

Abstract
The catalytic subunit of cAMP-dependent protein kinase A (PKA-C) is an exquisite example of a single molecule allosteric enzyme, where classical and modern views of allosteric signaling merge. In this chapter, we describe the mapping of PKA-C conformational dynamics and allosteric signaling in the free and bound states using a combination of NMR spectroscopy and molecular dynamics simulations. We show that ligand binding affects the enzyme’s conformational dynamics, shaping the free-energy landscape toward the next stage of the catalytic cycle. While nucleotide and substrate binding enhance the enzyme’s conformational entropy and define dynamically committed states, inhibitor binding attenuates the internal dynamics in favor of enthalpic interactions and delineates dynamically quenched states. These studies support a central role of conformational dynamics in many aspects of enzymatic turnover and suggest future avenues for controlling enzymatic function.
I. Introduction
Protein phosphorylation is a central signaling event in eukaryotes and is orchestrated by protein kinases that catalyze the transfer of the γ-phosphoryl group of a nucleoside triposphate (usually adenosine triphosphate, ATP) to an amino acid hydroxyl group (commonly serine, threonine, or tyrosine) of a protein substrate. The protein kinase family makes up ~2000 enzymes in humans, and ~30% of all proteins encoded by the human genome undergo reversible phosphorylation (Cohen, 2001). Since aberrant phosphorylation plays a role in many diseases, from cancer to cardiovascular disease, a complete elucidation of the molecular mechanisms that regulate reversible phosphorylation would have a major impact on designing innovative treatments (Cohen, 1999, 2002).
Protein kinase A (PKA, EC: 2.7.1.37) was the first protein kinase ever to be studied (Walsh, et al., 1968). PKA exists as a holoenzyme of two regulatory subunits and two catalytic subunits. An allosteric conformational change triggered by 3′,5′-cyclic adenosine monophosphate causes the regulatory subunits to release active catalytic subunits and phosphorylate protein substrates. The catalytic core of this enzyme (PKA-C, 350 residues, Fig. 1A) is highly conserved throughout the protein kinase superfamily and has thus been used as a model enzyme for the other members (Johnson, et al., 2001). PKA-C recognizes the amino acid sequence R-R-X-S/T-Y (where X is any residue and Y is a hydrophobic residue) present in hundreds of cellular substrates (Boeshans et al., 1999; Shabb, 2001). PKA-C undergoes several co- and posttranslational modifications such as deamidation and phosphorylation at the N-terminus (Johnson, et al., 2001), and myristoylation at the N-terminal glycine (Fig. 1A; Taylor et al., 2004). In addition to catalyzing phosphoryl transfer, PKA-C also serves as a hub for several interacting partners that constitute complex signaling machinery (Taylor et al., 2004). It has been hypothesized that co- and posttranslational modifications of PKA-C may constitute chemical signals for substrate recognition and localization within the cell, allowing this ubiquitous enzyme to select specific substrates (Taylor et al., 2004).
Fig. 1.

Architecture of PKA-C. (A) Secondary structure, domains, conserved motifs (see asterisks), and ligand-binding regions of PKA-C. (B) Conformational changes observed in PKA-C by X-ray crystallography. The major conformational states are defined by the angle between the large and small lobes and by the S53/G186Cα distance (open, intermediate, and closed conformational states). The average angles and distances are calculated from open (PDB: 3O7L, 1CMK, 1CTP, 1J3H, and 2QVS), intermediate (PDB: 1BKX, 1BX6, 1STC, 1JLU, 1RE8, 1REK, 3DND, 3DNE, 3IDB, and 3IDC), and closed (PDB: 1JBP, 1ATP, 1APM, 1YDS, 1YDR, and 1YDT) X-ray structures.
In spite of the plethora of structural and functional studies on this kinase, several key mechanistic questions on phosphorylation remain unanswered: What governs substrate recognition? What determines substrate specificity? What are the driving forces for substrate release? How can PKA-C recognize soluble and membrane bound substrates? Answering these central questions will help our understanding of the etiology of diseases such as cancer, diabetes, rheumatoid arthritis, and various cardiomyopathies (Cohen, 2001).
X-ray crystallography has offered an essential contribution to understanding the structural architecture of protein kinases. The pioneering work of Taylor and coworkers resulted in the first atomic resolution structure of PKA-C in the inhibited state (Zheng et al., 1993). Following this study, several crystal structures were obtained, which mimicked major conformational states of the enzyme along the reaction coordinates, by using inhibitors, pseudosubstrates, and nucleotide analogs (Johnson, et al., 2001; Taylor et al., 2004). Based on these structures and kinetic data on mutant forms of PKA-C, Taylor and coworkers hypothesized the existence of a sophisticated intramolecular allosteric network that controls substrate recognition, chemical step, and product release (Kornev et al., 2008). Although supported by spectroscopic and computational studies, the allosteric network of communication had never been probed experimentally until recently (Yang et al., 2005, 2009; Kornev et al., 2006; Masterson et al., 2008, 2010; Hyeon et al., 2009; Kornev and Taylor, 2009, 2010; Karginov et al., 2010; Gaffarogullari et al., 2011).
Our group has studied the structural dynamics of PKA-C using liquid-state NMR spectroscopy with the objective of mapping the allosteric regulation of the kinase along the catalytic cycle (Masterson et al., 2008, 2010, 2011a,b; Gaffarogullari et al., 2011; Ha et al., 2011). This chapter summarizes these studies, highlighting our most recent progress in connecting conformational dynamics (molecular motion) of the kinase with catalysis, defined here as the sum of three main steps: substrate recognition, chemical step, and product release. Using a combination of NMR studies and molecular dynamics (MD) simulations, we attempted to trace the conformational and dynamic states of the enzyme. We found that ligand binding affects the conformational equilibrium of the enzyme and defines the different dynamical states that regulate kinase activity: committed, uncommitted, and quenched states (Masterson et al., 2010, 2011a,b). Our studies emphasize the importance of both conformational and dynamic changes for allostery to take place.
II. Major Conformational States Identified by X-Ray Crystallography
A wealth of crystallographic data is available on PKA-C, which is a direct result of the ability to express this enzyme as a recombinant protein in Escherichia coli (Slice, L.W. and Taylor S.S., 1989). Well over a dozen structures have been deposited for PKA-C in the protein data bank (PDB) (Johnson, et al., 2001). The fold of PKA-C is shared throughout the protein kinase family (Johnson, et al., 2001; Fig. 1A,B). The enzyme is bean shaped, comprising a small lobe, containing mainly β-sheets, and a mostly helical large lobe. The small lobe includes the nucleotide binding site, glycine-rich loop, β-strand 3, and the C-helix (Johnson, et al., 2001; Taylor et al., 2004). The large lobe contains docking sites for substrates, regulatory subunits, and colocalization elements such as A-kinase anchoring proteins (Wong and Scott, 2004). Key motifs such as the catalytic, DFG, and peptide-positioning loops are situated at an active site cleft formed between the two lobes, where phosphoryl transfer takes place (Fig. 1B; Johnson, et al., 2001; Taylor et al., 2004). The crystallographic data also provided snapshots for the arrangement of the functional groups involved in the phosphorylation process, identifying the position of the γ-phosphate of ATP and the acceptor hydroxyl group of the phosphorylatable serine in the substrate (Taylor et al., 2004). These static representations of the enzyme, along with fluorescence anisotropy data, suggested that the enzyme toggles between three major conformational states: an open state (typically observed with no ligand bound), an intermediate state (typically observed with a singly bound ligand), and a closed state (observed in the ternary complexes) (Taylor et al., 2004; Fig. 1B). Because significant conformational changes are observed upon nucleotide binding, it has been hypothesized that the allosteric signal initiated upon nucleotide binding travels via the hydrophobic core from the small to the large lobe (Kornev et al., 2008). Structural changes are also observed for regions peripheral to the active site cleft. Based on these observations and computational studies, it has been proposed that two hydrophobic spines buried in the catalytic core may work as a transmission line for intramolecular allosteric signaling, thus modulating binding interactions and catalysis (Kornev et al., 2006, 2008; Kornev and Taylor, 2009, 2010). Analyzing how the signal is transmitted within the catalytic core is central to understanding enzyme function and regulation. To this extent, modern NMR spectroscopy methods provide atomic views of short- and long-range conformational and dynamic changes, offering a unique perspective on allostery (Kern and Zuiderweg, 2003; Boehr et al., 2006; Tzeng and Kalodimos, 2009; Kalodimos, 2011).
III. NMR Analysis of Ligand Binding Shows Positive Allosteric Cooperativity in PKA-C
A stepping stone for carrying out NMR studies is the optimization of functional samples that also behave well spectroscopically (i.e., provide well-resolved fingerprint resonances). Under our experimental conditions, PKA-C is functional as established by 31P NMR, with kinetics similar to those measured by coupled enzyme assays (Masterson et al., 2008). The assignment of the backbone resonances, however, was far from trivial. The line shapes of the amide fingerprint in the [1H,15N]-TROSY-HSQC spectra are inhomogeneous, due to intermediate exchange broadening in the chemical shift time scale. In addition to the classical triple resonance experiments, a combination of selectively labeled samples and new spectral-editing techniques (i.e., CCLS-HSQC (Tonelli et al., 2007)) was necessary to attain ~83% of the total backbone resonance assignments (Masterson et al., 2009). All of the key regions were identified in the enzyme fingerprint, which enabled us to trace the changes in the backbone amide resonances upon ligand binding. The NMR titrations carried out with 5′-adenylylimido-diphosphate (AMP-PNP) and a standard kinase substrate, Kemptide (LRRASLG), allowed us to map the effects of the ligands on both wild-type PKA-C (PKA-CWT) and PKA-CY204A, a mutant with a severely impaired catalytic efficiency (Fig. 2; Moore et al., 2003; Yang et al., 2005; Masterson et al., 2008). Since the formation of the ternary complex occurs via random sequential order (Kong and Cook, 1988; Adams, 2001), we performed the NMR titrations by adding AMP-PNP first followed by Kemp-tide and then reversed the order of ligands. The on-rate for nucleotide binding was approximately two orders of magnitude slower than the diffusion rate, suggesting that binding is likely to occur via structural rearrangements. These conformational changes are necessary for proper alignment and positioning of the nucleotide and substrate, along with shielding the active site from water. The PKA-CWT ligand-binding experiments revealed a positive K-type cooperativity (Table I), with dissociation constants similar to those reported previously (Taylor et al., 2004). Although weak (~fourfold), the K-type cooperativity is mutual between substrate and nucleotide: binding of the nucleotide enhances the substrate affinity, and substrate binding enhances the affinity for the nucleotide. Interestingly, titrations with the catalytically impaired mutant PKA-CY204A showed that the ligand-binding events are decoupled, that is, there is no binding cooperativity. The chemical shift perturbations observed in the enzyme fingerprint spectra revealed the presence of local and long-range (allosteric) changes in PKA-CWT. Based on the X-ray structures, these changes occur as far as ~20 Å from the ATP-binding site, including the N- and C-terminal regions of the small lobe and residues near the peptide-positioning loop of the large lobe (Fig. 2). Interestingly, the chemical shift changes for PKA-CY204A were substantially smaller than for the PKA-CWT. A few long-range perturbations were detected in the large lobe, although with a different pattern (Fig. 2), and substrate binding resulted in very small changes with no detectable allosteric effects. Overall, AMP-PNP binding did not produce the allosteric changes observed for PKA-CWT. Structurally, PKA-CY204A is almost superimposable to PKA-CWT as assessed by X-ray crystallography (Yang et al., 2004). However, the deletion of a key hydrogen bond between the hydroxyl group of Y204 and E230 of the F-helix in the mutant disrupts the interaction network at the interface of the N- and C-lobes, as well as the interaction with the P-2 site of the substrate (Moore et al., 2003). The NMR fingerprints of the wild-type and the mutant were nearly superimposable. However, the elimination of these key interactions dismantles the allosteric communication (i.e., no long-range effects were observed) and disrupts the K-type cooperativity between nucleotide and substrate observed for PKA-CWT.
Fig. 2.

Chemical perturbations upon ligand binding for PKA-CWT and PKA-CY204A. The binding of nucleotide and substrate indicates attenuated local and long-range (allosteric) perturbations for PKA-CY204A.
Table I.
Binding Constants (Kd, in units of micromolar) Measured from the NMR Titrations of PKA-C
| AMP-PNP first
|
Kemptide first
|
||||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||
| PKA-CWT | 39 ± 6 | 292 ± 61 | 980 ± 163 | 12 ± 3 | |||
| PKA-CY204A | 78 ± 20 | 834 ± 92 | 700 ± 105 | 68 ± 4 | |||
From these measurements, two spectroscopic features were prominent. First, the extent of chemical shift changes (Δδ) upon ligand binding was exiguous (~0.03 ppm), which was unexpected based on the structural differences observed in the X-ray structures (Johnson, et al., 2001; Taylor et al., 2004). The latter may be due to dynamic averaging of the backbone chemical shifts caused by the conformational equilibrium of the kinase in solution. Second, a number of resonances from highly conserved regions (glycine-rich, activation, and peptide-positioning loops) became exchanged broadened upon nucleotide binding, indicating that the enzyme is interconverting between at least two different conformational states in the microsecond to millisecond time scale. Although our initial NMR analysis was limited to chemical shift changes and warranted further investigation, we could infer that conformational dynamics played a key role in PKA-C function.
IV. Ligand Binding and Enzyme Motion
A. Thermodynamics and Kinetics of Ligand Binding
To understand the recognition process of the kinase, we compared the binding of the endogenous inhibitor of the kinase, protein kinase inhibitor (PKI), with that of a physiological substrate of PKA-C, phospholamban (PLN). PKI controls PKA-C function and cellular localization (Walsh and Van Patten, 1994; Dalton and Dewey, 2006) acting as a physiological break to enzyme activity. PKI’s recognition sequence occupies the active site and prevents phosphorylation of cellular targets. In contrast, PLN is a substrate of PKA-C and is reversibly phosphorylated during the heart muscle contraction-relaxation cycle (Traaseth et al., 2008). In both studies, we utilized either synthetic or recombinant peptides, each corresponding to either the 5–24 fragment of PKI (PKI5–24) or the 1–20 fragment of PLN (PLN1–20). Steady-state kinetic data showed that the PKA-C catalytic efficiency for the PLN1–20 peptide was comparable to that of full-length PLN (Masterson et al., 2010, 2011a,b) and had kinetics similar to Kemptide (Masterson et al., 2010, 2011a,b). The K-type cooperativity of PLN1–20 was slightly higher than that of Kemptide, with a fivefold increase in binding affinity in the presence of nucleotide. In contrast, PKI5–24 binding affinity was significantly higher (Kd ~ 2 μM for the apo enzyme). The latter was accompanied by ~40-fold increase in binding affinity when the nucleotide was present (Fig. 3; Masterson et al., 2011a,b). Thermocalorimetric measurements showed that while the binding of the PLN1–20 peptide substrate is entropically driven, the binding of the PKI5–24 peptide inhibitor binding is enthalpically driven. A substantial increase in the overall enzyme thermostability was observed upon binding PKI5–24, whereas the nucleotide-bound form, or nucleotide and substrate bound form, caused only a negligible increase. Stabilization effects were also present upon addition of excess amounts of Mg2+ ions. Under these conditions, Mg2+ behaves as a noncompetitive inhibitor (Cook et al., 1982; Adams and Taylor, 1993; Herberg et al., 1999; Zimmermann et al., 2008) and two Mg2+ ions bind the nucleotide binding cleft. The kinetic effects of the competitive (PKI5–24) and noncompetitive (secondary Mg2+ ion) inhibitors were synergistic, with an overall decrease of Kd to ~ 12 nM. The latter is in agreement with the additive changes observed for the two inhibitors by thermal melting curves (Zimmermann et al., 2008; Masterson et al., 2011a,b). From these data, it is possible to extrapolate that as the enzyme stability increases, reactivity decreases.
Fig. 3.

Thermocalorimetric parameters for the binding of the substrate PLN1–20 and inhibitor PKI5–24 to PKA-C. (A) Binding energy for substrate and inhibitor to the apo or nucleotide-bound PKA-C. PLN1–20 is entropy driven, while PKI5–24 binding is enthalpy driven. (B) Thermal melting of PKA-C complexes. A small increase in thermal stability (relative to the apo enzyme melting temperature, ΔT) is present upon nucleotide or substrate binding under low and high Mg2+ concentration. A substantial increase in stability is measured in the presence of PKI5–24 and high Mg2+.
B. X-Ray Structures of PKA-C in Complex with Substrate and Inhibitors
The crystal structures of the ternary complexes of PKA-C with AMP-PNP and PLN1–19 or PKI5–24 displayed striking differences between the substrate- and inhibitor-bound complexes (Zheng et al., 1993; Masterson et al., 2010). In spite of excess amounts of substrate and nucleotide, the PKA-C/AMP-PNP/PLN1–19 complex crystallized with two enzyme molecules in the asymmetric unit: an enzyme in a closed conformation bound to both ligands and an apo form enzyme in an open conformation. Interestingly, the apo form had lower B-factors than those in the ternary complex and, with the exception of the first few residues at the N-terminus, showed a well-defined electron density for all regions of the enzyme. On the other hand, the electron density of the complex with the substrate was undefined for the highly conserved glycine-rich loop and part of the C-terminal tail, which is believed to stabilize interactions with the substrate. These regions were observed in the crystal structures formed with PKI5–24 (Johnson, et al., 2001; Taylor et al., 2004), in which the inhibitor likely traps these regions into a stable conformational minimum. Qualitatively, there is an agreement between the thermodynamic data and the structural features observed in the crystals of the substrate versus the inhibitor; when the enthalpic contribution is dominant, the structure is more resolved (i.e., lower B-factors), whereas when the entropic contribution is dominant, static and dynamic disorder decrease the resolution (i.e., higher B-factors).
C. Dynamic States of PKA-C Define Catalysis and Inhibition
To follow the conformational changes along the enzymatic coordinates, we analyzed five different forms of PKA-C: (1) apo enzyme, (2) binary form (AMP-PNP bound), (3) ternary complex with low Mg2+ bound to PLN1–20 (which mimics the productive complex), (4) a ternary complex bound to PKI5–24 with low Mg2+ (nonproductive complex), and (5) a ternary complex bound to PKI5–24 with high Mg2+ (super-inhibited, nonproductive complex) (Masterson et al., 2011a,b). The overall trend in chemical shift changes in the enzyme fingerprint correlates qualitatively with the displacement of Cα atoms observed by X-ray crystallography for the apo, binary and ternary forms. Therefore, in agreement with the crystal structures (Fig. 1B), only minimal changes occur in the overall fold for the transitions from nucleotide-bound binary form to the ternary complexes, and the average conformation of the ternary complex with peptide inhibitor closely resembles the ternary complex with substrate.
Although small, the chemical shift changes affect the catalytically important regions of the enzyme that are conserved throughout all protein kinases, including the glycine-rich, activation, DFG, and peptide-positioning loops (Fig. 4). Many of the changes in the amide fingerprint followed linear progressions from the apo, to the intermediate, to the closed super-inhibited ternary complex (PKI5–24 plus two Mg2+, Fig. 4A). The linearity of chemical shifts revealed that the apo form exists as an ensemble of interchanging conformers (conformational equilibrium) that includes all of the different states of the kinase. Ligand binding acts on the enzyme equilibrium, shifting the populations of the conformational ensemble. Nucleotide binding shifts the conformational ensemble of open toward the intermediate state, and substrate or inhibitor peptide binding skews the population toward the closed state. To corroborate these data, we carried out NMR relaxation measurements, which are able to image ground and excited states of proteins (Palmer, 2001; Boehr et al., 2006, 2009; Popovych et al., 2006; Berlow et al., 2007; Henzler-Wildman et al., 2007; Loria et al., 2008; Baldwin and Kay, 2009; Korzhnev et al., 2010; Mittag et al., 2010; Villali and Kern, 2010; Kalodimos, 2011). In agreement with previous spectroscopic data (Taylor et al., 2004), our results show that the enzyme becomes more compact upon ligand binding. Small but significant differences in stability are observed between the ternary complexes, with the most stable complex (inhibited enzyme under high Mg2+) showing that the most compact structure has the shortest overall correlation time. These results follow the same trend as the thermocalorimetric data. The apo form of the enzyme displayed the expected flexibility in the nanosecond to picosecond (fast motion in the NMR time scale), with high mobility of residues in regions between structural elements such as the acidic cluster (residues 328–334) and the glycine-rich, catalytic, DFG, activation, and peptide-positioning loops. Surprisingly, using the conventional experiments for spin relaxation (Farrow et al., 1994), we could not detect microsecond to millisecond motions (slow motion in the NMR time scale) in the apo form. It should be noted, however, that the current NMR techniques are sensitive to a specific window of conformational dynamics within the microsecond to millisecond time scale and we cannot rule out any motions outside this range (Jarymowycz and Stone, 2006). Remarkably, upon nucleotide binding, we detected a noticeable increase in conformational dynamics, on both fast and slow time scales, for several key regions of the enzyme (Fig. 5). This was most apparent for the glycine-rich and the DFG loops, which are directly involved in nucleotide binding, as well as the activation loop, the peptide-positioning loop, and the acidic cluster (Fig. 6). Overall, the conformational dynamics of residues surrounding the active site were found to increase upon nucleotide binding. A diagnostic parameter of the conformational exchange is the relaxation rate of exchange (Rex), which quantifies motion in the microsecond to millisecond time scale (Palmer, 2001). We found Rex values greater than 25 Hz for several residues located in regions involved in catalysis. Although attenuated and interspersed, these conformational dynamics persist when PLN1–20 is bound. The latter indicates that conformational dynamics persists in the ternary complex and might be required for opening the active site and product release (Fig. 6). In fact, the linear correlation observed between Rex and Δω2, where Δω is the difference in chemical shifts between free and bound enzyme, confirms that several residues experiencing slow motions undergo synchronous conformational dynamics (i.e., opening and closing) (Massi et al., 2006). From these data, we estimated an average rate constant for opening the active site cleft of ~ 20–30 s−1, which agrees remarkably well with the catalytic constant (kcat ~ 23 s−1) (Adams and Taylor, 1992; Lew et al., 1997). Taken together, these data support that a global conformational change is the rate-determining step for enzyme turnover.
Fig. 4.

Linearity of chemical changes among different forms of PKA-C. (A) Linearly correlated chemical shift changes in different regions of the small and large lobes. The two extremes of the correlations are the apo and the super-inhibited ternary complex of PKA-C, suggesting a fast conformational exchange between closed and open conformational states. (B) The location of these representative residues included regions near the active site and allosteric sites as far as 20 Å (W302) away.
Fig. 5.

Free-energy landscape of PKA-C and ligand binding. Ligand binding defines the conformational equilibrium of the kinase, skewing the populations in a dynamically uncommitted (apo form) basin with motions not synchronous to turnover, dynamically committed basin (nucleotide-bound and ternary complex with substrate) with motions synchronous with turnover, and dynamically quenched basin (inhibited complexes) with attenuated motions and low free energy.
Fig. 6.
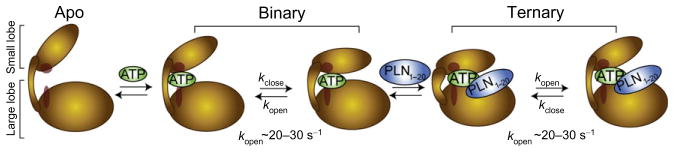
Schematic for the formation of the catalytically competent ternary complex. The apo enzyme constitutes a dynamically uncommitted state, with its C-spine (red) disengaged between the two lobes. The nucleotide acts as an allosteric effector which is sensed throughout the enzyme. This binding event completes the C-spine architecture and induces a dynamically committed state. Fluctuations between open and closed conformations take place at a time scale which is synchronous with turnover. These fluctuations persist in the ternary complex with substrate, limiting the rate of catalytic turnover.
From the above data, it is possible to speculate that the apo form of PKA-C experiences conformational dynamics outside the microsecond to millisecond time scale. This motion is not productive, that is, not synchronous with the opening and closing frequency necessary for catalysis. Nucleotide binding dynamically activates the enzyme, completing the C-spine architecture and engaging the small and large lobes (Kornev et al., 2006, 2008; Kornev and Taylor, 2009, 2010). The adenine group acts as a pivot point between the two major enzyme domains (Kornev and Taylor, 2009) and activates the microsecond to millisecond conformational dynamics necessary for catalysis to occur (Fig. 6). Changes in conformation and dynamics upon nucleotide binding prime the active site for catalysis; that is, while the active site may be preorganized for the chemical step, these internal motions initiated by the nucleotide may contribute to surmounting the free-energy barrier for phosphoryl transfer to take place (Nashine et al., 2010). Although direct experimental proof of the role of dynamics in the chemical step is lacking, it has been shown that several mutations that disrupt the C-spine region lead to a significant decrease in catalytic rate (Kornev and Taylor, 2009). The latter strongly supports the hypothesis that the C-spine is a necessary element for allosteric dynamic transmission of the signals from the small to the large lobe. The opening and closing of the active cleft persists in the ternary complex with the substrate. These motions are probably responsible for product release, enabling opening of the active site cleft after phosphoryl transfer and allowing the exit of ADP and the phosphorylated product.
This process described by the NMR data epitomizes a conformational selection mechanism (Csermely et al., 2010). Nucleotide binding skews the preexisting equilibrium toward the next step of the enzymatic cycle, increasing the enzyme affinity for the substrate (positive allosteric cooperativity) and enhancing substrate recognition (Figs. 3 and 5).
We also mimicked the inhibited and super-inhibited forms of the enzyme with ternary complexes of PKA-C bound to PKI5–24 under low and high Mg2+ concentrations, respectively. PKI5–24 binding to the enzyme quenched both fast and slow motions throughout the entire enzyme backbone, trapping it in a dynamically inert or quenched state (Fig. 5). The attenuation of the conformational dynamics is even more apparent when two Mg2+ ions are bound in the nucleotide binding cleft. In the presence of substrate, the kinase preserved most of the flexibility in the proximity of the enzyme active site. In contrast, the presence of the two inhibitors reduced these pervasive dynamics to a handful of residues located in the conserved loops. The linear chemical shifts progressed toward the closed state, suggesting a shift of the enzyme conformational ensemble toward a well-defined basin with quenched dynamics and unable to carry out catalysis (Fig. 4A).
D. Interpretation of Molecular Motions by MD Calculations
Although NMR can detect the rates of conformational motions, it cannot interpret how these motions occur. Therefore, we carried out MD simulations in explicit solvent with the different forms of PKA-C: apo, nucleotide-bound (with one or two bound Mg2+ ions), and ternary complexes with either substrate (PLN1–20) or inhibitor (PKI5–24 with one or two bound Mg2+ ions) (Masterson et al., 2011a,b). The analysis of the MD trajectories showed that the enzyme regions with the largest root mean square fluctuations (RMSF) coincide with the trends observed in the heteronuclear NOE measurements for both nucleotide-bound or ternary complex (Masterson et al., 2010, 2011a,b). Specifically, a good agreement between simulated and experimentally determined conformational dynamics was observed for the glycine-rich, activation, DFG, and peptide-positioning loops. Moreover, we observed marked differences between the motions experienced by the ternary complexes of PKA-C with nucleotide and substrate (PLN1–20) and those corresponding to the complex with nucleotide and inhibitor (PKI5–24). In particular, the RMSF for the complex with the substrate are on average higher than those with the inhibitors (Fig. 7A). Compared to the complex with PKI5–24, the ternary complex with the substrate displays significantly higher RMSF values for the glycine-rich loop, β3, B helix, the loop connecting the F and G helices, as well as the C-terminal region. On average, the RMSF values are higher for all of the catalytically important loops. Also, substantially higher RMSF values are obtained for both the nucleotide and the substrate in their respective binding sites. In sharp contrast, the average fluctuations of the inhibited complex are rather attenuated (Fig. 7D–F). The latter is apparent from the perspective of the enzyme, the inhibitor, and the nucleotide. Qualitatively, these data follow the same trend observed by the thermocalorimetric measurements as well as the NMR relaxation data. The analysis of intermolecular hydrogen bond and electrostatic interactions between the enzyme and bound peptides revealed strong interactions between the PKI5–24 (from the P+1 through P−6 sites) and several residues constituting the binding groove. In contrast, a lower occurrence of these types of interactions is detected for the substrate during the MD simulations (Fig. 7B–C). These results are in agreement with the enthalpy- and entropy-driven binding modes for inhibitor and substrate, respectively, determined by isothermal titration calorimetry.
Fig. 7.

MD simulations of PKA-C ternary complexes with PLN1–20 or PKI5–24. All plots indicate ternary complexes with PLN1–20 (red) or PKI5–24 (black). (A) Backbone sub-nanosecond RMSF of PKA-C ternary complexes. Strong (red) and weak (green) hydrogen bonding networks which persisted during the simulations when PKI5–24 (B) or PLN1–20 (C) was present. (D) RMSF of the peptide backbone of PLN1–20 or PKI5–24. (E) The structure and atom number of ATP, and (F) RMSF of ATP during simulations with the substrate or inhibitor.
The large amplitude motions were projected using principal component analysis (PCA) (Amadei, A. et al., 1993; Miyashita et al., 2003; Maragakis and Karplus, 2005; Henzler-Wildman et al., 2007). We found that the first two principal components accounted for ~ 60% of the variance in coordinates during the MD simulations (Fig. 8A). The first principal component (PC1) corresponded to the predicted opening and closing motion between the small and large lobes. The second component (PC2) corresponded to a shearing motion between these two lobes. This motion is favored by the greasy interface between the two major domains, conferring to the enzyme an “asymmetric bite” as monitored during MD simulations. A common parameter used in X-ray crystallography for the identification of the open and closed conformations is the distance between Ser53 and Gly186 (dS53–G186, Fig. 8B) in PKA-C, which was used to probe active site accessibility during the MD simulations. We combined PC1 with dS53–G186 to describe the relative motion of the two lobes. Based on 29 X-ray crystallographic structures, these two parameters are diagnostic of the kinase conformational state (Fig. 8C). The 2D plots reported in Fig. 8B display well-separated clusters, corresponding to the open, intermediate, and closed conformations. Among the different forms, the apo enzyme sampled a broad distribution of conformations that spanned all of the states: open, intermediate, and closed. The binary form with one Mg2+ ion bound had a distribution of conformations that sampled open and intermediates states, while addition of the substrate shifted the ensemble toward the closed state. The second Mg2+ ion narrowed the minima of the conformational landscape explored by the enzyme for both binary and ternary complexes. Interestingly, the simulations with PKI5–24 and two Mg2+ ions (super-inhibited complex) showed the narrowest conformational distribution, which converged toward the closed conformations identified by X-ray structures. Collectively, the three major conformational states hypothesized by the X-ray data correspond to well-defined dynamic states of the enzyme. Together with the NMR data, we can give a qualitative description of the energy landscape of the enzyme (Fig. 6), assigning the apo enzyme to a dynamically uncommitted state located in a broad basin, where motions are present but not on a timescale relevant for turnover; and to the nucleotide-bound or Michaelis complex with the substrate, a dynamically committed state, with a more defined basin and where motions are synchronous with the rate-limiting step of enzyme turnover; and finally to the inhibited and super-inhibited complexes a dynamically quenched state with a basin where the enzyme dynamics are hindered (Fig. 8). The latter state is more populated when two Mg2+ are bound, and conformational dynamics are substantially attenuated. Previous H/D exchange (Yang et al., 2005) and fluorescence measurements (Li et al., 2002) are consistent with our definitions of the energy landscape.
Fig. 8.

MD simulations of PKA-C. (A) PCA analysis of the MD trajectories indicated two orthogonal motions between the small and large lobes which described ~50% of the variance: an opening/closing motion (PC1) and a shearing between the lobes (PC2). (B) The occlusion of the active site was probed using the distance between S53 and G186 Cα atoms and monitored with PC1 to assess active site accessibility. (C) 2D plots indicated that inhibitors restrict the conformational space accessible by PKA-C, while the other forms accessed these states more frequently.
E. Mutual Conformational Selection Mechanism for Enzyme and Substrate
Studies of full-length PLN revealed a preexisting equilibrium between four major conformational states (Gustavsson et al., 2011) driven by the reversible binding of cytoplasmic domain Ia to membranes (Fig. 9): a highly populated T state (L-shaped configuration), with domain Ia adsorbed on the membrane surface; a T′ state, with domain Ib partially unfolded; an R′ state that is partially unfolded at the N-terminus and in the C-terminal part of domain Ia; and an R state, which has domain Ia and Ib unfolded. The T′, R′, and R states are less populated and represent excited states, with the T and R states representing two extremes of conformational dynamics in PLN. Kinetic and structural measurements using a series of PLN analogues showed a clear correlation between phosphorylation efficiency with the extent of R state (Masterson et al., 2011a,b). We found that in the ground state (T state), PLN is less accessible to the protein kinase, and this decreases the catalytic efficiency of phosphorylation. By shifting the conformational equilibrium toward the R state, PKA-C catalytic efficiency increases. A plausible explanation is that the enzyme binds to the R state more readily and selects the extended conformation for phosphoryl transfer to occur. NMR and sequence homology-driven molecular docking provided a view of the structural ensemble of the complex between PKA-C and PLN1–20 (Masterson et al., 2011a,b). These results corroborate the kinetic preference of the membrane detached and extended conformation (R state) of PLN. The structural ensemble of the complex agrees with the nuclear spin relaxation data. The substrate remains quite dynamic when bound in the active site, with the exception of the recognition sequence (Masterson et al., 2010, 2011a,b). On the other hand, PLN binding shifts the PKA-C conformational equilibrium toward the closed state of the enzyme (Masterson et al., 2010, 2011a,b). The energy landscape for both PLN and PKA-C affects one another, leading to mutual adaptation. This recognition process exemplifies the recently theorized mutual conformational selection model reported by Nussinov and coworkers (Csermely et al., 2010; Fig. 9). During the encounter (i.e., recognition process), the two binding partners affect each other’s energy landscape with a mutual adjustment in conformations. The binding process causes both proteins to reach a minimum in the free-energy landscape, allowing proper positioning of the chemical groups for catalysis to occur.
Fig. 9.

Proposed mutual conformational selection mechanism between PKA-C and PLN. Both PKA-C and PLN undergo preexisting equilibria between major conformational states. PKA-C fluctuates between open and closed conformations, induced by nucleotide binding. PLN undergoes conformational fluctuations between various degrees of folded and unfolded conformations. These equilibria influence one another upon interaction, leading to a mutual adaptation and allowing the proteins to reach a minimum in the free-energy landscape of the complex.
V. Conclusions and Future Perspectives
Our work highlights the prominent role of conformational dynamics in defining the energy landscape of PKA-C and its substrates. We found that local and long-range conformational dynamic changes contribute to the propagation of allosteric signals in this single protein enzyme. Both fast and slow dynamics (ps–ms) are modulated by ligand binding, emphasizing the necessity of high-frequency vibrations, rotations, and librations as well as low-frequency and large amplitude (i.e., breathing) motions for catalysis to occur. PKA-C internal motions increase upon nucleotide binding. Not only does ATP have an essential role for the chemistry occurring in the active site, but it also engages the two lobes of the enzyme structurally, completing the hydrophobic C-spine via its adenine ring. We propose that this event synchronizes the motions of small and large lobes, positioning the binding groove for substrate recognition, guiding it along the reaction coordinates. Taken with the kinetic measurements, the analysis of the enzyme and substrate structural dynamics within the Michaelis complex underscores the mutual dynamic adaptation of the two proteins upon binding. This mutual conformational selection mechanism is not surprising, given the large number of substrates targeted by PKA-C. Substrate and inhibitor binding have opposite effects. While substrate binding preserves the opening and closing of the active cleft, inhibitor binding traps the enzyme in a dynamically quenched state, where most of the motions are rather attenuated. This aspect opens up the possibility of tuning the structural dynamics of PKA-C via allosteric modulators, controlling enzyme function.
However, many aspects of allosteric propagation and signaling within PKA-C and other enzymes remain obscure. While most of the allosteric network can be clearly traced via contiguous residues (either in sequence or in space), there are many noncontiguous paths that are difficult to rationalize based on only physical contacts between atoms. Recent interpretations of allosteric signaling rely on statistical correlations of the chemical shifts to describe the propagation of allosteric signaling (Lipchock and Loria, 2010; Selvaratnam et al., 2011). Although state-of-the-art, these approaches still fall short for the complete description of allosteric phenomena. From a statistical thermodynamic point of view, allostery is a nonequilibrium relaxation process, where both entropy and enthalpy changes are concurrent and define the energetic states of a system (the protein and the environment) (Tsai et al., 2008; Vendruscolo, 2011). Indeed, the NMR analysis presented here is limited to the quantification of the conformational entropy of the protein backbone, while side chains dynamics and solvent effects remain uncharacterized. The combination of methyl group NMR spin relaxation methods with more sophisticated and longer time scale MD simulations promises to fill this apparent lacuna.
Acknowledgments
This work was supported by the NIH grants GM72701 and GM 100310 to G. V. and T32DE007288 to L. R. M. NMR data were collected at NMRFAM (NIH: P41RR02301, P41GM66326, RR02781, and RR08438; NSF: DMB-8415048, OIA-9977486, BIR-9214394) and the U. of Minnesota NMR Facility (NSF BIR-961477). Modeling and calculations were carried out at the Minnesota Supercomputing Institute.
References
- Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chem Rev. 2001;101:2271–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- Adams JA, Taylor SS. Energetic limits of phosphotransfer in the catalytic subunit of cAMP-dependent protein kinase as measured by viscosity experiments. Biochemistry. 1992;31:8516–8522. doi: 10.1021/bi00151a019. [DOI] [PubMed] [Google Scholar]
- Adams JA, Taylor SS. Divalent metal ions influence catalysis and active-site accessibility in the cAMP-dependent protein kinase. Protein Sci. 1993;2:2177–2186. doi: 10.1002/pro.5560021217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadei A, Linssen ABM, Berendsen HJC. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Baldwin AJ, Kay LE. NMR spectroscopy brings invisible protein states into focus. Nat Chem Biol. 2009;5:808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- Berlow RB, Igumenova TI, Loria JP. Value of a hydrogen bond in triosephosphate isomerase loop motion. Biochemistry. 2007;46:6001–6010. doi: 10.1021/bi700344v. [DOI] [PubMed] [Google Scholar]
- Boehr DD, Dyson HJ, Wright PE. An NMR perspective on enzyme dynamics. Chem Rev. 2006;106:3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeshans KM, Resing KA, Hunt JB, Ahn NG, Shabb JB. Structural characterization of the membrane-associated regulatory subunit of type I cAMP-dependent protein kinase by mass spectrometry: identification of Ser81 as the in vivo phosphorylation site of RI-alpha. Protein Sci. 1999;8:1515–1522. doi: 10.1110/ps.8.7.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. The development and therapeutic potential of protein kinase inhibitors. Curr Opin Chem Biol. 1999;3:459–465. doi: 10.1016/S1367-5931(99)80067-2. [DOI] [PubMed] [Google Scholar]
- Cohen P. The role of protein phosphorylation in human health and disease. Eur J Biochem. 2001;268:5001–5010. doi: 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Cook PF, Neville ME, Jr, Vrana KE, Hartl FT, Roskoski R., Jr Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry. 1982;21:5794–5799. doi: 10.1021/bi00266a011. [DOI] [PubMed] [Google Scholar]
- Csermely P, Palotai R, Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci. 2010;35:539–546. doi: 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Dewey WL. Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides. 2006;40:23–34. doi: 10.1016/j.npep.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, et al. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Gaffarogullari EC, Masterson LR, Metcalfe EE, Traaseth NJ, Balatri E, Musa MM, et al. A myristoyl/phosphoserine switch controls cAMP-dependent protein kinase association to membranes. J Mol Biol. 2011;411:823–836. doi: 10.1016/j.jmb.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M, Traaseth NJ, Karim CB, Lockamy EL, Thomas DD, Veglia G. Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum Ca(2+)-ATPase. J Mol Biol. 2011;408:755–765. doi: 10.1016/j.jmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha KN, Masterson LR, Hou Z, Verardi R, Walsh N, Veglia G, et al. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc Natl Acad Sci USA. 2011;108:2735–2740. doi: 10.1073/pnas.1013987108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- Herberg FW, Doyle ML, Cox S, Taylor SS. Dissection of the nucleotide and metal-phosphate binding sites in cAMP-dependent protein kinase. Biochemistry. 1999;38:6352–6360. doi: 10.1021/bi982672w. [DOI] [PubMed] [Google Scholar]
- Hyeon C, Jennings PA, Adams JA, Onuchic JN. Ligand-induced global transitions in the catalytic domain of protein kinase A. Proc Natl Acad Sci USA. 2009;106:3023–3028. doi: 10.1073/pnas.0813266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarymowycz VA, Stone MJ. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chem Rev. 2001;101:2243–2270. doi: 10.1021/cr000226k. [DOI] [PubMed] [Google Scholar]
- Kalodimos CG. NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 2011;20:773–782. doi: 10.1002/pro.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM. Engineered allosteric activation of kinases in living cells. Nat Biotechnol. 2010;28:743–747. doi: 10.1038/nbt.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Kong CT, Cook PF. Isotope partitioning in the adenosine 3′,5′-monophosphate dependent protein kinase reaction indicates a steady-state random kinetic mechanism. Biochemistry. 1988;27:4795–4799. doi: 10.1021/bi00413a032. [DOI] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS. Defining the conserved internal architecture of a protein kinase. Biochim Biophys Acta. 2009;1804:440–444. doi: 10.1016/j.bbapap.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS. Protein Kinases: evolution of a dynamic regulatory protein. Trends Biochem Sci. 2010;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev AP, Haste NM, Taylor SS, Ten Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci USA. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, Kay LE. A transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329:1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- Lew J, Taylor SS, Adams JA. Identification of a partially rate-determining step in the catalytic mechanism of cAMP-dependent protein kinase: a transient kinetic study using stopped-flow fluorescence spectroscopy. Biochemistry. 1997;36:6717–6724. doi: 10.1021/bi963164u. [DOI] [PubMed] [Google Scholar]
- Li F, Juliano C, Gorfain E, Taylor SS, Johnson DA. Evidence for an internal entropy contribution to phosphoryl transfer: a study of domain closure, backbone flexibility, and the catalytic cycle of cAMP-dependent protein kinase. J Mol Biol. 2002;315:459–469. doi: 10.1006/jmbi.2001.5256. [DOI] [PubMed] [Google Scholar]
- Lipchock JM, Loria JP. Nanometer propagation of millisecond motions in V-type allostery. Structure. 2010;18:1596–1607. doi: 10.1016/j.str.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria JP, Berlow RB, Watt ED. Characterization of enzyme motions by solution NMR relaxation dispersion. Acc Chem Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- Maragakis P, Karplus M. Large amplitude conformational change in proteins explored with a plastic network model: adenylate kinase. J Mol Biol. 2005;352:807–822. doi: 10.1016/j.jmb.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Massi F, Wang C, Palmer AG., 3rd Solution NMR and computer simulation studies of active site loop motion in triosephosphate isomerase. Biochemistry. 2006;45:10787–10794. doi: 10.1021/bi060764c. [DOI] [PubMed] [Google Scholar]
- Masterson LR, Mascioni A, Traaseth NJ, Taylor SS, Veglia G. Allosteric cooperativity in protein kinase A. Proc Natl Acad Sci USA. 2008;105:506–511. doi: 10.1073/pnas.0709214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Shi L, Tonelli M, Mascioni A, Mueller MM, Veglia G. Backbone NMR resonance assignment of the catalytic subunit of cAMP-dependent protein kinase A in complex with AMP-PNP. Biomol NMR Assign. 2009;3:115–117. doi: 10.1007/s12104-009-9154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Cheng C, Yu T, Tonelli M, Kornev AP, Taylor SS, et al. Dynamics connect substrate recognition to catalysis in protein kinase A. Nat Chem Biol. 2010;6:821–828. doi: 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Shi L, Metcalfe E, Gao J, Taylor SS, Veglia G. Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc Natl Acad Sci USA. 2011a;108:6969–6974. doi: 10.1073/pnas.1102701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Yu T, Shi L, Wang Y, Gustavsson M, Mueller MM, et al. cAMP-dependent protein kinase A selects the excited state of the membrane substrate phospholamban. J Mol Biol. 2011b;412:155–164. doi: 10.1016/j.jmb.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit. 2010;23:105–116. doi: 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- Miyashita O, Onuchic JN, Wolynes PG. Nonlinear elasticity, protein-quakes, and the energy landscapes of functional transitions in proteins. Proc Natl Acad Sci USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Adams JA, Taylor SS. Structural basis for peptide binding in protein kinase A. Role of glutamic acid 203 and tyrosine 204 in the peptide-positioning loop. J Biol Chem. 2003;278:10613–10618. doi: 10.1074/jbc.M210807200. [DOI] [PubMed] [Google Scholar]
- Nashine VC, Hammes-Schiffer S, Benkovic SJ. Coupled motions in enzyme catalysis. Curr Opin Chem Biol. 2010;14:644–651. doi: 10.1016/j.cbpa.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A., 3rd Nmr probes of molecular dynamics: overview and comparison with other techniques. Annu Rev Biophys Biomol Struct. 2001;30:129–155. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci USA. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabb JB. Physiological substrates of cAMP-dependent protein kinase. Chem Rev. 2001;101:2381–2411. doi: 10.1021/cr000236l. [DOI] [PubMed] [Google Scholar]
- Slice LW, Taylor SS. Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli. J Biol Chem. 1989;264:20940–20946. [PubMed] [Google Scholar]
- Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–269. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Tonelli M, Masterson LR, Hallenga K, Veglia G, Markley JL. Carbonyl carbon label selective (CCLS) (1)H-(15)N HSQC experiment for improved detection of backbone (13)C-(15)N cross peaks in larger proteins. J Biomol NMR. 2007;39:177–185. doi: 10.1007/s10858-007-9185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Ha KN, Verardi R, Shi L, Buffy JJ, Masterson LR, et al. Structural and dynamic basis of phospholamban and sarcolipin inhibition of Ca (2+)-ATPase. Biochemistry. 2008;47:3–13. doi: 10.1021/bi701668v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, del Sol A, Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J Mol Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng SR, Kalodimos CG. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- Vendruscolo M. Protein regulation: the statistical theory of allostery. Nat Chem Biol. 2011;7:411–412. doi: 10.1038/nchembio.603. [DOI] [PubMed] [Google Scholar]
- Villali J, Kern D. Choreographing an enzyme’s dance. Curr Opin Chem Biol. 2010;14:636–643. doi: 10.1016/j.cbpa.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DA, Perkins JP, Krebs EG. An adenonsine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763–3765. [PubMed] [Google Scholar]
- Walsh DA, Van Patten SM. Multiple pathway signal transduction by the cAMP-dependent protein kinase. FASEB J. 1994;8:1227–1236. doi: 10.1096/fasebj.8.15.8001734. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Yang J, Ten Eyck LF, Xuong NH, Taylor SS. Crystal structure of a cAMP-dependent protein kinase mutant at 1.26A: new insights into the catalytic mechanism. J Mol Biol. 2004;336:473–487. doi: 10.1016/j.jmb.2003.11.044. [DOI] [PubMed] [Google Scholar]
- Yang J, Garrod SM, Deal MS, Anand GS, Woods J, Virgil L, et al. Allosteric network of cAMP-dependent protein kinase revealed by mutation of Tyr204 in the P+1 loop. J Mol Biol. 2005;346:191–201. doi: 10.1016/j.jmb.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Yang J, Kennedy EJ, Wu J, Deal MS, Pennypacker J, Ghosh G, et al. Contribution of non-catalytic core residues to activity and regulation in protein kinase A. J Biol Chem. 2009;284:6241–6248. doi: 10.1074/jbc.M805862200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Knighton DR, ten Eyck LF, Karlsson R, Xuong N, Taylor SS, et al. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry. 1993;32:2154–2161. doi: 10.1021/bi00060a005. [DOI] [PubMed] [Google Scholar]
- Zimmermann B, Schweinsberg S, Drewianka S, Herberg FW. Effect of metal ions on high-affinity binding of pseudosubstrate inhibitors to PKA. Biochem J. 2008;413:93–101. doi: 10.1042/BJ20071665. [DOI] [PubMed] [Google Scholar]