

Abstract
Cancer chemotherapy is hampered by serious toxicity to healthy tissues. Conceivably, encapsulation of cytotoxic drugs in actively-targeted, biocompatible nanocarriers could overcome this problem. Accordingly, we used sterically stabilized mixed micelles (SSMM) composed of biocompatible and biodegradable phospholipids to solubilize paclitaxel (P), a hydrophobic model cytotoxic drug, and deliver it to breast cancer in rats. To achieve active targeting, the surface of SSMM was grafted with a ligand, human vasoactive intestinal peptide (VIP) that selectively interacts with its cognate receptors overexpressed on breast cancer cells. We found that even in vitro cytotoxicity of P-SSMM-VIP was 2-fold higher that that of free paclitaxel (p<0.05). Given the unique attributes of P-SSMM and P-SSMM-VIP, most notable small hydrodynamic diameter (~15nm) and stealth properties, biodistribution of paclitaxel was significantly altered. Accumulation of paclitaxel in breast tumor was highest for P-SSMM-VIP, followed by P-SSMM and Cremophor based paclitaxel (PTX). Importantly, bone marrow accumulation of paclitaxel encapsulated in both SSMM-VIP and SSMM was significantly less than that of PTX. Administration of clinically-relevant dose of paclitaxel (5mg/kg) as P-SSMM-VIP and P-SSMM eradicated carcinogen-induced orthotopic breast cancer in rats, whereas PTX decreased tumor size by only 45%. In addition, a 5-fold lower dose (1mg/kg) of paclitaxel in actively targeted P-SSMM-VIP was associated with ~80% reduction in tumor size while the response to PTX and P-SSMM was significantly less. Hypotension was not observed when VIP was grafted onto SSMM. Based on our findings, we propose further development of effective and safe VIP-grafted phospholipid micelle nanomedicines of anti-cancer drugs for targeted treatment of solid tumors in humans.
Keywords: phospholipid mixed micelles, targeted drug delivery, human vasoactive intestinal peptide, breast cancer, paclitaxel, MNU-induced breast cancer
Introduction
The efficacy of cancer chemotherapy is hampered by dose limiting toxicity to healthy tissues. Compounding this problem is the fact that potent cytotoxic drugs, such as paclitaxel, are sparsely water-soluble and are, therefore, formulated with cosolvents, such as Cremophor EL®, which are toxic themselves [1]. Hence, developing alternate therapeutic modalities, that increase selective anti-cancer efficacy of cytotoxic drugs while minimizing their systemic toxicity, is urgently needed. Given these issues, targeted delivery of cytotoxic drugs specifically to the site of action using safe biocompatible materials could represent an important means to overcome this problem.
Phospholipid micelles, specifically sterically stabilized mixed micelles (SSMMs) and sterically stabilized micelles (SSMs) are self-assembled nanoparticles that are very attractive for drug delivery due to multiple reasons. Phospholipid molecules, used to prepare these micelles are biodegradable, relatively non-toxic and already FDA approved for human use as components of the marketed parenteral pharmaceuticals [2,3]. Structurally, the phospholipid micelles are a multi-compartment system that consists of an internal lipophilic core where water-insoluble drugs can reside and an outer polyethylene glycol (PEG) corona where semipolar molecules can be accommodated. Poor water solubility of many cytotoxic agents substantially limits their clinical use. We have demonstrated that encapsulation of hydrophobic drugs, including taxanes, in phospholipid micelles enhances their aqueous solubility in some cases up to thousand-fold without using any toxic solvents or detergents [4–7]. In contrast to many other surfactant micelles, phospholipid micelles are relatively stable against dilution due to extremely low critical micellar concentration (~1μM) in aqueous media [8]. Moreover, our data suggest that phospholipid micelles can enhance stability of the drugs, protecting them from chemical or enzymatic degradation [4,9]. Nanoconstructs are thought to facilitate enhanced accumulation of entrapped drugs in areas with increased vascular permeability, such as in certain solid tumor malignancies [10]. More specifically, particles with size of ~15nm, similar to SSM and SSMM, were estimated to possess most favorable balance between systemic clearance and vascular extravasation, resulting in improved tumor accumulation [11].
Drug accumulation at the tumor site can be further improved by grafting the micelle surface with a targeting ligand specific to a cancer cell surface marker. In recent years the use of endogenous peptides, that are less immunogenic than large antibody fragments, for targeted drug delivery of therapeutics as well as imaging agents has received a lot of attention [12–14]. One of these attractive ligands is human vasoactive intestinal peptide (VIP) [15,16]. VIP receptors (VIP-R) have been shown to be overexpressed in many cancers, with about five times higher expression in all examined human breast cancer specimens [17,18], as well as in carcinogen-induced rat breast neoplasms [19]. Regardless of the histological site of origin, both ductal and lobular breast carcinomas overexpress VIP-Rs [20]. Furthermore, VIP-guided nanotherapy may also be useful for advanced cancers as evidenced by VIP-R expression in metastatic sites at the same or higher level than in primary neoplasms, including breast [21,22]. Moreover, the internalizing nature of VIP-R was shown to promote faster intracellular accumulation of the micelle cargo [23] and enhance sensitivity of resistant cells to anticancer agent by saturation or bypassing of the efflux pumps [24]. It is important to note, that VIP-Rs are only expressed in extravascular regions of microvasculature [25]. When VIP is associated with nanomicelles its extravasation to healthy tissues is minimized, hence off-target effect of the peptide is eliminated.
These attributes of phospholipid micelles as a drug delivery system and VIP as a specific ligand for cancer cell targeting, led us to test the cytotoxicity and safety of a novel, actively-targeted paclitaxel nanomedicine, P-SSMM-VIP, against breast cancer both in vitro and in vivo.
Materials and Methods
Materials
Lipids, egg phosphatidylcholine (PC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethyleneglycol)2000] (DSPE-PEG2000), were purchased from Lipoid GmbH (Ludwigshafen, Germany) and Northern Lipids Inc. (Vancouver, Canada), respectively. Activated lipid for peptide conjugation 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-n-[poly(ethylene glycol)]-N-hydroxysuccinamide (PE-PEG3400-NHS) was obtained from Shearwater Polymers Inc. (Huntsville, AL). Paclitaxel (>99% purity) from Sigma-Aldrich Co. (St. Louis, MO), 3H-labeled paclitaxel for in vivo studies from Moravek Biochemicals (Brea, CA), and paclitaxel injection (generic equivalent of Taxol®) from Bedford Laboratories (Bedford, OH) were used. Vasoactive intestinal peptide (VIP), with common sequence for human and rat His-Ser-Asp-Ala-Val-Phe-Thr-Asp-Asn-Tyr-Thr-Arg-Leu-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Asn-Ser-Ile-Leu-Asn-NH2, was synthesized by solid-phase synthesis and purified by Protein Research Laboratory at Research Resources Center, University of Illinois at Chicago. N-methyl nitrosourea (MNU) used for the induction of breast cancer was obtained from Ash Stevens Inc. (Detroit, MI). TS-2™ tissue solubilizer and Safety-Solve™ complete counting cocktail were obtained from Research Products International Corp. (Mount Prospect, IL). All other chemicals were of analytical grade from Fisher Scientific (Itasca, IL) or Sigma-Aldrich Co (St. Louis, MO).
Cell lines
MCF-7 human breast adenocarcinoma cells (ATCC # HTB-22) were kindly provided by Dr. William T. Beck (Department of Biopharmaceutical Sciences, University of Illinois at Chicago). The cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and 50 IU/ml penicillin-50 μg/ml streptomycin in a humidified atmosphere of 5% CO2 at 37 °C.
Preparation of the formulations
Dispersions of paclitaxel solubilized in sterically stabilized mixed micelles (P-SSMM) and blank SSMM were prepared by coprecipitation and rehydration method, as described previously [7]. Optimal lipid composition for DSPE-PEG2000 to PC in a molar ratio of 90:10 was used in these studies as previously determined [7]. VIP was conjugated to the distal end of activated PEGylated lipid (PE-PEG3400-NHS) as previously described [26]. This reaction takes place between NHS-ester and a primary amine yielding predominantly a 1:1 PE-PEG-VIP conjugate. Incorporation of PE-PEG3400-VIP construct into preformed P-SSMM or SSMM was achieved by coincubation for 1 h at 25°C to form P-SSMM-VIP or SSMM-VIP, respectively, alike our previously published protocols [4,23]. The saturation molar ratio of PE-PEG3400-VIP:SSMM was determined by isothermal titration calorimetry (ITC) by measuring total heat of the insertion as previously described [27,28], with modifications. Briefly, calorimetric analysis was carried out at 25°C in a 4200 ITC unit (Calorimetry Sciences Corporation, Provo, UT). Measuring cell was filled with 2mM SSMM in 0.01M HEPES buffer, pH 6.6 and PE-PEG3400-VIP (0.7mM) in same buffer was injected in 10 μl aliquots. Integrated heat change for each injection after subtracting the heat of dilution versus injection number was plotted and analyzed by Bindworks™software (Calorimetry Sciences Corporation, Provo, UT). Average values for K (binding constant) and H (reaction enthalpy) for the titration were computed with an aid of Bindworks™software based on the integrated modeling plots.
For in vivo tissue distribution studies, P-SSMM was prepared as described above containing lmg/ml paclitaxel spiked with 34μCi/ml 3H-paclitaxel (specific activity 5.5Ci/mmol).
Formulation characterization
Particle size distribution and mean particle diameter was determined by dynamic light scattering using a Nicomp 380 particle size analyzer (Particle Sizing Systems, Santa Barbara, CA). Paclitaxel concentration was evaluated by reversed-phase high performance liquid chromatography (RP-HPLC) as described previously [7]. Phospholipid content was determined by a modified Bartlett’s phospholipid assay [29]. VIP levels were assessed by enzyme-linked immunosorbent assay (ELISA) according to manufacturer instructions (Peninsula Laboratories, San Carlos, CA).
In vitro cytotoxicity
Human mammary adenocarcinoma MCF-7 cell line that was shown to overexpress VIP receptors [24,30] was used to evaluate cytotoxic activity of paclitaxel in SSMM-VIP and SSMM. A dimethyl sulfoxide (DMSO) solution of paclitaxel was used as a free drug control. Vehicle controls, SSMM-VIP and SSMM, were tested at same lipid concentration as the test solutions.
Samples were prepared as described above and serial dilutions were made to obtain paclitaxel concentrations ranging from 0.5–5 nM. Viable cells were seeded in 190 μl of the complete medium at a density of 12,000 cells/well in a 96-well plate. After that, 10 μl of test solutions and controls were added to each corresponding well. Final DMSO concentrations in each well did not exceed 0.5%. Each sample was evaluated in triplicate. The plates with test and control solutions were then incubated for 72 hours in a humidified atmosphere of 5% CO2 at 37°C. After the incubation period, cell viability was determined by sulforhodamine B assay [6,7]. The values were expressed as percent of survival and paclitaxel effective dose (ED50) was calculated using nonlinear regression analysis. Readings obtained for the solvent controls were used to define 100% growth.
Animals and breast cancer induction
All animal procedures were performed in compliance with UIC institutional guidelines and protocols approved by the Institutional Animal Care Committee.
Virgin female Sprague Dawley rats (45-day old, ~150 g) purchased from Harlan Laboratories (Indianapolis, IN) were housed and maintained in controlled temperature and humidity environment of Biological Recourse Laboratory of UIC on 12 hour light/dark cycle with free access to food and drinking water.
Breast cancer was induced by injection of carcinogen, MNU as previously described with some modifications [12,31]. Briefly, acclimatized virgin female Sprague Dawley rats were anesthetized with ketamine/xylazine (13.3/1.3 mg per 100g body weight, intraperitoneally). Each animal received a single intravenous injection of MNU (50 mg/kg body weight) in acidified saline (pH 5.0) via tail vein. Mammary tumors became palpable within 100–150 days after the injection. Once detected, two largest orthogonal dimensions were measured with electronic vernier calipers to estimate tumor size. The in vivo studies were initiated when the tumor size reached approximately 350 mm3.
In vivo biodistribution
These studies investigated the tissue distribution of paclitaxel delivered in SSMM-VIP in comparison with SSMM and commercial formulation paclitaxel for injection. The breast cancer bearing rats (16 animals per group) were anesthetized with ketamine/xylazine (13.3/1.3 mg per 100g body weight, intraperitoneally) and received a single intravenous injection of 5mg/kg paclitaxel in SSMM-VIP, SSMM or PTX each spiked with 3H-paclitaxel (34 μCi/ml) through the tail vein. The rats (4 animals/time point) were sacrificed at 15 min, 60 min, 4h, and 24h post drug administration with an overdose of pentobarbital (200 mg/kg) intraperitoneally. Breast tumor, liver, spleen, kidneys, heart, lungs, and bone marrow were excised, homogenized in phosphate-buffered saline, and mixed with TS-2™ tissue and gel solubilized. Aliquots of solubilized tissue samples were mixed with Safety-Solve™ complete counting cocktail and total radioactivity were counted using a Beckman Coulter LS 6500 scintillation counter (Fullerton, CA). Concentration of paclitaxel per gram of tissue was determined based on known specific activity of the administered 3H-paclitaxel.
In vivo efficacy
These studies aimed to determine if paclitaxel administered in SSMM resulted in improved efficacy in comparison with PTX and if actively targeting P-SSMM-VIP was able to improve the efficacy further. Rats with qualifying breast tumor size were randomly divided into groups of six. The assigned rats were then treated intravenously with paclitaxel at 5mg/kg (an equivalent clinical human dose) and 1 mg/kg in one of the following formulations: P-SSMM-VIP, P-SSMM, and PTX. Vehicle controls, SSMM-VIP and SSMM, corresponding to the highest treatment group were also tested. The dosing schedule was once every three days for a total of 5 cycles. The tumor size was monitored daily with electronic vernier calipers for a total of 30 days post treatment initiation.
Effect of P-SSMM-VIP on systemic arterial blood pressure
Rats with qualifying tumor size were randomly assigned into two groups of four animals. Rats were acclimatized to a heated restrainer for 10 min daily for five consecutive days. Thereafter, systemic arterial pressure was monitored using a non-invasive tail-cuff blood pressure system RTBP2000 (Kent Scientific Corporation, Torrington, CT) and recorded with the aid of the software provided by the manufacturer. On the day of the experiment, animals were placed in the heated restrainer for 10 min and baseline systolic pressure recorded. Once the pressure stabilized, the tail was numbed with 10% lidocaine jelly and P-SSMM-VIP or VIP alone (4.5 μmol/kg VIP; 5mg/kg paclitaxel corresponding to highest doses required for efficacy in vivo experiments) were injected intravenously through the tail vein. Systolic blood pressure of each animal was then monitored and recorded over 2 hours after the injection.
Data and statistical analyses
All in vitro experiments were performed in triplicate and data were expressed as mean ± standard deviation (SD). The data from each in vivo experiment were represented as mean ± standard error of the mean (SEM). Statistical analysis was performed using analysis of variance (ANOVA) followed by Tukey’s test. A value of p<0.05 was considered statistically significant.
Results and discussion
In vitro characterization and optimization of P-SSMM-VIP
Previously we have shown that the optimum molar ratio of DSPE-PEG2000 and PC for SSMM was 90:10 [8,32] and at this ratio paclitaxel was solubilized at 1.5 fold higher concentrations when compared to simple DSPE-PEG2000 micelles [7].
One of the new aspects of this study was to covalently bind VIP on the surface of SSMM for active targeting. This was achieved using the same technique that we previously developed and reported for VIP conjugation on stealth liposomes for imaging [26]. The reaction involved conjugation of the distal end of PEGylated lipid to the N-terminal of the peptide. This linking position does not interfere with the peptide receptor interaction [33]. A longer PEG spacer (PEG3400) was utilized to allow more flexibility to VIP when inserting into the PEG corona.
Isothermal titration calorimetry (ITC) was used to determine optimum ratio of the peptide- lipid conjugate (PE-PEG3400-VIP) insertion into SSMM. ITC has been widely used for the measurement of the binding equilibrium, which is directly computed from the heat evolved on association or physical insertion of ligands (e.g. peptides) with its binding partner (e.g. lipid surface) [28,34,35]. The heats of insertion of PE-PEG3400-VIP into SSMM after subtraction of the heat of dilution were integrated with the aid of Bindworks™ software (Fig. 1). Insertion of the conjugate into SSMM reached saturation at the molar ratio of ~17:1, total phospholipid: PE- PEG3400-VIP. Interaction between the binding partners was strong as indicated by the modeled binding constant (K), 45,583.73±4,605.70 and enthalpy (ΔH), −98.09±0.56kJ/mol. Based on these results, further SSMM-VIP formulations were prepared at the molar ratio corresponding to 15mM phospholipid to 0.9mM peptide.
Fig. 1.
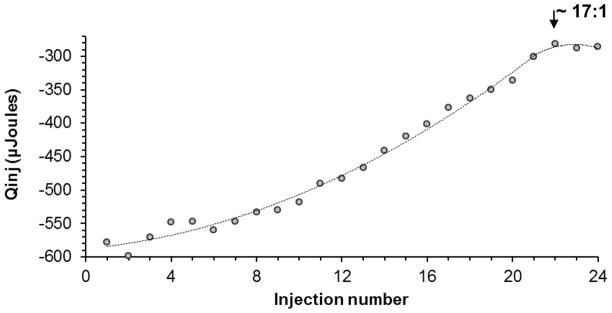
Integrated heat change per injection for the titration of PE-PEG3400-VIP and SSMM. Saturation of SSMM by PE-PEG3400-VIP was achieved between injection 22 and 24, corresponding to the molar ration of ~17:1 total lipid to the peptide conjugate
The optimized formulation of P-SSMM-VIP was characterized and compared to P-SSMM (Table 1). P-SSMM average hydrodynamic diameter was 14.2±0.26nm which did not change significantly on insertion of VIP, 14.5±1.19 nm. Concentration of encapsulated paclitaxel in both SSMM and SSMM-VIP was found to be around lmg/ml. The phospholipid content of P-SSMM and P-SSMM-VIP was not significantly different. The VIP content of P-SSMM-VIP was found to be 0.854±0.07mM, which translated to about five VIP molecules per micelle, computed based on the known micelle aggregation number of ninety [8].
Table 1.
Characteristics of optimized P-SSMM-VIP and P-SSMM formulations
| Formulation | Average diameter (nm) | Phospholipid content (mM) | Paclitaxel concentration (μg/ml) | VIP content (mM) |
|---|---|---|---|---|
| P-SSMM | 14.2±0.26 | 14.8±0.09 | 999.0±2.10 | |
| P-SSMM-VIP | 14.5±1.19 | 15.8±0.07 | 987.1±22.03 | 0.854±0.067 |
values are mean ± SD from three separate formulation preparations
In vitro cytotoxicity
To assess the effect of solubilization of paclitaxel in SSMM on cytotoxicity against breast cancer cell line, as well as to evaluate the influence of active VIP-targeting in this regard, we evaluated the in vitro cytotoxicity of the formulations.
The cytotoxic profiles of paclitaxel in various formulations tested against breast adenocarcinoma cell line MCF-7, which is VIP-R positive [24,30], are depicted on Fig. 2. Paclitaxel solubilized in SSMM showed similar cytotoxicity compared to a solution of paclitaxel in DMSO with ED50 values of 7.80±0.53nM and 8.17±1.08nM, respectively. Remarkably, a two fold increase in ED50 (3.76±0.26 nM) was observed in case of P-SSMM-VIP. This increase in cytotoxicity could be attributed, in part, to the accelerated rate of intracellular drug accumulation by VIP-R internalization [23]. Blank carriers, SSMM and SSMM-VIP showed no toxicity to the cells at the concentration range corresponding to the drug-containing test solutions (Fig. 2).
Fig. 2.

Cytotoxic activity of paclitaxel solubilized in SSMM-VIP, SSMM, and DMSO against human breast adenocarcinoma MCF-7 cell line. Drug free vehicles, SSMM-VIP and SSMM, were also tested at concentrations equivalent to the test solutions. Data represent the mean ± SD of triplicates (n=3/formulation)
Nanocarriers alter biodistribution of paclitaxel
The ultimate goal of targeted delivery is to achieve increased accumulation of drug at the cancer site and diminished exposure to systemic tissues, to reduce drug toxicity. Therefore, the biodistribution data for the drug administered in a nanocarrier in comparison with conventional formulation would provide an understanding of possible outcome on therapeutic application of targeted delivery system. Thus, in these studies biodistribution of P- SSMM-VIP, P-SSMM, and commercial PTX in rats bearing MNU-induced breast cancer was compared. The formulations were administered at a single clinical paclitaxel dose of 5mg/kg. The drug accumulation in various organs was evaluated up to 24 hours post injection.
Rapid distribution of P-SSMM and P-SSMM-VIP to the mammary tumor within first 15 min was significantly (p<0.05) higher (~2x) than for PTX (Fig. 3. a). In contrast, distribution of P-SSMM-VIP and P-SSMM to the bone marrow was significantly lower followed by rapid clearance in comparison with PTX (Fig. 3. b). The later could be attributed to the notorious propensity of molecular paclitaxel to accumulate in bone marrow causing myelosuppression [36]. In contrast, SSMM encapsulation minimized, to some extent, drug contact with marrow and contributed to rapid clearance along the concentration gradient back to the circulation through inherently “leaky” vascular fenestration (85–100 nm) of marrow [37].
Fig. 3.

Paclitaxel exposures over 24 hours in rats bearing MNU-induced breast cancer following intravenous administration of 5 mg/kg paclitaxel in P-SSMM-VIP, P-SSMM, and commercial product Paclitaxel for injections (PXT): a) Breast cancer; b) Bone marrow. Data represent the mean ± SEM; n=4 rats/formulation/time point
Moreover, P-SSMM-VIP accumulated significantly more at the tumor site (AUC0-t 87.8±9.5 μg-h/g; p<0.05) compared to both P-SSMM (45.8±2.3 μg-h/g) and PTX (26.7 ±3.2 μg-h/g) as shown on Fig. 4. This confirms our hypothesis that active VIP-targeting results in retention of the micelles at the tumor causing an increase in drug concentration with time in VIP-R positive tumor mass. Furthermore, binding to VIP-R promote cellular accumulation of the carrier with its entrapped load via internalization, as was previously shown by us [23], assisting in retention of the drug within the tissue. In contrast, non-actively targeted micelles have less potential to hold up in the tissue and can escape back into the circulation.
Fig. 4.

Comparative paclitaxel tissue distribution (AUC0–24h) in rats bearing MNU-induced breast cancer following intravenous administration of 5mg/kg paclitaxel formulated in SSMM-VIP, SSMM, and commercial product Paclitaxel for injections (PXT). Data represent the mean ± SEM; n=4 rats/formulation/time point; * p<0.05 compared to PXT; # p<0.05 compared to PXT; ^ p<0.05 compared to P-SSMM
Lower cumulative exposure to micellar paclitaxel in comparison with commercial PXT product was observed for other tissues, including heart, kidneys, lung, and spleen (Fig. 4). No statistically significant formulation-dependent differences were observed for the liver exposure (Fig. 4). This finding may be rationalized by the fact that paclitaxel metabolism and elimination takes place in the liver [38] and translocation of the drug from other organs to the liver with time may contribute to this matter. In addition, formulation dependant differences in paclitaxel distribution on the cellular level (hepatocytes vs. Kupffer cells) may exist, contributing to evident similarity of drug deposition in liver. Variances in mechanisms of body elimination for the particulates in comparison with molecular drug may, at least partially, play a role in observed biodistribution differences.
Based on several other reports on biodistribution studies for various nanoparticulate drug systems [39–41], we conclude that biodistribution strongly depends on the carrier properties, such as size, composition, steric stabilization, in addition to the features of the drug itself, such as hydrophobicity and the mechanisms of its elimination. Nevertheless, in most cases drug delivery in a carrier system usually results in a favorable tumor accumulation.
In vivo efficacy of P-SSMM-VIP against MNU-induced breast cancer in rats
MNU-induced rat mammary cancer is a well-established animal model, widely used in the study of carcinogenesis and efficacy evaluation of chemotherapeutic and chemopreventive agents [12,42,43]. It is an orthotopically developed model in which the carcinogen induces and promotes tumor formation in the mammary gland and mimics the multistage process of human mammary carcinogenesis. It differs from a xenograft model, derived from a single-cell clone that lacks the development of cellular architecture of the natural tumor environment. Furthermore, we have shown in MNU-induced breast cancer rat model that the VIP-Rs are overexpressed approximately 5 times more in the breast cancer tissue than in surrounding normal breast [19]. Hence, the MNU-induced cancer animal model, with many resemblances of the human disease, such as realistic anatomic location and targeting environment, was chosen for this study.
Tumor response profiles to each of the treatments are illustrated in Fig. 5. Both drug-free vehicle controls, SSMM and SSMM-VIP, did not show any anticancer action in vivo. Tumor burden in both cases was increased about 100-fold similar to untreated animals at the end of the observation period (data not shown). Treatment with P-SSMM-VIP at a low paclitaxel dose of lmg/kg resulted in a significant (80±3.6%) reduction of the tumor size, compared to both P-SSMM (40±3.6%) and PTX (26±4.5%) at the end of the observation period (day 30) as presented in Fig. 5. a. Treatment with the clinical paclitaxel dose of 5mg/kg resulted in a dramatic increase in efficacy with near complete cancer eradication for P-SSMM-VIP (95±0.3%) and P-SSMM (93±2.0%) compared to PTX which resulted in 45±4.5% tumor regression (Fig. 5. b). In addition, the rate of tumor regression was faster for P-SSMM and P-SSMM-VIP compared to PTX. However, at 5mg/kg paclitaxel no significant differences were observed between P-SSMM and P-SSMM-VIP. This may be due to the fact that at the dose of 5mg/kg paclitaxel the passive targeting was able to achieve the maximal response to the treatment and active targeting could not show any further improvement. However, it is likely that actively-targeted P-SSMM-VIP can be superior to P-SSMM at a dose lower than 5mg/kg paclitaxel and/or with a reduced dosing frequency may be sufficient to obtain complete cancer eradication. It is worth noting expression of VIP-R in metastatic sites at the same or higher level than in primary patient neoplasms, including breast cancer [21,22]. Therefore, actively VIP targeted carriers are capable of delivery of their cargo directly to the primary as well as metastatic tumor sites.
Fig. 5.

In vivo efficacy of paclitaxel against MNU-induced breast cancer in rats; rate of tumor regression on treatment with micellar formulations of paclitaxel, P-SSMM-VIP and P-SSMM, compared to commercial product Paclitaxel for injections (PXT): a) Paclitaxel dose 1 mg/kg. b) Paclitaxel dose 5 mg/kg. Data represent the mean ± SEM; n=6 rats/formulation; ↑ symbolize dosing. *-indicate statistical significance (p<0.05) between specified groups at the end of the study
Safety evaluation of P-SSMM-VIP
VIP is an endogenous peptide and its receptors are widely distributed in the body. Therefore, exogenously administered VIP may cause side effects such as hypotension due to its well-known vasodilatory action [44]. Unlike other receptors for ligands presently tested in clinical trials, such as folate and transferrin [45,46], VIP-Rs are not expressed on microvascular endothelial cells [25]. Therefore, VIP needs to extravasate in order to interact with its receptors expressed on subluminal smooth muscle cells and elicit side effects. A distinct advantage of targeting nanocarriers with VIP is a restriction of the extravasation of the entire construct, due to its size, only at the site of action and as consequent total elimination of VIP-caused hypotension.
In this study we evaluated the safety of VIP when used as a targeting ligand, conjugated to the surface of nanocarrier SSMM, encapsulating cytotoxic drug paclitaxel. Systolic blood pressure following single administration of either P-SSMM-VIP or aqueous VIP (equivalent to peptide dose 4.5 μmol/kg) to MNU-induced tumor bearing rats was determined by a non-invasive tail cuff method. We observed that administration of aqueous VIP evoked a rapid decrease in systemic arterial pressure (Fig. 6). By contrast, P-SSMM-VIP had no significant effects on systemic arterial pressure. These data suggest that P-SSMM-VIP did not extravasate from normal vasculature to interact with VIP-Rs. As mentioned above, the vasculature in breast tumors is leaky due to the discontinuous endothelial lining of the cancer neovasculature. Thus, this presents a unique and novel opportunity where VIP-conjugated SSMM preferentially extravasates only at tumor site.
Fig. 6.

Effect of P-SSMM-VIP and aqueous VIP on systemic arterial pressure of breast cancer bearing rats on intravenous administration at a single injection at the peptide dose corresponded to 4.5μmol/kg. Data represent the mean ± SEM; n=3 rats/formulation; * p<0.05 compared to P-SSMM-VIP
As additional safety measure, if necessary, all D-VIP (inactive enantiomer) rather than L-VIP (native peptide) could be used as an active targeting, but metabolically inert, moiety [47].
Abraxane® is a relatively new paclitaxel nanomedicine that is used for breast cancer treatment [41,48]. Vehicle toxicity of Taxol® is eliminated with this product, since paclitaxel is bound to albumin nanoparticles (130nm), instead of being solubilized in Cremophor EL and ethanol. Being a nanoparticulate system, Abraxane® may also utilize, at least in part, passive targeting mechanism to improve efficacy and reduce toxicity of the drug. However, Abraxane® lacks active targeting mechanism and steric stabilization property to avoid RES uptake. We believe our proposed paclitaxel formulation, P-SSMM-VIP that uses safe biocompatible excipients as the vehicle with steric stabilization and active targeting should be superior to currently available paclitaxel formulations with respect to the drug safety and efficacy.
Conclusions
In conclusion, these studies demonstrated for the first time the feasibility of in vivo active targeting using human vasoactive intestinal peptide surface conjugated to nanomicelles. We have successfully developed a nanomedicine for a model drug paclitaxel with surface grafted VIP, using biocompatible phospholipids. P-SSMM-VIP demonstrated improved in vitro cytotoxicity compared to non-targeted system. Furthermore, the in vivo studies demonstrated that paclitaxel delivered in the targeted carrier accumulated significantly more at the mammary tumors due to its size, long circulation and interaction with target receptors. In addition, targeted delivery resulted in reduced accumulation of paclitaxel in healthy tissues especially those associated with systemic toxicities such as bone marrow. Moreover, the increase in tumor accumulation due to targeting resulted in significantly enhanced drug activity in treating rats with MNU-induced breast cancer. Using the clinical dose of paclitaxel (5mg/kg), the complete tumor eradication was achieved by P-SSMM-VIP, whereas the commercial cremophore-based formulation of paclitaxel, at the same dose regimen showed only 45% tumor regression. Therefore, we propose further development of SSMM-VIP as a targeted delivery platform for paclitaxel and other anticancer drugs against cancers that overexpress VIP-R.
Supplementary Material
Acknowledgments
This study was supported, in part, by National Institutes of Health (NIH) grants CA121797 and AG024026, and Department of Veterans Affairs Merit Review Program. The investigation was conducted in a facility constructed with support from Research Facilities Improvement Grant CO6RR15482 from National Center for Research Resources NIH.
References
- 1.Singh S, Dash AK. Paclitaxel in cancer treatment: perspectives and prospects of its delivery challenges. Crit Rev Ther Drug Carrier Syst. 2009;26(4):333–72. doi: 10.1615/critrevtherdrugcarriersyst.v26.i4.10. [DOI] [PubMed] [Google Scholar]
- 2.Lim SB, Banerjee A, Onyuksel H. Improvement of drug safety by the use of lipid-based nanocarriers. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee A, Onyuksel H. Human Pancreatic Polypeptide in a Phospholipid-Based Micellar Formulation. Pharm Res. 2012;29(6):1698–711. doi: 10.1007/s11095-012-0718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koo OM, Rubinstein I, Onyuksel H. Actively targeted low-dose camptothecin as a safe, long-acting, disease-modifying nanomedicine for rheumatoid arthritis. Pharm Res. 2011;28(4):776–87. doi: 10.1007/s11095-010-0330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Onyuksel H, Mohanty PS, Rubinstein I. VIP-grafted sterically stabilized phospholipid nanomicellar 17-allylamino-17-demethoxy geldanamycin: a novel targeted nanomedicine for breast cancer. Int J Pharm. 2009;365(1–2):157–61. doi: 10.1016/j.ijpharm.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesur H, Rubinstein I, Pai A, Onyuksel H. Self-associated indisulam in phospholipid-based nanomicelles: a potential nanomedicine for cancer. Nanomedicine. 2009;5(2):178–83. doi: 10.1016/j.nano.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishnadas A, Rubinstein I, Önyüksel H. Sterically Stabilized Phospholipid Mixed Micelles: In Vitro Evaluation as a Novel Carrier for Water-Insoluble Drugs. Pharm Res. 2003;20(2):297–302. doi: 10.1023/a:1022243709003. [DOI] [PubMed] [Google Scholar]
- 8.Ashok B, Arleth L, Hjelm RP, Rubinstein I, Onyuksel H. In vitro characterization of PEGylated phospholipid micelles for improved drug solubilization: effects of PEG chain length and PC incorporation. J Pharm Sci. 2004;93(10):2476–87. doi: 10.1002/jps.20150. [DOI] [PubMed] [Google Scholar]
- 9.Kuzmis A, Lim SB, Desai E, Jeon E, Lee BS, Rubinstein I, Onyuksel H. Micellar nanomedicine of human neuropeptide Y. Nanomedicine. 2011;4(7):464–71. doi: 10.1016/j.nano.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–51. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Wittrup KD, Thurber GM, Schmidt MM, Rhoden JJ. Practical theoretic guidance for the design of tumor-targeting agents. Methods Enzymol. 2012;503:255–68. doi: 10.1016/B978-0-12-396962-0.00010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagar S, Krishnadas A, Rubinstein I, Blend MJ, Onyuksel H. VIP grafted sterically stabilized liposomes for targeted imaging of breast cancer: in vivo studies. J Control Release. 2003;91(1–2):123–33. doi: 10.1016/s0168-3659(03)00242-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhang XX, Eden HS, Chen X. Peptides in cancer nanomedicine: Drug carriers, targeting ligands and protease substrates. J Control Release. 2011 doi: 10.1016/j.jconrel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol Membr Biol. 2010;27(7):286–98. doi: 10.3109/09687688.2010.521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reubi JC. In vitro evaluation of VIP/PACAP receptors in healthy and diseased human tissues. Clinical implications. Ann N Y Acad Sci. 2000;921:1–25. doi: 10.1111/j.1749-6632.2000.tb06946.x. [DOI] [PubMed] [Google Scholar]
- 16.Ortner A, Wernig K, Kaisler R, Edetsberger M, Hajos F, Kohler G, Mosgoeller W, Zimmer A. VPAC receptor mediated tumor cell targeting by protamine based nanoparticles. J Drug Target. 2010;18(6):457–67. doi: 10.3109/10611860903508796. [DOI] [PubMed] [Google Scholar]
- 17.Reubi JC, Laderach U, Waser B, Gebbers JO, Robberecht P, Laissue JA. Vasoactive intestinal peptide/pituitary adenylate cyclase-activating peptide receptor subtypes in human tumors and their tissues of origin. Cancer Res. 2000;60(11):3105–12. [PubMed] [Google Scholar]
- 18.Schulz S, Rocken C, Mawrin C, Weise W, Hollt V, Schulz S. Immunocytochemical identification of VPAC1, VPAC2, and PAC1 receptors in normal and neoplastic human tissues with subtype-specific antibodies. Clin Cancer Res. 2004;10(24):8235–42. doi: 10.1158/1078-0432.CCR-04-0939. [DOI] [PubMed] [Google Scholar]
- 19.Dagar S, Sekosan M, Rubinstein I, Onyuksel H. Detection of VIP receptors in MNU-induced breast cancer in rats: implications for breast cancer targeting. Breast Cancer Res Treat. 2001;65(1):49–54. doi: 10.1023/a:1006406617497. [DOI] [PubMed] [Google Scholar]
- 20.Reubi JC. In vitro identification of vasoactive intestinal peptide receptors in human tumors: implications for tumor imaging. J Nucl Med. 1995;36(10):1846–53. [PubMed] [Google Scholar]
- 21.Reubi C, Gugger M, Waser B. Co-expressed peptide receptors in breast cancer as a molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2002;29(7):855–62. doi: 10.1007/s00259-002-0794-5. [DOI] [PubMed] [Google Scholar]
- 22.Raderer M, Kurtaran A, Yang Q, Meghdadi S, Vorbeck F, Hejna M, Angelberger P, Kornek G, Pidlich J, Scheithauer W, Virgolini I. Iodine-123-vasoactive intestinal peptide receptor scanning in patients with pancreatic cancer. J Nucl Med. 1998;39(9):1570–5. [PubMed] [Google Scholar]
- 23.Rubinstein I, Soos I, Onyuksel H. Intracellular delivery of VIP-grafted sterically stabilized phospholipid mixed nanomicelles in human breast cancer cells. Chem Biol Interact. 2008;171(2):190–4. doi: 10.1016/j.cbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onyuksel H, Jeon E, Rubinstein I. Nanomicellar paclitaxel increases cytotoxicity of multidrug resistant breast cancer cells. Cancer Lett. 2009;274(2):327–30. doi: 10.1016/j.canlet.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fahrenkrug J, Hannibal J, Tams J, Georg B. Immunohistochemical localization of the VIP1 receptor (VPAC1R) in rat cerebral blood vessels: relation to PACAP and VIP containing nerves. J Cereb Blood Flow Metab. 2000;20(8):1205–14. doi: 10.1097/00004647-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Dagar S, Sekosan M, Lee BS, Rubinstein I, Onyuksel H. VIP receptors as molecular targets of breast cancer: implications for targeted imaging and drug delivery. J Control Release. 2001;74(1–3):129–34. doi: 10.1016/s0168-3659(01)00326-1. [DOI] [PubMed] [Google Scholar]
- 27.Sou K, Endo T, Takeoka S, Tsuchida E. Poly(ethylene glycol)-modification of the phospholipid vesicles by using the spontaneous incorporation of poly(ethylene glycol)-lipid into the vesicles. Bioconjug Chem. 2000;11(3):372–9. doi: 10.1021/bc990135y. [DOI] [PubMed] [Google Scholar]
- 28.Seelig J. Titration calorimetry of lipid-peptide interactions. Biochim Biophys Acta. 1997;1331(1):103–16. doi: 10.1016/s0304-4157(97)00002-6. [DOI] [PubMed] [Google Scholar]
- 29.Zuidam NJ, de Vruch R, Crommelin DJA. Characterization of liposomes. In: Torchilin VP, Weissig V, editors. Liposomes. 2. Oxford University Press; 2003. p. 31. [Google Scholar]
- 30.Moody TW, Jensen RT. Breast cancer VPAC1 receptors. Ann N Y Acad Sci. 2006;1070:436–9. doi: 10.1196/annals.1317.058. [DOI] [PubMed] [Google Scholar]
- 31.Moon RC, Constantinou AI. Dietary retinoids and carotenoids in rodent models of mammary tumorigenesis. Breast Cancer Res Treat. 1997;46(2–3):181–9. doi: 10.1023/a:1005995925246. [DOI] [PubMed] [Google Scholar]
- 32.Arleth L, Ashok B, Onyuksel H, Thiyagarajan P, Jacob J, Hjelm RP. Detailed structure of hairy mixed micelles formed by phosphatidylcholine and PEGylated phospholipids in aqueous media. Langmuir. 2005;21(8):3279–90. doi: 10.1021/la047588y. [DOI] [PubMed] [Google Scholar]
- 33.Couvineau A, Tan YV, Ceraudo E, Lacapere JJ, Murail S, Neumann JM, Laburthe M. The human VPAC1 receptor: identification of the N-terminal ectodomain as a major VIP-binding site by photoaffinity labeling and 3D modeling. Ann N Y Acad Sci. 2006;1070:205–9. doi: 10.1196/annals.1317.015. [DOI] [PubMed] [Google Scholar]
- 34.Bhunia A, Domadia PN, Bhattacharjya S. Structural and thermodynamic analyses of the interaction between melittin and lipopolysaccharide. Biochim Biophys Acta. 2007;1768(12):3282–91. doi: 10.1016/j.bbamem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 35.Schote U, Ganz P, Fahr A, Seelig J. Interactions of cyclosporines with lipid membranes as studied by solid-state nuclear magnetic resonance spectroscopy and high-sensitivity titration calorimetry. J Pharm Sci. 2002;91(3):856–67. doi: 10.1002/jps.10071. [DOI] [PubMed] [Google Scholar]
- 36.Hagiwara H, Sunada Y. Mechanism of taxane neurotoxicity. Breast Cancer. 2004;11(1):82–5. doi: 10.1007/BF02968008. [DOI] [PubMed] [Google Scholar]
- 37.Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69(1):1–9. doi: 10.1016/j.ejpb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Spratlin J, Sawyer MB. Pharmacogenetics of paclitaxel metabolism. Crit Rev Oncol Hematol. 2007;61(3):222–9. doi: 10.1016/j.critrevonc.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Li X, Wang L, Xu Y, Cheng X, Wei P. Formulation and pharmacokinetic evaluation of a paclitaxel nanosuspension for intravenous delivery. Int J Nanomedicine. 2011;6:1497–507. doi: 10.2147/IJN.S21097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fetterly GJ, Straubinger RM. Pharmacokinetics of paclitaxel-containing liposomes in rats. AAPS PharmSci. 2003;5(4):E32. doi: 10.1208/ps050432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012;14(2):282–95. doi: 10.1208/s12248-012-9339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsubura A, Lai YC, Miki H, Sasaki T, Uehara N, Yuri T, Yoshizawa K. Review: Animal models of N-Methyl-N-nitrosourea-induced mammary cancer and retinal degeneration with special emphasis on therapeutic trials. In Vivo. 2011;25(1):11–22. [PubMed] [Google Scholar]
- 43.Nowfar S, Teplitzky SR, Melancon K, Kiefer TL, Cheng Q, Dwived PD, Bischoff ED, Moro K, Anderson MB, Dai J, Lai L, Yuan L, Hill SM. Tumor prevention by 9-cis-retinoic acid in the N-nitroso-N-methylurea model of mammary carcinogenesis is potentiated by the pineal hormone melatonin. Breast Cancer Res Treat. 2002;72(1):33–43. doi: 10.1023/a:1014912919470. [DOI] [PubMed] [Google Scholar]
- 44.Virgolini I, Raderer M, Kurtaran A, Angelberger P, Banyai S, Yang Q, Li S, Banyai M, Pidlich J, Niederle B, Scheithauer W, Valent P. Vasoactive intestinal peptide-receptor imaging for the localization of intestinal adenocarcinomas and endocrine tumors. N Engl J Med. 1994;331(17):1116–21. doi: 10.1056/NEJM199410273311703. [DOI] [PubMed] [Google Scholar]
- 45.Leamon CP. Folate-targeted drug strategies for the treatment of cancer. Curr Opin Investig Drugs. 2008;9(12):1277–86. [PubMed] [Google Scholar]
- 46.Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121(2):159–76. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 47.Rubinstein I, Ashok B, Tsueshita T, Onyuksel H. All D-VIP mitigates vasodilation elicited by L-VIP, micellar L-VIP and micellar PACAP1-38, but not PACAP1-38, in vivo. Peptides. 2005;26(3):509–15. doi: 10.1016/j.peptides.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 48.Montana M, Ducros C, Verhaeghe P, Terme T, Vanelle P, Rathelot P. Albumin-bound paclitaxel: the benefit of this new formulation in the treatment of various cancers. J Chemother. 2011;23(2):59–66. doi: 10.1179/joc.2011.23.2.59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.