

Abstract
A localized and effective innate immune response to pathogenic bacterial invasion is central to host survival. Identification of the critical local innate mediators of lung defense against such pathogens is essential for a complete understanding of the mechanism(s) underlying effective host defense. In an acute model of Streptococcus pneumoniae lung infection, deficiency in matrix metalloproteinase (MMP)2 and MMP9 (Mmp2/9−/−) conferred a survival disadvantage relative to wild-type mice treated under the same conditions. S. pneumoniae-infected Mmp2/9−/− mice recruited more polymorphonuclear leukocytes to the lung but had higher bacterial burdens. Mmp2/9−/− mice showed significantly higher levels of IL-17A, IP-10, and RANTES in the lung. Although MMP2-dependent cleavage partially inactivated IL-17A, MMP9 was critical for effective bacterial phagocytosis and reactive oxygen species generation in polymorphonuclear neutrophils. These data demonstrate critical nonredundant and protective roles for MMP2 and MMP9 in the early host immune response against S. pneumoniae infection.
Exposure of the lower respiratory tract to commensal and pathogenic microorganisms due to inhalation of infected droplets or aspiration occurs routinely yet is often asymptomatic because of the remarkably efficient surveillance mechanisms of the lung that include resident alveolar macrophages and rapid-responding innate immune cells such as polymorphonuclear neutrophils (PMNs) (1, 2). However, in the setting of immune compromise or at the extremes of age, such typically innocuous exposures to microorganisms can escape normal innate immunity and produce symptomatic pneumonia that, if untreated, would progress to serious complications such as sepsis syndrome (3).
Among the first cytokines secreted in response to pathogen-associated proinflammatory molecules is IL-17A, which induces the recruitment of activated PMNs to sites of infection (4–6). An essential role for PMNs in early defense against bacteria is illustrated by the invasive and highly lethal disease that is induced by typically innocuous strains of Streptococcus pneumoniae when administered to mice that have been depleted of PMNs (7). Conversely, in other settings, overwhelming recruitment of PMNs into the lung without effective bacterial killing can be equally deleterious (8). Thus, understanding how PMNs mediate efficient bacterial killing may hold the key to more effective treatments against pathogenic microbial infections.
The protective function of PMNs depends largely on the timely phagocytosis of invading organisms, which induces the release of reactive oxygen species (ROS) and numerous antimicrobial proteins (9, 10). Microbial phagocytosis is markedly enhanced by opsonization through numerous serum factors including Abs, complement proteins, C reactive protein, mannan binding lectin, and others (9). Moreover, the antimicrobial effects of PMNs have been shown to extend into the extracellular space through formation of potent antimicrobial structures termed neutrophil extracellular traps that are composed of threads of nuclear chromatin armed with diverse antimicrobial proteins that can extend the killing function of neutrophils beyond their typical life span (11, 12). Much of the opsonophagocytic killing of S. pneumoniae, an encapsulated Gram-positive bacterium, has been attributed to the action of three major PMN-associated serine proteinases: elastase, cathepsin G, and proteinase 3 (13). However, the role of other major classes of potentially antimicrobial enzymes such as matrix metalloproteinases (MMPs) in the clearance of S. pneumoniae has not previously been investigated.
The MMPs are a large family of zinc-dependent enzymes that play critical roles in innate and adaptive immunity in humans and animal models of human diseases (14). The diverse and highly prominent representation of this gene family within hematopoietic cells points to their central role in direct modulation of immune mediators implicated in host defense. Several MMPs have been shown to cleave directly and alter the function of cytokines and chemokines during acute inflammation (15–17). Further, despite the fact that MMPs have redundant substrate specificity, studies with animal models of acute and chronic infection, have shown distinct roles for several closely related members of this family. For instance, MMP2 truncates CCL7 (also known as MCP3), and the cleaved product serves as an antagonist of the parent molecule by blocking signaling through the cognate CCRs and inhibiting chemotaxis (18). In contrast, MMP9, a similar proteinase, cleaves CXCL8 (IL-8) to increase markedly its chemotactic activity and has been shown to be critical in host defense in a model of sepsis (19, 20).
MMP2 and MMP9 can establish the chemotactic gradients needed for effective clearance of inflammatory cells in allergic lung disease and are upregulated during the acute onset of bacterial pneumonia (21). However, whether their combined mechanism of action on the cytokines and chemokines produced during innate immune responses is beneficial or deleterious remains unknown. To elucidate further the role of these two closely related MMPs in innate immunity, we challenged double deficient (Mmp2/9−/−) mice with S. pneumoniae, a clinical isolate that causes pneumonia in humans.
Materials and Methods
Mice
MMP2 and MMP9 null mice (eight generations back-crossed to C57BL/6 background) were bred in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited transgenic animal facility at Baylor College of Medicine. MMP2/MMP9 double null (Mmp2/9−/−) mice were generated from F2 and F3 crosses of single null mice as we have described previously (17). Mice were euthanized by single i.p. injection of 0.1 ml Beuthanasia-D (Schering-Plough Animal Health Care, Kenilworth, NJ) solution. Gelatin gel zymography using 1 μl tail blood was used to determine the presence or absence of MMP2 or MMP9 and Mmp2/9−/− protein in experimental mice. Wild-type (WT) C57BL/6 mice were purchased from the Baylor College of Medicine vivarium. All experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine and followed the National Research Council Guide for the Care and Use of Laboratory Animals.
Reagents
Casein (C8654), Histopaque 1077 and 1119, p-aminophenylmercuric acetate (A9563), 1,10-phenanthroline (MMP inhibitors), Nonidet P-40 (NP-40), Luminol (09253), HRP (P8125), iodophenol (10201), and IgG from human serum (I2511) were purchased from Sigma-Aldrich (St. Louis, MO). Bacto Todd-Hewitt Broth, Bacto Yeast Extract, trypticase soy agar with 5% sheep blood agar (TSA II), FITC rat anti-mouse Ly-6G Ab, and Annexin V-FITC Apoptosis Detection Kit were obtained from BD Pharmingen (San Jose, CA). BacLight Red bacterial stain was purchased from Invitrogen (Carlsbad, CA). Mouse rMMP9, rMMP2, and rIL-17A/CF were purchased from R&D Systems (Minneapolis, MN). Polystyrene beads 3 μm in diameter were obtained from Polysciences (Warrington, PA).
Bacterial inoculation and mouse survival study
S. pneumoniae TIGR4 strain is a clinical isolate described previously (22, 23) and was grown in 0.5% THY broth at 37°C without shaking under anaerobic conditions for 8 h. Bacteria were prepared by centrifugation at 4000 rpm for 5 min and were washed once in 0.5% THY broth before resuspension in 10% glycerol for storage at −80°C or in PBS to the desired concentration using spectrophotometric estimation at an OD of 0.7 at 600 nM. Colony-forming units (CFU) of bacterial suspensions were determined by serial dilutions using TSA II plates that were incubated at 37° C overnight.
WT (C57BL/6), Mmp2−/−, Mmp9−/−, and Mmp2/9−/− mice were lightly anesthetized with isoflurane and infected intranasally with S. pneumoniae (1 × 107 CFU) in 50-μl suspensions in PBS using a protocol slightly modified from previously described protocols for S. pneumoniae (24, 25). The exact inoculum was determined from plating S. pneumoniae on TSA II on the day of inoculation. Survival was monitored over the subsequent 5 d every 12 h.
Bacterial dissemination/clearance
At specified time points after intranasal inoculation of S. pneumoniae, mice were euthanized, and bronchoalveolar lavage (BAL) cells were collected by instilling and withdrawing 1-ml aliquots of PBS (pH 7.2) twice. To determine bacterial burdens, lung and spleen were aseptically collected, weighed, and homogenized in 10 ml sterile PBS. A portion of the homogenate and BAL sample were serially diluted and spread on TSA II plates, and the bacterial burden was quantified and expressed as CFU per gram of tissue 24 h later.
Gelatin gel zymography
Zymography was used to analyze for MMP2 and MMP9 in BAL fluid using standard protocols (26). Briefly, 10 μl BAL fluid was separated in 10% SDS-polyacrylamide gels that contained 2% gelatin and washed in 2.5% Triton X-100 before overnight incubation at 37°C in developing buffer (50 mM Tris HCL at pH 8, 5 mM CaCl2, and 0.02% NaN3). Gels were then fixed and stained with 50% methanol and 10% acetic acid that contained 0.3% w/v Coomassie blue. MMP2 and MMP9 appear as clear bands at ~68 and 102 kDa, respectively (17).
Chemokine and cytokine measurements
Serum and BAL fluid samples collected from mice previously infected with S. pneumoniae were centrifuged for 5 min at 12,000 rpm and 800 × g, respectively, and the supernatants were stored at −80°C for batch cytokine/chemokine analysis using Mouse Cytokine/Chemokine Panel I Kits (Milliplex, Millipore, Billerica, MA). Cytokines/chemokines (IL-1β, IL-6, IL-10, IL-12, IL-15, IL-17, IFN-γ, IP-10, KC, MCP-1, MIP-1α, RANTES, and TNF-α) were measured in duplicate according to the manufacturers’ specifications using a Bio-Plex system (Bio-Rad, Hercules, CA) and the concentrations of each cytokine were obtained with Bio-Plex Manager 4.0 software (MiraiBio, Alameda, CA).
MMP activation and in vitro cleavage assay
Carrier-free mouse rIL-17A (5 μg; R&D Systems) was incubated for 4 h at 37°C with 0.5 μg 4-aminophenylmercuric acetate (APMA)-activated MMP2 and MMP9 in the presence or absence of the MMP inhibitor 1,10-phenanthroline (10 mM; Sigma). Equal volumes of activated MMPs/rIL-17A were loaded on 16.5% tricine gels and resolved using electrophoresis. Cleaved and uncleaved proteins were visualized with the Proteosilver Plus Silver Stain Kit according to the manufacturer’s instructions (Bio-Rad).
IL-17A functional studies
Mouse embryonic fibroblasts (MEFs) isolated from 14-d mouse embryos (C57BL/6) were grown in high-glucose DMEM supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA). After reaching confluence, serum was removed, and MEFs were then treated for 6 h or overnight with increasing concentrations of intact rIL-17A or MMP2-, MMP9-cleaved rIL-17A. Overnight treated cell culture supernatants and 6-h treated cells were collected separately for analysis of cytokines and gene expression using ELISA and quantitative real-time PCR, respectively.
Quantitative real-time PCR
Total RNA was extracted from homogenized MEFs with TRIzol (Invitrogen) following the manufacturer’s instructions. cDNA was synthesized using RNase HRT (Invitrogen) and analyzed by using iQ SYBR Green Supermix in an iCycler (both from Bio-Rad) in triplicate. The expression levels of each gene were normalized to the reference gene Actb expression level using a standard curve method. The following primer pairs were used: Actb, forward 5′-TCCTTCGTTGCCGGTCCAC-3′ and reverse 5′-ACCAGCGCAGCGATATCGTC-3′; Ccl20, forward 5′-ATGGCCTGCG-GTGGCAAGCGTCTG-3′ and reverse 5′-TAGGCTGAGGAGGTTCACA-GCCCT-3′; Ccl2, forward 5′-CTCAGCCAGATGCAGTTAACGCCC-3′ and reverse 5′-GGTGCTGAAGACCTTAGGGCAGAT-3′; Ccl7, forward 5′-CTCATAGCCGCTGCTTTCAGCATC-3′ and reverse 5′-GTCTAAGT-ATGCTATAGCCTCCTC-3′; Cxcl1, forward 5′-CGCTTCTCTGTGCAG-CGCTGCTGCT-3′ and reverse 5′-AAGCCTCGCGACCATTCTTGAGT-G-3′.
Neutrophil isolation
Mouse peritoneal PMNs were prepared as described (27). Briefly 1 ml ~10% casein dissolved in PBS was injected twice over 16 h into the peritoneal cavity of mice; 3 h after the second injection, PMNs were harvested by two lavages of the peritoneal cavity with 5 ml cold PBS/EDTA. Casein-induced peritoneal cells were treated with an ammonium chloride–potassium (ACK) lysis buffer to remove RBCs and were washed two times with PBS. Alternatively, PMNs were isolated by discontinuous density gradient centrifugation with Histopaque 1077 and 1119 (Sigma). Washed PMNs were resuspended in HBSS with 10 mM HEPES. PMN purity was >95%, as determined by HEMA 3-stained cytospin preparations, and the viability of cells was >95% as assessed by trypan blue dye exclusion. PMNs were kept at 4°C and used within 2 h of isolation for all in vitro assays.
In vitro PMN bacterial killing assay
PMNs (1 × 106) were incubated with S. pneumoniae (2 × 106 CFU) at a 1:2 ratio in 1 ml HBSS–10 mM HEPES buffer in a shaking incubator at 37°C for 1 h. At designated time points, 40-μl aliquots were removed, and equal volumes of 2% NP-40 were added on ice to lyse PMNs without affecting the bacterial viability (28). The suspension of bacteria and lysed PMNs was subsequently diluted and spread on TSA II plates. The plates were incubated overnight and bacterial CFU at each time point were measured. To determine intracellular killing function, PMNs (1 × 106 cells per tube) were resuspended in HBSS–10 mM HEPES, and S. pneumoniae (5 × 106 CFU) was allowed to incubate for 20 min; cells were then washed with PBS three times to remove the nonphagocytosed bacteria, and washed cells were lysed with 1% NP-40 at 0, 1, and 2 h of incubations, and CFU were determined as described above.
To determine direct antibacterial function of activated MMPs, S. pneumoniae (1 × 106 CFU) was incubated with APMA-activated MMP2 or MMP9 in HBSS–10 mM HEPES for 1 h at 37°C in a shaking incubator in increasing doses starting with 0.01–0.5 μg/ml. Activated MMP2, MMP9, and buffer containing APMA (Vehicle) were purified three times with Microcon centrifugal filter (Millipore) before coincubation; CFU were determined as described above.
Determination of bacterial phagocytosis
PMNs were resuspended at a working concentration of 2 ×106/ml in HBSS with 10 mM HEPES and incubated with S. pneumoniae at a ratio of 1:2 on culture glass slides for 30 and 60 min at 37°C in the presence or absence of 2.5% mouse serum and were fixed in 4% (v/v) formaldehyde in PBS and permeabilized with 0.5% Triton X-100 for 15 min. BacLight Red bacterial stain (Invitrogen), FITC rat anti-mouse LY-6G (BD Pharmingen), and DAPI (Vector Laboratories, Burlingame, CA) were used to detect bacteria, PMNs, and nuclei, respectively. Images were acquired on a microscope (Eclipse TE2000-E; Nikon, Melville, NY) at ×40 magnification (Plan Fluor 40×/0.75; Nikon). Digital images were obtained (CoolSNAP ES; Photometrics) and analyzed using imaging software (NIS elements AR; Nikon). Flow cytometric analyses of bacterial phagocytosis were performed using the same conditions as described above with PMN/bacterial cocultures in polystyrene tubes. Intracellular detection of bacteria was accomplished by permeabilizing cells with 0.5% saponin and staining with BacLight Red bacterial stain (Invitrogen) and FITC rat anti-mouse LY-6G (BD Pharmingen); cells were then analyzed on an LSR II flow cytometer (Becton Dickinson), and data were analyzed with FlowJo software (Tree Star; Ashland, OR).
Measurement of ROS
Measurement of the production of superoxide and other ROS in PMNs was determined by Luminol ECL (29). Briefly, Luminol (0.3 mM), HRP (15 μg/ml), and iodophenol (30 μM) were freshly prepared and added to PMNs (1 × 106 cells/ml) and plated onto 96-well plates. Cell suspensions were equilibrated to 37°C for 5–10 min before opsonized polystyrene beads or S. pneumoniae (coated with 3 mg/ml human IgG [Sigma]) were added at a 1:5 ratio of PMNs to beads or S. pneumoniae. Chemiluminescence was measured in a BMG Labtech NOVOstar microplate reader with an integrated pipettor system (BMG Labtech, Cary, NC) as relative units for 1.5 h continuously. The data processing was performed using MAT-LAB R2009b (The MathWorks, Natick, MA). The light intensity measured from chemiluminescence experiments provides the rate of production of superoxide. The integration of the light intensity gives the total amount of superoxide measured. The calibration of the chemiluminescence assay was performed using known quantities of potassium superoxide. All experiments were repeated five times.
Statistics
All data were analyzed with Prism 5 statistical analysis software. Kaplan–Meier survival curve analysis was performed for curve comparison with log-rank test. For all other experiments, the data are expressed as the mean value ± SEM. Statistical significance was determined using a t test, one-way ANOVA, or Bonferroni multiple-comparison test. A p value <0.05 was considered significant.
Results
Increased mortality and decreased bacterial clearance in Mmp2/9−/− mice
Lung mesenchymal cells constitutively express MMP2, whereas MMP9 is stored within the granules of many inflammatory cells (14); these enzymes are secreted rapidly in the lung alveolar lining fluid after inflammation and in response to acute infection (Supplemental Fig. 1). Mice deficient in MMP2 and MMP9 (Mmp2/9−/−) inoculated intranasally with S. pneumoniae showed increased mortality relative to WT (p = 0.0054), whereas we did not detect a difference in increased mortality in Mmp2−/− and Mmp9−/− mice relative to WT treated under the same conditions (p = 0.2469 and p = 0.1358, respectively) (Fig. 1A–C). Consistently we found a markedly higher bacterial burden in lung and spleen of Mmp2/9−/− mice compared with that of WT mice under the same conditions (Fig. 1D, Supplemental Fig. 2).
FIGURE 1.

MMP2/9 provide survival advantage in Gram-positive S. pneumoniae infection. A–C, Kaplan–Meier survival curves for (A) Mmp2−/−, (B) Mmp9−/−, and (C) Mmp2/9−/− compared with WT mice intranasally infected with 1 × 107 CFU S. pneumoniae with equal number of mice in each group; increased mortality of Mmp2/9−− mice was statistically significant (p = 0.0054, log-rank test). D, Bacterial burden (CFU) in lung and spleen of WT and Mmp2/9−/− mice 24 h after intranasal infection with 5 × 107 CFU S. pneumoniae (n = 5 mice per group). Data are representative of three independent experiments (mean ± SEM). *p < 0.05 (t test). SP, S. pneumoniae.
Aberrant innate immune function in the absence of MMP2 and MMP9
The preceding findings revealed failure to control properly bacterial infection in Mmp2/9−/− mice compared with that in single-deficiency or WT mice treated under the same conditions. To gain further insight into the innate immune function, and because the phenotype was most pronounced in the absence of MMP2 and MMP9, we examined BAL fluid of WT and Mmp2/9−/− mice that were inoculated with S. pneumoniae. This analysis revealed that the increase in lung bacterial burdens seen in Mmp2/9−/− mice was accompanied by an increase in total BAL fluid PMNs and macrophages in the BAL fluid by 24 h postinfection (Fig. 2A–C). Consistent with these findings, histological examination of the lung tissue revealed increased inflammatory cells within the lung parenchyma of Mmp2/9−/− mice, reflecting that the differential bacterial burdens that result in exaggerated innate cellular immunity were generated in the absence of these MMPs (Fig. 2D).
FIGURE 2.
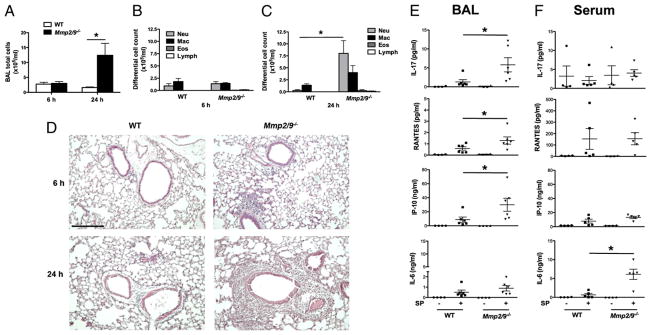
Mmp2/9−/− mice have an ineffective innate immune function. A–C, Total cell counts at 6 h and 24 h (A); neutrophils (Neu), macrophages (Mac), eosinophils (Eos), and lymphocytes (Lymph) counts 6 h (B) and 24 h (C) in BAL fluid of WT and Mmp2/9−/− mice with S. pneumoniae (5 × 107 CFU) intranasal infection (n = 5 mice per each group). Increased PMNs were detected in BAL fluid of Mmp2/9−/− mice, which peaked at 24 h postinfection (p = 0.02). Data are representative of three independent experiments. *p < 0.05 (t test). D, Lung histology. Lung sections stained with H&E (n = 5 mice per group) were prepared 6 h and 24 h postinfection with 5 × 107 CFU S. pneumoniae. Scale bar, 100 μm. Data are representative of three independent experiments. E and F, Luminex assay of cytokines/chemokines in BAL fluid (E) and serum fluid (F) of WT and Mmp2/9−/− mice (n = 5 mice per group) 24 h after S. pneumoniae (5 × 107 CFU) intranasal infection. Luminex assay data in serum showed increase in IL-6 and trends for increase in other proinflammatory cytokines such as TNF-α, IP-10, and IL-1β in infected Mmp2/9−/− mice compared with infected WT mice. Data are representative of three independent experiments (mean ± SEM). *p < 0.05 (t test). SP, S. pneumoniae.
The observed increase in lung inflammation was accompanied by enhanced secretion of cytokines and chemokines, including elevated serum IL-6 and increased IL-17A, CXCL10 (IP-10), and CCL5 (RANTES) in the BAL fluid of Mmp2/9−/− mice infected with S. pneumoniae (Fig. 2E, 2F). Whereas both KC (CXCL1) and MCP-1 (CCL2) were upregulated in the BAL and serum of S. pneumoniae-infected mice, there were no differences noted in WT and Mmp2/9−/− mice (Supplemental Fig. 3). These findings suggest that the increased systemic and local cytokines and chemokines promote the enhanced recruitment of inflammatory cells to the lung of infected Mmp2/9−/− mice.
MMP2 cleaves and inactivates IL-17A
Our results suggest that there is an ineffective innate cellular response against S. pneumoniae airway challenge in Mmp2/9−/− mice. To understand this aberrant response further, we focused on the known fundamental properties of MMPs, which have been shown previously to alter the biological activity of cytokines and chemokines during acute and chronic inflammation (30). Because IL-17A coordinates the recruitment of PMNs to sites of infection (31), the enhanced expression of this cytokine in parallel with airway PMNs suggested to us that IL-17A might be inactivated by either MMP2 or MMP9. Indeed, to varying degrees, both activated MMP2 and MMP9 cleaved rIL-17A at multiple sites (Fig. 3A). As expected from our prior studies (32), full-length rIL-17A induced proinflammatory cytokine (IL-6) and chemokine (KC and CCL20) expression from MEFs. However, these responses were markedly attenuated when MMP2-cleaved rIL-17A and, to a lesser extent, MMP9-cleaved rIL-17A were used in these assays (Fig. 3B). Together, these findings demonstrate that MMP2 and to a lesser degree MMP9 limit the action of IL-17A. The lack of both MMP2 and MMP9 in Mmp2/9−/− mice most likely results in prolonged IL-17A activity that in part explains the enhanced neutrophilia seen.
FIGURE 3.

MMP2 cleaves IL-17A and inactivates proinflammatory function. A, Silver staining of rIL-17A (5 μg) left untreated and incubated for 4 h at 37°C with APMA-activated MMP2 and MMP9 (0.5 μg; + MMP2, + MMP9) and then separated by electrophoresis through a 16.5% tricine gel. Arrowheads indicate MMP2- and MMP9-cleaved fragments of rIL-17A. Results are representative of at least three independent experiments. B, ELISA of IL-6 and KC in supernatants of MEFs stimulated overnight in the presence of equal amounts of native rIL-17A (20 ng/ml and 100 ng/ml), MMP2-cleaved rIL17A (20 ng/ml and 100 ng/ml), or MMP9-cleaved rIL-17A (20 ng/ml and 100 ng/ml). Medium alone serves as a control. Data are representative of three different experiments (mean ± SEM; n = 3 samples). *p < 0.05 (one-way ANOVA and t test). C, Real-time RT-PCR analysis of the expression of CCL20, CCL2, CCL7, and KC by MEF culture stimulated with MMP2- and MMP9-cleaved and uncleaved rIL-17A (20 ng/ml) for 6 h treated as described in B. Data are representative of three independent experiments. *p < 0.05.
Decreased killing and phagocytic ability of Mmp2/9−/− PMNs
Although MMP2 and MMP9 inhibit PMN recruitment by inhibiting IL-17A activity, these findings cannot explain the decreased bacterial clearance observed in Mmp2/9−/− mice that have enhanced neutrophilia. We therefore asked whether MMP-deficient PMNs possessed impaired bactericidal activity due to either reduced phagocytic or killing activity, or possibly both. Accordingly, we isolated and cocultured peritoneal PMNs with S. pneumoniae in different ratios. We found that relative to WT PMNs, PMNs isolated from Mmp2/9−/− mice were markedly impaired in their ability to kill bacteria (Fig. 4A). The difference in bacterial killing ability persisted over several hours (data not shown), and we speculate that the early PMN response to lung bacterial challenge may contribute significantly to pathogen clearance in vivo.
FIGURE 4.

Decreased bacterial killing and phagocytosis by Mmp2/9−/− PMNs. A, In vitro PMNs bacterial killing assay. Mmp2/9+/+ and Mmp2/9−/− peritoneal PMNs were incubated with S. pneumoniae in HBSS–10 mM HEPES for 60 min at 37°C. After incubation, coincubation aliquots were lysed, diluted, and plated on TSA II plate; CFU were counted after overnight incubation. Data are representative of two independent experiments (n = 3 mice per group). *p < 0.05 (t test). B, Numbers of Mmp2/9+/+ and Mmp2/9−/− peritoneal PMNs attached to S. pneumoniae after 30- and 60-min incubation. Coincubation aliquots were cyto-spun on the glass slides and visualized after H&E staining (n = 3 mice). Data are representative of at least three independent experiments (mean ± SEM). *p < 0.05 (t test). C, Fluorescence microscopy image of phagocytic function of Mmp2/9+/+ and Mmp2/9−/− peritoneal PMNs after 60-min coincubation with S. pneumoniae in HBSS–10 mM HEPES at 37°C. Bacteria stain positive with BacLight Red bacterial stain (red), neutrophils are indicated in green (FITC rat anti-mouse Ly-6G), and nuclei are shown in blue (DAPI staining). Data are representative of three independent experiments (n = 3 mice per group). D, Flow cytometric assay of phagocytic function of Mmp2/9+/+ and Mmp2/9−/− peritoneal PMNs. S. pneumoniae stained with BacLight Red was incubated with peritoneal PMNs at a 2:1 ratio in the conditions described in C, followed by Ly-6G staining. Numbers in quadrants indicate percentage of cells in each. BacLight Red expression based on WT and Mmp2/9−/− PMNs coincubation with S. pneumoniae. Data are representative of three independent experiments (n = 4 mice per group). SP, S. pneumoniae.
We next evaluated the phagocytic function of PMNs under the same conditions using fluorescently labeled S. pneumoniae. We found that significantly fewer bacteria were associated with Mmp2/9−/− PMNs at different time points compared with WT PMNs (Fig. 4B, 4C). Moreover, as assessed by flow cytometry, fluorescent S. pneumoniae was markedly less associated with PMNs deficient in MMP2 and MMP9, confirming a defect in phagocytosis (Fig. 4D).
MMP9 is required for phagocytic function of PMNs
Whereas cleavage of IL-17A reflects the actions of MMP2 and MMP9, our studies indicate that the phagocytic defect shown in elicited Mmp2/9−/− PMNs may reflect the effects of the absence of MMPs. Mmp2/9−/− mice infected with S. pneumoniae fail to control bacterial dissemination, further supporting an intrinsic defect in PMN-associated function of MMPs. To determine the relative importance of these enzymes to PMN phagocytosis, we first determined the expression of MMP2 and MMP9 in PMNs at baseline and after exposure to S. pneumoniae. This analysis revealed that MMP9, but not MMP2, was expressed in mouse PMNs regardless of activation status (Supplemental Fig. 4). Moreover, Mmp9−/− PMNs phenocopied Mmp2/9−/− PMNs with respect to phagocytic ability, whereas Mmp2−/− PMNs did not show any phagocytic defects; addition of Mmp2−/− serum partially rescued the phagocytic defect in Mmp2/9−/− PMNs (Fig. 5A, 5B and data not shown). Notably, the phagocytic defect in MMP9-deficient PMNs could be partially rescued in the presence of serum containing MMP9 (i.e., with WT, or Mmp2−/− serum) but not serum from Mmp9−/− or Mmp2/9−/− mice (Fig. 5A, 5B). Adding activated mouse rMMP9, but not rMMP2, to Mmp9−/− PMNs enhanced phagocytosis of S. pneumoniae (Fig. 5C). Together, these data demonstrate that MMP9, but not MMP2, is required for optimum phagocytosis of S. pneumoniae by PMNs.
FIGURE 5.

MMP9 is required for proper phagocytic function in PMNs. A, Flow cytometry of the phagocytosis assay of Mmp9+/+ and Mmp9−/− peritoneal PMNs coincubation with S. pneumoniae in HBSS–10 mM HEPES, or plus WT serum (2.5%), or plus Mmp2−/− serum (2.5%), or plus Mmp9−/− serum (2.5%), or plus Mmp2/9−/− serum (2.5%) for 60 min at 37°C, followed by staining with BacLight Red and Ly-6G. Numbers beside outlined areas indicate percentage BacLight Red+Ly-6G+ live cells not gated dead cells. B, Phagocytic ability is indicated based on A. Data are representative of two independent experiments (n = 4 mice per group). *p < 0.05 (versus Mmp9+/+ PMNs and S. pneumoniae coincubation without serum media), **p < 0.05 (versus Mmp9−/− PMNs and S. pneumoniae coincubation without serum media) (one-way ANOVA and t test). C, Phagocytic ability based on flow cytometry after coincubation of S. pneumoniae and Mmp9+/+ and Mmp9−/− peritoneal PMNs with APMA-activated MMP2 and MMP9 for 60 min at 37°C, followed by staining with BacLight Red and Ly-6G. Data are representative of three independent experiments (mean ± SEM). *p < 0.05 (versus Mmp9+/+ PMNs and S. pneumoniae coincubation in media only), **p < 0.05 (versus Mmp9−/− PMNs and S. pneumoniae coincubation in media only) (one-way ANOVA and t test).
PMNs require MMP9 for intracellular bacterial killing
MMP12 and serine proteinases such as neutrophil elastase possess direct antimicrobial activity (13, 33). However, when added to S. pneumoniae in increasing amounts, activated rMMP2 and rMMP9 failed to kill, indicating that, unlike MMP12, these MMPs do not possess significant intrinsic bactericidal activity (Fig. 6A). To elucidate the indirect role that MMPs may play in facilitating PMN bactericidal activity, we tested the function of PMNs isolated from WT, Mmp9−/−, and Mmp2−/− mice. We found that PMNs deficient in MMP9 but not MMP2 showed significantly reduced bactericidal activity compared with WT PMNs (Fig. 6B). Furthermore, removal of extracellular bacteria through serial washes in PMNs cocultured with S. pneumoniae showed higher numbers of S. pneumoniae recovered in lysed WT PMNs, indicating that WT PMNs were able to phagocytose bacteria more efficiently compared with MMP9-deficient PMNs (Fig. 6C). Time-course study of the same coculture experiments showed that within 2 h, the concentration of intracellular bacteria decreased within WT PMNs, consistent with the efficient bactericidal function of these cells. In contrast, Mmp9−/− PMNs phagocytosed S. pneumoniae inefficiently, but also failed to inhibit bacterial growth over 2 h (Fig. 6C). Together, these studies suggest that MMP9 is critical for the efficient phagocytosis and killing of S. pneumoniae.
FIGURE 6.

PMNs require MMP9 for intracellular antimicrobial activity. A, S. pneumoniae was incubated with APMA-activated MMP2 and MMP9 in HBSS–10 mM HEPES for 60 min at 37°C. Bacteria alone (Input) and APMA-containing buffer used to activate MMP2 and MMP9 (Vehicle) serve as controls. Data are expressed as CFU (representative of three independent experiments; mean ± SEM). B, Peritoneal PMNs (Mmp9−/−, Mmp2−/−, and WT) were incubated with S. pneumoniae at a 1:2 ratio at 37°C for 60 min, and S. pneumoniae CFU were determined as described in Fig. 4 (n = 4 mice per group; mean ± SEM). *p = 0.0006 (WT and Mmp9−/−); NS, p = 0.3 (Mmp2−/− and WT) (Student t test). C, Peritoneal PMNs (Mmp9+/+ and Mmp9−/−) were incubated with S. pneumoniae in HBSS–10 mM HEPES without serum for 20 min at 37°C and washed with PBS three times (at time 0). S. pneumoniae (CFU) in lysed PMNs were determined at indicated time points (0, 1, and 2 h). Data are representative of three independent experiments (n = 3 mice per group; mean ± SEM). *p < 0.05. D and E, Superoxide production as a function of time (D) and total superoxide production (E) for Mmp9+/+ and Mmp9−/− PMNs were measured after incubation with IgG opsonized S. pneumoniae and IgG opsonized latex beads (3 μM) in PBS without antibiotics at 37°C for 1.5 h. Data are representative of three independent experiments (n = 4 mice per group; mean ± SEM). *p < 0.05 (versus Mmp9+/+ PMNs with IgG opsonized S. pneumoniae incubation), **p < 0.001 (versus Mmp9+/+ PMNs with IgG opsonized latex beads incubation) (t test). F, Flow cytometry of apoptosis analysis after 60-min coincubation of S. pneumoniae and peritoneal PMNs of WT and Mmp9−/− mice at 37°C; assay with annexin V/PI staining to measure the percentage of early apoptotic cells (annexin V+/PI−) and late apoptotic death/necrotic cells (annexin V+/PI+). Numbers in quadrants indicate percentage of cells in each. G, Average frequency of late apoptotic death/necrotic (annexin V+/PI+) Mmp9−/− PMN cells with S. pneumoniae incubation. Data are representative of three independent experiments (n = 4 mice per group; mean ± SEM). *p < 0.05 (versus Mmp9+/+ PMNs coincubation with S. pneumoniae). SP, S. pneumoniae.
Innate immune cells commonly produce ROS as part of a critically important mechanism for bacterial killing, although this mechanism may be less important for the killing of S. pneumoniae (1, 34). When we analyzed production of ROS by PMNs exposed to S. pneumoniae, we observed significantly decreased superoxide production, a measure of ROS generation, in Mmp9−/− PMNs compared with that of WT (Fig. 6D). This finding was not unique to S. pneumoniae, as exposure of PMNs to inert beads resulted in a qualitatively similar, but quantitatively exaggerated, enhanced oxidative burst in WT relative to Mmp9−/− PMNs (Fig. 6E).
Recent evidence points to phagocytosis and the production of ROS as critical mediators of PMN apoptosis that is in turn essential for the resolution of both infection and inflammation (12). Because we found reduced phagocytosis and ROS generation by MMP-deficient PMNs, we next investigated the effect of S. pneumoniae on PMN apoptosis. In the absence of S. pneumoniae, WT and Mmp9−/− PMNs showed similar viability in vitro (Supplemental Fig. 5). S. pneumoniae exhibited increased necrosis (annexin V+/propidium iodide [PI]+) and decreased apoptosis (PI−/annexin V+) compared with WT PMNs treated under the same conditions (Fig. 6F, 6G). Thus, these data demonstrate that in addition to the requirement of MMP9 for PMN intracellular killing and ROS generation, MMP9 critically influences the rate at which PMNs enter necrotic and apoptotic pathways in response to bacterial challenge.
Discussion
MMP2 and MMP9 have previously been linked to the regulation of adaptive immune processes through the degradation of extracellular matrix proteins and the proteolytic modification of chemokines. We have now demonstrated that both proteinases inactivate the cytokine IL-17A, a function that is critical for terminating innate immune events such as neutrophilia. We show that mouse PMNs express only MMP9, which is required for both PMN phagocytosis and ROS production against S. pneumoniae. Because of their similarities in substrate specificity, we predicted that the absence of either enzyme would be compensated for when mice were challenged with S. pneumoniae. Consistent with this, we found no significant differences in survival in Mmp2−/− and Mmp9−/− mice exposed to S. pneumoniae. Significantly, impaired survival was only observed in Mmp2/9−/− animals. Decreased survival in Mmp2/9−/− mice infected with S. pneumoniae further coincided with higher numbers of bacteria isolated from the lung and spleen 24 h postinfection. Furthermore, while similar numbers of inflammatory cells were recruited into the lungs of both WT and Mmp2/9−/− mice at an early point (6 h) after intranasal challenge with bacteria, by 24 h bacterial dissemination was apparent in these mice and was accompanied by more pronounced neutrophilia. Together, these findings reveal an essential and unexpectedly complex role for MMP2 and MMP9 in innate host defense.
In our search for the contributing factors that could drive the increase in lung PMN recruitment, we found a significant increase in secretion of IL-17A, an early effector cytokine that is responsible for activation and recruitment of PMNs (35, 36). In response to IL-17A, fibroblasts express GM-CSF, IL-6, and IL-8, which are critical for the proliferation of hematopoietic progenitor cells and the maturation of PMNs (32). It is unclear whether IL-17A expression is linked to the dysregulated MMP expression, but microarray analysis of IL-17A–induced genes has found upregulation of several chemokines such as CXCL1 (KC), CXCL10, CCL2 (MCP-1), CCL7, CCL20, and MMP3 and MMP13 (37, 38). We did not find increase in CXCL1, but many proinflammatory cytokines and chemokines display a dynamic and transient expression during an acute infection; as such, a limitation of our study is that we only report one time point after bacterial challenge, which could have missed the acute changes in chemokine concentration. Similarly, transgenic mice overexpressing IL-17A in the lung over the age of 10 mo exhibit spontaneous airway inflammation and mucus hyperplasia that is associated with increased expression of several chemokines and MMPs (38).
Notably, we found that MMP2 and to a lesser degree MMP9 efficiently cleave and inactivate IL-17A, which we reasoned would be a necessary counter to the potentially deleterious effects, including acute lung injury, of recruited PMNs. We also found that the proteolytic processing of MMP2 was critical for IL-17A function because after cleavage by MMP2, IL-17A less efficiently induced expression of proinflammatory genes in MEFs. Therefore, the increased lung neutrophilia observed in Mmp2/9−/− mice is in part explained by the absence of the immunoregulatory effect of MMP2 and MMP9, but we cannot exclude the possibility that the failure to clear bacteria efficiently also contributed to this finding. Similarly, the early demise of Mmp2/9−/− mice is likely also multifactorial, being attributable to excess PMN recruitment to the lungs and bacterial dissemination.
To address the mechanisms underlying the failure of PMNs to eradicate Gram-positive bacteria in the absence of MMP2 and MMP9, we examined the individual contribution of these proteinases using an in vitro model system. We found that although both MMP2 and MMP9 are upregulated in lung tissue of infected mice, only MMP9 is present in activated PMNs. With regard to essential PMN functions, activated recombinant MMP9 partially rescued the phagocytic defect in MMP9-deficient PMNs. In agreement with our report, Mmp9−/− mice were more susceptible to Escherichia coli infection and showed much higher cytokine and chemokine levels than WT mice under the same conditions; however, in contrast to our findings, phagocytosis of this Gram-negative organism was not impaired in Mmp9−/− PMNs, which points out the differences in PMN function that are governed by the characteristics of the invading pathogens, as further evidenced by the finding that MMP9 activity enhances host susceptibility to pulmonary infection with Francisella tularensis (39, 20).
Most likely, MMP2 and MMP9 are not unique in their capacity to modulate the innate immune responses, and in fact a role for other MMP members has previously been examined. MMP28 (epilysin), a protease that is constitutively expressed in bronchial epithelial cells, can control PMN recruitment into the lung in a model of Pseudomonas aeruginosa pneumonia (40). Acute intranasal challenge of Mmp28−/− mice with bacteria results in an early increase in macrophage recruitment into the lungs and enhanced bacterial clearance (40). Notably, these mice have significantly reduced PMN recruitment into their lungs, a finding that is dissimilar to the response to bacterial infection that we observed in Mmp2/9−/− mice. Further, macrophage depletion in Mmp28−/− mice results in impaired clearance of P. aeruginosa and increased early PMN recruitment to the lung, indicating that MMP28 may be responsible for restraining macrophage recruitment, which, similar to the need for PMNs, is necessary for early defense against invading pathogens. The mechanism of Gram-negative bacterial killing differs greatly from that of Gram-positive organisms. As such, whether Mmp28−/− mice will have the same response to S. pneumoniae remains unknown. Further, whether MMP2 and MMP9 also act to restrain macrophage recruitment in response to Gram-positive infection is less likely as both macrophages and PMNs were increased in infected mice.
Unlike the report of direct killing activity of serine proteinases expressed in the granules of PMNs or MMP12 (macrophage elastase), an enzyme predominately expressed in mature tissue macrophages (13, 33), we found that MMP2 and MMP9 possess no direct bactericidal activity. Our data suggest that much like an indirect effect of MMP7 (matrilysin), an epithelial cell-specific MMP that has been shown to cleave pro–α-defensin into a bactericidally active peptide (41), the combined action of MMP2 and MMP9 may be critical for PMN function to eliminate pathogenic organisms in vivo. We found that MMP9, but not MMP2, aids in efficient bacterial phagocytosis, and consistent with this finding, we found that phagocytosis-mediated induction of ROS is reduced in PMNs isolated from Mmp9−/− mice. Although the mechanism of effective eradication of Gram-positive organisms is thought to be primarily independent of the action of ROS (1), nonetheless, in vivo, most likely contributions from multiple complementary antimicrobial pathways provide a global protection against invading pathogens.
Although PMNs have a relatively short half-life, exposure to bacterial components is known to extend significantly their longevity (9). Notably, we found that infected Mmp9−/− PMNs undergo a more rapid apoptotic death compared with WT infected mice under similar conditions. This reduced half-life could further contribute to the ineffective function of PMNs in S. pneumoniae infection. Our results support the idea that MMP2 and MMP9 serve as anti-inflammatory mediators that specifically attenuate the function of IL-17, which is required to avoid excessive accumulations of PMNs in the lung. We report that MMP9 aids PMNs directly by modulating phagocytosis and possibly through ROS-mediated bacterial killing. Finally, we found that PMNs deficient in MMP9 have decreased survival and undergo bacterial-induced apoptotic cell death.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants HL082487 and HL72419 (to F.K.) and HL075243, AI057696, and AI070973 (to D.B.C.), by an American Heart Association fellowship (to S.-H.L.), by a National Research Service Award training grant (AI053831), and by the Alpha One Foundation.
We thank Dr. James Musser at the Methodist Hospital Research Institute in Houston for comments.
Abbreviations used in this article
- APMA
4-aminophenylmercuric acetate
- BAL
bronchoalveolar lavage
- MEF
mouse embryonic fibroblast
- MMP
matrix metalloproteinase
- NP-40
Nonidet P-40
- PI
propidium iodide
- PMN
polymorphonuclear neutrophil
- ROS
reactive oxygen species
- TSA II
trypticase soy agar with 5% sheep blood agar
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- 2.Silva MT. Neutrophils and macrophages work in concert as inducers and effectors of adaptive immunity against extracellular and intracellular microbial pathogens. J Leukoc Biol. 2010;87:805–813. doi: 10.1189/jlb.1109767. [DOI] [PubMed] [Google Scholar]
- 3.Weiser JN. The pneumococcus: why a commensal misbehaves. J Mol Med. 2010;88:97–102. doi: 10.1007/s00109-009-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang SH, Dong C. IL-17F: regulation, signaling and function in inflammation. Cytokine. 2009;46:7–11. doi: 10.1016/j.cyto.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125:281–288. doi: 10.1111/j.1365-2567.2008.02950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolls JK. Th17 cells in mucosal immunity and tissue inflammation. Semin Immunopathol. 2010;32:1–2. doi: 10.1007/s00281-010-0198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthias KA, Roche AM, Standish AJ, Shchepetov M, Weiser JN. Neutrophil-toxin interactions promote antigen delivery and mucosal clearance of Streptococcus pneumoniae. J Immunol. 2008;180:6246–6254. doi: 10.4049/jimmunol.180.9.6246. [DOI] [PubMed] [Google Scholar]
- 8.Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation. 2009;120:2012–2024. doi: 10.1161/CIRCULATIONAHA.108.771170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soehnlein O, Weber C, Lindbom L. Neutrophil granule proteins tune monocytic cell function. Trends Immunol. 2009;30:538–546. doi: 10.1016/j.it.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 13.Standish AJ, Weiser JN. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol. 2009;183:2602–2609. doi: 10.4049/jimmunol.0900688. [DOI] [PubMed] [Google Scholar]
- 14.Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;87:69–98. doi: 10.1152/physrev.00022.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, Overall CM. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 16.McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 17.Greenlee KJ, Corry DB, Engler DA, Matsunami RK, Tessier P, Cook RG, Werb Z, Kheradmand F. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol. 2006;177:7312–7321. doi: 10.4049/jimmunol.177.10.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- 19.Van Den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J, Opdenakker G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur J Biochem. 2003;270:3739–3749. doi: 10.1046/j.1432-1033.2003.03760.x. [DOI] [PubMed] [Google Scholar]
- 20.Renckens R, Roelofs JJTH, Florquin S, de Vos AF, Lijnen HR, van’t Veer C, van der Poll T. Matrix metalloproteinase-9 deficiency impairs host defense against abdominal sepsis. J Immunol. 2006;176:3735–3741. doi: 10.4049/jimmunol.176.6.3735. [DOI] [PubMed] [Google Scholar]
- 21.Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, Werb Z, Kheradmand F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aaberge IS, Eng J, Lermark G, Løvik M. Virulence of Streptococcus pneumoniae in mice: a standardized method for preparation and frozen storage of the experimental bacterial inoculum. Microb Pathog. 1995;18:141–152. doi: 10.1016/s0882-4010(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 23.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001;293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- 24.Park PW, Pier GB, Hinkes MT, Bernfield M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature. 2001;411:98–102. doi: 10.1038/35075100. [DOI] [PubMed] [Google Scholar]
- 25.Bubeck Wardenburg J, Patel RJ, Schneewind O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect Immun. 2007;75:1040–1044. doi: 10.1128/IAI.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corry DB, Rishi K, Kanellis J, Kiss A, Song LZ, Xu LzJ, Feng L, Werb Z, Kheradmand F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo Y, Dorf ME. Isolation of mouse neutrophils. Curr Protoc Immunol. 2001 May;Chapter 3(Unit 3.20) doi: 10.1002/0471142735.im0320s22. [DOI] [PubMed] [Google Scholar]
- 28.Clemens RA, Newbrough SA, Chung EY, Gheith S, Singer AL, Koretzky GA, Peterson EJ. PRAM-1 is required for optimal integrin-dependent neutrophil function. Mol Cell Biol. 2004;24:10923–10932. doi: 10.1128/MCB.24.24.10923-10932.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- 30.Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 31.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 32.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 33.Houghton AM, Hartzell WO, Robbins CS, Gomis-Rüth FX, Shapiro SD. Macrophage elastase kills bacteria within murine macrophages. Nature. 2009;460:637–641. doi: 10.1038/nature08181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest. 2009;119:1899–1909. doi: 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007;17:435–440. doi: 10.1038/cr.2007.35. [DOI] [PubMed] [Google Scholar]
- 36.Kolls JK, McCray PB, Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. 2008;8:829–835. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J Leukoc Biol. 2005;77:388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 38.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malik M, Bakshi CS, McCabe K, Catlett SV, Shah A, Singh R, Jackson PL, Gaggar A, Metzger DW, Melendez JA, et al. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J Immunol. 2007;178:1013–1020. doi: 10.4049/jimmunol.178.2.1013. [DOI] [PubMed] [Google Scholar]
- 40.Manicone AM, Birkland TP, Lin M, Betsuyaku T, van Rooijen N, Lohi J, Keski-Oja J, Wang Y, Skerrett SJ, Parks WC. Epilysin (MMP-28) restrains early macrophage recruitment in Pseudomonas aeruginosa pneumonia. J Immunol. 2009;182:3866–3876. doi: 10.4049/jimmunol.0713949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, López-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.