

Abstract
This report demonstrates that a repeated “challenge” high-dose methamphetamine (METH) injection regimen rapidly decreases striatal K+-stimulated dopamine (DA) release concurrent with decreases in both synaptosomal membrane-associated (referred to herein as membrane-associated) and previously-reported decreases in non-synaptosomal membrane-associated (presumably cytoplasmic) vesicular DA uptake and content. Resembling previously reported effects involving cytoplasmic vesicles wherein uptake was decreased 48 h after treatment, the decrease in membrane-associated uptake persisted 72 h. Cytoplasmic and membrane-associated vesicular DA uptakes were decreased 7 d after the challenge regimen. A single METH injection also rapidly decreased K+-stimulated DA release, membrane-associated DA content, and membrane-associated DA uptake; however, unlike after the challenge regimen, the decrease in uptake recovered by 24 h. Pretreatment with the D2 receptor antagonist, eticlopride, did not attenuate the decrease in membrane-associated uptake as assessed 1 h after either a single or challenge treatment. However, eticlopride attenuated the decrease in membrane-associated uptake caused by the challenge regimen as assessed 24 h later. These data reveal complex effects of METH on vesicular function that vary according to the vesicle population under study, dosing regimen, and time after treatment. These may contribute to both the decrease in K+-stimulated DA release and the persistent dopaminergic deficits caused by METH.
Keywords: dopamine, methamphetamine, vesicular monoamine transporter-2, D2 receptor
Introduction
Methamphetamine (METH) is a highly addictive central nervous system stimulant. Repeated high-dose METH administrations cause long-term dopaminergic system deficits, including reductions in dopamine (DA) content (Kogan et al. 1976; for review, see Gibb et al. 1994), tyrosine hydroxylase activity (Hotchkiss et al. 1979; Kogan et al. 1976) and DA transporter (DAT) activity and/or levels (Nakayama et al. 1993; Volkow et al. 2001; Wagner et al. 1980; Wilson et al. 1996; Woolverton et al. 1989) in rodents, non-human primates, and/or humans. The mechanisms underlying these deficits are unclear. However, DA, per se, likely contributes to this neuronal damage, as it has been suggested that aberrant cytoplasmic DA levels consequent to its displacement by METH from synaptic vesicles promote oxidative stress (Cubells et al. 1994; LaVoie and Hastings 1999; for review, see Fleckenstein et al. 2000). The transport of DA into synaptic vesicles is solely mediated by the vesicular monoamine transporter-2 (VMAT-2). Therefore, VMAT-2 plays an important role in regulating DA distribution and its aberrant function likely contributes to METH-induced long-term dopaminergic deficits (Fumagalli et al. 1999; Guillot et al. 2008; Thomas et al. 2008; for review, see Fleckenstein et al. 2009).
Previous studies demonstrated that amphetamine inhibits potassium- and stimulation-dependent DA release in both striatal slices (Bowyer et al. 1987; Schmitz et al. 2001 and references cited therein) and synaptosomes (Bowyer et al. 1987). In addition, in vivo studies demonstrated that K+-stimulated DA release in rat striatum is decreased following repeated METH administrations (Bustamante et al. 2002). These decreases in exocytotic release are likely, at least in part, a result of well-characterized amphetamine/METH-induced depletions of vesicular DA content (Sandoval et al. 2003; for review, see Sulzer et al. 2005). As it is known that VMAT-2 mediates DA uptake and thereby determines quantal size, (Pothos et al. 2000; for review, see Gasnier 2000; Sulzer and Pothos 2000), a decrease in VMAT-2 activity may contribute to a decrease in K+-stimulated DA release. This possibility is supported by findings that METH treatment decreases VMAT-2-mediated DA uptake into at least some striatal vesicle populations (Brown et al. 2000; Chu et al. 2008; Hogan et al. 1999; Sandoval et al. 2003).
Others and we have described two different VMAT-2-containing subcellular fractions; specifically, one that co-fractionates with striatal synaptosomal membranes after osmotic lysis (referred to herein as membrane-associated (VMAT-2M) vesicles) and the second that does not (referred to herein as cytoplasmic (VMAT-2C) vesicles; Volz et al. 2007; see also Eyerman and Yamamoto 2007). Volz et al. (2007) reported that the sequestration potentials of these populations differ. Further, these populations respond differentially to treatment with the DA re-uptake inhibitor, methylphenidate (Volz et al. 2007). It was heretofore unknown if these populations respond differentially to the DA-releasing agent, METH. Whereas effects of METH on striatal VMAT-2C-associated vesicles have been largely characterized (for review, see Fleckenstein et al. 2009), the impact of METH on VMAT-2M-containing vesicles has received little attention. Accordingly, this study investigated the impact of METH on VMAT-2M-associated vesicles. Taken together with previous reports, results revealed a complex series of effects on VMAT-2-containing vesicles that vary according to the vesicle population under study, the METH dosing regimen, and the time after METH administration. The decreases in VMAT-2 function in both populations likely underlie the decrease in K+-stimulated DA release caused by the stimulant, and may contribute to the neurotoxic potential of METH.
Materials and Methods
Animals
Male Sprague-Dawley rats (300–380 g; Charles River Laboratories, Raleigh, NC) were maintained under controlled light and temperature conditions, with food and water provided ad libitum. All animal procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals and approved by the University of Utah Institutional Animal Care and Use Committee.
Drugs and Chemicals
METH was supplied by the Research Triangle Institute (Research Triangle Park, NC). Eticlopride [S-(-)-3-chloro-5-ethyl-N-[(1-ethyl-2-pyrrolidinyl) methyl]-6-hydroxy-2-methoxybezamide] hydrochloride was purchased from Sigma-Aldrich (St. Louis, MO). All drugs, including eticlopride, were administered at doses and times used previously to elucidate effects of psychostimulants in rat brain (Brown et al. 2002; Broening et al. 2005; Hadlock et al. 2009). Drug doses were calculated as the free base, dissolved in 0.9% (w/v) saline, and administered at 1 ml/kg. 7,8-[3H]DA (40, 54.2, 56.8 Ci/ mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA).
Subcellular Fractionation
Fresh striatal tissues were homogenized in ice-cold sucrose buffer (320 mM sucrose, 3.8 mM NaH2PO4, 12.7 mM Na2HPO4; pH 7.4) and then centrifuged (800 × g, 12 min, 4°C). The resulting supernatant was centrifuged (22,000 × g, 15 min, 4°C) to obtain the synaptosomal pellet. This synaptosomal pellet was then resuspended and homogenized in ice-cold water (2 ml) to lyse the synaptosomal membranes. Ice-cold 25 mM HEPES and 100 mM potassium tartrate (pH 7.5) were then added to the homogenate and centrifuged (20,000 × g, 20 min, 4°C) to obtain the VMAT-2M-associated vesicle pellet. To isolate VMAT-2C-associated vesicles, 1 mM ice-cold MgSO4, pH 7.5, was added to the supernatant, and the resulting mixture was centrifuged (100,000 × g, 45 min, 4°C) to obtain the VMAT-2C-associated vesicle pellet.
Vesicular DA Uptake
The VMAT-2 assay buffer contained 25 mM HEPES, 100 mM potassium tartrate, 1.7 mM ascorbic acid, 2 mM MgSO4, 0.05 mM EGTA, and 0.1 mM EDTA with a final pH of 7.5. DA uptake in membrane-associated vesicles was performed by incubating 100 μl of the resuspended membrane-associated vesicular pellet at 30°C for 3 min in VMAT-2 assay buffer with 2 mM ATP-Mg2+ in the presence of 300 nM DA (e.g., a mixture of 30 nM [3H]DA and 270 nM non-radioisotope-labeled DA) final concentration except in kinetic analyses, wherein 0.0125–2.0 μM DA (obtained from a mixture of 0.0125-0.10 μM [3H]DA and 0-1.90 μM DA non-radioisotope-labeled). The reaction was terminated by incubating on ice and rapid filtration through Whatman GF/F filters (Whatman, Clifton, NJ), soaked previously in 0.5% polyethylenimine. Filters were washed three times with ice-cold VMAT-2 assay buffer using a Brandel filtering manifold (Brandel, Inc., Gaithersburg, MD). Radioactivity trapped in filters was counted using a liquid scintillation counter. Nonspecific values were determined by measuring DA uptake at 4°C in VMAT-2 assay buffer. DA uptake in cytoplasmic vesicles were measured as described above but in the presence of 30 nM [3H]DA. Protein concentrations were determined by using the Bradford protein assay (Bio-Rad, Hercules, CA; Bradford 1976).
K+-Stimulated DA Release
Rotating disk electrode (RDE) voltammetry was used to measure K+ -stimulated DA release in striatal suspensions prepared from treated rats as described previously (Volz et al. 2007). Each sample consisted of one striatum that was placed on an ice-cold watch glass and chopped by hand with an ice-cold razor blade for about 30 s. The chopped striatum was then placed in 500 μl of DAT assay buffer (126 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 16 mM sodium phosphate, 1.4 mM MgSO4, 11 mM glucose; pH 7.4) inside the RDE glass chamber and was resuspended by repetitive pipetting for approximately 1 min. The resulting striatal suspension was allowed to stand for 12 min and was then washed by the addition and subsequent removal of 250 μl of fresh DAT assay buffer six times. The washes were done without disrupting the settled striatal tissue. A detection current baseline was obtained in approximately 18 min, and then a small quantity of DAT assay buffer containing an elevated KCl concentration (for a final concentration of 40 mM K+ inside the RDE glass chamber) was added to the striatal suspension to stimulate DA release. The initial velocity of K+ -stimulated DA release (measured from the first 3 s of release), the magnitude of K+ -stimulated DA release (the maximum amount of DA released), and the duration of K+ -stimulated DA release (the amount of time for the maximal amount of DA to be released) were calculated and normalized to striatal wet weight as described previously (Volz et al. 2007).
Vesicular DA content
Vesicular DA content was measured using high-performance liquid chromatography as described previously (Chu et al. 2008; Volz et al. 2007). The membrane-associated and cytoplasmic vesicle pellets were prepared as described above and resuspended in ice-cold tissue buffer (50 mM sodium phosphate, 30 mM citric acid, 10% (v/v) methanol; pH 2.5) at 50 and 100 mg original striatal wet weight/ml tissue buffer, respectively. The resuspended vesicle preparations were then sonicated for approximately 10 s and centrifuged (22,000 × g, 15 min, 4°C). A 50 μl aliquot of the resulting supernatant from each sample was then injected into a high-performance liquid chromatography system (4.6 × 250 mm; Whatman, Maidstone, UK; Partisphere C18 column) that was coupled to an electrochemical detector (+730 mV relative to an Ag/AgCl reference electrode). The pH 2.86 mobile phase contained 50 mM sodium phosphate, 30 mM citric acid, 0.16 mM EDTA, 1.5 mM sodium octyl sulfate, and 10% (v/v) methanol (Chapin et al. 1986; Volz et al. 2007).
VMAT-2 Immunoreactivity
SDS-polyacrylamide gel electrophoresis and Western blot analysis were completed on the membrane-associated vesicle samples as described previously (Riddle et al. 2002; Volz et al. 2007). Twelve micrograms of protein (as determined with a Bradford protein assay) from each membrane-associated vesicle sample was loaded onto the 4~12 % gel. Bound VMAT-2 antibody was visualized with horseradish peroxidase-conjugated rabbit secondary antibody (BioSource International, Camarillo, CA). Bands on all Western blots were quantified by densitometry using a FluorChem SP Imaging System from Alpha Innotech (San Leandro, CA).
Statistics
All statistical analysis was performed on raw data for each treatment group by one-way analysis of variance, followed by a Newman-Keuls post hoc test or Student’s t-test using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA). Differences among groups were considered significant if the probability of error was less than or equal to 5%.
Results
Effect of a “challenge” METH administration
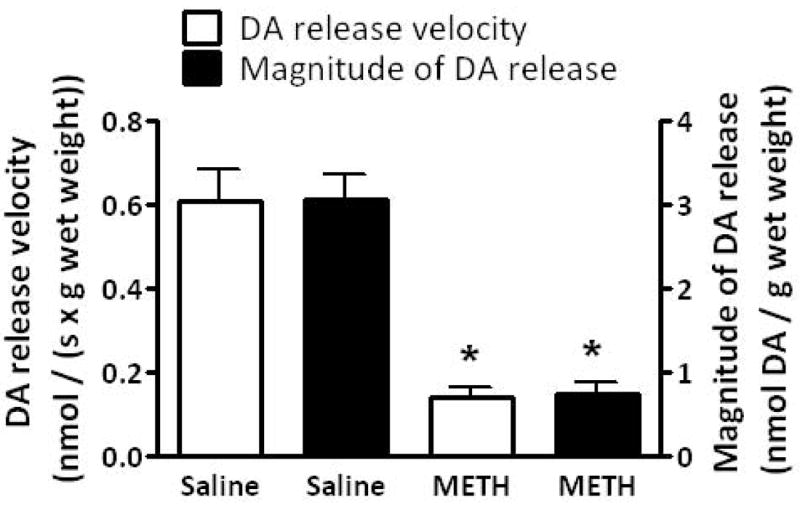
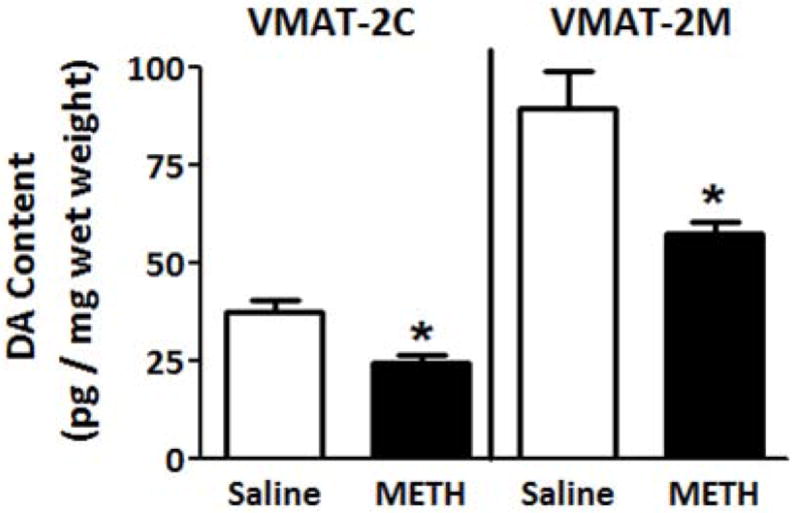
Results presented in Fig. 1A demonstrate that multiple high-dose administrations of METH (4 × 7.5 mg/kg/injection, s.c., 2-h intervals; a regimen referred to herein as a “challenge” regimen and selected because it is well-established to cause persistent dopaminergic neuronal deficits; Chu et al. 2008; Sandoval et al. 2003) rapidly decreased the velocity and magnitude of K+-stimulated DA release, as assessed in striatal suspensions 1 h after the final treatment. Concurrent with METH-induced decrease in K+-stimulated DA release, this treatment decreased both membrane-associated (e.g., VMAT-2M-mediated) and cytoplasmic (e.g., VMAT-2C-mediated) vesicular DA content (Fig. 1B).
Fig. 1.
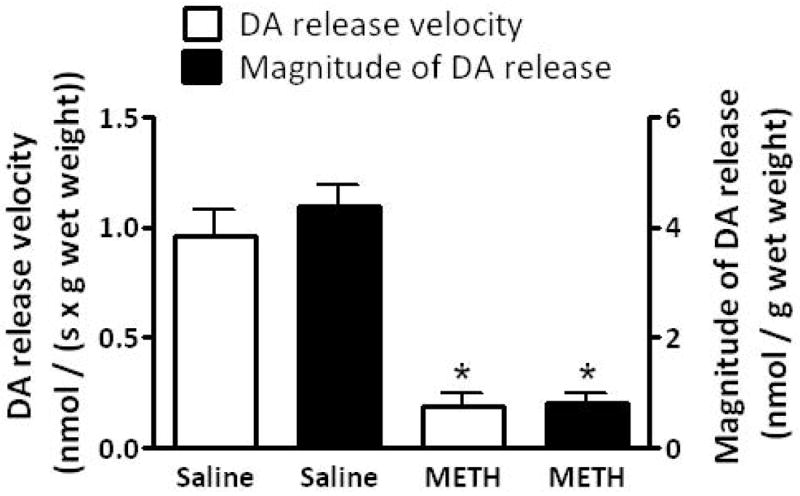
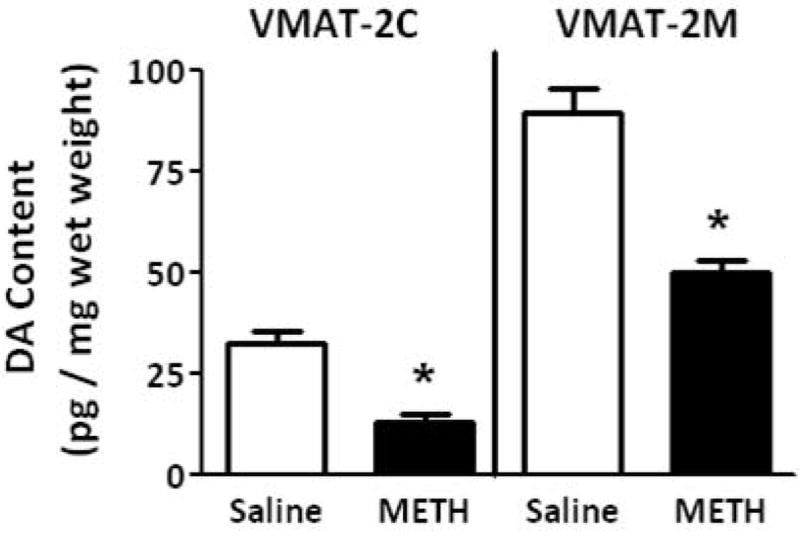
A “challenge” METH regimen decreases both (A) the velocity and magnitude of K+-stimulated DA release from striatal suspensions and (B) DA content in the VMAT-2M- and VMAT-2C-associated vesicle fractions (B). Rats received four injections of METH (7.5 mg/kg/injection, s.c., 2-h intervals) or saline vehicle (1 ml/kg/injection, s.c., 2-h intervals) and were killed 1 h later. Columns represent the mean ± 1 S.E.M. of 6 to 8 independent determinations. *, indicates a statistical difference between saline- and METH-treated animals.
To investigate mechanisms underlying the METH-induced decrease in K+-stimulated DA release and vesicular DA content, the impact of the stimulant on vesicular DA uptake was assessed. VMAT-2M-containing vesicles were the focus of study for comparison with previous work describing effects on VMAT-2C-associated vesicles (for review, see Fleckenstein et al. 2009 and also Discussion below). As reported for VMAT-2C-associated vesicles (Brown et al. 2000), results presented in Fig. 2A demonstrate that the challenge METH regimen under study rapidly (e.g., within 1 h) decreased vesicular [3H]DA uptake into VMAT-2M-associated vesicles. The decrease in VMAT-2C, and presumably VMAT-2M, function was likely not due to residual METH introduced by the original subcutaneous injections, as previous studies have demonstrated that isolation of synaptosomes, and thus VMAT-2C and VMAT-2M, “washes” METH to levels of < 6 nM (Fleckenstein et al. 1997; Kokoshka et al. 1998), a concentration that is below the concentration necessary to affect DA uptake in preparation. In particular, VMAT-2C- and VMAT-2M-associated DA uptake is unaffected by direct application of METH at concentrations of up to approximately 0.5 μM (Brown et al. 2000) and 1 μM, respectively. Noteworthy, this decrease was due to a decrease in Vmax (199.0 ± 7.6 fmol/μg for 3 min saline vs. 164.2 ± 9.8 fmol/μg for 3 min METH; p < 0.05) without a change in Km (162.3 ± 8.1 nM saline; 239.2 ± 46.9 nM METH; n = 3, Fig. 2B). Similar to that which was reported previously for VMAT-2C-mediated DA uptake wherein decreases persisted 48 h (Chu et al. 2008; later time points were not assessed); this decrease in VMAT-2M-mediated uptake persisted 72 h after treatment (Fig. 2A). Both VMAT-2C- and VMAT-2M-mediated activity were decreased 7 d after the challenge METH regimen (Fig. 3).
Fig. 2.
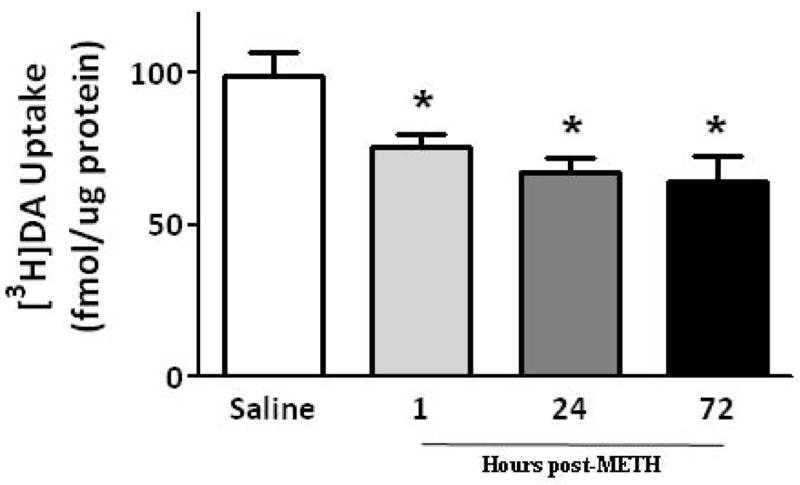
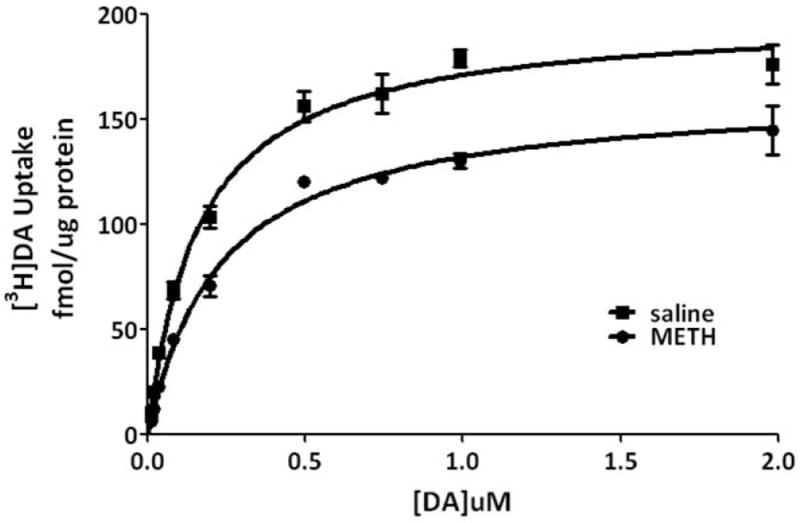
(A) A “challenge” METH regimen rapidly decreases VMAT-2M-mediated DA uptake. Rats received four injections of METH (7.5 mg/kg/injection, s.c., 2-h intervals) and were killed 1, 24 or 72 h later. Other rats received four injections of saline vehicle (1 ml/kg/injection, s.c., 2-h intervals) and were killed 1 h later. Columns represent the mean ± 1 S.E.M. of 6 to 8 independent determinations. *, indicates a statistical difference from saline-treated animals. (B) Kinetic analysis of DA uptake via VMAT-2M-associated vesicles. Values represent the mean ± 1 S.E.M. of 3 independent determinations. Rats received four injections of METH (7.5 mg/kg/injection, s.c., 2-h intervals) or saline vehicle (1 ml/kg/injection, s.c., 2-h intervals) and were killed 1 h later.
Fig. 3.
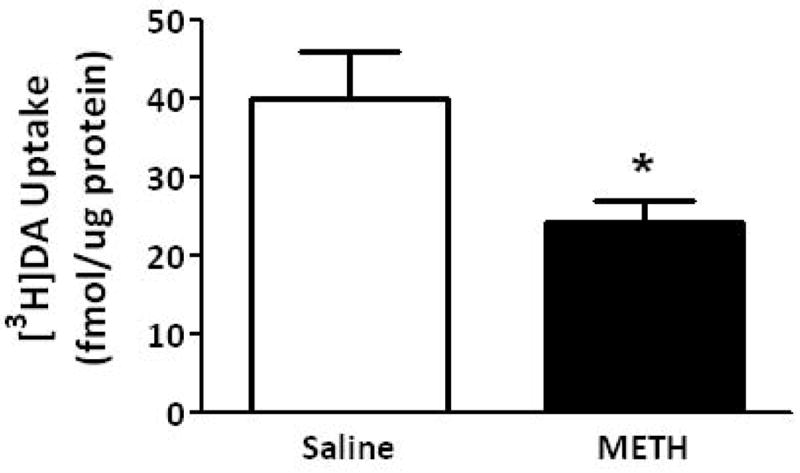
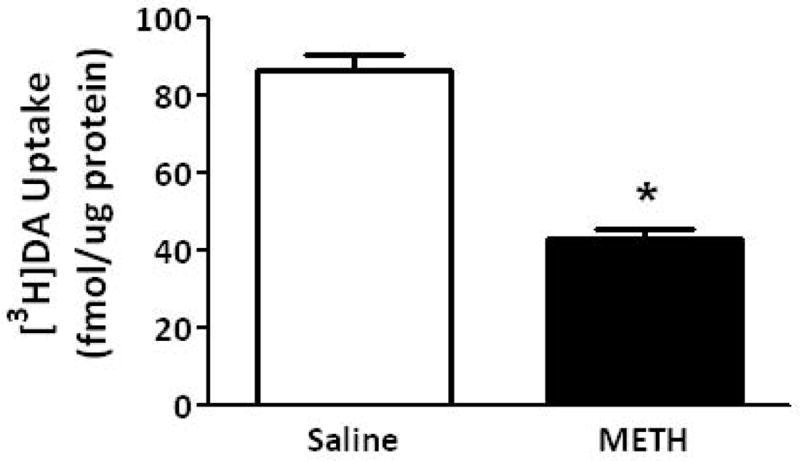
A “challenge” METH regimen decreases both VMAT-2M- and VMAT-2C-mediated DA uptake. Rats received four injections of METH (7.5 mg/kg/injection, s.c., 2-h intervals) or saline vehicle (1 ml/kg/injection, s.c., 2-h intervals) and were killed 7 d later. Columns represent the mean ± 1 S.E.M. of 6 to 8 independent determinations. *, indicates a statistical difference between DA uptake in saline- and METH-treated animals.
Effect of a single METH administration
Results presented in Figure 4 demonstrate that as after multiple administrations, a single METH injection (15 mg/kg s.c.; Brown et al. 2002) rapidly decreased both the velocity and magnitude of K+-stimulated DA release, as well as membrane-associated vesicular DA content (Fig. 4A and B). VMAT-2M immunoreactivity was not altered 1 h after a single METH treatment (data not shown). This treatment rapidly (within 0.5 h) decreased VMAT-2M-mediated vesicular DA uptake as well; an effect that persisted at least 3 h (Fig. 5A). However, unlike effects of a challenge METH regimen, this decrease in uptake recovered by 24 h (Fig. 5B).
Fig. 4.
A single METH administration decreases both the velocity and magnitude of K+-stimulated DA release from striatal suspensions (A) and DA content in the VMAT-2M- and VMAT-2C-containing vesicle fractions (B). Rats received an injection of METH (15 mg/kg, s.c.) or saline vehicle (1 ml/kg, s.c.) and were killed 1 h later. Each column represents the mean ± S.E.M. of 6 to 8 independent determinations. *, indicates a statistical difference between saline- and METH-treated animals.
Fig. 5.
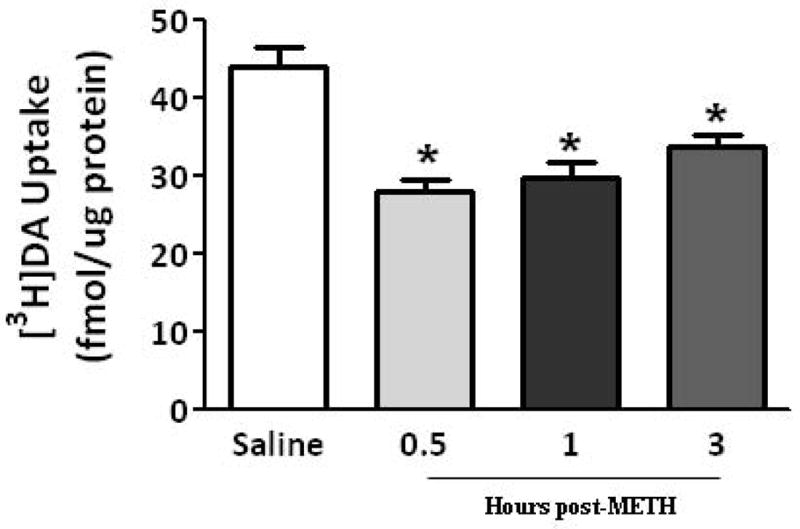
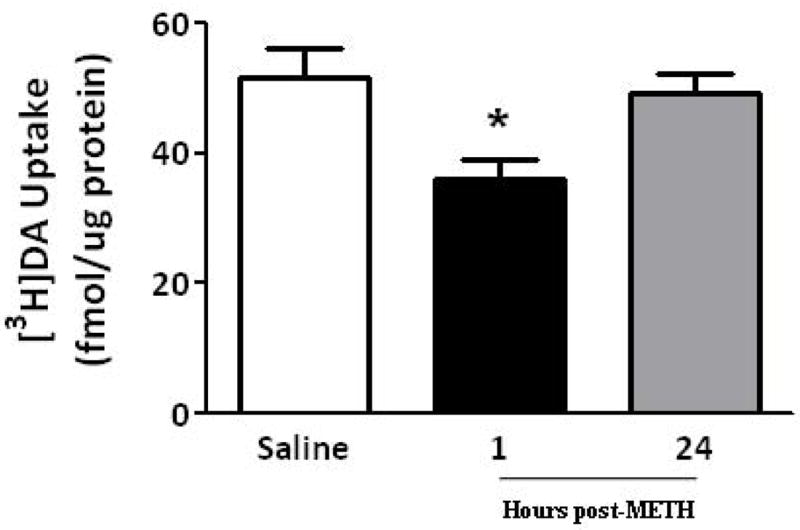
A single METH administration rapidly decreases (within 0.5 h) VMAT-2M-mediated DA uptake; an effect that persists at least 3 h (A). Rats received an injection of METH (15 mg/kg, s.c.) and were killed 0.5, 1 or 3 h later. Other rats received an injection of saline vehicle (1 ml/kg, s.c.) and were killed 0.5 h later. Each column represents the mean ± S.E.M. of 6 to 8 independent determinations. *, indicates a statistical difference from saline-treated animals. A single METH-induced VMAT-2M-mediated DA uptake decrease recovers 24 h later (B). Rats received an injection of METH (15 mg/kg, s.c.) and were killed 1 or 24 h later. Other rats received an injection of saline vehicle (1 ml/kg, s.c.) and were killed 1 h later. Each column represents the mean ± S.E.M. of 6 to 8 independent determinations. *, indicates a statistical difference from saline-treated animals.
Eticlopride pretreatment attenuated METH-induced DA uptake decrease in VMAT-2M-mediated vesicular DA uptake at 24 h after treatment
Pretreatment with the D2 receptor antagonist, eticlopride (0.5 mg/kg/injection 30 min prior to each injection; Brown et al., 2002; Hadlock et al., 2009) did not attenuate the decrease in VMAT-2M-mediated uptake as assessed 1 h after either a challenge (Fig. 6) or a single (data not shown) administration of the stimulant. However, eticlopride treatment attenuated the decrease in VMAT-2M-associated uptake caused by a challenge METH treatment as assessed 24 h later (Fig. 7).
Fig. 6.
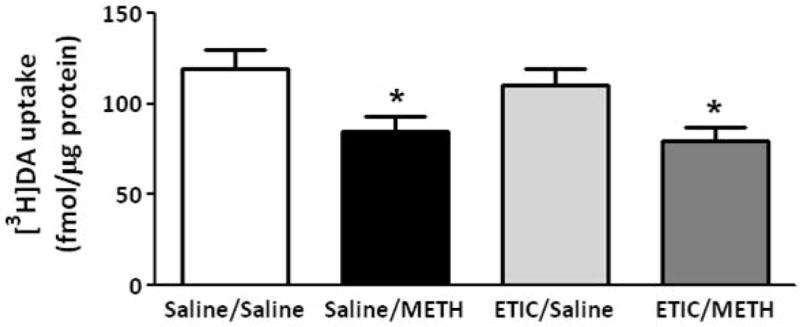
Eticlopride pretreatment does not attenuate the METH-induced decrease in VMAT-2M-associated vesicles as assessed 1 h after METH treatment. Rats received four injections of METH (7.5 mg/kg/injection, s.c., 2-h intervals) or saline vehicle (1 ml/kg/injection, s.c., 2-h intervals) and were killed 1 h later. Thirty minutes before each injection, rats were pretreated with eticlopride (ETIC; 0.5 mg/kg/injection, i.p.) or saline vehicle (1 ml/kg/injection, i.p.). Columns represent the mean ± 1 S.E.M. of 10 to 12 independent determinations. *, indicates a statistical difference from saline/saline treated animals.
Figure 7.
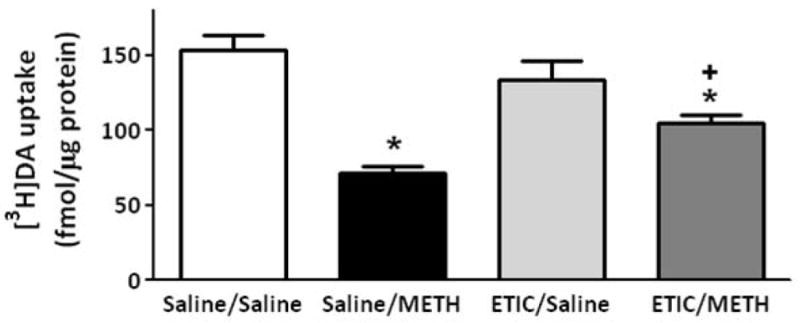
Eticlopride pretreatment attenuates the METH-induced DA uptake decrease in VMAT-2M vesicles as assessed 24 h after METH treatment. Rats received four injections of METH (7.5 mg/kg/injection, s.c., 2-h intervals) or saline vehicle (1 ml/kg/injection, s.c., 2-h intervals) and were killed 24 h later. Thirty minutes before each injection, rats were pretreated with eticlopride (ETIC; 0.5 mg/kg/injection, i.p.) or saline vehicle (1 ml/kg/injection, i.p.). Each column represents the mean ± S.E.M. of 12 independent determinations. *, indicates a statistical difference from saline/saline treated animals; +, indicates a statistical difference from saline/METH treated animals.
Discussion
As noted above, previous studies have demonstrated that amphetamine and its analog, METH, inhibit K+-stimulated DA release (Bowyer et al. 1987; Bustamante et al. 2002) as assessed in striatal slices, synaptosomes, or in vivo microdialysis. Our findings confirm and extend these data by demonstrating that a single METH treatment rapidly decreases both the magnitude and velocity of K+-stimulated DA release. Further, administration of a challenge METH regimen used commonly to cause persistent dopaminergic neuronal deficits (Chu et al. 2008; Eyerman and Yamamoto 2007; Sandoval et al. 2003) decreases K+-stimulated DA release. Present and past data permit speculation that these decreases are caused by diminished vesicular DA content due to reduction in VMAT-2 function. In particular, previous studies from our laboratory have demonstrated that METH treatment decreases VMAT-2C-mediated DA uptake and cytoplasmic DA content (Brown et al. 2000; Sandoval et al. 2003). Further, the present study demonstrates the novel finding that both a single and challenge METH regimen decrease DA content within, and the activity of, VMAT-2M-associated vesicles.
As noted above, others and we have described two different VMAT-2-containing subcellular fractions; specifically, the VMAT-2M-associated and VMAT-2C-associated vesicles. In 2007, Volz et al. reported that the sequestration potentials of these populations differ. In particular, VMAT-2M-associated vesicles are capable, in the presence of high DA concentrations, of sequestering significantly greater quantities of DA in total than the cytoplasmic vesicles. Because of this greater sequestration capacity, it has been suggested that at least a subpopulation of the VMAT-2M-associated vesicles may serve as a “DA sink” to capture DA and prevent cytoplasmic accumulation. Interestingly, previous studies demonstrated that the ready-releasable vesicle pool marker, piccolo, was detected in VMAT-2M- but not in VMAT-2C-associated fractions suggesting the presence of the ready-releasable vesicle pool in VMAT-2M-associated vesicles (Volz et al. 2007).
In addition to the differences in VMAT-2M- and VMAT-2C-associated vesicles described above, psychostimulants differentially affect VMAT-2C and VMAT-2M function. For example, administration of both the DA reuptake inhibitors, cocaine and methylphenidate, decrease VMAT-2 immunoreactivity in the VMAT-2M-associated fraction, and increase VMAT-2 immunoreactivity in the VMAT-2C-associated fraction, without altering VMAT-2 immunoreactivity in whole synaptosomes (Riddle et al. 2002; Volz et al. 2007; Farnsworth et al. 2009). Of particular interest is the unique finding that in addition to trafficking VMAT-2 and associated vesicles away from membranes, methylphenidate also kinetically up-regulates the decreased number of VMAT-2M-associated vesicles such that a larger quantity of DA is transported (Volz et al. 2007). In contrast, cocaine redistributes VMAT-2-associated vesicles from membranes but does not kinetically up-regulate DA transport into these vesicles (Farnsworth et al. 2009). Importantly, previous studies have focused on the effect of the VMAT-2C-associated vesicles after METH treatment; thus, the present report is the first publication to elucidate the effect of METH on VMAT-2M-associated function.
The mechanisms whereby METH exerts its effects on VMAT-2-associated vesicles are complex and vary according to the vesicle population under study. For example, earlier reports revealed that the challenge METH regimen under study rapidly (within 1 h) decreases VMAT-2C-mediated DA uptake concurrently with a decrease in VMAT-2C-associated immunoreactivity (Riddle et al. 2002; Sandoval et al. 2003; Eyerman and Yamamoto 2007; Chu et al. 2008). This finding was attributed to a redistribution of VMAT-2C and associated vesicles to a population of vesicles that was not retained in the preparation of the synaptosomes (Riddle et al. 2002; Sandoval et al. 2003; Chu et al. 2008; see also Hogan et al. 2000). In contrast, the present study demonstrates that the challenge treatment under study decreased VMAT-2M-mediated uptake 1 h after METH treatment (Fig 2A), despite a lack of effect on immunoreactivity (Sandoval et al. 2003; Chu et al. 2008). Thus, mechanisms beyond a redistribution of vesicles underlie the acute effects of the present challenge METH regimen on VMAT-2M-associated vesicles at this time point.
The mechanisms underlying the decrease in VMAT-2M-mediated activity 1 h after a challenge METH treatment are not known. Considerable evidence from studies involving cultured midbrain dopaminergic neurons indicates that amphetamine and its analogs reduce pH gradients resulting in decreased VMAT-2-mediated uptake and disrupted DA storage (Sulzer and Rayport 1990; Sulzer et al. 1992). This mechanism is difficult to test using the ex vivo model system under study as VMAT-2-containing vesicles comprise only a small fraction of the acidic vesicles in striatal tissue and cannot be readily sorted in order to selectively assess intravesicular pH. Noteworthy, the METH-induced decrease in VMAT-2M-mediated uptake was attributable to a reduction in Vmax, but not Km, of vesicles displaying Michaelis-Menten kinetics. This kinetic profile of the VMAT-2M-associated vesicles differs from the sigmoidal profile reported previously (Volz et al. 2007), perhaps owing to methodological issues (e.g., assessing total uptake capacity over 3 min using [3H]DA uptake vs. assessing initial DA transport velocity using RDE voltammetry).
The complex nature of the effect of METH on VMAT-2 not only varies as a function of the vesicle population under study, but also the time after exposure to the stimulant. For example, and as noted above, administration of a METH challenge regimen decreases VMAT-2M-associated activity, but not immunoreactivity as assessed 1 h after treatment. In contrast, both VMAT-2M-associated immunoreactivity (Eyerman and Yamamoto 2007) and activity are decreased 24 h after a challenge treatment. These distinctions indicate that different mechanisms underlie the 1- and 24-h phenomena. Further evidence for distinct mechanisms at these time points comes from findings that only the 24-h (and not the 1-h) effect of a challenge METH treatment on VMAT-2M activity is attenuated by pretreatment with the D2 receptor antagonist, eticlopride.
As with the repeated, high-dose challenge METH regimen under study, a single METH injection decreased both K+-stimulated DA uptake and vesicular DA content. These effects are likely consequent to the rapid decrease in both VMAT-2C- (Brown et al. 2002) and VMAT-2M- (Fig. 4A) mediated uptake. Unlike effects of the challenge treatment, the effects of a single METH administration recover within 24 h (Fig. 4B, Brown et al. 2002). Whereas a small component of the decrease in VMAT-2C-mediated uptake 1 h after a single METH injection may be D2 receptor-mediated (Brown et al. 2002), the present study did not reveal a role for D2 receptors in mediating the effect of a single METH on VMAT-2M-associated uptake assessed at this time point (data not shown).
Noteworthy, the D2 (vs. the D1) receptor was a focus of study because of its presynaptic localization relative to synaptic vesicles. This work builds on studies suggesting that amphetamine inhibits vesicular release by activating D2 autoreceptors (Schmitz et al. 2001; for review see Sulzer et al. 2005). Future studies will assess the impact of post-synaptic circuitry (including D1, GABA and glutamatergic receptors) on VMAT-2 function.
As noted above, the reduction in K+-stimulated DA release caused by METH is likely due to the diminished vesicular DA uptake and content caused by the stimulant. An additional potential consequence of the decrease in vesicular DA uptake is its contribution to the persistent dopaminergic deficits caused by the stimulant. In particular, it has been suggested that diminished VMAT-2 function promotes intracellular DA accumulation (for review, see Fleckenstein et al. 2009). Extra vesicular DA, in turn, can be readily oxidized and form highly reactive species (for review, see Stokes et al. 1999) that promote long-term damage to dopaminergic neurons. Support for this hypothesis comes from findings by Cubells and coworkers (1994) that METH promotes aberrant accumulation of DA and subsequently reactive species formation in dopaminergic neuronal cultures. Further, Fumagalli et al. (1999) reported that heterologous VMAT-2 knockout mice were more susceptible to METH-induced deficits compared with wild-type mice. In addition, Thomas et al. (2008) demonstrated that inhibition of VMAT-2 with reserpine worsens the persistent DA deficits caused by METH. Of relevance, D2 receptor antagonist pretreatment not only prevents the deficits in VMAT-2M function 24 h after treatment, but also the persistent dopaminergic deficits caused by METH treatment (O’Dell et al. 1993; Sonsalla et al. 1986; Hadlock et al. submitted) suggesting an association between these phenomena.
In conclusion, the present study, taken together with previous reports, revealed a complex series of effects on VMAT-2-containing vesicles that vary according to the vesicle population under study, the METH dosing regimen, and the time after METH administration. The decreases in VMAT-2 function in both populations likely contribute to the persistent dopaminergic deficits caused by the stimulant and the decrease in K+-stimulated DA release caused by METH.
Acknowledgments
This work was supported by grants DA 00869, DA 04222, DA 13367, DA 11389, DA 019447, and DA 00378 from the National Institute on Drug Abuse.
Abbreviations
- METH
methamphetamine
- DA
dopamine
- VMAT-2
vesicular monoamine transporter-2
- DAT
dopamine transporter
- RDE
rotating disk electrode
References
- Bowyer JF, Masserano JM, Weiner N. Inhibitory effects of amphetamine on potassium-stimulated release of [H3]dopamine from striatal slices and synaptosomes. J Pharmacol Exp Ther. 1987;240:177–186. [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Broening HW, Morford LL, Vorhees CV. Interactions of dopamine D1 and D2 receptor antagonists with D-methamphetamine-induced hyperthermia and striatal dopamine and serotonin reductions. Synapse. 2005;56:84–93. doi: 10.1002/syn.20130. [DOI] [PubMed] [Google Scholar]
- Brown JM, Hanson GR, Fleckenstein AE. Methamphetamine rapidly decreases vesicular dopamine uptake. J Neurochem. 2000;74:2221–2223. doi: 10.1046/j.1471-4159.2000.0742221.x. [DOI] [PubMed] [Google Scholar]
- Brown JM, Riddle EL, Sandocal V, Weston RK, Hanson JE, Crosby MJ, Ugarte YV, Gibb JW, Hanson GR, Fleckenstein AE. A single methamphetamine administration rapidly decreases vesicular dopamine uptake. J Pharmacol Exp Ther. 2002;302:497–501. doi: 10.1124/jpet.302.2.497. [DOI] [PubMed] [Google Scholar]
- Bustamante D, You Z-B, Castel M-N, Johansson S, Goiny M, Terenius L, Hokfelt T, Herrera-Marschitz M. Effect of single and repeated methamphetamine treatment on neurotransmitter release in substantia nigra and neostriatum of the rat. J Neurochem. 2002;83:645–654. doi: 10.1046/j.1471-4159.2002.01171.x. [DOI] [PubMed] [Google Scholar]
- Chapin DS, Lookingland KJ, Moore KE. Effects of LC mobile phase composition on retention times for biogenic amines, and their precursors and metabolites. Curr Sep. 1986;7:68–70. [Google Scholar]
- Chu P-W, Seferian KS, Birdsall E, Truong JG, Riordan JA, Metcalf CS, Hanson GR, Fleckenstein AE. Differential regional effects of methamphetamine on dopamine transport. Eur J Pharmacol. 2008;590:105–110. doi: 10.1016/j.ejphar.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyerman DJ, Yamamoto BK. A rapid oxidation and persistent decrease in the vesicular monoamine transporter 2 after methamphetamine. J Neurochem. 2007;103:1219–1227. doi: 10.1111/j.1471-4159.2007.04837.x. [DOI] [PubMed] [Google Scholar]
- Farnsworth SJ, Volz TJ, Hanson GR, Fleckenstein AE. Cocaine alters vesicular dopamine sequestration and potassium-stimulated dopamine release: the role of D2 receptor activation. J Pharmacol Exp Ther. 2009;328:807–812. doi: 10.1124/jpet.108.146159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckenstein AE, Gibb JW, Hanson GR. Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur J Pharmacol. 2000;406:1–13. doi: 10.1016/s0014-2999(00)00639-7. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. J Pharmacol Exp Ther. 1997;282:834–838. [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Hanson GR. Psychostimulant-induced alterations in vesicular monoamine transporter-2 function: Neurotoxic and therapeutic implications. Neuropharmacology. 2009;56:133–138. doi: 10.1016/j.neuropharm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasnier B. The loading of neurotransmitters into synaptic vesicles. Biochime. 2000;82:327–337. doi: 10.1016/s0300-9084(00)00221-2. [DOI] [PubMed] [Google Scholar]
- Gibb JW, Hanson GR, Johnson M. Neurochemical mechanisms of toxicity. In: Cho AK, Segal DS, editors. Amphetamine and its analogs. Academic Press; San Diego: 1994. pp. 269–295. [Google Scholar]
- Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li Y, Emson PC, Miller GW. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlock GC, Baucum AJ, II, King JL, Horner KA, Cook GA, Gibb JW, Wilkins DG, Hanson GR, Fleckenstein AE. Mechanisms underlying methamphetamine-induced dopamine transporter complex formation. J Pharmacol Exp Ther. 2009;329:169–174. doi: 10.1124/jpet.108.145631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan KA, Staal RG, Sonsaalla PK. Vesicular uptake of DA, but not [3H]-DHTB binding to VMAT-2, is compromised 24 h after methamphetamine treatment. Soc Neurosci Abstr. 1999;25:1965. [Google Scholar]
- Hogan KA, Staal RG, Sonsaalla PK. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: disparity between homogenates and vesicle preparations. J Neurochem. 2000;74:2217–2220. doi: 10.1046/j.1471-4159.2000.0742217.x. [DOI] [PubMed] [Google Scholar]
- Hotchkiss AJ, Morgan ME, Gibb JW. The long-term effects of multiple doses of methamphetamine on neostriatal tryptophan hydroxylase, tyrosine hydroxylase, choline acetyltransferase, and glutamate decarboxylase activities. Life Sci. 1979;25:1373–1378. doi: 10.1016/0024-3205(79)90414-4. [DOI] [PubMed] [Google Scholar]
- Kogan FJ, Nichols WK, Gibb JW. Influence of methamphetamine on nigral and striatal tyrosine hydroxylase activity and on striatal dopamine levels. Eur J Pharmacol. 1976;36:363–371. doi: 10.1016/0014-2999(76)90090-x. [DOI] [PubMed] [Google Scholar]
- Kokoshka JM, Metzger RR, Wilkins DG, Gibb JW, Hanson GR, Fleckenstein AE. Methamphetamine treatment rapidly inhibits serotonin, but not glutamate, transporters in rat brain. Brain Res. 1998;799:78–83. doi: 10.1016/s0006-8993(98)00472-7. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Koyama T, Yamashita I. Long-lasting decrease in dopamine uptake sites following repeated administration of methamphetamine in the rat striatum. Brain Res. 1993;601:209–212. doi: 10.1016/0006-8993(93)91712-2. [DOI] [PubMed] [Google Scholar]
- O’Dell SJ, Weihmuller FB, Marshall JF. Methamphetamin-induced dopamine overflow and injury to striatal dopamine terminals: attenuation by dopamine D1 or D2 antagonists. J Neurochem. 1993;60:1792–1799. doi: 10.1111/j.1471-4159.1993.tb13405.x. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle EL, Topham MK, Haycock JW, Hanson GR, Fleckenstein AE. Differenrial trafficking of the vesicular monoamine transporter-2 by methamphetamine and cocaine. Eur J Pharmacol. 2002;449:71–74. doi: 10.1016/s0014-2999(02)01985-4. [DOI] [PubMed] [Google Scholar]
- Sandoval V, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J Pharmacol Exp Ther. 2003;304:1181–1187. doi: 10.1124/jpet.102.045005. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonsalla PK, Gibb JW, Hanson GR. Role of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J Pharmacol Exp Ther. 1986;238:932–937. [PubMed] [Google Scholar]
- Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Rev. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Pothos EN. Regulation of quantal size by presynaptic mechanisms. Rev Neurosci. 2000;11:159–212. doi: 10.1515/revneuro.2000.11.2-3.159. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Pothos E, Sung HM, Maidment NT, Hobel BG, Rayport S. Weak base model of amphetamine action. Ann NY Acad Sci. 1992;654:525–528. doi: 10.1111/j.1749-6632.1992.tb26020.x. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog in Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2007.05155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang G-J, et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psych. 2001;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Volz TJ, Farnsworth SJ, King JL, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate administration alters vesicular monoamine transporter-2 function in cytoplasmic and membrane-associated vesicles. J Pharmacol Exp Ther. 2007;323:738–745. doi: 10.1124/jpet.107.126888. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in humans, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Woolverton WL, Ricaurte GA, Forno LS, Seiden LS. Long-term effects of chronic methamphetamine administration in rhesus monkeys. Brain Res. 1989;486:73–78. doi: 10.1016/0006-8993(89)91279-1. [DOI] [PubMed] [Google Scholar]