

Abstract
Huntington's disease (HD) is a devastating neurodegenerative disorder with no disease-modifying treatments available. The disease is caused by expansion of a CAG trinucleotide repeat and manifests with progressive motor abnormalities, psychiatric symptoms, and cognitive decline. Expression of an expanded polyglutamine repeat within the Huntingtin (Htt) protein impacts numerous cellular processes, including protein folding and clearance. A hallmark of the disease is the progressive formation of inclusions that represent the culmination of a complex aggregation process. Methylene blue (MB), has been shown to modulate aggregation of amyloidogenic disease proteins. We investigated whether MB could impact mutant Htt-mediated aggregation and neurotoxicity. MB inhibited recombinant protein aggregation in vitro, even when added to preformed oligomers and fibrils. MB also decreased oligomer number and size and decreased accumulation of insoluble mutant Htt in cells. In functional assays, MB increased survival of primary cortical neurons transduced with mutant Htt, reduced neurodegeneration and aggregation in a Drosophila melanogaster model of HD, and reduced disease phenotypes in R6/2 HD modeled mice. Furthermore, MB treatment also promoted an increase in levels of BDNF RNA and protein in vivo. Thus, MB, which is well tolerated and used in humans, has therapeutic potential for HD.
Introduction
Huntington's disease (HD) is caused by an abnormal CAG repeat expansion leading to an expanded polyglutamine repeat in the Huntingtin protein (Htt) (The Huntington's Disease Collaborative Research Group, 1993; Gusella and MacDonald, 1995). No treatment currently exists that modifies onset or progression of the disease. A pivotal set of studies described nuclear inclusions that accumulate in both diseased mouse and human patient brain tissues, providing the first suggestion that HD involves protein misfolding and aggregation (Davies et al., 1997; DiFiglia et al., 1997) with a repeat dependence to protein aggregation (Scherzinger et al., 1997). Later studies suggested that inclusions may sequester toxic soluble mutant Htt species and thereby lower the risk of neuronal death in primary neurons (Arrasate et al., 2004). Indeed, a tight correlation between an Htt antibody specific for monomers and small oligomers, 3B5H10, predicts neuronal toxicity (Miller et al., 2011). Therefore, the identification of aggregation intermediates and the use of small molecules that modulate aggregation intermediates may define the desired outcomes for treatments.
The drug methylene blue (MB) has been reported to successfully complete a Phase IIb clinical trial for the treatment of mild to moderate Alzheimer's disease (AD), showing an improvement in cognitive function after 6 months and slowing the progression of AD by 81% over a 1 year period (Gura, 2008). A Phase 2 MB clinical trial is also currently in progress for enhancing extinction learning in posttraumatic stress disorder (www.clinicaltrials.gov). MB has desirable properties for drug candidates that act in the CNS, including high solubility in aqueous media (Riha et al., 2005), ability to cross the blood–brain barrier (Peter et al., 2000) and act in the CNS (Küpfer et al., 1994), and low toxicity (Küpfer et al., 1994). It also is used in humans for other indications (DiSanto and Wagner, 1972), making it a candidate that could be quickly used.
MB can affect numerous cellular processes; however, its role as a modulator of aggregation is thought to underlie its potential use in AD and other dementias. MB prevented aggregation of recombinant tau and amyloid β (Aβ), yet when added to filamentous tau increased aggregate number (Taniguchi et al., 2005). It also prevented in vitro Aβ oligomer formation by enhancing fibril formation (Necula et al., 2007). In vivo, MB reduced soluble Aβ levels and rescued early cognitive deficits in 3xTg-AD mice (Medina et al., 2010). In studies of polyglutamine repeat disorders, MB increased degradation androgen receptor polypeptides (Wang et al., 2010) and prevented mutant Htt aggregation in zebrafish (van Bebber et al., 2010). Together, these and other data suggest that MB may be a potent modulator of protein aggregation that could influence HD onset or progression.
Based on a screen of aggregation modulating compounds on recombinant polyglutamine repeat polypeptide aggregation, we investigated whether MB could modulate mutant Htt aggregation and neurotoxicity in vitro and in vivo. MB can reduce oligomer formation and insoluble mutant Htt aggregation in vitro and in mouse primary cortical neurons and reduce aggregation and neurodegeneration in a Drosophila model of HD. Treatment also delayed motor deficits and restored BDNF levels in R6/2 mice.
Materials and Methods
Recombinant protein purification
GST-Httex1Q53 was purified as described (Muchowski et al., 2000; Wacker et al., 2004; Behrends et al., 2006; Lotz et al., 2010). Freshly prepared, unfrozen Httex1Q53 was used for all experiments.
Analysis of mutant Htt aggregation by filter-trap analyses and atomic force microscopy
Before each experiment, recombinant purified Httex1Q53 was centrifuged (20,000 × g) for 30 min at 4°C to remove aggregates. Httex1Q53 proteins were incubated at 6 μm in 20 mm Tris-HCl, pH 7.5, 150 mm KCl, and 1 mm dithiothreitol at 37°C with shaking at 800 rpm. PreScission Protease (4 units/100 μg of fusion protein; GE Healthcare) were added at time 0 to initiate the aggregation. Methylene blue was added in specific concentrations (1, 10, and 100 μm) to reaction mixtures at time point 0, 4, or 24 h. Two micrograms of mixtures were removed for filter-trap assay. According to PubChem (NCBI records of compounds) and the Hazardous Substances Data Bank (National Library of Medicine), the solubility of MB in water at room temperature is 43.6 g/L, which is equivalent to 136 mm. This is significantly higher than our highest concentration of 300 μm.
Filter-trap assay.
Aggregation of Httex1Q53 was initiated as described above. Httex1Q53 (2 μg) was removed from the 6 μm reaction, boiled in SDS loading buffer, and applied to a nitrocellulose membrane (0.45 μm pore size; Schleicher and Schuell) through a slot-blot manifold. The membranes were washed with 3% SDS buffer by vacuum filtering, incubated in blocking buffer, and treated as described previously (Wacker et al., 2004). Quantification was performed by densitometry with ImageJ. Relative insoluble mutant Htt aggregates were normalized to insoluble control aggregates with no addition of methylene blue.
Atomic force microscopy.
All images were collected in tapping mode with an MFP-3D atomic force microscope system (Asylum Research). The aggregation reactions were performed as described for the filter-trap assay. Before imaging, 10 μl of the reaction solution was deposited onto freshly cleaved mica (Ted Pella) and incubated for 1 min. The substrate was washed with 200 μl of ultrapure water and dried under a gentle stream of air. Images were taken with silicon cantilevers (Veeco Instruments) with a nominal spring constant of 40 N/m and resonance frequency of ∼300 kHz. Typical imaging parameters were a drive amplitude of 150–500 kHz with set points of 0.7–0.8 V, scan frequencies of 2–4 Hz, an image resolution of 512 × 512 points, and a scan size of 2–10 μm.
Primary mouse neurons
Mouse cortical neuronal cultures were prepared from embryonic day 16 (E16)–E18 mice (C57/BL6 strain). Cerebral cortices were isolated and dissociated by 0.125% (v/v) trypsin-EDTA (Life Technologies) digestion and trituration with a fire-polished Pasteur pipette. Cortical neurons were plated at 1 × 106 cells per well, in poly-l-lysine-coated (Sigma) six-well plates (Corning) in Neurobasal medium supplemented with B27, 2 mm GlutaMax, and streptomycin/amphotericin B (Life Technologies). The neuronal cultures were transduced with lentiviral vectors at ratio of 100 ng p24 antigen (multiplicity of infection, 10) per 106 cells 1 d after plating (1 DIV). The transfer vectors for the lentivirus used in this study are pFUGW genome vectors containing either GFP or Htt-Exon-1-Q103-GFP (provided by A. Khoshnan and P. Patterson, California Institute of Technology, Pasadena, CA) (Southwell et al., 2009). One day after transduction (2 DIV), neurons were treated with MB (0 nm, 10 nm and 100 nm). Treatment was repeated every 3 d by replacing 50% of the medium with fresh medium containing the same concentration of methylene blue for each well, respectively. Neuronal cultures were harvested at 7–10 DIV for Western blot and filter-retardation analysis.
Analysis of oligomer formation: SDS-agarose gel electrophoresis
Primary neurons were lysed in RIPA buffer and analyzed for Htt oligomer formation by SDS-agarose gel electrophoresis (AGE) as described previously (Weiss et al., 2008). Briefly, for 1% agarose gels, 2 g agarose (Fisher) was dissolved in 200 ml 375 mmol/L Tris–HCl, pH 8.8, brought to boiling in a microwave oven. After melting, SDS was added to a final concentration of 0.1%. Samples were diluted 1:1 into nonreducing Laemmli sample buffer (150 mmol/L Tris–HCl, pH 6.8, 33% glycerol, 1.2% SDS, and bromophenol blue). Purified ferritin (440 kDa) was taken as high-molecular-weight size markers (Sigma). After loading, gels were run in Laemmli running buffer (192 mmol/L glycine, 25 mmol/L Tris base, 0.1% SDS) at 100 V until the bromophenol blue running front had migrated 12 cm. Semidry electroblotter model HEP-1 (OWL Scientific) was used to blot the gels on PDVF membranes (Millipore) at 200 mA for 1 h with a Shelton Scientific SH300 power supply (transfer buffer: 192 mmol/L glycine, 25 mmol/L Tris base, 0.1% SDS, and 15% methanol). As the thickness of the 1% gels decreased substantially during the 1 h transfer, the electroblotter top was periodically tightened to guarantee constant and even contact between the gel and the electroblotter when blotting these gels. After transfer, the membrane was blocked in 5% milk solution and incubated in a 1:2000 dilution of anti-GFP antibody (Clontech) overnight at 4°C. Horseradish peroxidase-conjugated anti-rabbit secondary (Jackson ImmunoResearch) was used at a 1:10,000 dilution followed by incubation for 45 min at room temperature, and the signal was detected using Supersignal West Pico (Pierce). Quantification was performed by densitometry with ImageJ. Relative insoluble mutant Htt aggregates were normalized to insoluble control aggregates with no addition of methylene blue. The distance of the peak signal intensity to the running front was calculated using ImageJ. The peak distance of the untreated sample was set to 100 and the treated samples were compared to this value.
Filter-retardation assay
Filter-retardation assays on primary neuron lysates were performed as described previously (Wanker et al., 1999). Samples were boiled in 2% SDS and added to cellulose acetate membrane (0.22 μm pore size; Schleicher and Schuell) through a dot-blot manifold. The membranes were washed with 2% SDS buffer by vacuum filtering, incubated in blocking buffer, and treated as described above for the SDS-AGE membranes.
Primary rat neuron toxicity assay
Primary cortical neuron cultures were prepared from rat embryos (E18–E20). Cortical tissue was dissected from embryos in dissection media kynurenic acid (DMKY) on ice, rinsed with DMKY, and incubated with papain in DMKY media plus cysteine for 15 min at 37°C. Cortical tissue was rinsed with DMKY and then incubated with trypsin inhibitor in DMKY media for 15 min at 37°C. Cortical tissue was rinsed three times with Opti-MEM media containing glucose and then dissociated into single neurons by trituration in Opti-MEM/glucose media. Approximately 500,000 cells were plated in each well of poly-d-lysine/laminin-coated 24-well tissue culture plates. Opti-MEM/glucose media was replaced with Neurobasal media containing B27 supplement, penicillin/streptomycin, and l-glutamine 2–3 h after plating. Neurons were maintained in 37°C incubators with 5% CO2. Rat primary cortical neurons (E18–E20) were transfected in 24-well plates at 4–5 d in vitro using Lipofectamine 2000 (Invitrogen) according to standard protocol (Arrasate and Finkbeiner, 2005). Briefly, for cotransfections, 2 μg each of pGW1-mCherry (survival marker) and pGW1-Htt-ex1-Q72-GFP plasmids were mixed with 8 μl Lipofectamine 2000 in Opti-MEM media and added to media in wells containing ∼500,000 neurons. Cells were incubated with the plasmid/Lipofectamine mixture for 2 h, and media was replaced with Neurobasal media. Neurons were treated with vehicle (PBS) or with MB at a 100 nm final concentration 6–12 h after transfection. Fluorescent images of individual neurons were captured at 24 h after transfection and at ∼24 h intervals thereafter with a robotic microscope. Survival of individual neurons was measured by longitudinal observation of mCherry fluorescence, by which cell death correlates with the disappearance of detectable fluorescence.
Drosophila, crosses, pseudopupil assay, and aggregate analysis
The polyglutamine-expressing transgenic stock used was w;uas-Httex1pQ93 (P463) (Steffan et al., 2001). Males of these flies were mated with females expressing the pan-neuronal elav driver w;P[w_mW.hs_GawB]elavC155 (#6923; Bloomington Stock Center, Bloomington, IN). Cultures were raised at 25°C on standard cornmeal molasses food. MB was added to a final concentration of 8 μm.
Pseudopupil analysis.
Seven-day-old adult flies were decapitated and their heads mounted in nail polish on a microscope slide. Each head was then covered with immersion oil and examined under a Nikon EFD-3 Optiphot-2 microscope with a 50× oil objective. At least 250 ommatidia in five to eight flies were examined, and the number of visible rhabdomeres was counted for each.
Immunochemistry.
Eye antennal imaginal discs were dissected in 1× PBS. The tissue was fixed in 4% formaldehyde at room temperature for 20 min and thoroughly washed with PBT (0.2% Triton X-100 in 1× PBS). After blocking (10% bovine serum albumin in 0.2% Triton X-100 in PBS; 1 h at room temperature), tissues were incubated with primary antibody in blocking solution overnight at 4°C. After washing in PBS, secondary antibody was applied for 1 h at room temperature. The primary antibodies were a sheep anti-Htt S830 (1:1000; a kind gift from G. Bates, King's College London, London, UK) and rat anti-elav (1:200; Developmental Studies Hybridoma Bank). Secondary antibodies were from Jackson ImmunoResearch (1:200). Every picture is a merged image of a series of 1 μm confocal Z-slices (Zeiss LSM510) that bracket the Htt staining region. For quantifying aggregation size, aggregates were counted in bins ranging from 1–16 μm as detected by immunostaining. The number of aggregates in the 16 bins was averaged over four independent discs, and the average size is given as the average percentage of aggregates larger than 1 μm in the four discs.
Animal subjects and treatments
Twenty R6/2 HD mice with 115 ± 5 CAG repeats were obtained from The Jackson Laboratory. Twenty nontransgenic littermates were used as age-matched controls. All mice were housed in a barrier animal facility at the University of California, Irvine, housed five per cage with food and water available ad libitum, and kept on a 12 h light/dark cycle. All studies and procedures were done in accordance with the guidelines of the University of California, Irvine, Institutional Animal Care and Use Committee. Treatments were started at 5 weeks of age. Treatment chow was created by blending chow pellets with 25 mg of MB per 100 g of chow. Water was mixed in at 10 ml per 100 g of chow to pack down the chow. Ten R6/2 mice and 10 nontransgenic mice were given MB in the chow, the remaining 10 R6/2 mice and 10 nontransgenic mice were given vehicle control chow.
Animal testing
All behavioral tasks were performed and analyzed by investigators blinded to experimental treatments. Body weights were obtained daily at 6–9 weeks of age at the same time each day.
Clasping.
Mice were suspended by the tail for 10 s daily at 6–9 weeks of age as described previously (Mangiarini et al., 1996). The presence or absence of hindlimb clasping was noted, and significance was determined. Power analysis reveals that this study was only at 65% power; however, to achieve 95% power, the study would require >100 animals, which is impractical in an in vivo setting.
Rotarod.
Rotarod evaluations were performed using an accelerating rotarod apparatus (Dual Species Economex Rota-Rod; 0207-003M; Columbus Instruments) as described previously (Carter et al., 1999) at 7 and 9 weeks of age. Briefly, the rotarod assay consisted of a rotating rod that mice were trained to walk on at a fixed speed of 16 rpm (four times with a 5 min rest interval) and tested using an accelerating speed protocol that examined their ability to stay on the rod for 60 s (4–40 rpm in 1 min four times, with a 5 min rest interval). The lowest value from all four tests was discarded, and the remaining three trials were averaged and analyzed for significance.
Pole test.
Mice were also examined for performance on a vertical pole (1 cm in diameter, 60 cm high) as described previously (Hickey et al., 2008) at 6 and at 8 weeks of age. The mice were tested with a modification of this protocol by placing them facing down on the vertical pole, and total time to descend measured. Mice were habituated to the task 1 d before testing. On the test day, the total time taken to descend was measured on four trials per mouse. These values were then averaged and analyzed for significance.
Biochemical analysis of mouse tissue
The same treatment procedure used in the behavioral testing was performed on a different cohort of mice, both R6/2 and nontransgenic as before. The mice were killed at 8 weeks of age (3 weeks of treatment), as most of the behavior analysis revealed a difference in phenotype at that age and duration of treatment. The mice were killed by nembutol overdose (Donovan and Brown, 2006). Both striatum and cortical tissues were harvested for biochemical analysis. Half of the cohort was used for protein analysis, and the other half of the cohort was used for qRT-PCR analysis. Samples used for protein analysis were homogenized in a modified RIPA buffer (10 mm Tris, pH7.2, 158 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, and 1× Complete protease inhibitors; Roche). Samples were sonicated three times for 30 s each on ice at a power of 2. The amount of protein was quantified by the Lowry method. SDS-AGE and filter-retardation assays were performed as described above. SDS-PAGE was performed using a NuPAGE 4–12% Bis-Tris polyacrylamide gel (Invitrogen) and run until the dye front reached the bottom of the gel. The proteins were then electroblotted onto a nitrocellulose membrane (Bio-Rad). This blot was blocked for 1 h in Starting Block buffer (Pierce) at room temperature. The blot was then probed with anti-BDNF antibody (Santa Cruz Biotechnology; 1:5000) and anti-actin antibody (Sigma; 1:5000). Peroxidase-conjugated AffiniPure goat anti-rabbit secondary antibody (Jackson ImmunoResearch) was used at 1:15,000 for 1 h at room temperature. Proteins on blots were detected using Supersignal West Pico detection reagent (Pierce).
qRT-PCR analysis of mouse tissue
Frozen cortical samples were homogenized in trizol, and RNA was run through the Qiagen RNeasy column with on-column DNase I digestion. cDNA was prepared from 0.5 μg of RNA using Reverse Transcription Supermix from Bio-Rad. The resulting cDNA was diluted 1:5 in water and used for quantitative PCR by the SYBR Green method (Bio-Rad). Primers for quantitative PCR are as follows: mouse actin sense, 5′AGGTATCCTGACCCTGAAG3′; mouse actin antisense, 5′GCTCATTGTAGAAGGTGTGG3′; mouse BDNF sense, 5′TCGTTCCTTTCGAGTTAGCC3′; mouse BDNF antisense, 5′TTGGTAAACGGCACAAAAC3′.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5.04 software. All data are expressed as mean ± SEM, and a value of p < 0.05 was considered to be statistically significant. Statistical comparisons of densitometry results were performed by one-way ANOVA followed by Bonferroni's multiple comparison tests. Kaplan–Meier curves were used to estimate cell survival in the primary neuron assay, and differences in the curves were assessed with the log-rank (Mantel–Cox) test. The Drosophila pseudopupil assay was analyzed by ANOVA followed by Bonferroni's multiple comparison tests. Student's t tests were used for aggregate size and number comparisons from the Drosophila study. One-way ANOVA followed by Bonferroni's multiple comparison tests was used to establish significance of the body weight analysis from the R6/2 mouse study. Significance in the clasping assay was determined by Fisher's exact probability test. The rotarod and pole test data were analyzed by two-way repeated-measures ANOVA followed by Bonferroni's multiple comparisons tests. Power analysis was performed using GraphPad StatMate 2.00.
Results
MB decreases in vitro aggregation of monomers, oligomers, and fibrils of recombinant Httex1p
It has been shown that MB can modulate aggregation of amyloidogenic proteins including Aβ and tau (Doh-Ura et al., 2000; Korth et al., 2001; Taniguchi et al., 2005; Necula et al., 2007; van Bebber et al., 2010) and polyglutamine repeat peptides in vitro (C. Glabe, unpublished observations). Therefore, we first determined whether MB could also prevent mutant Htt aggregation in vitro. Aggregation of an N-terminal purified fragment of Htt encoded by the first exon of the HD gene containing 53 glutamines (Httex1Q53) and fused to GST was induced by incubation with a site-specific protease at 37°C, resulting in the removal of the GST moiety, release of the Htt fragments, and the rapid aggregation of the fragments into oligomers and fibrillar aggregates (Lotz et al., 2010). Since MB can have opposing effects on aggregation when treating monomers versus fibrils (Taniguchi et al., 2005), we determined whether MB could alter insoluble Htt formation starting with either monomeric Htt or preformed oligomers and fibrils. Effective concentration range were determined by titration. Either 1, 10, or 100 μm MB was added at specific time points during the aggregation reaction [0 h (monomer), 4 h (oligomer), or 24 h (fibril)], and the amount of insoluble Httex1Q53 retained by filter-trap assay (an assay that detects large SDS-insoluble aggregates, >0.45 μm) was quantified relative to the amount of control insoluble aggregates with no addition of MB (Wacker et al., 2004; Legleiter et al., 2010). When added to monomeric mutant Htt, MB addition significantly decreased insoluble Htt present at 48 h after induction (one-way ANOVA, F = 30.08, p = 0.0001). Interestingly, MB also reduced levels of insoluble Htt when added to preformed oligomers (one-way ANOVA, F = 12.67, p = 0.002) and even fibrillar Htt (one-way ANOVA, F = 14.39, p = 0.001) (Fig. 1B,C). These results demonstrate that MB can block the aggregation of monomeric, oligomeric, and fibrillar Htt into insoluble aggregates in vitro, with the greatest effect when added to monomers.
Figure 1.
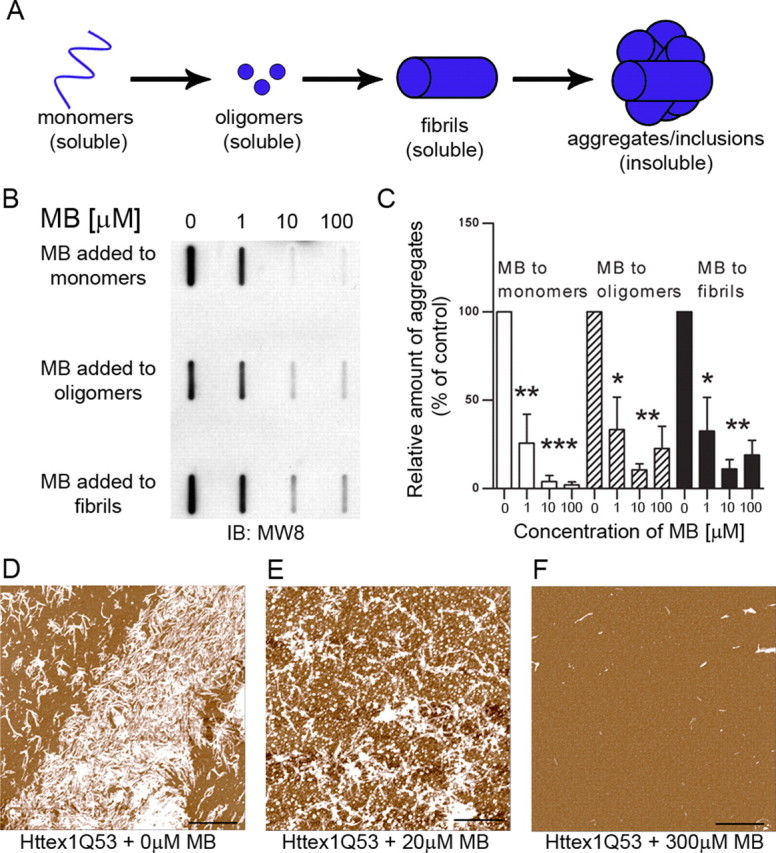
MB decreases aggregation when treating monomers, oligomers, and fibrils of Httex1Q53. A, Schematic presentation of Htt species formed during aggregation. B, MB significantly reduces the formation of insoluble Htt as seen by filter-trap assay. MB can reduce insoluble Htt when added to monomeric Htt as well as oligomeric and fibrillar Htt. C, Relative insoluble mutant Htt aggregates were normalized to insoluble control aggregates with no addition of methylene blue (Bonferroni's multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001). Error bars represent group means ± SEM. D–F, MB reduced the formation of fibrillar structures present when analyzed by atomic force microscopy. Scale bars: 2 μm.
Atomic force microscopy (AFM) analysis was next performed at 24 h after induction and revealed mostly fibrillar structures in the absence of MB with a small amount of globular structures present (Fig. 1D). However, with low-dose MB (20 μm) added at 0 h, there is a decrease in the amount of fibrillar Htt and an increase in the number of globular structures (Fig. 1E). A high dose of MB (300 μm) reduced the overall production of higher Htt aggregates (note that AFM cannot detect soluble monomers), prevented the formation of the fibrillar structures, and decreased the size of the globular species (Fig. 1F). These concentrations effectively altered aggregation in a similar experiment using polyglutamine repeat peptides (C. Glabe, unpublished observations). These data indicate that MB has a direct inhibitory effect on mutant Httex1Q53 aggregation and decreases formation of insoluble aggregates in vitro.
MB decreases oligomeric and insoluble Htt aggregate formation and toxicity in the primary cortical neuron model of HD
We next evaluated whether either oligomer and/or visible aggregates could be modulated by MB in cell-based mutant Httex1-expressing systems. Primary cortical neurons were isolated and transduced with lentiviral constructs encoding a control construct (EGFP) and expanded toxic (103Q) Httex1 protein tagged with EGFP (Southwell et al., 2009). SDS-AGE was used to examine oligomer formation in these cells (Weiss et al., 2008; Legleiter et al., 2010; Miller et al., 2011). This assay, which uses low SDS and no reducing agent, allows the visualization of large (>400 kDa), SDS-soluble oligomeric species. The species fractionated by the SDS-AGE technique were originally described as “aggregates”; however, since SDS-AGE can resolve soluble, nonsedimentable oligomers (Legleiter et al., 2010) and these proteins resolved by SDS-AGE are also recognized by the antifibrillar oligomer antibody OC (Millipore; AB2286) (Sontag et al., 2012), these species are defined as oligomers. This technique is valuable as it not only delineates the levels (intensity) but also the apparent molecular weight of the oligomers (distance of the peak signal to the running front). We found that 100 nm MB treatment for 10 d decreased oligomer formation by 46% (one-way ANOVA, F = 16.74, p = 0.004) in mouse primary cortical neurons expressing Httex1Q93 tagged with EGFP (Fig. 2A,B). By measuring the distance from the running front to the peak signal intensity, we were able to determine the relative size of the oligomers. The distance of the untreated sample was set to 100, and the distance of the peak intensity of each treated sample was compared to that of the untreated sample. Using this calculation, 100 nm MB decreased the relative size of the oligomers by 40% (one-way ANOVA, F = 36.05, p = 0.0005) in these same samples compared to no-drug controls at approximately equal intensities (Fig. 2C). This effect is not due to a difference in levels of oligomeric species present; as other compounds alter the levels of oligomers without altering the size of the oligomers present (Sontag et al., 2012; E. M. Sontag, unpublished results). To determine the effect of MB on insoluble Htt, we used a filter-retardation assay (Wanker et al., 1999) to quantitate the amount of SDS-insoluble aggregates that do not pass through a cellulose acetate membrane (0.22 μm pore size). The amount of insoluble Htt was decreased by 58% (one-way ANOVA, F = 15.29, p = 0.004) when treating with 100 nm MB (Fig. 2D,E); higher concentrations were toxic. This reduction was not a result of reduced expression of Httex1Q103, as determined by qRT-PCR analysis (data not shown).
Figure 2.
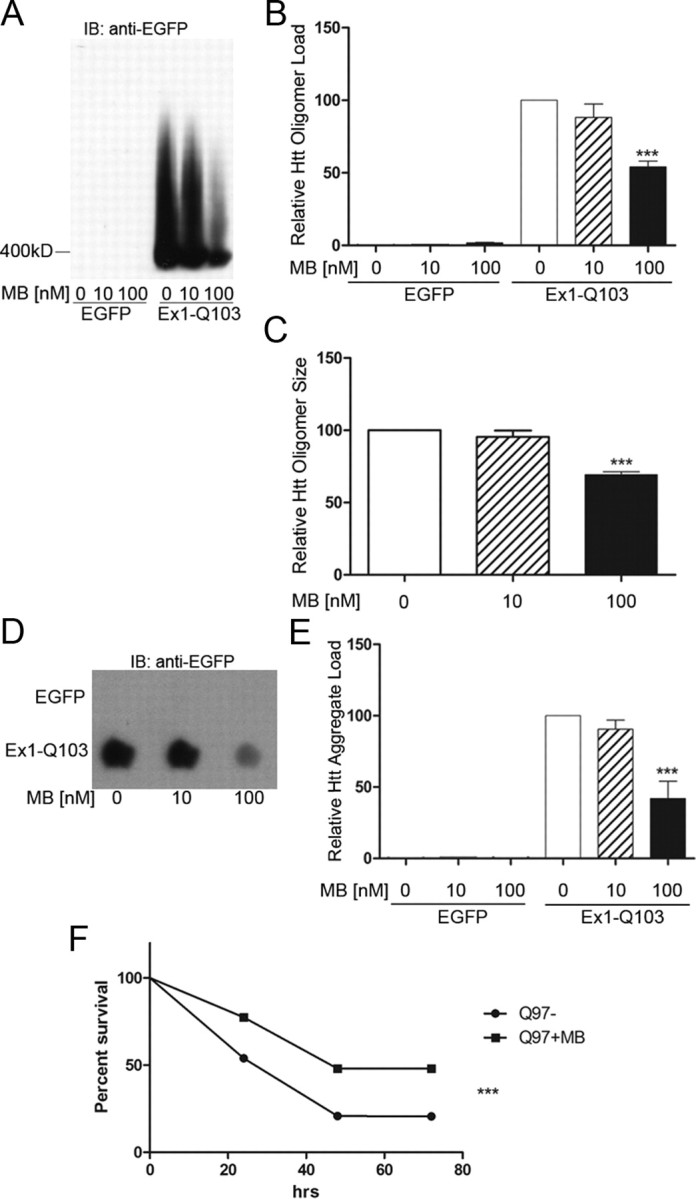
MB reduces oligomeric and insoluble Httex1Q103 as well as increasing survival in primary neurons expressing Httex1Q103. A, MB reduces the amount of oligomeric Htt resolved by SDS-AGE analysis. B, Relative oligomeric mutant Htt aggregates were normalized to oligomeric control aggregates with no addition of MB (Bonferroni's multiple comparisons, ***p < 0.001). C, Relative size of oligomeric mutant Htt aggregates were normalized to oligomeric control aggregates with no addition of MB (Bonferroni's multiple comparisons, ***p < 0.001). D, MB decreases the amount of insoluble Htt retained on the filter-retardation assay. E, Relative insoluble mutant Htt aggregates were normalized to insoluble control aggregates with no addition of MB (Bonferroni's multiple comparisons, ***p < 0.001). Error bars represent group means ± SEM. F, MB increases cell survival in primary neurons expressing Httex1Q97 over the course of 72 h (log-rank test, χ2 = 47.19, ***p < 0.0001). Experiment 1 monitored 572 total neurons [395 MB−, 177 MB+; p < 0.0001 (log rank); hazard ratio, 2.638 (2.00–3.479)], and Experiment 2 monitored 859 total neurons [523 MB−, 336 MB+; p = 0.0008 (log rank); hazard ratio, 1.455 (1.168–1.811)]. Together, Experiment 1 and Experiment 2 monitored 1431 total neurons [918 MB−, 513 MB+; p < 0.0001 (log rank); hazard ratio, 1.801 (1.518–2.138)].
Since MB reduces the levels of oligomers and insoluble aggregates, we next tested whether treatment with MB translated to improved survival of individual rat primary cortical neurons expressing Httex1Q103-EGFP. To observe cell survival, a robotic microscope system was used to perform direct longitudinal visualization of cell survival and quantitation of cell fate (Arrasate and Finkbeiner, 2005). Using this system, MB treatment (100 nm final concentration) increased cell survival by 25% (log-rank test, χ2 = 47.19, p < 0.0001) over a time period of 72 h (Fig. 2F). Two independent experiments were performed where each experiment had three to six replicates per condition. Experiment 1 monitored 572 total neurons [395 MB−, 177 MB+; p < 0.0001 (log rank); hazard ratio, 2.638 (2.00–3.479)], and Experiment 2 monitored 859 total neurons [523 MB−, 336 MB+; p = 0.0008 (log rank); hazard ratio, 1.455 (1.168–1.811)]. Together, Experiment 1 and Experiment 2 monitored 1431 total neurons [918 MB−, 513 MB+; p < 0.0001 (log rank); hazard ratio, 1.801 (1.518–2.138)].
Together, these data signify that MB can decrease the presence of potentially toxic aggregation species in cells and increase survival of neurons.
MB reduces aggregation and prevents neurodegeneration in a Drosophila model of HD
To test the effect of MB in vivo, Drosophila expressing an expanded exon I fragment of human Htt were fed food containing 8 μm MB. Neurodegeneration is readily observed in the compound eye of Drosophila (Steffan et al., 2001), where a trapezoidal arrangement of seven visible rhabdomeres (light-gathering structures of the photoreceptor cells) can be evaluated for loss of photoreceptor neurons. Expression of Httex1Q93 leads to a progressive loss of rhabdomeres from the normal seven visible rhabdomeres to approximately five rhabdomeres per ommatidium (Steffan et al., 2001) (Fig. 3A). Treating flies with MB revealed a significant increase in the number of rhabdomeres over vehicle controls. When Drosophila were treated with MB at an early (larval) stage before visible aggregate formation, the number of rhabdomeres increased from 4.83 to 5.54 at day 7 after eclosion (Student's t test, p = 0.0005), consistent with a decrease in neurodegeneration (Fig. 3A). However, when adult flies were fed immediately following eclosion, neurodegeneration was not delayed or prevented by day 7 (Student's t test, p = 0.591) (Fig. 3A). In contrast to the ability to quantitate the formation of aggregates following the gradient of expression in larvae, this aggregation process has already occurred in adult flies; therefore, aggregation was not quantified at this time point. This effect was not due to a requirement for increased amounts of MB in adult flies, as increasing the concentration of MB did not improve outcome (data not shown). On the other hand, treating Htt challenged Drosophila in the larval stages significantly decreased the number of aggregates by 87% (Student's t-test, p = 0.005) (Fig. 3B) and reduced the size of the aggregates (Student's t test, p = 0.03) (Fig. 3C). Representative images of this analysis are shown in Figure 3D. Together, these data support the hypothesis that MB could be beneficial in treating mutant Htt-mediated neurodegeneration, potentially through the modulation of aggregation.
Figure 3.
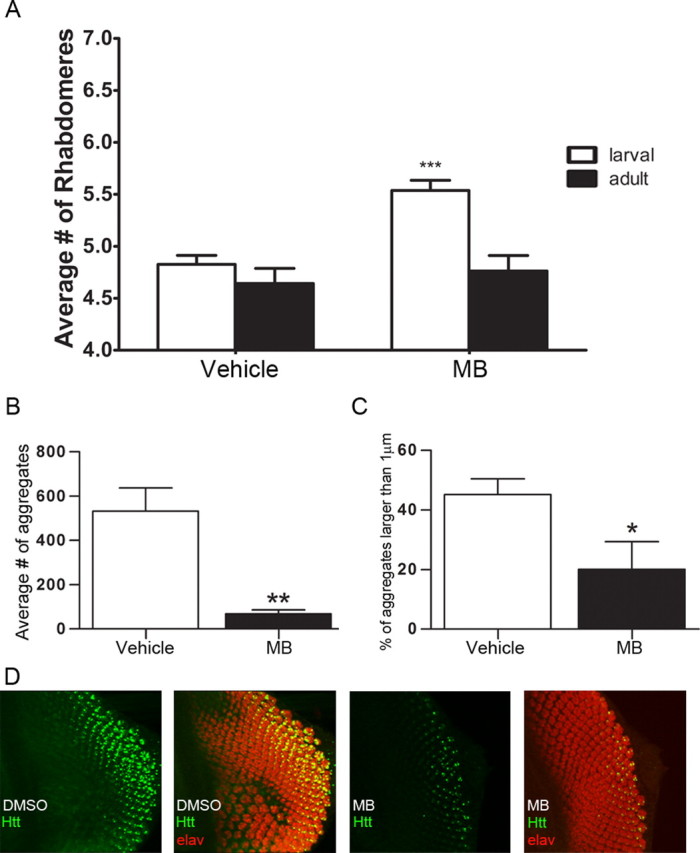
MB decreases neurodegeneration and aggregation in a Drosophila model of HD expressing Httex1Q93. A, Drosophila were fed MB beginning at the larval (n = 5) or adult (n = 5) stage and analyzed for the number of rhabdomeres per omatidium. MB treatment beginning at the larval stage resulted in an increase in rhabdomeres per omatidium (Student's t test, ***p = 0.0005). The adult fed flied did not exhibit a rescue in neurodegeneration (Student's t test, p = 0.591). B, Treating Drosophila at the larval stage (n = 4) resulted in a decrease in the average number of aggregates (Student's t test, **p = 0.005). C, Treating at the larval stage (n = 4) also resulted in decreased size of the aggregates (Student's t test, *p = 0.03). Error bars represent group means ± SEM. D, Representative photomicrographs of eye antennal imaginal discs (larval) immunolabeled with anti-Htt (S830, green) and anti-elav (red) used for quantitation of aggregate number and size.
MB reduces behavioral phenotypes and may delay disease progression in the R6/2 mouse model of HD
Because MB altered neurodegeneration in Drosophila, in vivo studies were extended to determine whether MB could alter behavioral phenotypes associated with HD-like symptoms in a mouse model of the disease. R6/2 mice, which express the first exon of human Htt with a highly expanded repeat (115Q for this study), display reproducible and aggressive phenotypes, beginning at ∼5–6 weeks. Mice show progressive loss of body weight, exhibit dyskinesia of the limbs when held by the tail (also referred to as clasping) (Mangiarini et al., 1996), and exhibit progressive loss of motor coordination and balance that can be measured by maintaining balance while running on a spinning rod (rotarod) (Carter et al., 1999). These mice also display a behavioral abnormality of a decrease in sensorimotor performance observed when mice climb down a vertical pole (Hickey et al., 2008). R6/2 mice were treated with 25 mg of MB per 100 g of chow beginning at 5 weeks of age for 4 weeks, based on MB treatment in 3xTg-AD mice (Medina et al., 2010). The mice were assayed daily for body weight and clasping phenotypes. The ability to perform the pole test was assessed at 6 and 8 weeks of age and performance on the rotarod was evaluated at 7 and 9 weeks of age (Fig. 4A).
Figure 4.

MB treatment ameliorates behavioral phenotypes in the R6/2 mouse model of HD. A, Timeline of the study illustrating the age of the mice when treatment commenced and the timing of the behavioral testing (n = 10 per group). B, Treating R6/2 mice (n = 10) results in higher body weight compared to vehicle controls (n = 10; repeated-measures ANOVA, F = 45.71, p < 0.0001; Bonferroni's multiple comparison, p < 0.01). MB-treated R6/2 mice (n = 10) were not significantly different from nontransgenic littermates (n = 10; Bonferroni's multiple comparison: p > 0.05). C, MB treatment did not alter the clasping phenotype of the R6/2 mice (n = 10 in each group; Fisher's exact probability test, p > 0.05). D, Improvements in rotarod performance were also observed when treating R6/2 mice with MB (n = 10 in each group; ANOVA, F(3,36) = 12.49, p < 0.0001; Bonferroni's multiple comparison, *p < 0.05 at 7 weeks, p > 0.05 at 9 weeks). E, Treatment with MB resulted in improved performance (two-way ANOVA, treatment, F(3,36) = 4.64, p = 0.008; time, F(3,36) = 10.48, p = 0.003) on the pole-climbing task (n = 10 in each group). Bonferroni's multiple comparison, *p < 0.05, ****p < 0.0001. Error bars represent group means ± SEM.
Differences in body weights between the groups (repeated-measures ANOVA, F = 45.71, p < 0.0001) were observed with MB administration. Treating R6/2 mice with MB resulted in a statistically significant (Bonferroni's multiple comparison, p < 0.0001) increase in body weight over the vehicle-treated R6/2 controls. Body weights of mice treated with MB were similar to those of nontransgenic littermates (Bonferroni's multiple comparison, p > 0.05), whereas the R6/2 mice treated with vehicle had significantly (Bonferroni's multiple comparison, p < 0.0001) reduced body weights compared to nontransgenic controls (Fig. 4B). MB did not significantly alter the onset of dyskinesia (clasping) (Fig. 4C), although at 8 weeks of age there is a trend toward a significant difference between the MB-treated R6/2 mice and vehicle-treated R6/2 mice (Fisher's exact probably test, p = 0.07). Power analysis reveals this study was only at 65% power for the 8 week clasping experiment with 10 mice each; therefore, with higher numbers of animals this trend may become significant.
Both rotarod and pole tests showed significantly delayed progression of phenotypic deficits in the R6/2 mice. MB-treated R6/2 mice performed better on the rotarod behavioral test at 7 weeks of age than vehicle-treated R6/2 mice (Bonferroni's multiple comparison, p < 0.05) (Fig. 4D), demonstrating better forelimb and hindlimb motor coordination and balance. However, by 9 weeks of age there is no significant difference between the MB-treated R6/2 mice and vehicle controls (Bonferroni's multiple comparison, p > 0.05). Two-way repeated-measures ANOVA indicates differences in both treatment (two-way ANOVA, F(3,36) = 12.49, p < 0.0001) and time (two-way ANOVA, F(3,36) = 8.50, p = 0.006) in the rotarod analysis. Improvements in the ability of the mice to climb down a vertical pole were also observed when treating R6/2 mice with MB (two-way ANOVA, treatment, F(3,36) = 4.64, p = 0.008; time, F(3,36) = 10.48, p = 0.003) (Fig. 4E). MB-treated R6/2 mice were significantly faster at climbing down the pole than the vehicle-treated R6/2 controls at 8 weeks of age (Bonferroni's multiple comparison, p < 0.05). In addition, the vehicle-treated R6/2 mice became significantly impaired at the pole test task between 6 and 8 weeks of age (Bonferroni's multiple comparison, P<0.0001), whereas MB-treated R6/2 mice did not show a similar progression during that time (Bonferroni's multiple comparison, p > 0.05). These results indicate that MB might delay the onset of motor coordination loss and possibly HD phenotype progression in the R6/2 mice.
MB decreases levels of insoluble Htt and increases BDNF in R6/2 mice
Given that MB modulates aggregation in vitro and in Drosophila, we examined whether the protective effect of MB in R6/2 mice correlated with altered aggregation properties. R6/2 and nontransgenic mice treated with MB or vehicle control were killed at 8 weeks of age and analyzed for the presence of aggregated Htt. No difference was detected in the levels of oligomeric Htt between MB-treated R6/2 mice and vehicle controls by SDS-AGE (data not shown), potentially reflecting timing of killing. In contrast, the levels of insoluble Htt in the striatum decreased with MB treatment measured by filter-retardation assay (one-way ANOVA, F = 29.18, p = 0.0001) (Fig. 5A,B), consistent with the direct impact of MB on mutant Htt aggregation in vitro.
Figure 5.
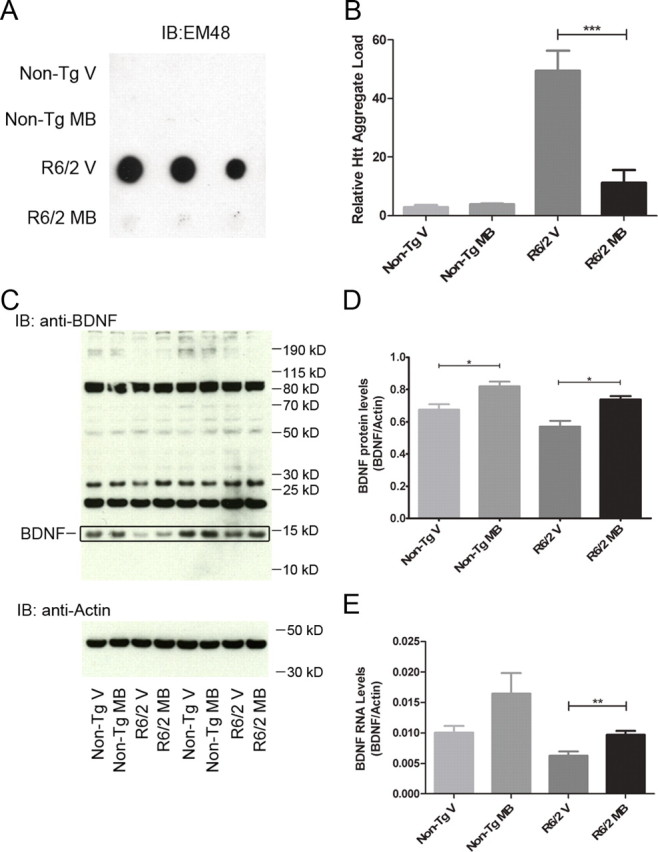
MB decreases Htt aggregation and increases levels of BDNF in R6/2 mouse model of HD. A, Striatal samples from nontransgenic littermates and R6/2 mice with vehicle or MB treatment were homogenized and analyzed for levels of insoluble Htt by filter-retardation assay. B, Densitometry of the filter-retardation assay reveals a significant decrease in the levels of insoluble Htt in the striatum of R6/2 mice when treated with MB (ANOVA, F = 29.18, p = 0.0001; Bonferroni's multiple comparison, ***p < 0.001). C, Cortical samples from nontransgenic littermates and R6/2 mice with vehicle or MB treatment were homogenized and analyzed for BDNF protein by SDS-PAGE assay and Western blot analysis. The BDNF is indicated. D, Densitometry of the SDS-PAGE analysis shows a significant increase in BDNF protein levels with MB treatment in both nontransgenic littermates and R6/2 mice (ANOVA, F = 11.35, p = 0.0002; Bonferroni's multiple comparison, *p < 0.05). Samples were normalized to actin as a control. E, Cortical samples from nontransgenic littermates and R6/2 mice with vehicle or MB treatment were homogenized and levels of BDNF mRNA quantified by qRT-PCR (ANOVA, F = 5.609, p = 0.0097; Bonferroni's multiple comparison, **p < 0.01). Error bars represent group means ± SEM. Samples were normalized to actin as a control.
While MB appears to decrease aggregation in vivo, we also evaluated whether this compound resulted in neurotrophic effects that could also be involved in its protective effects. Chronic expression of mutant Htt causes decreased neurotrophic expression, particularly of BDNF in mouse models and human patient brain (Zuccato et al., 2001). In parallel, increased BDNF often corresponds to preclinical trials in HD mouse models that show efficacy (Apostol et al., 2008; Peng et al., 2008). We therefore investigated levels of BDNF in R6/2 following MB treatment at 8 weeks. BDNF protein levels (one-way ANOVA, F = 11.35, p = 0.0002) (Fig. 5C,D) are increased in both treated nontransgenic and R6/2 mice, suggesting that MB enhances BDNF levels independent of the presence of mutant Htt, potentially contributing to the improvement in behavior. Additionally, the levels of BDNF mRNA are increased in MB-treated R6/2 mice and nontransgenic mice (ANOVA, F = 5.609, p = 0.0097) (Fig. 5E). The increase seen in the nontransgenic mice does not quite reach statistical significance, but does indicate a trend toward higher levels of BDNF mRNA (p = 0.09). This appears to be a direct cellular effect, as BDNF levels were also increased in mouse primary neurons expressing mutant Httex1 (data not shown).
Discussion
Aggregation of mutant Htt protein is a hallmark of HD. Because this phenotype is consistently observed in models of HD and human patient brains, it was among the first therapeutic targets for which chemical compound screens were performed. Recent studies using longitudinal imaging suggest that the inclusions per se are not toxic in culture, but rather that soluble monomeric or oligomeric species may be primary pathogenic forms (Arrasate et al., 2004; Miller et al., 2011). Given the ability of MB to alter aggregation in other systems (Doh-Ura et al., 2000; Korth et al., 2001; Taniguchi et al., 2005; Necula et al., 2007; van Bebber et al., 2010), we examined its ability to modulate both insoluble and soluble oligomeric mutant Htt and show that this compound does indeed reduce levels of both under cell free conditions. In addition to modulating the process of mutant Htt aggregation and formation of oligomeric species, MB also reduced neurotoxicity in primary neurons, altered photoreceptor neurodegeneration in Drosophila, and improved behavioral phenotypes in R6/2 mice, suggesting that this biochemical readout induced by MB correlates with protection from neurotoxicity.
To evaluate possible direct effects of MB on the propensity of the Htt protein to aggregate, a recombinant protein system was used and showed that not only could MB prevent the aggregation of monomeric Htt, but also decreased further aggregation of preformed mutant Htt oligomers and fibrils. From this assay, it is not possible to discern whether MB destabilizes the aggregated forms as observed previously for tau paired helical filaments (Wischik et al., 1996), or whether MB alters the equilibrium between monomers and Htt aggregates. These results have implications for therapeutic development as MB can slow aggregation of the mutant Htt protein even when aggregation has already started and aggregated species are present in vitro. Importantly, these cell-free assays also suggest that MB can have a direct effect on the mutant Htt protein independent of its previously described cellular effects on mitochondria (Atamna et al., 2008; Atamna and Kumar, 2010), the proteasome (Medina et al., 2010), or autophagic flux (Wang et al., 2010).
It is known that MB self-associates to form dimers and potentially higher-order aggregates in the range of 10 μm and above with an equilibrium of ∼100 μm (Moreno-Villoslada et al., 2010). MB is likely dimerized in the cell-free assay given the concentrations of 100 and 300 μm; however, we see the same results in the cell-free system as in the cell assays (decreased insoluble Htt) using nanomolar levels of MB that will not self-associate. Therefore, it is unlikely that the dimer is negatively impacting the mechanism of action.
MB also reduced the formation of both oligomeric and insoluble Htt in primary neurons expressing mutant Htt. In these studies, MB not only decreased the amount, but also the size of oligomers compared to vehicle-treated controls.
Longitudinal assessment of single cells using imaging allowed the visualization of the survival of only those cells expressing mutant Htt, providing a much more accurate and sensitive assessment of cell fate. Using this approach, MB increased the survival of primary neurons, correlating with a decrease in oligomeric species. In this same system, monomeric Htt and small oligomers are associated with decreased cell survival (Miller et al., 2011); therefore, reducing the levels and size of oligomers with MB could lead to a functionally relevant outcome. These results are in contrast to a previous study of MB in zebrafish embryos injected with mRNA expressing mutant Htt polypeptide (van Bebber et al., 2010), where visible aggregates were altered, but there was no effect on total embryo viability. The Htt construct expressed in zebrafish did not contain the proline-rich region of Httex1p, the lack of which also altered the outcome of MB on aggregation in stably expressing truncated Httex1p PC12 cells (E. M. Sontag, unpublished observations).
To evaluate whether increased survival in primary neurons could translate to increased neuronal survival in vivo, we examined the effect of MB on the number of photoreceptor neurons in the compound eye of Drosophila melanogaster expressing human mutant Httex1p. The progressive loss of rhabdomeres in this model (Steffan et al., 2001) was partially restored by MB treatment. Importantly, we found this protective effect only when treatment began at the larval stage versus in adult flies, suggesting that early treatment before formation of visible aggregates may be more effective than when aggregates have already formed. This idea is supported by positive results on cognition in a human clinical trial for AD in individuals with mild or moderate AD (Gura, 2008). MB decreased aggregate number and size in Drosophila treated at the larval stage, confirming that (1) MB modulates mutant Htt aggregation in vivo, and (2) the size and levels of oligomeric Htt may be pathologically relevant.
Previous studies have determined that orally administered small molecule aggregation modulators can alter aggregation and improve motor performance in R6/2 mice, including C2-8 (Chopra et al., 2007) and trehalose (Tanaka et al., 2004). We therefore hypothesized that MB might also modulate behavioral phenotypes in these mice. R6/2 mice progressively lose body weight over the course of the disease (Mangiarini et al., 1996), and MB treatment prevented the characteristic reduction, with R6/2 mice maintaining weight at nontransgenic littermate control levels. R6/2 mice also exhibit dyskinesia of the limbs when held by the tail (clasping) (Mangiarini et al., 1996), and MB-treated mice trended toward control phenotypes; however, this altered clasping phenotype did not achieve statistical significance. Future optimization of powering, dosing, formulation of MB, and assessment of bioavailability may yield even stronger beneficial effects from MB treatment. Both of these outcomes are important, as weight loss and dyskinesia are reported in patients with HD (Harper, 1991).
Loss of coordination and an unsteady gait are characteristic of symptomatic HD (Harper, 1991). R6/2 mice also exhibit a loss of motor coordination and balance that can be measured by assessing balance on a rotarod (Carter et al., 1999). MB-treated R6/2 mice were able to maintain balance and coordination significantly better on the rotarod behavioral test at 7 weeks of age than vehicle-treated R6/2 mice; however, by 9 weeks of age, the benefit was no longer observed, suggesting that while symptoms in this highly aggressive model were not completely prevented, they were delayed.
A decrease in sensorimotor performance can also be detected by a delay in the ability to climb down a vertical pole (Hickey et al., 2008). The MB-treated R6/2 mice were significantly faster in this assay than the vehicle-treated R6/2 controls at 8 weeks of age, and indeed the progressive decline between 6 and 8 weeks of age observed for vehicle-treated mice was not observed in MB-treated mice during that time. This result could indicate that MB is delaying disease progression in the R6/2 mice. Data from the R6/2 experiments showing that MB can alter the time course of several behavioral phenotypes in R6/2 mice correlate with the results of a human MB clinical trial for the treatment of Alzheimer's disease; in this trial, MB slowed the progression of AD by 81% over the course of one year (Gura, 2008). These results are significant as currently there are no Food and Drug Administration (FDA)-approved HD-modifying therapies for altering weight loss, dyskinesia, and loss of motor coordination. The only FDA-approved therapy for HD is tetrabenezine, which is palliative and reduces the symptoms of chorea in HD patients (Yero and Rey, 2008).
One possible mechanism contributing to improved behavior is increased levels of BDNF, which could also represent a pharmacodynamic marker for MB treatment. Although we do not yet know the precise mechanisms involved, MB appears to increase levels of BDNF independent of the presence of mutant Htt in both primary neuron and mouse models. This effect may be selective, as testing levels of DARPP32, synaptophysin, and phosphor-ERK levels did not reveal differences between treated and untreated groups (data not shown). Downregulation of total BDNF mRNA and protein and reduction in anterograde BDNF transport down corticostriatal axons are observed in HD (Gauthier et al., 2004; Zuccato et al., 2005). Additionally, BDNF mRNA levels increase after treatment of R6/2 mice with compounds such as CEP-1347 and sertraline (Conforti et al., 2008; Peng et al., 2008), consistent with the idea that increasing BDNF levels may be protective in HD.
MB is used to treat a variety of human conditions such as malaria (Ehrlich and Guttman, 1891; Atamna et al., 1996), methemoglobinemia (Kristiansen, 1989; Mansouri and Lurie, 1993), and septic shock (Faber et al., 2005; Kwok and Howes, 2006), and in preventing some side effects of chemotherapy (Pelgrims et al., 2000; Patel, 2006). MB also has a very high absolute bioavailability in humans (Walter-Sack et al., 2009). Maximum blood concentrations of MB were reached within 1–2 h after oral administration in humans, with a half-life of 5–6.5 h (Peter et al., 2000). MB shows efficacy in the brain as well, having been used for the treatment of depression (Naylor et al., 1986) and improvement of cognitive function (Callaway et al., 2004), memory (Callaway et al., 2004; Riha et al., 2005), and discriminative learning in rats (Wrubel et al., 2007). Furthermore, cerebellar brain concentrations of MB are higher than plasma levels following oral administration in mice (O'Leary et al., 2010). While these clinical findings lend promise to moving forward to clinical trials, testing MB in HD patients would require integration of its activity as an MAO inhibitor as well.
Together, our results with MB showing decreases of multiple aggregation species and increased BDNF, and its known positive effects in brain and well-tolerated clinical use in humans, offer a unique opportunity to move this compound forward to large-scale preclinical tests and potentially rapid clinical application to HD patients.
Footnotes
This work was supported by the Huntington's Disease Society of America (L.M.T., J.L.M.), CHDI Inc. (L.M.T., C.G., and J.L.M.), and NIH Grants NS52789 (L.M.T.), NS45284 (J.L.M.), NS054753 (P.J.M.), and PN2EY016525 (subaward, L.M.T.). The MW8 monoclonal antibody developed by Paul Patterson was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by the Department of Biological Sciences, University of Iowa (Iowa City, IA). The lentiviral Huntingtin plasmids used in the aggregation study were kindly provided by Paul Patterson and Ali Khoshnan from the California Institute of Technology, and virus preparations were obtained from the University of California, Los Angeles Vector Core, which is supported by NIH Grants JCCC/P30 CA016042 and CURE/P30 DK041301.
References
- Apostol BL, Simmons DA, Zuccato C, Illes K, Pallos J, Casale M, Conforti P, Ramos C, Roarke M, Kathuria S, Cattaneo E, Marsh JL, Thompson LM. CEP-1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice. Mol Cell Neurosci. 2008;39:8–20. doi: 10.1016/j.mcn.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Finkbeiner S. Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A. 2005;102:3840–3845. doi: 10.1073/pnas.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Atamna H, Kumar R. Protective role of methylene blue in Alzheimer's disease via mitochondria and cytochrome c oxidase. J Alzheimers Disease. 2010;20(Suppl 2):S439–S452. doi: 10.3233/JAD-2010-100414. [DOI] [PubMed] [Google Scholar]
- Atamna H, Krugliak M, Shalmiev G, Deharo E, Pescarmona G, Ginsburg H. Mode of antimalarial effect of methylene blue and some of its analogues on Plasmodium falciparum in culture and their inhibition of P. vinckei petteri and P. yoelii nigeriensis in vivo. Biochemical Pharmacol. 1996;51:693–700. doi: 10.1016/s0006-2952(95)02258-9. [DOI] [PubMed] [Google Scholar]
- Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, Ames BN. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008;22:703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- Behrends C, Langer CA, Boteva R, Bottcher UM, Stemp MJ, Schaffar G, Rao BV, Giese A, Kretzschmar H, Siegers K, Hartl FU. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F. Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacol Biochemistry Behav. 2004;77:175–181. doi: 10.1016/j.pbb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra V, Fox JH, Lieberman G, Dorsey K, Matson W, Waldmeier P, Housman DE, Kazantsev A, Young AB, Hersch S. A small-molecule therapeutic lead for Huntington's disease: preclinical pharmacology and efficacy of C2–8 in the R6/2 transgenic mouse. Proc Natl Acad Sci U S A. 2007;104:16685–16689. doi: 10.1073/pnas.0707842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti P, Ramos C, Apostol BL, Simmons DA, Nguyen HP, Riess O, Thompson LM, Zuccato C, Cattaneo E. Blood level of brain-derived neurotrophic factor mRNA is progressively reduced in rodent models of Huntington's disease: restoration by the neuroprotective compound CEP-1347. Mol Cell Neurosci. 2008;39:1–7. doi: 10.1016/j.mcn.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- DiSanto AR, Wagner JG. Pharmacokinetics of highly ionized drugs. II. Methylene blue—absorption, metabolism, and excretion in man and dog after oral administration. J Pharm Sci. 1972;61:1086–1090. doi: 10.1002/jps.2600610710. [DOI] [PubMed] [Google Scholar]
- Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol. 2000;74:4894–4897. doi: 10.1128/jvi.74.10.4894-4897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J, Brown P. Euthanasia. Curr Protoc Immunol. 2006;73:1.8.1–1.8.4. doi: 10.1002/0471142735.im0108s73. [DOI] [PubMed] [Google Scholar]
- Ehrlich P, Guttman P. Ueber die wirkung des methylenblau bei malaria. Berlin Klin Wochenschr. 1891;28:953–956. [Google Scholar]
- Faber P, Ronald A, Millar BW. Methylthioninium chloride: pharmacology and clinical applications with special emphasis on nitric oxide mediated vasodilatory shock during cardiopulmonary bypass. Anaesthesia. 2005;60:575–587. doi: 10.1111/j.1365-2044.2005.04185.x. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Gura T. Hope in Alzheimer's fight emerges from unexpected places. Nat Med. 2008;14:894. doi: 10.1038/nm0908-894. [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME. Huntington's disease. Semin Cell Biol. 1995;6:21–28. doi: 10.1016/1043-4682(95)90011-x. [DOI] [PubMed] [Google Scholar]
- Harper PS. Huntington's disease. London: W.B. Saunders; 1991. [Google Scholar]
- Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, Chesselet MF. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington's disease mice. Neuroscience. 2008;157:280–295. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A. 2001;98:9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen JE. Dyes, antipsychotic drugs, and antimicrobial activity. Fragments of a development, with special reference to the influence of Paul Ehrlich. Danish Med Bull. 1989;36:178–185. [PubMed] [Google Scholar]
- Küpfer A, Aeschlimann C, Wermuth B, Cerny T. Prophylaxis and reversal of ifosfamide encephalopathy with methylene-blue. Lancet. 1994;343:763–764. doi: 10.1016/s0140-6736(94)91839-2. [DOI] [PubMed] [Google Scholar]
- Kwok ES, Howes D. Use of methylene blue in sepsis: a systematic review. J Intensive Care Med. 2006;21:359–363. doi: 10.1177/0885066606290671. [DOI] [PubMed] [Google Scholar]
- Legleiter J, Mitchell E, Lotz GP, Sapp E, Ng C, DiFiglia M, Thompson LM, Muchowski PJ. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J Biol Chem. 2010;285:14777–14790. doi: 10.1074/jbc.M109.093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz GP, Legleiter J, Aron R, Mitchell EJ, Huang SY, Ng C, Glabe C, Thompson LM, Muchowski PJ. Hsp70 and Hsp40 functionally interact with soluble mutant huntingtin oligomers in a classic ATP-dependent reaction cycle. J Biol Chem. 2010;285:38183–38193. doi: 10.1074/jbc.M110.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Mansouri A, Lurie AA. Concise review: methemoglobinemia. Am J Hematol. 1993;42:7–12. doi: 10.1002/ajh.2830420104. [DOI] [PubMed] [Google Scholar]
- Medina DX, Caccamo A, Oddo S. Methylene blue reduces aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2010;21:140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, Widjaja K, Shaby BA, Lotz GP, Newhouse Y, Mitchell EJ, Osmand A, Gray M, Thulasiramin V, Saudou F, Segal M, et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7:925–934. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Villoslada I, Torres-Gallegos C, Araya-Hermosilla R, Nishide H. Influence of the linear aromatic density on methylene blue aggregation around polyanions containing sulfonate groups. J Phys Chem B. 2010;114:4151–4158. doi: 10.1021/jp909105r. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor GJ, Martin B, Hopwood SE, Watson Y. A two-year double-blind crossover trial of the prophylactic effect of methylene blue in manic-depressive psychosis. Biol Psychiatry. 1986;21:915–920. doi: 10.1016/0006-3223(86)90265-9. [DOI] [PubMed] [Google Scholar]
- Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, Glabe CG. Methylene blue inhibits amyloid Aβ oligomerization by promoting fibrillization. Biochemistry. 2007;46:8850–8860. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- O'Leary JC, 3rd, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, Jinwal UK, Koren J, 3rd, Jones JR, Kraft C, Peters M, Abisambra JF, Duff KE, Weeber EJ, Gestwicki JE, Dickey CA. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PN. Methylene blue for management of Ifosfamide-induced encephalopathy. Ann Pharmacother. 2006;40:299–303. doi: 10.1345/aph.1G114. [DOI] [PubMed] [Google Scholar]
- Pelgrims J, De Vos F, Van den Brande J, Schrijvers D, Prové A, Vermorken JB. Methylene blue in the treatment and prevention of ifosfamide-induced encephalopathy: report of 12 cases and a review of the literature. Br J Cancer. 2000;82:291–294. doi: 10.1054/bjoc.1999.0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Masuda N, Jiang M, Li Q, Zhao M, Ross CA, Duan W. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington's disease mouse model. Exp Neurol. 2008;210:154–163. doi: 10.1016/j.expneurol.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter C, Hongwan D, Küpfer A, Lauterburg BH. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur J Clin Pharmacol. 2000;56:247–250. doi: 10.1007/s002280000124. [DOI] [PubMed] [Google Scholar]
- Riha PD, Bruchey AK, Echevarria DJ, Gonzalez-Lima F. Memory facilitation by methylene blue: dose-dependent effect on behavior and brain oxygen consumption. Eur J Pharmacol. 2005;511:151–158. doi: 10.1016/j.ejphar.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Sontag EM, Lotz GP, Yang G, Sontag CJ, Cummings BJ, Glabe CG, Muchowski PJ, Thompson LM. Detection of mutant Huntingtin aggregation conformers and modulation of SDS-soluble fibrillar oligomers by small molecules. J Hunting Dis. 2012;1:119–132. doi: 10.3233/JHD-2012-129004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell AL, Ko J, Patterson PH. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington's disease. J Neurosci. 2009;29:13589–13602. doi: 10.1523/JNEUROSCI.4286-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- van Bebber F, Paquet D, Hruscha A, Schmid B, Haass C. Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiol Dis. 2010;39:265–271. doi: 10.1016/j.nbd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- Walter-Sack I, Rengelshausen J, Oberwittler H, Burhenne J, Mueller O, Meissner P, Mikus G. High absolute bioavailability of methylene blue given as an aqueous oral formulation. Eur J Clin Pharmacol. 2009;65:179–189. doi: 10.1007/s00228-008-0563-x. [DOI] [PubMed] [Google Scholar]
- Wang AM, Morishima Y, Clapp KM, Peng HM, Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Inhibition of hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J Biol Chem. 2010;285:15714–15723. doi: 10.1074/jbc.M109.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999;309:375–386. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- Weiss A, Klein C, Woodman B, Sathasivam K, Bibel M, Régulier E, Bates GP, Paganetti P. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington's disease. J Neurochem. 2008;104:846–858. doi: 10.1111/j.1471-4159.2007.05032.x. [DOI] [PubMed] [Google Scholar]
- Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrubel KM, Riha PD, Maldonado MA, McCollum D, Gonzalez-Lima F. The brain metabolic enhancer methylene blue improves discrimination learning in rats. Pharmacol Biochem Behav. 2007;86:712–717. doi: 10.1016/j.pbb.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yero T, Rey JA. Tetrabenazine (Xenazine), an FDA-approved treatment option for Huntington's disease-related chorea. P T. 2008;33:690–694. [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M, Valenza M, Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]