

Abstract
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins of the syntaxin, SNAP-25, and VAMP families mediate intracellular membrane fusion through the formation of helical bundles that span opposing membranes. Soluble SNARE domains that lack their integral membrane anchors inhibit membrane fusion by forming nonfunctional complexes with endogenous SNARE proteins. In this study we investigate the dependence of membrane fusion on the concentration of a soluble SNARE coil domain derived from VAMP2. The increase in the inhibition of fusion observed with increasing concentration of inhibitor is best fit to a function that suggests three SNARE complexes cooperate to mediate fusion of a single vesicle. These three complexes likely contribute part of a protein and lipidic fusion pore.
Vesicle trafficking is a ubiquitous cellular process by which proteins and lipids are transported between organelles of the secretory pathway. Vesicles derived from a donor compartment deliver their cargo through fusion with the acceptor compartment membrane. Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins of the syntaxin, VAMP (also called synaptobrevin), and SNAP-25 families mediate this membrane fusion process (1, 2). In the nerve terminal, syntaxin and VAMP proteins are anchored in bilayers of the plasma membrane and vesicle membrane respectively by hydrophobic C-terminal domains. Fusion occurs when SNARE proteins on opposing membranes form four-helix bundles that bring the membrane in close opposition (3–6). The four-helix bundle that results from the fusion reaction is extremely stable and does not unfold until heated to 95°C (7). In vivo, SNARE complexes are dissociated through the action of α-SNAP and the ATPase N-ethylmaleimide-sensitive factor (NSF) so that they can participate in another round of membrane fusion (8). There are at least 35 SNARE proteins in mammals, and most are specifically localized to distinct membrane compartments (9). Formation of specific SNARE complexes likely contributes to the specificity of membrane fusion that underlies the organization of membrane compartments.
According to the current hypothesis of SNARE function, after docking, SNARE proteins form loosely associated trans complexes, which have been suggested as being either N-terminally zippered or oscillating between N-terminally zippered and further zippered states (10, 11) (Fig. 1, reaction 1). In this paper we refer to such loosely associated trans SNARE complexes as “trans SNARE complexes.” The set of SNARE proteins made up of one molecule each of syntaxin, VAMP, and SNAP-25 positioned next to each other before forming trans SNARE complex is referred to as “unassembled SNAREs.” Ca2+ may induce rapid, full zippering of trans SNARE complexes (6). Formation of this extremely stable complex brings the vesicle and cell membranes into close opposition, and may provide the driving force for fusion (Fig. 1, reaction 2). A well-established permeablized cell assay makes use of the fusion of morphologically docked dense core vesicles (DCVs) with the PC12 cell membrane. This reaction has been divided into two stages (12), of which the first “priming” stage is ATP dependent, and the second “triggering” stage is Ca2+ dependent.
Figure 1.

A model of SNARE coil domain mediated inhibition of norepinephrine (NE) release from PC12 cells. SNARE monomers located at a fusion site after vesicle docking undergo two conformational changes leading to membrane fusion. The first (reaction 1) is the formation of a loosely associated trans SNARE complex. The second (reaction 2) is a zippering of the trans SNARE complex that leads to fusion. The monomers are the substrates for binding of the SNARE coil domain inhibitors (reaction 3). The complex formed by reaction 3 does not lead to a fusion event and thus blocks exocytosis. The SNAP-25 loop has been omitted for simplicity.
Permeabilization of PC12 cells permits the entry of exogenous molecules into the cell while the integrity of the vesicle fusion machinery is retained. Bacterially expressed soluble coil domains of neuronal VAMP and syntaxin inhibit DCV release in this “cracked cell assay” (13, 14). Western blotting showed that the recombinant proteins added to permeabilized PC12 cells form SDS-resistant complexes with endogenous SNARE proteins (14), but such complexes cannot trigger fusion because the membrane anchor required for mediating fusion is absent from the recombinant proteins (15) (Fig. 1, reaction 3). The SNARE complexes formed by binding of recombinant SNARE coil domains with endogenous proteins are referred to as “inhibitory complexes” in this paper. It is worth noting that although the formation of trans SNARE complexes between endogenous proteins requires overcoming an energy barrier due to the repulsion and hydration of the two lipid bilayers, such an energy barrier does not exist for formation of the fully zippered inhibitory complexes. Therefore the configuration of inhibitory complexes resembles the more stable cis SNARE complexes formed by endogenous proteins after membrane fusion, and it is likely that dissociation of the inhibitory complexes is NSF and α-SNAP dependent.
The similarities between SNARE-mediated membrane fusion and viral membrane fusion are striking (2). Both involve the formation of very stable helical bundles. Both require conformational changes that bring membranes close together, likely disrupting the bilayers and promoting their fusion. The hemagglutinin fusion protein of influenza is proposed to function as a trimer (16). The gp64 viral fusion protein of baculovirus also oligomerizes into multimers to mediate fusion (17). Although it has been suggested that SNARE complexes may form a ring around the fusion site, there are little or no data in support of this hypothesis. SNARE proteins do form higher order complexes in vitro (18–20); however, these complexes may form during or after detergent solubilization, and the stoichiometry of these complexes is not well defined. Because SNAP-25 contributes two coils to the helical bundle, it has been proposed that each coil could be part of a separate complex and that this could be a mechanism of tying together a set of SNAREs, perhaps in a circular configuration. However, single SNAP-25 coils can function to mediate fusion when separated from each other (6, 14), and some intracellular fusion events use four independent SNARE proteins (21). So, linking of complexes by a multivalent SNARE component is clearly not a requirement for fusion.
Thus, we are left with a central question of how many SNARE complexes are required to mediate a membrane fusion event. In an attempt to answer this question, we studied the concentration dependence of the inhibition of DCV fusion with the plasma membrane in PC12 cells by a soluble SNARE coil domain. Fitting of experimental data to a theoretical function suggests that three SNARE complexes mediate a membrane fusion event.
Materials and Methods
Cracked PC12 Cell Assay.
PC12 cells were maintained, [3H]-norepinephrine (NE) labeled, cracked, and EGTA extracted as described (12). Cracked cell assay was carried out essentially as described in Scales et al. (14). Briefly, priming reactions containing ≈106 cracked cells, MgATP, cytosol, and recombinant proteins in 160 μl of KGlu buffer [50 mM Hepes (pH 7.2)/105 mM potassium glutamate/20 mM potassium acetate, 2 mM EGTA] were incubated at 30°C for the indicated priming times, then chilled on ice. Then 40 μl of 8 mM Ca2+ in KGlu buffer was added to adjust the final concentration to ≈1 μM free Ca2+, 2 mM MgATP, ≈0.5 mg/ml cytosol, and the indicated concentration of recombinant proteins. Release was triggered by incubating the reactions at 30°C for the indicated triggering times and then chilling them on ice. NE release was calculated as a percentage of the total 3H in the 2500 × g supernatant. Data were analyzed by using DELTAGRAPH 4.0 software.
Preparation of Recombinant Proteins.
Rat GST-VAMP2 coil (amino acids 25–94) was expressed in Escherichia coli BL21 (RIL) cells and purified by using glutathione-agarose as described (14). Soluble proteins were liberated by thrombin cleavage of the glutathione S-transferase (GST) tags immobilized on glutathione-agarose (Sigma), dialyzed into KGlu buffer, and assayed for concentration with the BCA (bicinchoninic acid) kit (Pierce), and the purity was verified by gel analysis.
Results
A Mathematical Model for the Inhibition of NE Release from PC12 Cells by a SNARE Coil Domain.
We propose that the dose–response of inhibition of vesicle release by a soluble SNARE coil domain is an indicator of the cooperativity of SNARE proteins in mediating fusion. If each vesicle fusion event requires several pairs of trans SNARE complexes to cooperate, and is prevented by formation of inhibitory complexes by only a subset of these SNARE proteins, then the fraction of vesicles inhibited from fusion in the presence of a SNARE coil domain inhibitor will exceed the fraction of the SNARE proteins diverted into inhibitory complexes from the fusion-competent SNARE pool. Starting with this hypothesis we sought to determine the SNARE cooperativity value, n, by empirical curve fitting of the inhibitor dose versus vesicle release response. To this end, we primed [3H]NE-loaded, cracked PC12 cells in the presence of the SNARE coil domain at 30°C for 15 min, then chilled the cells on ice, adjusted the Ca2+ concentration to ≈1 μM free Ca2+, and triggered release by incubating the cells at 30°C for 1.5, 2, or 3 min. Because the H3 domain of syntaxin 1A has been reported to self-oligomerize at micromolar concentration (22), we used the soluble VAMP2 coil domain peptide in all experiments. This peptide presents as a monomeric form when assayed by gel filtration. We made the following assumptions about the assay: (i) during the 15-min priming period, equilibrium is reached between unassembled SNAREs, trans SNARE complexes, and inhibitory complexes as shown in Fig. 1; (ii) during the 1.5- to 3-min triggering period, the reaction rate for inhibitory complex dissociation, k−3, is sufficiently low that the concentration of inhibitory complexes remains relatively constant.
Tests for these assumptions are presented later in results. To define the relationship of inhibitor concentration and release using cooperativity n as a parameter, we model the kinetics of fusion as follows:
The reactions that occur in the absence of inhibitor are
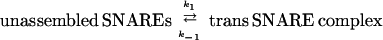 |
1 |
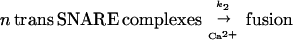 |
2 |
The additional reaction that occurs in the presence of VAMP2 coil inhibitor is
 |
3 |
Because the term “unassembled SNAREs” refers to a single set of SNARE proteins before complex assembly, reaction 1 is a first order reaction in both forward and reverse directions, and the units of k1, k−1 are both s−1. Reaction 3 is second order in the forward direction and first order in the reverse direction. Because the unit of VAMP2 concentration used in this paper is μM, the units of k3 and k−3 are μM−1⋅s−1 and s−1, respectively. Because n trans SNARE complexes cooperate to mediate fusion, they should be regarded as a unit in reaction 2, so 2 is first order and the unit of k2 is s−1. Under assumption i, equilibrium is reached for reactions 1 and 3 during the priming stage. Referring to the concentration of unassembled SNAREs, trans SNARE complex, inhibitory complex and VAMP2 coil domain as [U], [T], [I], and [V] respectively, the equilibrium can be described by equations
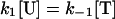 |
4 |
and
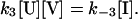 |
5 |
For the equilibrium of reaction 1 the ratio of trans SNARE complexes to unassembled SNAREs has previously been estimated to be about 20 in chromaffin cells (5). As the inhibitor concentration increases the concentration of unassembled SNAREs further decreases. Unassembled SNAREs are expected to mediate fusion at a slower rate than trans SNARE complexes because reaction 1 needs to take place first. However, to simplify the kinetic scheme we consider SNARE proteins in both states to mediate fusion at the rate of the trans SNARE complex, k2, and refer to both unassembled SNAREs and trans SNARE complexes as fusion-competent SNARE complexes. Given the proportion of unassembled SNAREs, such a simplification should give rise to <5% error in our predicted values of releases and n.
During triggering the equilibrium for reactions 1 and 3 is disturbed because the trans SNARE complexes are consumed in reaction 2. This drives reaction 1 in the forward direction and reaction 3 in the reverse direction. Assumption ii states that the rate of inhibitory complex dissociation is sufficiently slow that the concentration of inhibitory complexes remains approximately constant during a 3-min triggering period. Therefore only the SNARE proteins in fusion-competent SNARE complexes at the beginning of the triggering period contribute to fusion. The VAMP2 coil domain inhibitor is in large stoichiometric excess, with at least 2,000,000 peptide molecules per vesicle, thus its concentration during the fusion process can be taken as a constant. For the sake of simplification, concentration of fusion-competent SNARE complexes, unassembled SNAREs, trans SNARE complexes and inhibitory complexes in the presence of x μM of VAMP2 coil domain inhibitor are referred to as [F]x, [U]x, [T]x, [I]x. Combining Eqs. 4 and 5,
 |
and
 |
gives
 |
6 |
 |
where
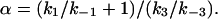 |
7 |
The unit of α is μM.
Eq. 6 states that the number of fusion-competent SNARE complexes in the presence of x μM VAMP2 coil domain is α/(α + x) fraction of that in the absence of inhibitor. Therefore in the presence of the VAMP2 coil domain each endogenous SNARE complex has α/(α + x) probability of being a fusion-competent SNARE complex. In the simplest scenario, if there are n pairs of SNARE proteins between the vesicle and cell membranes, and membrane fusion requires cooperative activity of n SNARE complexes, then the probability that a vesicle is able to fuse with cell membrane is
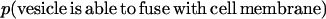 |
8 |
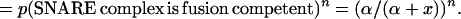 |
Therefore (α/(α + x))n fraction of the vesicles should be able to fuse with cell membrane in the presence of inhibitor according to the Poisson distribution. Because release is proportional to the number of vesicles able to fuse with the cell membrane, by normalizing maximum release without inhibitor to 1, the relationship of normalized release, y, and inhibitor concentration, x, can be expressed by
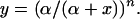 |
9 |
The triggering time used in our experiments was sufficiently short as to approximate linear release (Fig. 2).
Figure 2.

The time course of NE release from permeabilized PC12 cells. Cells were primed by incubating at 30°C for 15 min in the presence of ATP and cytosol. Then release was triggered by adjusting the free [Ca2+] to ≈1 μM and was stopped at the indicated time points by chilling the reaction on ice. The data are fitted to function y = b⋅(1 − e(−kx)), assuming that the triggering reaction is a first-order reaction. The range of triggering times used for our experiments is marked by the black bar. Regardless of the particular function used to fit the data the release increases approximately linearly over the triggering times used for our experiments.
In a more complicated scenario, it is possible that m pairs of SNARE proteins rest between the vesicle and cell membranes, but a subset of n pairs is sufficient to trigger fusion. If this were the case, however, then when (m−n)/m of the SNARE proteins are in inhibitory complexes, most vesicles would still have n competent SNARE complexes to mediate fusion. The inhibition curve should therefore show an initial plateau with an experimentally observable size. For example, if m equals 4 and n equals 1, the size of the plateau would be about half of the inhibitor concentration needed to inhibit 75% of release. We observed no sigmoidal shape in the inhibition curve in our experiments (Fig. 3A), so we consider such a scenario to be unlikely.
Figure 3.

Release at high levels of inhibition is optimal for estimating cooperativity. One set of experimental data corresponding to 15-min priming, 2-min triggering is plotted on linear plot (A) and double log plot (B). (A) Experimental data are fit to y = (α/(α + x))n, the best-fit curve shown is y = (17.8/(17.8 + x))2.9. (B) The same set of data are fit to functions y = (α/(α + x))n, where n are fixed values of 1, 2, 3, 4, and 5. The best-fit curves shown are y = 3.26/(3.26 + x); y = (10.6/(10.6 + x))2; y = (18.5/(18.5 + x))3; y = (26.5/(26.5 + x))4; y = (34.6/(34.6 + x))5. y = (18.5/(18.5 + x))3 corresponds to the darkened curve. Positions of the curves corresponding to y = 3.26/(3.26 + x) and y = (34.6/(34.6 + x))5 are indicated in the graph.
Experimental Data Suggests That Three SNARE Complexes Cooperate to Mediate Membrane Fusion.
Based on the model stated above, we wondered if the value of the parameters n and α could be determined by fitting experimental data to equation 9. As shown in Fig. 3B, one set of experimental data corresponding to 2-min triggering time was fitted to functions y = α/(α + x); y = (α/(α + x))2; y = (α/(α + x))3; y = (α/(α + x))4; and y = (α/(α + x))5. The value of α was determined by curve fitting. At low levels of inhibition curves of different n values are close to each other. At high levels of inhibition, however, the differences between curves are significant on a double-log plot. Therefore, accurately measuring release at high levels of inhibition should allow us to determine the n value unequivocally. In practice, the optimum release in absence of inhibitor measured in our experiments range from 15% to 30% of the total [3H]NE loaded into the cells, and the standard deviation of release is about 0.5% of total [3H]NE, so y values above 0.01–0.03, depending on the optimum release value, can be measured without being obscured by experimental error.
In Fig. 4, data from three typical experiments with different triggering times corresponding to 1.5, 2, and 3 min are shown. Baseline release in presence of 4 mM EGTA was subtracted from all data points, release without inhibitor was normalized to 1, and the resultant data points were fitted to Eq. 9. Table 1 is a summary of the results. The data suggest that formation of 3 trans SNARE complexes is both essential and sufficient for mediating Ca2+ induced vesicle fusion with the plasma membrane.
Figure 4.

Inhibition of NE release from PC12 cells by the VAMP 2 coil domain. Normalized inhibition is plotted versus concentration of the inhibitor in μM and the data are fit to the function y = (α/(α + x))n. Results from three representative experiments are shown with triggering times of 1.5, 2, and 3 min, respectively. The triggering time used in the assay and cooperativity value n obtained by curve fitting are indicated in each panel. The best fit curves shown are y = (9.0/(9.0 + x))2.7 in A; y = (15.2/(15.2 + x))3.2 in B; and y = (11.4/(11.4 + x))2.8 in C.
Table 1.
SNARE cooperativity determined by fitting SNARE coil domain inhibition curve
| Triggering time, min | No. of expts | n | α |
|---|---|---|---|
| 1.5 | 3 | 2.9 ± 0.2 | 10.9 ± 1.7 |
| 2 | 4 | 3.1 ± 0.3 | 16.9 ± 1.2 |
| 3 | 6 | 2.9 ± 0.4 | 13.4 ± 3.7 |
The values of n and α are given along with the SD.
The data shown in Table 1 also allow a test of assumption ii made in building the kinetic model. If ii does not hold, i.e., a significant fraction of inhibitory complexes dissociate during triggering and the resultant unassembled SNAREs participate in mediating fusion, then in the presence of inhibitor, the normalized release would increase with triggering time. This would shift the inhibition curve, such that the n value would change over time. Using the standard Student t test, we observed no statistically significant difference in the n value obtained at 1.5-, 2-, and 3-min triggerings. Thus assumption ii appears to be valid within the 3-min triggering limit.
Standard deviations for α are higher than those for n, as is shown in Table 1. One reason is that experimental error in the measurement of the VAMP2 coil domain concentration results in proportional error in the determination of α value, whereas it does not result in error for the determination of n value. VAMP2 coil domain peptides denature and lose vesicle release inhibition activity during storage. Such inactivation is not detectable by the bicinchoninic acid assay we used to measure peptide concentration and may cause up to 10% error in protein concentration determination. Alternatively, because α/(α + x) represents the fraction of SNARE proteins in fusion-competent SNARE complexes, there could be limited inhibitory complex dissociation occurring during the first 3 min of triggering. If this is true, then the extent of dissociation is likely too low to be reflected in the value of n. Substituting α = 10 μM and k1/k−1 = 20 into Eq. 7 gives an approximate estimation of k3/k−3 as 2 μM−1. Therefore Kd for inhibitory complex is 0.5 μM. At first sight k3 seems to be quite low given that k−3 is proposed to be small. However, in the cell endogenous SNARE proteins are likely placed in close proximity to each other by docking proteins, whereas soluble SNARE domains need to diffuse in three dimensions while searching for their binding partners, and their path is limited by the position of the lipid membranes and other proteins clustered at the fusion site. Such spatial limitations are likely reflected by a low coil domain-binding rate and the large excess of VAMP2 coil domain needed to inhibit release.
As a partial test of assumption i, we compared the inhibition curves generated from 15-min priming, 2-min triggering and 30-min priming, 2-min triggering (Fig. 5). If equilibrium was not reached for reactions 1 and 3 after 15-min priming, then an extra 15-min priming should change the concentration of SNARE proteins in each state, and this should be reflected by a difference between the two inhibition curves. Because we did not observe significant difference between the two curves, it is likely that during the 15-min priming period equilibrium was reached between unassembled SNAREs, trans SNARE complexes, and inhibitory complexes. During priming period the rate approaching equilibrium is determined by forward reaction rate of 3, whereas during triggering period the rate approaching equilibrium is determined by reverse reaction rate of 3. Therefore it is not surprising that the initial equilibrium is reached within 15 min but does not shift significantly toward new equilibrium after 3 min of triggering.
Figure 5.

Increasing priming time does not change the shape of the dose-dependent inhibition curve. Data from a 30-min priming, 2-min triggering experiment (×, n = 2.6) is compared with that of a 15-min priming, 2-min triggering experiment (▿, n = 3.2). The curve plotted shows the best-fitting curve for the 30-min priming dataset.
Discussion
We previously examined the issue of SNARE cooperativity by using a SNAP-25 rescue assay (23). In this assay, bacterially expressed SNAP-25 C-terminal domain was used to rescue vesicle release from cracked PC12 cells pretreated with botulinum neurotoxin E, which cleaves the C terminus of endogenous SNAP-25 proteins (6), and no obvious cooperativity between SNARE proteins was observed. However, the method of analysis and the accuracy of the data in our previous experiments would not have revealed a cooperativity of 3.
The major practical limit in the experiments presented here is the short time window within which the release can be analyzed by using our model. At shorter triggering time points the release signal is too low to determine release at a high level of inhibition accurately, whereas at longer time points the release is significantly influenced by dissociation of inhibitory complexes. Nevertheless, the control tests and the consistency of our determination of the n value as 3 support our conclusions.
To our knowledge, no data directly measuring SNARE cooperativity have been reported, although several groups have observed higher-order complexes formed from individual SNARE proteins carrying the transmembrane domain (24) or from the SDS-resistant core domains in detergent solutions (7, 18, 20) and on solid surface (19). Equilibrium between monomers and trimers of the core complex in detergent solution has been observed (7). Furthermore, motifs located in the transmembrane domains of VAMP2 and syntaxin 1A have been reported to mediate homo- and hetero- oligomerization of SNARE proteins (25). If such higher-order SNARE complexes exist in a physiological environment, one might imagine that at the fusion site a cluster of three t-SNAREs is positioned opposite to a cluster of three v-SNAREs. When fully zippered SNARE complexes form, the hetero-oligomerization might then extend to the transmembrane domains of the SNARE proteins. Such SNARE pairing is proposed both to bring the lipid bilayers close enough for lipid mixing to occur and to contribute to fusion by disturbing the conformation of lipid bilayers, or perhaps even guide lipid rearrangements through the fusion event. After SNARE pairing, the complexes may separate from each other laterally, promoting formation and dilation of a fusion pore. Our results do not rule out the possibility that the three pairs of SNARE proteins at the fusion pore are organized through interaction with other proteins. However, we do not find our data consistent with the hypothesis of SNAP-25 serving as a physical linker between complexes. In addition to the evidence against such a hypothesis discussed above, this mechanism would require extremely rapid dissociation of the resultant cis SNARE complexes to avoid limiting the expansion of the fusion pore, and it is unlikely that the NSF concentration at the fusion site would be high enough to fulfill such a task.
Alternatively, the organization of SNARE proteins into higher-order complexes might depend on other proteins involved in the late steps of fusion. Self-oligomerization in synaptotagmin has been well documented (26, 27). As a potential Ca2+ sensor this protein is well positioned to coordinate the organization of SNARE complexes. Intriguingly, a recent paper (28) reported a reduced Ca2+ cooperativity in Drosophila syntaxin mutants. In the context of our work, this result argues for both SNARE cooperativity and direct, one-to-one coupling of a Ca2+ sensor with individual SNARE complexes. The similarity of the Ca2+ cooperativity (n = 3–4) and the SNARE cooperativity (n = 3) suggests that there is a mechanistic link between these processes. Other candidates for modulating SNARE cooperativity include complexin (20), cysteine string protein (29), and nsec1/munc18 (30).
In summary, based on a current hypothesis for the mechanism of SNARE-mediated membrane fusion, we have built a model describing the relationship of NE release, SNARE coil domain concentration, and SNARE cooperativity in the cracked cell assay. We collected data by using a two-step priming–triggering protocol, and estimated the SNARE cooperativity to be 3. We propose the existence of a fusion pore that is likely to be composed of lipid, three VAMP2 transmembrane domains, and three syntaxin 1A transmembrane domains. Expansion of the fusion pore likely occurs when the SNARE proteins dissociate in the plane of the membrane. This study is an attempt to model intricate biological processes with a simple kinetic scheme, and we hope it will inspire novel quantitative approaches to remaining questions in the field.
Acknowledgments
We are grateful to Drs. Daniel Herschlag, Erwin Neher, Richard W. Tsien, and Lino Gonzalez, Jr., for insightful discussions, Dr. Richard S. Lewis, Dr. Suzie Scales, Mala Murthy, Dr. Cary Austin, and Dr. Jagath Junutula for critical reading of the manuscript, and Brian Y. Yoo for technical assistance. Y.Y.H. was supported by a Stanford Graduate Fellowship.
Abbreviations
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- NSF
N-ethylmaleimide-sensitive factor
- NE
norepinephrine
- DCV
dense core vesicle
References
- 1.Chen Y A, Scheller R H. Nat Rev Mol Cell Biol. 2001;2:98–106. doi: 10.1038/35052017. [DOI] [PubMed] [Google Scholar]
- 2.Jahn R, Sudhof T C. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- 3.Sutton R B, Fasshauer D, Jahn R, Brunger A T. Nature (London) 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 4.Weber T, Zemelman B V, McNew J A, Westermann B, Gmachl M, Parlati F, Söllner T H, Rothman J E. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 5.Xu T, Rammner B, Margittai M, Artalejo A R, Neher E, Jahn R. Cell. 1999;99:713–722. doi: 10.1016/s0092-8674(00)81669-4. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y A, Scales S J, Patel S M, Doung Y C, Scheller R H. Cell. 1999;97:165–174. doi: 10.1016/s0092-8674(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 7.Fasshauer D, Eliason W K, Brunger A T, Jahn R. Biochemistry. 1998;37:10354–10362. doi: 10.1021/bi980542h. [DOI] [PubMed] [Google Scholar]
- 8.Söllner T, Bennett M K, Whiteheart S W, Scheller R H, Rothman J E. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 9.Bock J B, Matern HT, Peden A A, Scheller R H. Nature (London) 2001;409:839–841. doi: 10.1038/35057024. [DOI] [PubMed] [Google Scholar]
- 10.Lin R C, Scheller R H. Neuron. 1997;19:1087–1094. doi: 10.1016/s0896-6273(00)80399-2. [DOI] [PubMed] [Google Scholar]
- 11.Hua S Y, Charlton M P. Nat Neurosci. 1999;2:1078–1083. doi: 10.1038/16005. [DOI] [PubMed] [Google Scholar]
- 12.Hay J C, Martin T F. J Cell Biol. 1992;119:139–151. doi: 10.1083/jcb.119.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong P, Chen Y A, Tam D, Chung D, Scheller R H, Miljanich G P. Biochemistry. 1997;36:4317–4326. doi: 10.1021/bi9625408. [DOI] [PubMed] [Google Scholar]
- 14.Scales S J, Chen Y A, Yoo B Y, Patel S M, Doung Y C, Scheller R H. Neuron. 2000;26:457–464. doi: 10.1016/s0896-6273(00)81177-0. [DOI] [PubMed] [Google Scholar]
- 15.McNew J A, Weber T, Parlati F, Johnston R J, Melia T J, Söllner T H, Rothman J E. J Cell Biol. 2000;150:105–117. doi: 10.1083/jcb.150.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danieli T, Pelletier S L, Henis Y I, White J M. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markovic I, Pulyaeva H, Sokoloff A, Chernomordik L V. J Cell Biol. 1998;143:1155–1166. doi: 10.1083/jcb.143.5.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poirier M A, Xiao W, Macosko J C, Chan C, Shin Y K, Bennett M K. Nat Struct Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- 19.Hohl T M, Parlati F, Wimmer C, Rothman J E, Söllner T H, Engelhardt H. Mol Cell. 1998;2:539–548. doi: 10.1016/s1097-2765(00)80153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokumaru H, Umayahara K, Pellegrini L L, Ishizuka T, Saisu H, Betz H, Augustine G J, Abe T. Cell. 2001;104:421–432. doi: 10.1016/s0092-8674(01)00229-x. [DOI] [PubMed] [Google Scholar]
- 21.Antonin W, Holroyd C, Fasshauer D, Pabst S V, Mollard G F, Jahn R. EMBO J. 2000;19:6453–6464. doi: 10.1093/emboj/19.23.6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Misura K M, Scheller R H, Weis W I. J Biol Chem. 2000;276:13273–13282. doi: 10.1074/jbc.M009636200. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y A, Scales S J, Scheller R H. Neuron. 2001;30:161–170. doi: 10.1016/s0896-6273(01)00270-7. [DOI] [PubMed] [Google Scholar]
- 24.Laage R, Rohde J, Brosig B, Langosch D. J Biol Chem. 2000;275:17481–17487. doi: 10.1074/jbc.M910092199. [DOI] [PubMed] [Google Scholar]
- 25.Margittai M, Otto H, Jahn R. FEBS Lett. 1999;446:40–44. doi: 10.1016/s0014-5793(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 26.Chapman E R, Desai R C, Davis A F, Tornehl C K. J Biol Chem. 1998;273:32966–32972. doi: 10.1074/jbc.273.49.32966. [DOI] [PubMed] [Google Scholar]
- 27.Littleton J T, Bai J, Vyas B, Desai R, Baltus A E, Garment M B, Carlson S D, Ganetzky B, Chapman E R. J Neurosci. 2001;21:1421–1433. doi: 10.1523/JNEUROSCI.21-05-01421.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart B A, Mohtashami M, Trimble W S, Boulianne G L. Proc Natl Acad Sci USA. 2000;97:13955–13960. doi: 10.1073/pnas.250491397. . (First Published November 28, 2000; 10.1073/pnas.250491397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graham M E, Burgoyne R D. J Neurosci. 2000;20:1281–1289. doi: 10.1523/JNEUROSCI.20-04-01281.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher R J, Pevsner J, Burgoyne R D. Science. 2001;291:875–878. doi: 10.1126/science.291.5505.875. [DOI] [PubMed] [Google Scholar]