

Abstract
One of the most promising strategies to increase the efficacy of standard chemotherapy drugs is by combining them with low doses of histone deacetylases inhibitors (HDACis). Regarded as chemosensitizers, the addition of well-tolerated doses of HDACis to platinum-based chemotherapeutics has been proven in vitro and in vivo in recent studies for many cancer types and stages. In this review, we discuss the most commonly used combinations of histone deacetylase inhibitors and platinum based drugs in the context of their possible mechanisms, efficiency, efficacy, and related drawbacks in preclinical and clinical studies.
Keywords: Histone deacetylase, cisplatin, cancer, chemotherapy, HDAC inhibitor, combination therapy, platinum drug
1. Introduction
Cancer can result from defects in genes such as mutations, deletions, and chromosomal abnormalities. These changes may result in the hyper-activation of oncogenes and/or the loss of function of tumor suppressor genes, allowing uncontrolled growth of malignant cells [1,2,3]. Epigenetic modifications, reversibly alter gene expression and can contribute to the initiation and progression of cancer. The reversible nature of epigenetic modifications in contrast to genetic alterations can permit malignant cells to return to a normal state. Recent advancements in the rapidly evolving field of cancer epigenetics show the extensive involvement of epigenetic machinery components in cancer [4,5,6,7,8,9]. Epigenetic abnormalities have been linked not only to cancer, but also to degenerative disorders, neuronal disorders, pediatric syndromes, autoimmune diseases and aging [10,11,12].
Chemotherapy is an often-effective treatment strategy that is designed to kill cancer cells in individuals with certain forms of cancers [13,14]. Many cancer therapies, such as Pt-based drugs, nitrogen-mustards, and drugs like Temozolomide, are based on DNA alkylation [15, 16]. Despite their popularity, the widespread application and efficacy of DNA alkylating agents is hindered by their toxic side effects, their limited activity against many types of cancer, and their susceptibility to acquired drug resistance. Platinum-based drugs such as cisplatin are used for the treatment of many cancers. While cisplatin is known to exert anticancer effects by multiple mechanisms, the best understood mode of action is via the generation of DNA lesions, initiation of the DNA damage response, and induction of mitochondrial apoptosis. Cisplatin treatment very often leads to the development of chemoresistance, thereby causing therapeutic failure. Therefore, an initial therapeutic success, associated with partial response or disease stabilization, is very common for cisplatin monotherapy. Understanding cisplatin resistance mechanism remains a key goal for improving anticancer therapy.
Cisplatin exerts anticancer effects via two intertwined signaling pathways: nuclear and cytoplasmic. Inert cisplatin is intracellularly activated by spontaneous aquation (hydrolysis) reactions in the cytoplasm. The resulting highly reactive molecules interact with a wide variety of cytoplasmic substrates, particularly nucleophiles such as reduced glutathione (GSH), methionine, metallothioneins and proteins. Therefore, cisplatin has the potential to deplete and tilt the redox balance toward oxidative stress and facilitate DNA damage. At the same time, cisplatin is susceptible to inactivation by a number of cytoprotective antioxidant pathways. Aquated cisplatin can also directly bind DNA to generate protein–DNA complexes and DNA–DNA adducts. These DNA-DNA adducts are known to form cisplatin-induced DNA lesions and account for most of the drug’s cytotoxicity. The inactivation of cisplatin by cytoplasmic scavengers limits the amount of reactive cisplatin in the cytoplasm, thus leading to elevated levels of GSH. This phenomenon has been observed in the context of cisplatin resistance, both in vitro and in vivo [17]. Most of the time, the inhibition of single pathways that sustain cisplatin resistance fails to restore sensitivity to normal levels. Effective strategies to both increase efficacy and reduce the adverse side effects of cisplatin chemotherapy treatment are being pursued. Combining classic chemotherapy with epigenetic drugs shows promise as they have been proven to have anti-cancer activity both in vitro and in vivo [18,19,20].
The principal mechanisms by which epigenetic modifications affect tissue-specific gene expression and gene silencing are DNA methylation, chromatin remodeling, microRNA post-transcriptional silencing, and covalent histone modification. Epigenetic changes via covalent modification of DNA and core histones create molecular landmarks that differentially activate or inactivate chromatin. Covalent histone modifications are conserved among eukaryotic organisms and serve as heritable marks for active or inactive chromatin. The regulation of chromosomal structure is mediated in part by modification of the histone core protein amino termini. These covalent modifications which include acetylation, methylation, phosphorylation, and ubiquitination, are a part of normal physiology, but can play a major role in carcinogenesis, with far-reaching implications for human biology and health [21,22]. Drugs that target specific enzymatic processes involved in epigenetic regulation are emerging as effective approaches for the treatment and prevention of certain cancer [23,24,25,26].
Histone acetylation, particularly for histones H3 and H4, is one of the most studied and best understood covalent histone modifications [27,28]. Acetyl groups are transferred to and from lysine residues on the N-terminal tail and on the surface of the nucleosome core. Dynamic regulation of acetylation is facilitated by two enzyme classes: histone acetyltransferases (HAT) and histone deacetylases (HDAC). The opposing actions of HAT and HDAC allow gene expression to be regulated in response to the environment. It has been shown that the changes in transcription due to altered expression levels of HAT and HDAC are closely associated with cancer [29,30]. Reduced levels of histone acetylation as a result of aberrant HDAC activity have been detected in several human tumors and appear to repress tumor suppressor genes, thereby contributing to tumor onset and progression. HDAC inhibitors (HDACis) can reactivate gene expression and inhibit the growth and survival of cancer cells. Several HDACis are currently FDA-approved for the treatment of certain types of cancers, diseases such as bipolar disorder, epilepsy, and depression, but they are finding a multitude of uses in other diseases such as diabetes and HIV [31,32,33]. The specificity of these HDACi compounds for cancer cells and their potency in vitro and in vivo highlight the potential of HDACis as potent agents for the treatment of cancer. HDACis have been shown to sensitize tumor cells to the induction of apoptosis by ionizing radiation, ultraviolet (UV) radiation, and several DNA-damaging agents [34,35,36,37,38].
Over the past several years, targeted cancer drugs have been considered among the most promising developments for obtaining high therapeutic effect with negligible toxicity. Compared to traditional chemotherapy drugs that can affect both healthy and cancer cells, targeted chemotherapy produces fewer adverse effects without impacting efficacy. There are numerous opportunities for targeted therapies because cancer cells utilize multiple intersecting and redundant biochemical pathways to remain viable. Modulating multiple pathways improves upon existing single-target agents, which generally realize only modest improvement and sub-therapeutic effect on solid tumors. Preclinical evidence has shown that adding HDACis to a diverse range of standard chemotherapeutic agents leads to synergistic induction of cancer cell death, demonstrating the utility of a multi-targeted approach involving HDAC modulation in the treatment of cancer. In this review, we focus on HDAC inhibition and platinum-based drug combined therapy for cancer.
2. HDAC inhibition for cancer therapy
Aberrant histone acetylation is observed in a multitude of disorders and cancers, altering gene transcription and ultimately affecting normal cellular behavior. Many malignant conditions demonstrate heightened activity of HAT, exposing transcription factors previously repressed by histone acetylation and altering the association of certain proteins with DNA. Many of the deleterious effects of unregulated HAT activity are mediated by changes in cell cycle control, affecting the function of checkpoint regulators such as p53, E2F, and GATA1. Unregulated HAT activity can also affect chromatin remodeling and promote uncontrolled replication [39]. HDACis are thought to possess anti-cancer activity because of their ability to halt the cell cycle and induce expression of pro-apoptotic proteins that may correct the proliferative state of cancer cells.
While HDAC inhibition alters less than 2% of transcriptional regulation of the entire genome, many important cell cycle regulators become upregulated with HDACis [40]. In a number of cancer cell lines, it was found that the CDKN1A gene encoding p21 was upregulated independently of p53 after treatment with HDAC inhibitors [41,40,42,43]. P21 causes growth arrest, usually in response to injurious stimuli, by inhibiting Cdk2 and halting the cell cycle at G1. Inhibition of Cdk activates retinoblastoma protein (pRb), inhibiting E2F, which is necessary for promotion of the cell cycle to the S phase and preventing further tumor growth. The mechanism of HDACi-induced apoptosis is not currently understood but may involve the re-establishment of tumor suppression machinery such as p53, upregulation of pro-apoptotic proteins, and downregulation of anti-apoptotic signaling [44]. Additionally, the apoptotic effect of HDACis may be drug-specific. Several studies report the activation of the intrinsic (mitochondrial) pathway of apoptosis following administration of SAHA, possibly through a combination of oxidative stress producing reactive oxygen species (ROS) and a reduction in the ability to counter free radicals via downregulation of thioredoxin [45,46]. HDACis may also promote apoptosis by reversing the silencing of pro-apoptosis proteins, including caspase-8, FAS, and APAF1 [47]. In response to the anti-proliferative and pro-apoptotic effects of HDACis, a number of structurally diverse small molecule inhibitors have been developed as cancer therapies [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. In preclinical studies, several classes of HDACis have shown broad and potent anti-tumor activity with low toxicity in normal cells [64,65,61]. Clinically, many HDAC inhibitors have been used as monotherapies for the treatment of cancer (Table 1). PXD101 (Belinostat) has been utilized in clinical trials for hepatocellular carcinoma, multiple myeloma, various ovarian cancers, B-cell non-Hodgkin’s lymphoma, T-cell lymphoma, AML, and several other types of malignant cancers [66,67,68,69,70,71].
Table 1.
HDACi mono-therapy for cancer
| HDACi | Cancer target | Mechanism | Results | Refs |
|---|---|---|---|---|
| Vorinostat/SAHA (Zolinza) | Cutaneous T-cell lymphoma (CTCL) | DAC Class I, IIa, V | USFDA approval for CTCL | [55,72,73] |
| Romidepsin (FK228 and FR901228) | Cutaneous T-cell lymphoma (CTCL) Peripheral T-cell lymphoma (PTCL) |
pan-HDAC Class I, Class II and Class IV HDACs | USFDA approval for CTCL Clinical Phase II |
[55] [74] |
| CRA-024781/(PCI 24781) | Refractory advanced solid tumors | Class I, IIb | Clinical phase I | [55, 75] |
| R306465/(JNJ- 16241199) | Advanced tumors | Class I | Clinical phase I | [55, 76] |
| LBH589 (panobinostat) | Advanced solid tumors or NHL, Advanced solid tumors or lymphoma | Class I, IIa, IIb, IV | Clinical phase III | [55,77,78] |
| Belinostat/PXD101 | Peripheral T-cell lymphoma (PTCL) Unresectable hepatocellular carcinoma multiple myeloma, various cancer, B-cell non-Hodgkin’s lymphoma, T-cell lymphoma, AML | Class I, IIa, IIb, IV | Clinical phase III | [55], [79], [66], [67], [68], [69], [70], [71] |
| VPA | Refractory advanced cancer | Class I, IIa | Clinical Phase II | [55] |
| SNDX- 275/entinostat (MS-275) | Advanced leukemia/MDS | HDAC 1, 2, 3, 9 | Clinical Phase II | [55,62] |
| MGCD0103 | Advanced leukemia/MDS Relapsed and or refractory classical HL |
HDAC 1, 2, 3, 11 | Clinical Phase I Clinical Phase I |
[57,62] [62] |
The FDA has approved two HDACis for hematological and solid tumors, and several other HDAC-targeted drug candidates are being evaluated clinical trials for other forms of cancer [18,26]. The HDACi suberoylanilide hydroxamic acid (SAHA) was the first HDACi to be approved by the FDA in 2006 for the treatment of cutaneous T-cell lymphoma (CTCL) [20]. The second HDACi a cyclic peptide, romidepsin, received FDA approval in 2009 to treat CTCL in patients who have received at least one prior systemic therapy [80]. While the chemotherapeutic effect of monotherapy with HDAC inhibitor in vitro is obvious, they are known to be effective on only a defined subset of hematological tumors. Their efficacy against solid tumors is significantly less than their efficacy against these hematological tumors; therefore, the combination of HDACis with other cancer drugs is a promising strategy to obtain the full therapeutic potential of these inhibitors in cancer [81].
3. Combined treatment: classic chemotherapy and HDAC inhibitors
Although chemotherapy and HDACis target different sites, the close connection and functional importance of DNA and chromosome structure in cancer development suggests an interaction between these two therapeutic methods [82,83]. The formation of DNA adducts by chemotherapy drugs requires accessibility to the DNA sequence, which can be regulated by altering DNA-associated proteins such as histones. Indeed, HDACis have been regarded as “sensitizer drugs” that display synergistic activity with other agents that alter DNA structure, such as DNA methyltransferase inhibitors and retinoic acids [44]. In addition, the activation of gene expression and induction of apoptosis have been reported in these combinative strategies [84,85].
Although the nature of the molecular mechanisms underpinning combination therapy is largely unknown, higher concentrations of histone deacetylases are believed to exert cell cycle redistribution, induction of apoptosis, and downregulation of survival signals [86,87]. The effect of HDAC inhibition has been attributed at least partly to histone acetylation leading to altered double strand break (DSB) formation and repair. The open chromatin structure induced by histone hyperacetylation renders the DNA more accessible to cellular processes, including damage. Chemotherapeutic agents such as cisplatin bind with DNA to form platinum-DNA adducts; such structural changes may lead to impaired DNA replication and site-specific recombination in cancer cells.
Cisplatin is eliminated from cells by glutathione, the most critical cellular antioxidant. Resistance develops from over-expression of anti-apoptotic proteins Bcl-2 and XIAP, which are associated with increased glutathione synthesis. A proposed mechanism suggests that chromatin relaxation by HDACis may promote accessibility of DNA to transcription factors, thereby downregulating the expression of genes such as Bcl-2 and XIAP and facilitating the formation of platinum-DNA adducts (Fig. 1) [88]. Relaxed chromatin structure and downregulated gene expression help enhance cytotoxicity by giving cisplatin better access to DNA while preventing cells from mitigating the resulting DNA damage. In practice, HDACis (SAHA and Sirtinol in particular) are effective in causing HeLa cells to succumb to the damage caused by platinum-based chemotherapeutics (Fig. 1) [88].
Fig. 1.

Proposed mechanisms for synergistic effect of HDACi and cisplatin combined therapy HDACi induces hyperacetylation of histones, relaxing chromatin and rendering it more accessible for cisplatin. Treatment with low doses of HDACi sensitizes cells to further damage by cisplatin by initiating a pro-apoptotic state defined by increased caspase activity and decreased anti-apoptotic proteins. Glutathione, a major inactivator of cisplatin, is synthesized in lower quantities in HDACi-treated cells.
A number of HDAC inhibitors have been explored in combination with chemotherapeutics, including SAHA, FK-228, MS-275, CI-994, and TSA [89,90,64,82,91,63]. Most of these studies have focused on hematologic cancers, however there are a few that have also examine efficacy in solid tumors [90]. While there are many different treatment combinations of HDACi and Pt drugs for cancer targets awaiting clinical investigation, the drugs most frequently used for combination therapy research with platinum-based chemotherapy are SAHA and valproic acid (VPA).
Cisplatin and various analogues are currently the standard of care for oral squamous cell carcinoma (OSCC). However, cisplatin treatment can result in acquired resistance of cancer cells and dose-dependent systemic toxicity. Many studies have demonstrated the benefit of co-administration of oral SAHA, with cell viability and apoptotic assays showing synergistic induction of cytotoxicity and apoptosis in human tongue, OSCC, Tca8113 and KB cell lines [92]. Furthermore, diverse apoptosis-associated proteins, including p53, BID, cytochrome C and caspase-3, were involved in the induction of apoptosis when treated with cisplatin and subtoxic doses of SAHA. This combinatory treatment is regarded as a potential novel strategy for treatment of OSCC [92].
Recent studies in OSCC show that combination of cisplatin with SAHA renders 60% of cells apoptotic after 48 hours of treatment and is associated with a number of elevated markers related to endoplasmic reticulum (ER) stress. Caspase-4 and -12 were significantly elevated, with caspase-12-related apoptosis being modulated via the caspase inhibitor z-VAD-fmk [88,94,95,21]. Additionally, SAHA alone rapidly induced sustained phosphorylation of eukaryotic translation initiation factor-2 (eIF2α), which is often observed in ER stress. This pathway appears to be particularly important in sensitizing cells to cisplatin because this effect is strongly challenged by administration of Salubrinal, a selective eIF-2α phosphatase inhibitor that prevents ER stress-related apoptosis [21].
The levels of phospho-Akt are reduced in SAHA-treated cells and are associated with increased activity of protein phosphatase 1 (PP1), a stimulator of retinoblastoma protein-mediated apoptosis [102]. These findings indicate that the induction of ER stress and upregulation of its associated pathways are integral to the mechanism by which SAHA enhances apoptosis, particularly by PP1 upregulation and Akt dephosphorylation [21]. Although cisplatin remains a vital mainstay of chemotherapy, the frequency of recurrence and drug resistance leads to poor survival rates for head and neck cancer patients and underscores the need to develop effective modulators of platinating agents [21]. This study has shown that SAHA enhances the efficacy of cisplatin by activating the atypical ER stress pathway to increase apoptosis. Further investigation is required to yield information about the targets of cisplatin and SAHA in this pathway.
Carboplatin is an analog of cisplatin with less nonhematologic toxicity than the parent molecule. A combination of carboplatin and paclitaxel, a non-Pt based mitotic inhibitor, is commonly used to treat solid malignancies, resulting in less platelet toxicity than treatment with carboplatin alone [103,104]. This combination has a toxicity profile that does not overlap with that of SAHA. A phase I clinical study by Ramalingam et al. sought to determine dosing guidelines for SAHA administered on two different schedules with carboplatin and paclitaxel [63]. Further studies on the same combination showed that SAHA improved the efficacy of carboplatin and paclitaxel in patients with advanced non–small-cell lung cancer (NSCLC) [91,63].
Cisplatin is also considered to be an effective tool in chemotherapy for cervical cancer [88]. Severe side effects, such as nephrotoxicity and ototoxicity, limit the dose of cisplatin despite excellent anti-neoplastic activity. In a study performed by Jin et al., a combination treatment of cisplatin and SAHA was tested on HeLa cervical cancer cells [88]. According to this study, low concentrations of SAHA worked synergistically with cisplatin to induce cytotoxicity to a greater extent than with either agent alone. The combination of cisplatin and SAHA induces a marked reduction in cell viability, as determined by Western blot analysis and DAPI staining, showing significantly increased caspase-3 activation and apoptosis in HeLa cells treated with both drugs compared to treatment with cisplatin alone.
Valproic acid (VPA), commonly used to treat epilepsy and bipolar mania, is the second most studied HDACi in combination therapy with platinum-based drugs. It has been reported that the VPA/cisplatin duo amplifies anticancer activity over cisplatin alone in many cancer cell lines, such as head and neck squamous cell carcinoma (HNSCC), human ovarian cancer (SK-OV-3, OVCAR-3, and TOV-21G), and melanoma (M14) [98,97,99].
Vandermeers et al. evaluated the anticancer effect of combining valproate treatment with pemetrexed and cisplatin on mesothelioma cells, a first-line regimen for this malignancy [96]. Present chemotherapeutic regimens are marginally efficacious in malignant pleural mesothelioma, as these tumor cells are particularly resistant to radio- and chemotherapy. Vandermeers utilized cell cultures and xenograft mouse models showing that the inclusion of HDAC inhibitors in the cisplatin and pemetrexed combination may improve the response to treatment. Based on these results, the potential benefit of adding valproate to standard chemotherapy in malignant pleural mesothelioma patients is being evaluated in a phase II clinical trial in refractory and recurrent cases [96].
HDAC inhibitors MS-275, TSA, and FK-228 have also been shown to enhance activity together with cisplatin on different cancer cell lines [22,100,95,101]. FK228, MS-275 and TSA enhanced the induction of cell death by cisplatin in an ovarian cancer cell line noted for its cisplatin resistance (OVCAR-8). PCl-24781, a highly potent HDAC class I and II inhibitor in various cancer cells at nanomolar concentrations, has shown synergistic anti-cancer effect in soft tissue sarcoma (STS) when combined with low dose conventional chemotherapy. PCI-24781 co-administered with cisplatin has been evaluated in STS cell lines and in vivo mouse studies [19]. Both models show that combined HDAC inhibition and low-dose chemotherapy exhibits anti-STS activity in vitro and in vivo. Increased cancer cell toxicity has been observed when combining cisplatin and TSA in human glioblastoma cell line D54 and breast cancer cell line MCF-7 [22]. The same combination of drugs re-sensitizes human bladder cancer cells that have become resistant to cisplatin [101]. Enhanced cytotoxicity, chemosensitization, and apoptosis are observed in treatment with MS-275 and cisplatin in human oral squamous cell carcinoma [95].
4. Future directions: Bifunctional molecules containing HDACi and DNA-Pt binding properties
It is well established that certain cancer cells exhibit a higher level of HDAC expression [105,106,107,108,109,110,111,112]. Furthermore, HDAC inhibitors are known to show preferential selectivity for malignant cells in many cancers. By coupling these effects in a single molecule, it is possible to target multiple pathways simultaneously, as shown in the work by Gleason et al (2008), which combined an HDACi with a nuclear vitamin D receptor ligand [113]. More recently, bifunctional molecules have been designed to improve selectivity by tethering an HDAC inhibitor to a platinum-based DNA binding agent [114], [115], [116], [117]. The HDAC inhibitor, with its known affinity for cancer cells, confers selectivity to the drug and is expected to deliver the chemotherapeutic DNA binding agent alongside the HDAC inhibitor to the tumor [118], [119], [61]. These molecules are thought to have a different mechanism from that of classic DNA binding platinum based drugs and are expected to be active against a broader spectrum of human cancer cells compared to classic platinating drugs.
Bifunctional molecules combining HDAC inhibitory and DNA alkylating activity were pioneered by Celine J. Marmion et al. [115,116,117]. As shown in Figure 2, a SAHA derivative was designed to contain a platinum DNA-binding moiety in such a way that its respective actions would ideally remain unchanged. This bifunctional molecule showed marked selectivity for tumor cells relative to non-tumorigenic cells, offering a distinct advantage over treatments involving cisplatin alone. It has been observed that the presence of the malonate linker on the SAHA moiety reduced the anticipated dual functionality once inside the cell nucleus [120]. As such, covalent linker groups should be carefully selected so as not to affect activity. Bifunctional molecules trans-[Pt(VPA–1H)2(NH3)(py)] and trans-[Pt(VPA–1H)2(py)2] containing trans-Pt complexes of the HDACi valproic acid (VPA) were recently reported by the same research group, and treatment with these drugs resulted in apoptosis in A2780 (parental) and A2780 cisR (cisplatin resistant) ovarian cancer cells [116].
Fig. 2.
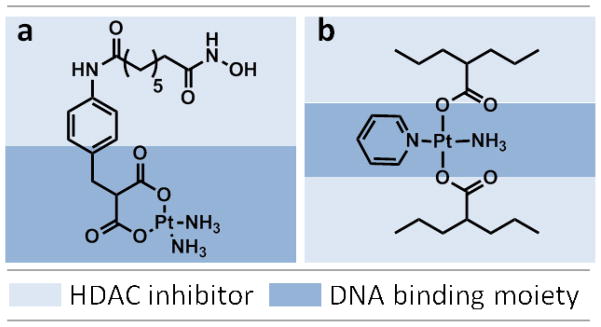
Bifunctional molecules with dual capability of HDAC inhibition and Platinum-DNA binding. a) cis-[Pt(NH3)2(malSAHA–2H)], b) trans-[Pt(VPA–1H)2(NH3)(py)]
5. Conclusion and outlook
Therapy that induces DNA damage is currently the most popular treatment modality for cancer, manifesting a substantial increase in survival of cancer patients. Unfortunately, some cancers do not respond to the treatment or develop resistance to DNA damaging agents. The utility of HDAC inhibition in chemosensitizers that increase the efficacy of Pt-based chemotherapeutics has been explored in several studies, and HDAC inhibitors were found to benefit even cross-resistant strains of malignancies at very low doses. Preclinical studies carried out in vitro and in vivo by combination therapy with HDACis and standard-of-care platinum-based agents have shown synergistic improvement in cytotoxicity to cancer cells.
Despite evidence showing that HDAC inhibition can enhance anti-cancer efficacy of a diverse range of traditional chemotherapeutics in vitro, the underpinning mechanism remains unverified. By identifying this mechanism of action, it would be possible to rationalize these treatment combinations and optimize them for maximal effect in a variety of cancer types. The effect of any combination regimen using HDACis and platinum drugs can be analyzed by integrating functional cell-based assays with preclinical testing using animal models of cancer to obtain details on mechanisms of action, therapeutic efficacy, and toxicity.
In many cases, issues of selectivity and the emergence of adverse effects have undermined the use of HDACis with classic chemotherapy against many cancers. More recently, novel drugs that complex HDAC-binding agents with platinum-based chemotherapeutics have been synthesized that would ideally reduce the toxic effect these drugs have on normal, healthy cells. Most of these studies are in preclinical phase I testing, awaiting further investigation to evaluate their curative potential. Future work in the field should similarly focus on improving selectivity of these drugs to amplify accumulation in cancer cells at lower, presumably less toxic, doses. Most importantly, understanding the mechanisms by which combination therapy suppresses tumors in each type and stage of cancer is a major challenge that needs to be overcome to advance treatment development.
Table 2.
Pre-clinical studies on combined treatment of HDACi with platinum-based chemotherapeutics
| HDACi | Cancer target | Results | Refs |
|---|---|---|---|
| SAHA w/cisplatin | oral squamous cell carcinoma | cisplatin and SAHA increase cytotoxicity and sensitize OSCC cells to apoptosis | [92] |
| SAHA w/cisplatin | oral squamous cell carcinoma | increases endoplasmic reticulum stress-mediated apoptosis | [93] |
| SAHA w/cisplatin | oral squamous cell carcinoma cells | Chemosensitization and apoptotic enhancement | [94,95] |
| SAHA Carboplatin (with Paclitaxel) | advanced solid malignancies (head and neck) | increase anticancer activity (now in clinical trial for advanced solid malignancies | [91] |
| SAHA w/cisplatin | human glioblastoma (D54), breast cancer (MCF-7) | increases cytotoxicity of cisplatin | [82] |
| SAHA w/cisplatin | Cervical cancer (HeLa cells) | has synergistic effect on the HeLa cell viability | [88] |
| VPA w/cisplatin and pemetrexed | mesothelioma (cell cultures & tumor xenografts) | increases antitumor efficacy; being evaluated in phase II trial for refractory/recurrent malignant pleural mesothelioma | [96] |
| VPA w/cisplatin | head and neck squamous cell carcinoma (HNSCC) | enhances the anticancer activity of cisplatin in HNSCC cell lines | [97] |
| VPA w/cisplatin | human ovarian cancer (SK- OV-3, OVCAR-3, and TOV- 21G) | synergistic cytotoxicity with cisplatin; enhances cisplatin- mediated cytotoxicity in resistant cancer cells | [98] |
| VPA w/cisplatin | M14 melanoma cells | sensitizes M14 cells to chemotherapy treatment | [99] |
| FK-228 MS-275, TSA | human ovarian & colon cancer cell lines | enhances cytotoxic effects reduced dose of chemo agent | [100] |
| PCI-24781 | soft tissue sarcoma | enhances cytotoxic effects | [19] |
| TSA | human glioblastoma (D54) breast cancer (MCF-7) | increases cytotoxicity of cisplatin | [82] |
| TSA | Human Bladder Cancer Cell Line | resensitizes cisplatin- resistant human bladder cancer cells | [101] |
| MS-275 | human oral squamous carcinoma cells | enhanceds cytotoxicity, chemosensitization, and apoptosis | [95] |
Table 3.
Clinical trials of HDACi and Pt drugs combination therapy
| HDACi | Cancer target | Purpose | Gov. registration # | Patient population | Results | Clinical phase |
|---|---|---|---|---|---|---|
| SAHA (CDDP+Capecitabine) | Metastatic or Recurrent Gastric Cancer | Exploring the role of SAHA | NCT01045538 | 45 | CR | I/II |
| VPA(Hydralazine) | Cervical cancer | Determining response rate, safety, biological effects | NCT00404326 | 17 | Completed | II |
| Belinostat (CDDP+Etoposide) | Small cell lung carcinoma | Determining a safe, tolerable phase II dose | NCT00926640 | 39 | CR | I |
| CUDC-101 (CDDP) | Head and neck | A dose escalation study | NCT01384799 | 22 | CR | I |
| SAHA | Non-small cell lung cancer (non-SCLC) | determining the maximum tolerated dose | NCT01059552 | 22 | CR | I |
| SAHA(CDDP+Isotrti noin+Combined chemotherapy) | Embryonal tumors (Brain and Central Nervous System Tumors) | Side effects of giving SAHA isotretino in together with chemotherapy | NCT00867178 | 62 | CR | I |
| SAHA(CDDP+ Pemetrexed) | Advanced solid tumors | safety and tolerability | NCT00106626 | 52 | Safety issues: Yes | I |
| MK0683 (CDDP+Gemcitabine) | Non-SCLC | safety and tolerability | NCT00423449 | 61 | MTD 400 mg for up to 10 days in 21- day cycles Safety issues: Yes | I |
Ref source: Clinicaltrials.gov
CDDP: Cisplatin, CR: Current, MTD: Maximum tolerated dose, SCLC: Small cell lung cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alberghina L, Chiaradonna F, Vanoni M. Systems biology and the molecular circuits of cancer. Chem bio chem. 2004;5:1322–1333. doi: 10.1002/cbic.200400170. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER. Human cancer syndromes: clues to the origin and nature of cancer. Science. 1997;278:1043–1050. doi: 10.1126/science.278.5340.1043. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature medicine. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 4.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature Reviews Genetics. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 5.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 6.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawan C, Vaissière T, Murr R, Herceg Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2008;642:1–13. doi: 10.1016/j.mrfmmm.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Feinberg AP, Tycko B. The history of cancer epigenetics. Nature Reviews Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 9.Laird PW. Cancer epigenetics. Human molecular genetics. 2005;14:R65–R76. doi: 10.1093/hmg/ddi113. [DOI] [PubMed] [Google Scholar]
- 10.Rodenhiser D, Mann M. Epigenetics and human disease: translating basic biology into clinical applications. Canadian Medical Association Journal. 2006;174:341–348. doi: 10.1503/cmaj.050774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Molecular pharmacology. 2010;77:126–135. doi: 10.1124/mol.109.061333. [DOI] [PubMed] [Google Scholar]
- 12.Gray SG. Epigenetic treatment of neurological disease. Epigenomics. 2011;3:431–450. doi: 10.2217/epi.11.67. [DOI] [PubMed] [Google Scholar]
- 13.DeVita VT, Chu E. A history of cancer chemotherapy. Cancer research. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 14.Chabner BA, Roberts TG. Chemotherapy and the war on cancer. Nature Reviews Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 15.Olszewski U, Hamilton G. A better platinum-based anticancer drug yet to come? Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry. 2010;10:293–301. doi: 10.2174/187152010791162306. [DOI] [PubMed] [Google Scholar]
- 16.Sherman SE, Lippard SJ. Structural aspects of platinum anticancer drug interactions with DNA. Chemical Reviews. 1987;87:1153–1181. [Google Scholar]
- 17.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 18.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. The oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 19.Lopez G, Liu J, Ren W, Wei W, Wang S, Lahat G, Zhu QS, Bornmann WG, McConkey DJ, Pollock RE. Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clinical cancer research. 2009;15:3472–3483. doi: 10.1158/1078-0432.CCR-08-2714. [DOI] [PubMed] [Google Scholar]
- 20.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer research. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 21.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 22.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Young H. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nature medicine. 2001;7:437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 23.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nature Reviews Drug Discovery. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 24.Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJM, Anzalone L, Pezzani L, Di Giacomo AM, Fonsatti E. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. Journal of cellular physiology. 2007;212:330–344. doi: 10.1002/jcp.21066. [DOI] [PubMed] [Google Scholar]
- 25.PAPI A, FERRERI AM, ROCCHI P, GUERRA F, ORLANDI M. Epigenetic modifiers as anticancer drugs: effectiveness of valproic acid in neural crest-derived tumor cells. Anticancer research. 2010;30:535–540. [PubMed] [Google Scholar]
- 26.Balch C, Fang F, Matei DE, Huang THM, Nephew KP. Minireview: epigenetic changes in ovarian cancer. Endocrinology. 2009;150:4003–4011. doi: 10.1210/en.2009-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 28.Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–392. doi: 10.1016/s0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- 29.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Molecular cancer therapeutics. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 30.Hendrich B, Bickmore W. Human diseases with underlying defects in chromatin structure and modification. Human molecular genetics. 2001;10:2233–2242. doi: 10.1093/hmg/10.20.2233. [DOI] [PubMed] [Google Scholar]
- 31.DeVane C. Pharmacokinetics, drug interactions, and tolerability of valproate. Psychopharmacology bulletin. 2003;37:25–42. [PubMed] [Google Scholar]
- 32.Dinarello CA, Fossati G, Mascagni P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Molecular Medicine. 2011;17:333–352. doi: 10.2119/molmed.2011.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wightman F, Ellenberg P, Churchill M, Lewin SR. HDAC inhibitors in HIV. Immunology and Cell Biology. 2012;90:47–54. doi: 10.1038/icb.2011.95. [DOI] [PubMed] [Google Scholar]
- 34.Ververis K, Karagiannis TC. Overview of the Classical Histone Deacetylase Enzymes and Histone Deacetylase Inhibitors. ISRN Cell Biology. 2012;2012:1–12. [Google Scholar]
- 35.Touma SE, Goldberg JS, Moench P, Guo X, Tickoo SK, Gudas LJ, Nanus DM. Retinoic acid and the histone deacetylase inhibitor trichostatin a inhibit the proliferation of human renal cell carcinoma in a xenograft tumor model. Clinical cancer research. 2005;11:3558–3566. doi: 10.1158/1078-0432.CCR-04-1155. [DOI] [PubMed] [Google Scholar]
- 36.Kwa FAA, Balcerczyk A, Licciardi P, El–Osta A, Karagiannis TC. Chromatin modifying agents-the cutting edge of anticancer therapy. Drug discovery today. 2011;16:543–547. doi: 10.1016/j.drudis.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clinical cancer research. 2009;15:3970–3977. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
- 38.Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 39.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nature Reviews Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 40.Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene expression. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- 41.Kim YB, Ki SW, Yoshida M, Horinouchi S. Mechanism of cell cycle arrest caused by histone deacetylase inhibitors in human carcinoma cells. The Journal of antibiotics. 2000;53:1191–1200. doi: 10.7164/antibiotics.53.1191. [DOI] [PubMed] [Google Scholar]
- 42.Rosato RR, Grant S. Histone deacetylase inhibitors in cancer therapy. Cancer Biology and Therapy. 2003;2:30–37. doi: 10.4161/cbt.190. [DOI] [PubMed] [Google Scholar]
- 43.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proceedings of the National Academy of Sciences. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer letters. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 45.Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, Richon VM. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proceedings of the National Academy of Sciences. 2002;99:11700–11705. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, Smyth MJ, Johnstone RW. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proceedings of the National Academy of Sciences. 2001;98:10833–10838. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis-A Link between Cancer Genetics and Chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 48.Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Molecular pharmacology. 2005;68:917–932. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- 49.Botrugno OA, Santoro F, Minucci S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer letters. 2009;280:134–144. doi: 10.1016/j.canlet.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 50.Cang S, Ma Y, Liu D. New clinical developments in histone deacetylase inhibitors for epigenetic therapy of cancer. J Hematol Oncol. 2009;2:22–33. doi: 10.1186/1756-8722-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical Development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 52.Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 2009;69:1911–1934. doi: 10.2165/11315680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Manero G, Issa JP. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer investigation. 2005;23:635–642. doi: 10.1080/07357900500283119. [DOI] [PubMed] [Google Scholar]
- 54.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nature Reviews Drug Discovery. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 55.Lee MJ, Kim YS, Kummar S, Giaccone G, Trepel JB. Histone deacetylase inhibitors in cancer therapy. Current opinion in oncology. 2008;20:639. doi: 10.1097/CCO.0b013e3283127095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marchion D, Munster P. Development of histone deacetylase inhibitors for cancer treatment. Expert review of anticancer therapy. 2007;7:583–598. doi: 10.1586/14737140.7.4.583. [DOI] [PubMed] [Google Scholar]
- 57.Marks P. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 58.Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Current opinion in oncology. 2001;13:477. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 59.McLaughlin F, La Thangue NB. Histone deacetylase inhibitors open new doors in cancer therapy. Biochemical pharmacology. 2004;68:1139–1144. doi: 10.1016/j.bcp.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 60.Muller S, Kramer OH. Inhibitors of HDACs-Effective Drugs Against Cancer? Current cancer drug targets. 2010;10:210–228. doi: 10.2174/156800910791054149. [DOI] [PubMed] [Google Scholar]
- 61.Takai N, Narahara H. Preclinical studies of chemotherapy using histone deacetylase inhibitors in endometrial cancer. Obstetrics and gynecology international. 2010;2010:1–8. doi: 10.1155/2010/923824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clinical cancer research. 2009;15:3958–3969. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 63.Ramalingam SS, Parise RA, Ramananthan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clinical cancer research. 2007;13:3605–3610. doi: 10.1158/1078-0432.CCR-07-0162. [DOI] [PubMed] [Google Scholar]
- 64.Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochemical pharmacology. 2007;74:659–671. doi: 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 65.Marks P, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. Journal of cellular biochemistry. 2009;107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cashen A, Juckett M, Jumonville A, Litzow M, Flynn P, Eckardt J, LaPlant B, Laumann K, Erlichman C, DiPersio J. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS) Annals of hematology. 2011:1–6. doi: 10.1007/s00277-011-1240-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giaccone G, Rajan A, Berman A, Kelly RJ, Szabo E, Lopez-Chavez A, Trepel J, Lee MJ, Cao L, Espinoza-Delgado I. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. Journal of Clinical Oncology. 2011;29:2052–2059. doi: 10.1200/JCO.2010.32.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, Buhl-Jensen P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. European journal of haematology. 2008;81:170–176. doi: 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 69.Mackay HJ, Hirte H, Colgan T, Covens A, MacAlpine K, Grenci P, Wang L, Mason J, Pham PA, Tsao MS. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. European Journal of Cancer. 2010;46:1573–1579. doi: 10.1016/j.ejca.2010.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramalingam SS, Belani CP, Ruel C, Frankel P, Gitlitz B, Koczywas M, Espinoza-Delgado I, Gandara D. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. Journal of Thoracic Oncology. 2009;4:97. doi: 10.1097/JTO.0b013e318191520c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steele NL, Plumb JA, Vidal L, Tjørnelund J, Knoblauch P, Rasmussen A, Ooi CE, Buhl-Jensen P, Brown R, Evans TRJ. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clinical cancer research. 2008;14:804–810. doi: 10.1158/1078-0432.CCR-07-1786. [DOI] [PubMed] [Google Scholar]
- 72.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. Journal of Clinical Oncology. 2007;25:3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 73.Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piekarz R, Frye R, Wright J, Figg W, Allen S, Kirschbaum M, Zain J, Hutchins L, Showe L, Fojo T. Update of the NCI multiinstitutional phase II trial of romidepsin, FK228, for patients with cutaneous or peripheral T-cell lymphoma. J Clin Oncol. 2007;25:8027. [Google Scholar]
- 75.Undevia S, Janisch L, Schilsky R, Loury D, Balasubramanian S, Mani C, Sirisawad M, Buggy J, Miller R, Ratain M. Phase I study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of the histone deacetylase inhibitor (HDACi) PCI-24781. J Clin Oncol. 2008;26:14514S. [Google Scholar]
- 76.Fong P, Settatree S, Sinha R, Hardcastle A, Hellemans P, Arts J, Brown K, Janicot M, Aherne W, De Bono J. A first-in-man phase I study of R306465, a histone deacetylase (HDAC) inhibitor exploring pharmacokinetics (PK) and pharmacodynamics (PD) utilizing an electrochemiluminescent immunoassay in patients (p) with advanced tumours. J Clin Oncol. 2007;25:3578. [Google Scholar]
- 77.Prince H, George D, Patnaik A, Mita M, Dugan M, Butterfoss D, Masson E, Culver K, Burris H, Beck J. Phase I study of oral LBH589, a novel deacetylase (DAC) inhibitor in advanced solid tumors and non-hodgkin’s lymphoma. J Clin Oncol. 2007;25:3500. [Google Scholar]
- 78.Sharma S, Vogelzang N, Beck J, Patnaik A, Mita M, Dugan M, Hwang A, Masson E, Culver K, Prince H. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of LBH589, a novel deacetylase (DAC) inhibitor given intravenously on a new once weekly schedule. J Clin Oncol. 2007;25:14019. [Google Scholar]
- 79.Yeo W, Lim R, Ma B, Hui P, Chan L, Mo F, Yu S, Ho S, Koh J, Chan A. A phase I/II study of belinostat (PXD101) in patients with unresectable hepatocellular carcinoma. J Clinical Oncol. 2007;25:15081. [Google Scholar]
- 80.Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, Gardner ER, Figg WD, Bates SE. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert review of anticancer therapy. 2010;10:997–1008. doi: 10.1586/era.10.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rangwala S, Zhang C, Duvic M. HDAC inhibitors for the treatment of cutaneous T-cell lymphomas. Future. 2012;4:471–486. doi: 10.4155/fmc.12.6. [DOI] [PubMed] [Google Scholar]
- 82.Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer research. 2003;63:7291–7300. [PubMed] [Google Scholar]
- 83.Nelson SM, Ferguson LR, Denny WA. DNA and the chromosome–varied targets for chemotherapy. Cell & chromosome. 2004;3:2–28. doi: 10.1186/1475-9268-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Whang YM, Choi EJ, Seo JH, Kim JS, Yoo YD, Kim YH. Hyperacetylation enhances the growth-inhibitory effect of all-trans retinoic acid by the restoration of retinoic acid receptor β expression in head and neck squamous carcinoma (HNSCC) cells. Cancer chemotherapy and pharmacology. 2005;56:543–555. doi: 10.1007/s00280-004-0970-3. [DOI] [PubMed] [Google Scholar]
- 85.Zhu WG, Lakshmanan RR, Beal MD, Otterson GA. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer research. 2001;61:1327–1333. [PubMed] [Google Scholar]
- 86.Mehnert JM, Kelly WK. Histone deacetylase inhibitors: biology and mechanism of action. The Cancer Journal. 2007;13:2329. doi: 10.1097/PPO.0b013e31803c72ba. [DOI] [PubMed] [Google Scholar]
- 87.Xu W, Parmigiani R, Marks P. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 88.Jin KL, Park JY, Noh EJ, Hoe KL, Lee JH, Kim JH, Nam JH. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. Journal of Gynecologic Oncology. 2010;21:262–268. doi: 10.3802/jgo.2010.21.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blumenschein GR, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, Chiao JH, Chen C, Frankel SR. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza™, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Investigational new drugs. 2008;26:81–87. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 90.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature Reviews Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 91.Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, Thomas S, Espinoza-Delgado I, Vokes EE, Gandara DR. Carboplatin and Paclitaxel in Combination With Either Vorinostat or Placebo for First-Line Therapy of Advanced Non–Small-Cell Lung Cancer. Journal of Clinical Oncology. 2010;28:56–62. doi: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shen J, Huang C, Jiang L, Gao F, Wang Z, Zhang Y, Bai J, Zhou H, Chen Q. Enhancement of cisplatin induced apoptosis by suberoylanilide hydroxamic acid in human oral squamous cell carcinoma cell lines. Biochemical pharmacology. 2007;73:1901–1909. doi: 10.1016/j.bcp.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 93.Suzuki M, Endo M, Shinohara F, Echigo S, Rikiishi H. Enhancement of cisplatin cytotoxicity by SAHA involves endoplasmic reticulum stress-mediated apoptosis in oral squamous cell carcinoma cells. Cancer chemotherapy and pharmacology. 2009;64:1115–1122. doi: 10.1007/s00280-009-0969-x. [DOI] [PubMed] [Google Scholar]
- 94.Rikiishi H, Shinohara F, Sato T, Sato Y, Suzuki M, Echigo S. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. International journal of oncology. 2007;30:1181–1188. [PubMed] [Google Scholar]
- 95.Sato T, Suzuki M, Sato Y, Echigo S, Rikiishi H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. International journal of oncology. 2006;28:1233–1241. [PubMed] [Google Scholar]
- 96.Vandermeers F, Hubert P, Delvenne P, Mascaux C, Grigoriu B, Burny A, Scherpereel A, Willems L. Valproate, in combination with pemetrexed and cisplatin, provides additional efficacy to the treatment of malignant mesothelioma. Clinical cancer research. 2009;15:2818–2828. doi: 10.1158/1078-0432.CCR-08-1579. [DOI] [PubMed] [Google Scholar]
- 97.Erlich RB, Rickwood D, Coman WB, Saunders NA, Guminski A. Valproic acid as a therapeutic agent for head and neck squamous cell carcinomas. Cancer chemotherapy and pharmacology. 2009;63:381–389. doi: 10.1007/s00280-008-0747-1. [DOI] [PubMed] [Google Scholar]
- 98.Bloemink M, Heetebrij R, Inagaki K, Kidani Y, Reedijk J. Reactions of unsymmetrically substituted derivatives of cisplatin with short oligodeoxynucleotides containing a-GpG-sequence: H-bonding interactions in pGpG moieties cross-linked by an asymmetric platinum complex enhancing the formation of one geometrical isomer. Inorganic Chemistry. 1992;31:4656–4661. [Google Scholar]
- 99.Valentini A, Gravina P, Federici G, Bernardini S. Valproic Acid Induces Apoptosis, p16INK4A Upregulation and Sensitization to Chemotherapy in Human Melanoma Cells. Cancer Biology and Therapy. 2007;6:185. doi: 10.4161/cbt.6.2.3578. [DOI] [PubMed] [Google Scholar]
- 100.Ozaki K, Kishikawa F, Tanaka M, Sakamoto T, Tanimura S, Kohno M. Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross-resistance to a wide range of DNA-damaging drugs. Cancer science. 2008;99:376–384. doi: 10.1111/j.1349-7006.2007.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yoon CY, Park MJ, Lee JS, Lee SC, Oh JJ, Park H, Chung CW, Abdullajanov MM, Jeong SJ, Hong SK. The Histone Deacetylase Inhibitor Trichostatin A Synergistically Resensitizes a Cisplatin Resistant Human Bladder Cancer Cell Line. The Journal of urology. 2011;185:1102–1111. doi: 10.1016/j.juro.2010.10.034. [DOI] [PubMed] [Google Scholar]
- 102.Wang RH, Liu C, Avramis VI, Berndt N. Protein phosphatase 1alpha-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene. 2001;20:6111–6122. doi: 10.1038/sj.onc.1204829. [DOI] [PubMed] [Google Scholar]
- 103.Bali P, Pranpat M, Swaby R, Fiskus W, Yamaguchi H, Balasis M, Rocha K, Wang HG, Richon V, Bhalla K. Activity of suberoylanilide hydroxamic acid against human breast cancer cells with amplification of her-2. Clinical cancer research. 2005;11:6382–6389. doi: 10.1158/1078-0432.CCR-05-0344. [DOI] [PubMed] [Google Scholar]
- 104.DeGeest, Hartenbach EM, Baergen R. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. Journal of Clinical Oncology. 2003;21:3194–3200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 105.Choi JH, Kwon HJ, Yoon BII, Kim JH, Han SU, Joo HJ, Kim DY. Expression profile of histone deacetylase 1 in gastric cancer tissues. Cancer science. 2001;92:1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fritzsche F, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC cancer. 2008;8:381–391. doi: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ishihama K, Yamakawa M, Semba S, Takeda H, Kawata S, Kimura S, Kimura W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. Journal of clinical pathology. 2007;60:1205–1210. doi: 10.1136/jcp.2005.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin KL, Pak JH, Park JY, Choi WH, Lee JY, Kim JH, Nam JH. Expression profile of histone deacetylases 1, 2 and 3 in ovarian cancer tissues. Journal of Gynecologic Oncology. 2008;19:185–190. doi: 10.3802/jgo.2008.19.3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, Rice VM. Drug-Induced Inactivation or Gene Silencing of Class I Histone Deacetylases Suppresses Ovarian Cancer Cell Growth. Cancer Biology & Therapy. 2007;6:795–801. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
- 110.Waltregny D, North B, Van Mellaert F, De Leval J, Verdin E, Castronovo V. Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. European Journal of Histochemistry. 2009;48:273–290. [PubMed] [Google Scholar]
- 111.Wang L, Zou X, Berger AD, Twiss C, Peng Y, Li Y, Chiu J, Guo H, Satagopan J, Wilton A. Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. American journal of translational research. 2009;1:62–71. [PMC free article] [PubMed] [Google Scholar]
- 112.Zhu P, Martin E, Mengwasser J, Schlag P, Janssen KP, Göttlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–463. doi: 10.1016/s1535-6108(04)00114-x. [DOI] [PubMed] [Google Scholar]
- 113.White Incorporation of histone deacetylase inhibition into the structure of a nuclear receptor agonist. Proceedings of the National Academy of Sciences. 2008;105:8250. doi: 10.1073/pnas.0709279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen Y. WO Patent 2,010,085,377. Hydroxamic acid derivatives. 2010
- 115.Griffith D, Morgan MP, Marmion CJ. A novel anti-cancer bifunctional platinum drug candidate with dual DNA binding and histone deacetylase inhibitory activity. Chemical Communications. 2009;6:105, 6735–6737. doi: 10.1039/b916715c. [DOI] [PubMed] [Google Scholar]
- 116.Griffith DM, Duff B, Suponitsky K, Kavanagh K, Morgan MP, Egan D, Marmion CJ. Novel trans-platinum complexes of the histone deacetylase inhibitor valproic acid; synthesis, in vitro cytotoxicity and mutagenicity. Journal of Inorganic Biochemistry. 2011:793–799. doi: 10.1016/j.jinorgbio.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 117.Marmion CJ, Griffith D. WO Patent WO/2011/006,908. Metal complexes having dual histone deacetylase inhibitory and DNA-binding activity. 2011
- 118.Dokmanovic M, Perez G, Xu W, Ngo L, Clarke C, Parmigiani RB, Marks PA. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Molecular cancer therapeutics. 2007;6:2525–2534. doi: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]
- 119.Lee JH, Choy M, Ngo L, Foster S, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proceedings of the National Academy of Sciences. 2010;107:14639–14644. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brabec V, Griffith DM, Kisova A, Kostrhunova H, Zerzankova L, Marmion CJ, Kasparkova J. Valuable Insight into the Anticancer Activity of the Platinum-Histone Deacetylase Inhibitor Conjugate, cis-[Pt (NH3) 2malSAHA–2H)] Molecular Pharmaceutics. 2012;9:1990–1999. doi: 10.1021/mp300038f. [DOI] [PubMed] [Google Scholar]