeric product. With a turnover number of approximately 30 s−1, the first catalytic cycle should be completed in about 0.03 s. The timecourse for product formation extending through the first several cycles revealed a burst of trimer formation, indicating that AlgL acts at the end of its polysaccharide substrate. Dimers were also observed, but at much lower concentrations.
Figure 4.

Pre-steady state product formation by AlgL. Products were separated from the quenched reaction by HPLC and quantitated as described in the text.
Discussion
The presence of a gene encoding an enzyme that degrades alginate in the operon for alginate synthesis seems paradoxical, although it is not unusual. The biosynthetic operons for other secreted polysaccharides produced by Pseudomonas aeruginosa also contain genes for a lyase or hydrolase (6). Nevertheless, assignment of functions to these enzymes remains speculative. Genetic studies have demonstrated that AlgL is required by P. aeruginosa; in its absence, alginate or a precursor, accumulates in the periplasm leading to rupture of the cells (7). To gain further insight into the role of AlgL in synthesis and secretion of alginate, we have examined its substrate specificity. In the alginate biosynthetic pathway the first polymeric product is mannuronan, a homopolymer of β-D-mannuronate residues (polyM). AlgG, a periplasmic enzyme, converts some mannuronate residues into α-L-guluronate residues through epimerization at the C5 position. Mature alginate is acetylated at O2 and O3 on some mannuronate residues, but the sequence in which epimerization and acetylation occur has not been established, so any of the polysaccharides that were examined in the present study are potential natural substrates for AlgL. If the primary role of AlgL were to degrade extracellular alginate, then the enzyme would be expected to show a preference for acetylated alginate. In fact, the opposite is observed; kcat/Km values for unacetylated polyM and alginate are approximately 10-fold higher than the values for the acetylated polysaccharides. Since the unacetylated polysaccharides are not found outside the periplasmic space, it seems likely that the primary substrates for AlgL are alginate or polyM that are within the periplasmic space. A ΔalgL mutant strain with an inducible alg operon was constructed by Ohman to investigate the role of AlgL (7). Following induction, the bacteria lysed after several hours, and electron micrographs suggested that a polymer accumulated in the periplasm until cell lysis occurred. These observations formed the basis for the proposal that AlgL is a component of a multiprotein scaffold that serves to shepherd alginate through the periplasm to AlgE, a porin-like protein through which alginate is secreted. The kinetic data reported here are consistent with the suggestion that the function of AlgL is to degrade mislocalized alginate or polyM (7). The difference in the kinetic parameters for polyM vs alginate are not significant, indicating that AlgL can act with equal facility on the polysaccharide before and after epimerization has occurred.
The variation in the kinetic parameters for the AlgL reaction as a function of substrate length suggests that the enzyme is optimized for acting on polymeric substrates. Although kcat varies only slightly with polymer length, kcat/Km increases with increasing polymer length and Km decreases. The relationship between kcat/Km and polymer length is particularly striking; kcat/Km increases linearly with no indication that a plateau value is being approached. A consequence of this behavior is that Km decreases for longer and longer substrates.
The cause of this phenomenon is interesting to consider. Although Km is routinely considered as a surrogate measure of substrate affinity, it makes little sense to consider the present results in that light. A linear polymer of alginate or polyM longer than 10 residues exceeds the dimensions of AlgL; and although alginate and polyM adopt somewhat structured conformations in solution, it is still difficult to envision how binding interactions between the substrate and enzyme could be distinct for a 150 residue polymer versus a 250 residue polymer. However, the observed kinetic data can be interpreted by considering the meaning of kcat/Km. Northrop has provided a particularly compelling explanation of kcat/Km, pointing out that all the microscopic rate constants in the mechanism appear as ratios except for k1, the bimolecular rate constant for association of substrate with the enzyme (18). As a consequence, kcat/Km is the product of k1 and a complex ratio that varies between 0 and 1. Thus, kcat/Km is seen to be a measure of the frequency with which the substrate is productively captured by the enzyme, where productive capture refers to binding events that lead to product formation. The ratio of rate constants that moderates k1 has a maximum value of 1 in the case that binding of one substrate molecule gives rise to one product. However, if a single substrate binding event can lead to formation of more than one equivalent of product, kcat/Km can increase in proportion to the number of equivalents of product formed per substrate binding event. In the case of AlgL, a ready explanation for the increase of kcat/Km with increasing substrate length is provided if the enzyme behaves in a processive manner. For an enzyme in which the processivity, that is the number of turnovers that occur before the enzyme dissociates from the substrate, is limited by the length of the polymeric substrate, kcat/Km should increase with the length of the substrate, because each substrate capture event leads to formation of multiple equivalents of product in proportion to the number of monomeric units in the polymer. The behavior of kcat as a function of substrate length depends on the relative rates of substrate binding and product release. The case in which kcat reaches a plateau value, as was observed with AlgL, occurs when product release is rapid. When substrate is limiting (kcat/Km conditions) the rate of the reaction is limited by binding of the substrate to the enzyme; longer substrates generate more product once they are bound, and so kcat/Km varies directly with substrate length. When the substrate is saturating, binding is not rate-limiting and the amount of product formed per unit time is independent of whether the product comes from a single long substrate or multiple equivalents of shorter substrate, and the result is that kcat does not vary with substrate length.
Exhaustive digestion of alginate or polyM with AlgL leads to a mixture of dimeric and trimeric products, which we have characterized by 1H NMR spectroscopy and mass spectrometry. To determine whether the products arise from sequential removal of dimers and trimers from the end of the polysaccharide substrate, or from repeated cleavage at internal sites until dimers and trimers are formed, we examined the time course for product formation in the pre-steady state. If AlgL cleaved at internal sites several catalytic cycles would have to occur before small products appeared and the timecourse would exhibit a lag in the appearance of dimers and trimers. On the other hand, removal of dimers and trimers from the end of the substrate would yield a timecourse in which dimers and trimers were immediately apparent. The results of rapid-mixing chemical quench studies of AlgL-catalyzed turnover of alginate are shown in Figure 4. The alginate used in this experiment was converted to mostly trimers by AlgL, and it is clear that there is no lag in trimer production. These data argue that AlgL acts at the end of the alginate molecule, removing successive dimers and trimers as the reaction proceeds.
Since AlgL acts with equal facility on alginate and polyM, we sought to determine whether it cleaves between mannuronate residues exclusively, or between mannuronate and guluronate residues. Early work on AlgL established that it did not cleave between adjacent guluronate residues (8), but did not address whether it could catalyze scission of mannuronate(β1*4)guluronate or guluronate(α1*4) mannuronate linkages. Because the reaction is a β-elimination arising from abstraction of the proton at C5 on the residue nearer the reducing end, and mannuronate and guluronate are C5 epimers, the same unsaturated residue is formed from enzyme action at mannuronate and guluronate residues. However, the residues from which protons were abstracted can be inferred by comparison of the relative proportions of mannuronate and guluronate in the starting polysaccharide and in the products. The protocol for determining the composition of alginate using 1H NMR spectroscopy is well-established (19). The composition of the dimeric and trimeric products can be determined by 1H NMR spectroscopy as well, because the chemical shift of H4 on the unsaturated terminal residue is sensitive to whether the residue is adjacent to mannuronate or guluronate (17). It is clear that AlgL can cleave between adjacent mannuronate residues, because polyM is a substrate. If AlgL cleaved mannuronate and guluronate-containing alginate by abstracting mannuronate protons exclusively, no guluronate residues would be converted to unsaturated residues, and the fractional guluronate composition of the products would increase. However, as shown in Figure 3, the guluronate content in the products was lower than in the starting material, implying that cleavage occurred at guluronate residues as well as mannuronates.
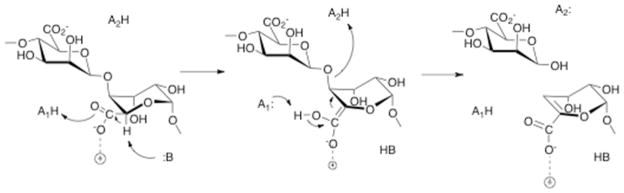
The finding that AlgL catalyzes a syn elimination, by proton abstraction from a mannuronate residue, and an anti elimination, by proton abstraction from a guluronate residue, is not unprecedented for a polysaccharide lyase (20), but is unusual from a mechanistic point of view. In addition to the fact that absolute stereochemical specificity is the norm in enzyme-catalyzed reactions, it has been suggested that the very high pK of the proton adjacent to the carboxylate in the substrate would lead to a mechanism requiring a general base and two general acids, and anti elimination is expected in order to avoid futile proton transfer between the general base and general acid (21). Gerlt and coworkers have proposed a general chemical mechanism for β-elimination reactions that addresses the thermodynamic difficulties inherent in abstraction of a proton alpha to a carboxylate; transposed onto the AlgL reaction, this mechanism consists of concerted neutralization of the carboxylate and general base-catalyzed abstraction of the C5 proton, followed by elimination of the substituent at C4 (Scheme 2). Alginate lyase from Sphingomonas sp. A1 catalyzes the syn elimination required for cleavage between adjacent mannuronate residues; structural studies revealed a critical tyrosine residue at the active site, Y246, that was proposed to serve as both general base and general acid in a reaction in which the proton is abstracted from C5 and transferred to the leaving group in a single step (22). The active site residues are conserved between the Sphingomonas enzyme and AlgL, and mutation of the homologous residue Y256 in AlgL does cause a substantial decrease in activity (E. Farrell, unpublished results). Whether Y256 is the only residue involved in proton transfers in AlgL-catalyzed cleavage at mannuronate residues remains to be established; the anti elimination that occurs during cleavage between mannuronate and guluronate cannot be mediated by a single residue. Two scenarios can be envisioned by which AlgL could catalyze both syn and anti eliminations. In the first, the enzyme would have two different general bases, one for each epimeric substrate. Alternatively, two binding modes would be available, so that each epimer could be acted on by the same general base. Chondroitin lyase ABC catalyzes cleavage of chondroitin sulfate and dermatan sulfate, which requires abstraction of the C5 proton from the epimers glucuronate and iduronate. It appears that the enzyme has two overlapping active sites, with residues at one site required for cleavage of both substrates, but a pair of required histidine residues at the second site that are required only for cleavage of dermatan sulfate (23). Heparinase II has an active site with the same constellation of amino acids that are found in the AlgL active site, and it also is capable of catalyzing syn and anti β-eliminations. It has been proposed that the activity results from the substrates binding in different orientations at the same active site (24). Mutagenesis studies have identified some critical active site residues, but the identities of the general acid and general base have not been determined with certainty. Mechanistic and structural studies of AlgL are underway to explore further details of acid-base catalysis in alginate cleavage.
Scheme 2.
Acknowledgments
The authors are grateful to Professors Michael Toney and Lynne Howell for useful discussions.
Abbreviations
- DTT
dithiothreitol
- PMSF
phenylmethylsulfonyl fluoride
- polyM
homopolymer of mannuronate residues, also called mannuronan
- Tris
tris(hydroxymethyl)aminomethane base
- MES
2-(N-morpholino)ethane sulfonic acid
- dp
degree of polymerization
Footnotes
Alginate isolated from seaweed is widely used in a variety of industrial and pharmaceutical applications and is not acetylated. Pseudomonas alginate is acetylated; however, for purposes of clarity in the present manuscript, we will use the term alginate to refer to deacetylated material, and acetylated alginate to refer to material that has not been chemically deacetylated.
This research was supported by a grant from the National Institutes of Health (GM081419 to PAT).
References
- 1.Yu H, Head NE. Persistent infections and immunity in cystic fibrosis. Frontiers in Bioscience. 2002;7:442–457. doi: 10.2741/a787. [DOI] [PubMed] [Google Scholar]
- 2.Song Z, Wu H, Ciofu O, Kong K-F, Hoiby N, Rygaard JR, Kharazmi A, Mathee K. Pseudomonas aeruginosa alginate is refractory to Th1 immune response and impedes host immune clearance in a mouse model of acute lung infection. J Med Microbiol. 2003;52:731–740. doi: 10.1099/jmm.0.05122-0. [DOI] [PubMed] [Google Scholar]
- 3.Naught LE, Gilbert S, Imhoff R, Snook C, Beamer L, Tipton P. Allosterism and cooperativity in Pseudomonas aeruginosa GDP-mannose dehydrogenase. Biochemistry. 2002;41:9637–9645. doi: 10.1021/bi025862m. [DOI] [PubMed] [Google Scholar]
- 4.Naught LE, Regni C, Beamer LJ, Tipton PA. Roles of active site residues in Pseudomonas aeruginosa phosphomannomutase/phosphoglucomutase. Biochemistry. 2003;42:9946–9951. doi: 10.1021/bi034673g. [DOI] [PubMed] [Google Scholar]
- 5.Shinabarger D, Berry A, May TB, Rothmel R, Fialho A, Chakrabarty AM. Purification and characterization of phosphomannose isomerase-guanosine diphospho-D-mannose pyrophosphorylase. A bifunctional enzyme in the alginate biosynthetic pathway of Pseudomonas aeruginosa. Journal of Biological Chemistry. 1991;266:2080–2088. [PubMed] [Google Scholar]
- 6.Franklin MJ, Nivens DE, Weadge JT, Howell PL. Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Frontiers in Microbiology. 2011;2:1–16. doi: 10.3389/fmicb.2011.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain S, Ohman DE. Role of an alginate lyase for alginate transport in mucoid Pseudomonas aeruginosa. Infect & Immun. 2005;73:6429–6436. doi: 10.1128/IAI.73.10.6429-6436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linker A, Evans LR. Isolation and characterization of an alginase from mucoid strains of Pseudomonas aeruginosa. J Bacteriol. 1984;159:958–964. doi: 10.1128/jb.159.3.958-964.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolfram F, Arora K, Robinson H, Neculai AM, Yip P, Howell PL. Expression, purification, crystallization and preliminary X-ray analysis of Pseudomonas aeruginosa AlgL. Acta Crystallographica Section F. 2012;68:584–587. doi: 10.1107/S1744309112012808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chitnis CE, Ohman DE. Cloning of Pseudomonas aeruginosa algG, which controls alginate structure. J Bacteriol. 1990;172:2894–2900. doi: 10.1128/jb.172.6.2894-2900.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.May TB, Chakrabarty AM. Isolation and assay of Pseudomonas aeruginosa alginate. Methods Enzymol. 1994;235:295–304. doi: 10.1016/0076-6879(94)35148-1. [DOI] [PubMed] [Google Scholar]
- 13.Anthon GE, Barrett DM. Determination of reducing sugars with 3-methyl-2-benzothiazolinonehydrazone. Anal Biochem. 2002;305:287–289. doi: 10.1006/abio.2002.5644. [DOI] [PubMed] [Google Scholar]
- 14.Ertesvag H, Skjak-Braek G. Modification of alginate using mannuronan C-5-epimerases. In: Bucke C, editor. Carbohydrate Biotechnology Protocols. Humana Press; Totowa, NJ: 1999. pp. 71–78. [Google Scholar]
- 15.Iwamoto Y, Araki R, Iriyama K, Oda T, Fukuda H, Hayashida S, Muramatsu T. Purification and characterization of bifunctional alginate lyase from Alteromonas sp. strain no. 272 and its action on saturated oligomeric substrates. Biosci Biotechnol Biochem. 2001;65:133–142. doi: 10.1271/bbb.65.133. [DOI] [PubMed] [Google Scholar]
- 16.Heyraud A, Gey C, Leonard C, Rochas C, Girond S, Kloareg B. NMR spectroscopy analysis of oligoguluronates and oligomannuronates prepared by acid or enzymatic hydrolysis of homopolymeric blocks of alginic acid. Application to the determination of the substrate specificity of Haliotis tuberculate alginate lyase. Carbohydr Res. 1996;289:11–23. doi: 10.1016/0008-6215(96)00060-2. [DOI] [PubMed] [Google Scholar]
- 17.Grasdalen H. High-field 1H n. m.r spectroscopy of alginate: sequential structure and linkage conformations. Carbohydr Res. 1983;118:255–260. [Google Scholar]
- 18.Northrop DB. Rethinking fundamentals of enzyme action. Adv Enzymol and Rel Areas Mol Bio. 1999;73:25–55. doi: 10.1002/9780470123195.ch2. [DOI] [PubMed] [Google Scholar]
- 19.Grasdalen H, Larsen B, Smidsrod O. A P.M.R. study of the composition and sequence of uronate residues in alginate. Carbohydrate Res. 1979;68:23–31. [Google Scholar]
- 20.Garron ML, Cygler M. Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology. 2010;20:1547–1573. doi: 10.1093/glycob/cwq122. [DOI] [PubMed] [Google Scholar]
- 21.Gerlt JA, Gassman PG. Understanding enzyme-catalyzed proton abstraction from carbon acids: details of stepwise mechanisms for beta-elimination reactions. J Am Chem Soc. 1992;114:5928–5934. [Google Scholar]
- 22.Mikami B, Ban M, Suzuki S, Yoon H-J, Miyake O, Yamasaki M, Ogura K, Maruyama Y, Hashimoto W, Murata K. Induced-fit motion of a lid loop involved in catalysis in alginate lyase A1-III. Acta Crystallographica Section D. 2012;68:1207–1216. doi: 10.1107/S090744491202495X. [DOI] [PubMed] [Google Scholar]
- 23.Shaya D, Hahn B-S, Bjerkan TM, Kim WS, Park NY, Sim J-S, Kim Y-S, Cygler M. Composite active site of chondroitin lyase ABC accepting both epimers of uronic acid. Glycobiology. 2008;18:270–277. doi: 10.1093/glycob/cwn002. [DOI] [PubMed] [Google Scholar]
- 24.Shaya D, Zhao W, Garron M-L, Xiao Z, Cui Q, Zhang Z, Sulea T, Linhardt RJ, Cygler M. Catalytic Mechanism of Heparinase II Investigated by Site-directed Mutagenesis and the Crystal Structure with Its Substrate. Journal of Biological Chemistry. 2010;285:20051–20061. doi: 10.1074/jbc.M110.101071. [DOI] [PMC free article] [PubMed] [Google Scholar]