

Abstract
We propose a mechanistic model for three-stage dehydrogenation of ammonia borane (AB) catalyzed by Shvo’s cyclopentadienone-ligated ruthenium complex. We provide evidence for a plausible mechanism for catalyst deactivation, the transition from fast catalysis to slow catalysis, and relate those findings to the invention of a second-generation catalyst that does not suffer from the same deactivation chemistry.
The primary mechanism of catalyst deactivation is borazine-mediated hydroboration of the ruthenium species that is the active oxidant in the fast catalysis case. This transition is characterized by a change in the rate law for the reaction and changes in the apparent resting state of the catalyst. Also, in this slow catalysis situation, we see an additional intermediate in the sequence of boron, nitrogen species, aminodiborane. This occurs with concurrent generation of NH3, which itself does not strongly affect the rate of AB dehydrogenation.
Keywords: Shvo’s Catalyst, Ammonia Borane, Hydrogen Storage, Catalyst Deactivation, Hydroboration
Introduction
Hydrogen is an attractive alternative transportation fuel that has appeal because it is carbon-free, easily oxidized in fuel cells, and potentially available from water electrolysis.1 Although pressurized hydrogen gas currently has some use in vehicles, its practicality in cars is limited by fuel range, convenience, and safety concerns.2 Particularly, hydrogen has low volumetric energy density (5.6 MJ/L at 700 bar) despite high mass energy density (120 MJ/kg for hydrogen).3 Thus, a highly weight-efficient strategy to store hydrogen as condensed matter might enable its translation more broadly into transportation applications and consumer products.
Ammonia borane (AB) is a promising material from which to build a practical hydrogen storage system, because it has high hydrogen density (19.6 wt%, ca. 9.9 MJ/L, 12.6 MJ/kg) and it can release hydrogen under mild conditions (thermolysis, hydrolysis, and catalysis, Figure 1).4 Transition metal catalyzed dehydrogenation of ammonia borane, particularly, is an area of active research interest because it may enable a more efficient fuel cycle than the well-known catalytic hydrolysis reaction that forms ammonia, which is poisonous to fuel cells, and strong B-O bonds, which are energetically costly to re-reduce. 5 Several active transition metal-based catalysts have been reported for AB dehydrogenation reactions, in either heterogeneous or homogeneous systems; these involve rhodium,6 iridium,7 ruthenium,8,9,10 nickel,11 palladium,12 and iron catalysts,13 among others14 (Figure 2).
Figure 1.

Ammonia Borane dehydrogenation reactions and possible products.
Figure 2.

Transition metal catalysts for AB dehydrogenation.
Our lab’s studies on catalytic AB dehydrogenation have been focused on the reactivity of Shvo’s catalyst 12 (Scheme 1)15 and its relatives. By analogy to the established mechanism for alcohol oxidation with 12, we presumed that the coordinative saturation of the reduced form of the Shvo system would preclude coordination of aminoborane, NH2BH2, to the catalyst, and thus disfavor the formation of insoluble oligomers, for [NH2BH2]n, which limits the hydrogen production of some catalysts7, 8 ammonia borane dehydrogenation to 1 equivalent.16 Although this proposal seems to hold true,9 the catalyst begins to deactivate after ca. 25% conversion in the first pass, which renders this system irrelevant to practical implementation.
Scheme 1.
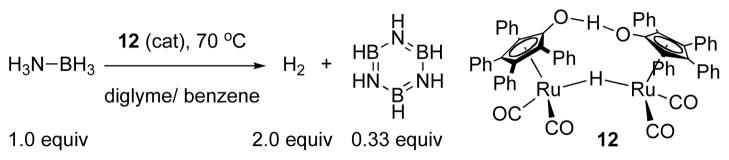
Dehydrogenation of AB with Shvo’s Catalyst, 12.
This text examines the molecular events that deactivate the Shvo catalyst in ammonia borane dehydrogenation. These include hydroboration of the active, oxidizing form of the catalyst by borazine, which involves the borylation of the catalyst’s ligand oxygen atom so that the turnover-limiting H-H bond-forming step is no longer accessible. Ultimately, we addressed this problem by designing a second-generation system, 13,17 that does not rely on an oxygen center as a proton acceptor in the same way as the Shvo catalyst. This second-generation system then enables access to high weight-efficiency dehydrogenation of ammonia borane.10
Results and Discussion
The kinetic profile of AB dehydrogenation with 12 shows complicated behavior that we deconvoluted into a sequence of three limiting cases (Figure 3).9 In our prior work we described cases 1 and 2 in detail,9 catalyst initiation and fast catalysis, respectively. The former is the case of low conversion and zero [borazine] with ruthenium beginning in its dimeric form. This can be easily studied in isolation, because these are the conditions at the beginning of the reaction and because initiation occurs quickly at 55 °C, where the catalysis is slow. The second case is the one in which AB conversion and [borazine] are low and the ruthenium in the system is no longer in the form of its dimeric precursor. This case can easily be studied in isolation by either (1) allowing the reaction to incubate at room temperature until it is initiated, i.e. 12’s characteristic μ-H peak (1H δ = −17.7 ppm) is consumed, or (2) delivering the ruthenium as dimer 18 (vide infra, Scheme 2). The third case, slow catalysis, occurs in the condition that [borazine] ~ [Ru atoms]. In this case the rate of dehydrogenation catalysis drops precipitously, and the catalyst “dies”. This case can be generated in isolation by adding a catalytic portion (1 eq. versus [Ruatom]) of borazine to the reaction mixture at the outset of dehydrogenation.9
Figure 3.
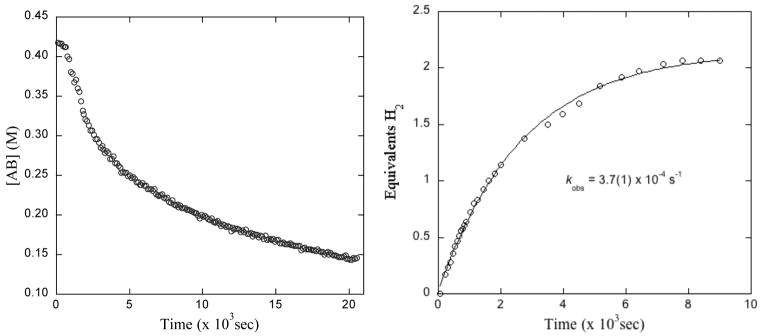
(left) 11B NMR data showing consumption of AB in the presence of 2.5 mol% 12 in a sealed J-Young NMR tube. (right) Eudiometer data showing production of hydrogen gas in the presence of 5.0 mol% 12 and 2.0 mol% ethanol in 2:1 diglyme/benzene-d6 at 70 °C.
Scheme 2.
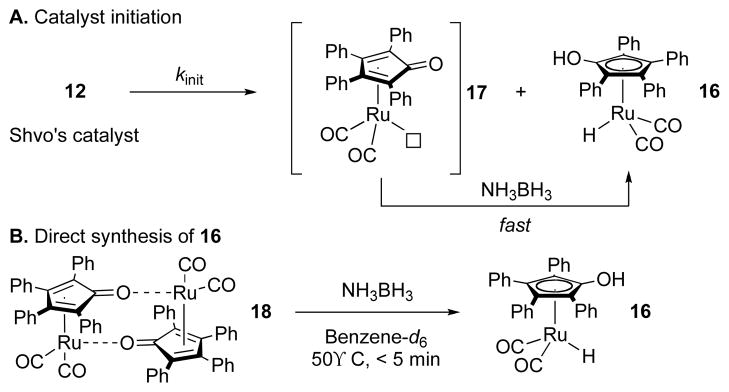
Catalyst Initiation.
A: Scheme for catalyst initiation. B: Rapid formation of 16 from 18. [AB] is 0.42 M in benzene-d6 solution, [Ru]2 is 5 mol% to AB.
Figure 3 shows (left) AB consumption and (right) H2 generation as functions of time for trials of dehydrogenation that cycle through all three of its mechanistic cases.9 This is particularly evident from AB consumption: AB is consumed slowly as the catalyst initiates (case 1) and then it is consumed more quickly once dimer 12 is cleaved. Ultimately, [borazine] increases and the catalysis slows down.
Understanding the mechanism of dehydrogenation in the high [borazine] case and the catalyst deactivation process is particularly important for the following reasons: 1. the kinetic profile of the AB consumption in the high [borazine] case is the one observed when the catalyst is re-used, and reusability is essential for practical applications. 2. Borazine is an unavoidable intermediate in AB dehydrogenation if ≥ 2 molar equivalents of hydrogen are released, yet the coordination chemistry of borazine is not well studied in the context of catalytic AB dehydrogenation systems.3 Understanding the mechanism of this deactivation is important for the design of more efficient catalytic systems and, ultimately, a broadly useful solution to reversible hydrogen storage on ammonia borane.
I. Catalyst Initiation (Case 1)
A short period in the beginning of the reaction (ca. 2% conversion) is an initiation period in which AB consumption is slow. This situation can be studied in isolation by monitoring the reaction at a temperature well below that needed for fast catalysis, 70 °C. Thus, upon heating to 55 °C, the bridging hydride in 12 (1H δ = −17.7 ppm) is replaced by the hydride of monomeric species 16 (1H δ = −10.0 ppm) at a rate of 7.96(21) × 10−4 s−1.9 This indicates that Shvo’s catalyst, 12, dissociates to its reduced monomer 16 and (presumably) oxidized monomer 17 (Scheme 2A). 1H NMR integrations show that 2 equivalents of 16 are formed with consumption of 12, so the reduction of 17 is rapid relative to dissociation of 12.
The finding of rapid reduction of 17 by ammonia borane is supported by the relatively rapid rate of reduction of 18, a stable dimer of 17, under analogous conditions (Scheme 2B). In this experiment we see that conversion of 18 to 16 reaches completion within 5 min at 50 °C (see Supporting Information). This corresponds to a rate constant of > 10−2 s−1 at 50 °C, which is faster than the rate of catalyst initiation (10−3 s−1 at 55 °C). Thus, the dissociation of dimer 12 is rate limiting in catalyst initiation. This result is in accordance with Shvo’s considerable dissociation enthalpy, 28.8 kcal/mol in toluene in the absence of AB.18b
II. Fast Catalysis (Case 2)
After the catalyst initiation, the kinetic profile of AB consumption displays fast, linear kinetics through ca. 20–30% conversion. In these conditions, the reaction has a zero-order dependence on [AB] and first order on catalyst’s [Ru] as determined by the respective zero and unity slopes of plots of ln [kobs] versus ln [AB] and ln [Ru].9 Throughout this case, 1H NMR shows a persistent monomeric ruthenium hydride at 1H δ = −10 ppm, which is plausibly the resting state of the catalyst. This is consistent with H-H bond formation as the turnover-limiting step in fast catalysis. This assignment is consistent with the observed kinetic dependencies of [AB]0 and [Ru]1. We further observe first-order dependence on [EtOH], which is consistent with a transition state model established by Casey for stoichiometric hydrogen loss from 16.18 Thus, we adopt Casey and Cui’s geometry for ethanol-mediated H-H bond formation from 16 as the turnover-limiting transition state of this catalysis (Scheme 3). In sum, the observed rate law in this case of the reaction is −d[AB]/dt = kobs[Ru][EtOH].
Scheme 3.

Proposed Mechanism of Fast Catalysis (Case 2).
III. Slow Catalysis (Case 3)
Kinetics of Ammonia Borane Dehydrogenation in the Slow Catalysis Case
Onset of the slow catalysis conditions, i.e. catalyst deactivation, is characterized by the appearance of curvature in the time course plot of [AB] and it becomes more likely as [borazine] rises. The conditions of slow catalysis cause the emergence of multiple κ1-Ru—H hydride peaks (from 1H δ = −9 to −10 ppm), which occurs simultaneously with exponential decay behavior in [AB]. We believe that these correspond, respectively, to (a) new resting state(s) of the catalyst and ammonia borane’s role in a new turnover-limiting step. Understanding this mechanism is essential to our studies on catalyst reuse and, we infer, spent fuel regeneration.
An essential feature of the high [borazine] case of the reaction is that the catalyst is re-usable within the limits of its kinetics. Thus, if a completed reaction mixture is treated with a new aliquot of ammonia borane, dehydrogenation will recommence upon heating, and the reaction’s kinetic profile will follow slow catalysis behavior wherein the rate of hydrogen production is too slow to be useful.9 It is easy to believe that, at the conclusion of the reaction, the concentration of [17] is very small, so dimer 12 is not re-formed, and it is unnecessary to repeat catalyst initiation (case 1) in catalyst re-use experiments. However, the absence of fast catalysis must result from a practically irreversible deactivation of the catalyst during the transition from fast catalysis to slow catalysis in the first run.
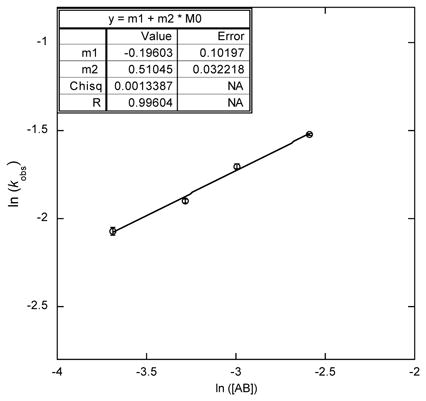
The exponential decay shape of the kinetic profile of AB consumption suggests that the reaction is now first order in [AB] in the slow catalysis regime (Table 1, left), as verified by a plot of ln [kobs] versus ln [AB] with a slope of 1.03(4) as recorded in the case of high [borazine]. This draws a significant contrast to fast catalysis where such a plot was of zero slope, and indicates that, unlike fast catalysis, once the catalyst deactivates, the turnover-limiting step for further turnover involves conversion of the resting species by one equivalent of ammonia borane.
Table 1.
Ammonia Borane Consumption as a Function of [AB] and [Ru] in Slow Catalysis.a
[AB] Dependence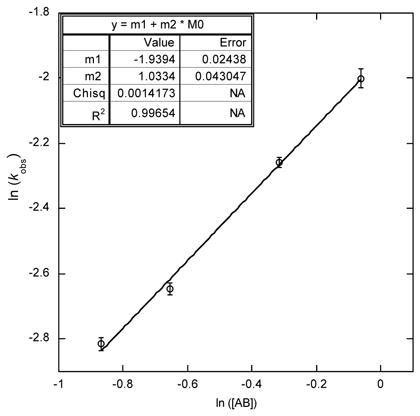
|
[Ru] Dependence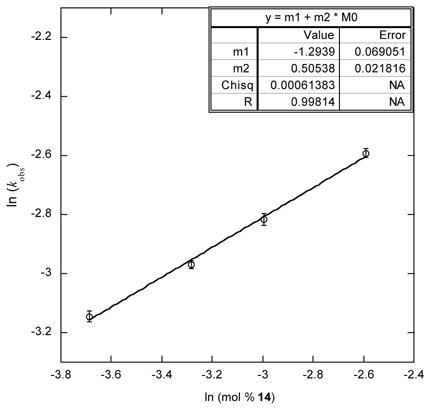
|
||
|---|---|---|---|
|
| |||
| AB (M) | Rate (kobs, s−1)b | 12 (mol%) | Rate (kobs, s−1)c |
| 0.42 | 5.99(12) × 10−5 | 2.5 | 4.31(7) × 10−5 |
| 0.52 | 7.09(19) × 10−5 | 3.75 | 5.13(6) × 10−5 |
| 0.73 | 1.05 (2) × 10−4 | 5.0 | 5.99(12) × 10−5 |
| 0.94 | 1.35(4) × 10−4 | 7.5 | 7.49(12) × 10−5 |
Data calculated from 11B NMR-monitored kinetic studies at 70 °C.
[Ruatom]0 is 42.0 mM.
[AB]0 is 0.42 M.
The kinetic order in ruthenium also changes upon the onset of slow catalysis conditions. [Ru] is first order in the fast catalysis case, but it becomes half order in the high [borazine] case: rate constants were measured for a series of [Ru] concentrations in AB dehydrogenation in the presence of borazine, which gave a plot of ln ([kobs]) versus ln ([Ru]) with a slope of 0.50(2) (Table 1, right). This is analogous to 12-catalyzed alcohol oxidation,19 wherein apparent half-order dependence on [Ru] is a result of equilibrium between 16 + 17 and dimer 12, for which a plot of ln (kobs) versus ln ([Ruatom]) has a slope of 0.40(6).20
Isotope Effects
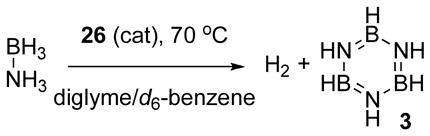
Kinetic isotope effects were determined for dehydrogenation of selectively deuterium-labeled ammonia borane isotopologs 21 ND3BH3, NH3BD3, and ND3BD3 in high [borazine] conditions. Comparison of measured rate constants for parallel runs at 70 °C gave kinetic isotope effects of kNHBH/kNDBH = 1.46(3), kNHBH/kNHBD = 1.07(5), and kNHBH/kNDBD = 2.30(4) (see Supporting Information). The KIE in NH is suggestive of a catalyst reactivation involving participation of the NH in its turnover-limiting step. This might be akin to a protonation of the resting state of the catalyst by an acidic NH proton. These data present a conundrum, however, which is that the product of the two single-label KIEs should equal the double-label KIE; in this case we have 1.46(3) × 1.07(5) = 1.56(6), which is well below the observed value of 2.30(4). Along these lines, Casey’s group has reported H/D exchange of the Ru—H group in Shvo’s catalyst with D2 (or vice-versa) of 16-Tol in THF without substituting the corresponding ligand O—H.18 Similarly, in two parallel runs of ND3BH3 dehydrogenation, similar portions of HD and H2 were formed. The presence of H2 (and by symmetry D2) implies the availability of a mechanism for proton/hydride exchange under our catalytic conditions. Based on our observations and their result, we conducted an experiment of ND3BD3 dehydrogenation with 1 atm H2 gas applied to the solution. HD was formed during this reaction (Scheme 4A), which necessitates an H/D crossover mechanism involving the final product, H2. We believe that the mechanism of this is the same as Casey’s H/D exchange mechanism, except that we suggest that this mechanism is available to the resting state(s) of our catalyst. To test this latter hypothesis, we treated borylated ruthenium complex 26 with 1 atm D2 in conditions analogous to our catalytic reactions. We observed the formation of HD at room temperature in 5 minutes and complete deuteration in the hydride position of 26 within 1 hour at 60 °C (Scheme 4B). This shows us that there is a mechanism for H/D exchange of the ruthenium hydride in an O-borylated homolog of the Shvo system. This result provides an explanation for the small experimental kNHBH/kNHBD value and the mismatch between our observed kNHBH/kNDBD and the value predicted by the separate values for proton and hydride: because there’s a facile mechanism for H/D exchange, an isotopic kinetic resolution is possible.
Scheme 4.

H/D Exchange Experiments.
Mechanistic Proposal
We propose, based on NMR observations, that fast catalysis ends (i.e. catalyst deactivaiton occurs) because borazine undergoes a hydroboration with ruthenium intermediate 17 to give the deactivated complex 21, which further converts to other derivatives (Scheme 5).9 Analogous reactions to the addition of 3 to 17 are known from the Casey18a and Clark22 labs (Scheme 6). Casey has shown hydrosilylation of the Shvo scaffold by triethylsilane. This adduct, 24-Tol, has a 1H δ of −9.20 in benzene-d6. Similarly, Clark has shown hydroboration of the Shvo complex with pinacol and catecholboranes in high yield at mild temperature. These adducts have 1H δ of −9.33 and −9.26 in benzene-d6, respectively.
Scheme 5.

Proposed Borazine-Mediated Hydroboration.
Scheme 6.

Hydroboration and Hydrosilylation of Tol-18.
We propose that this hydroborated species can dimerize to form a O—B—O and Ru—H—Ru bridged dimer (31, Scheme 5) akin to the parent Shvo complex and Clark’s [(μ-((cat)B(C4Ar4O)2)Ru2(CO)4(μ-H)] dimer, which accounts for the observed half-order kinetic dependence on [Ru]. These species can re-enter the catalytic cycle if the B—O bond affixing borazine to the catalyst is cleaved in the presence of ammonia borane, which accounts for its first order kinetic dependence. We do not know the mechanism of ammonia borane’s involvement in this step.
Catalyst Deactivation
We conducted a series of experiments directly to interrogate our proposal for the mechanism of slow catalysis, yet we observe that the proposed complex 21 is not stable to isolation. Borazine was added to dimer 18 at room temperature, and a bridging hydride peak formed at the beginning of the reaction (1H δ = −18.3) was then consumed in 2 minutes (Scheme 5). This was replaced by a set of 13 hydride peaks from 1H δ = −9.3 – −10.0 ppm, which correspond to κ1-Ru—H groups such as 22. We suspect that these signals correspond to multiple hydroboration events on a single borazine or ring-opened borazine derivatives.
We can create slow catalysis case conditions at the beginning of a dehydrogenation reaction very simply by adding 1 molar equivalents of borazine relative to [Ruatom] to the reaction mixture prior to heating. In these conditions the kinetic profile of the reaction does not show any properties of initiation or fast catalysis, but proceeds directly to the rate and rate law of slow catalysis.9 This is strong evidence indicating that borazine is the agent that causes catalyst deactivation and aptly accounts for the instant slow catalysis situation that is observed in catalyst reuse experiments. Because of their self-reactive nature, we are unable to isolate these complexes directly, but when a mixture of these materials is collected and excess borazine is quantitatively removed under reduced pressure, the resulting material can be isolated through aqueous work-up. This treatment cleaves any borazine rings remaining in the borazine-catalyst complex(es) and affords a ruthenium-containing adduct, ammonia complex 23,23 which can be isolated in 42% yield. This observation gives strong evidence that the deactivated catalyst, the one present in the slow catalysis case, is covalently bound to a borazine moiety because NH3 could not have been delivered in any other plausible way.
An Analog of the Deactivated Catalyst
Because our efforts to isolate and characterize our proposed deactivated catalyst were frustrated by its reactivity, we set about to devise a borazine analogue with which we could hydroborate 18 and generate a stable surrogate of the catalyst of the slow catalysis case. The premise of this design was our hypothesis that multiple equivalents of 17 are hydroborated by a single equivalent of borazine to yield multiple κ1-Ru—H signals in the 1H NMR spectrum. Along these lines, we prepared diazaborane 29 and treated it with dimer 18. The result was near-quantitative formation of 12 under rigorously anhydrous conditions (Scheme 6). We infer from this result that proposed dimer 27, if formed, apparently loses a borabenzoimidazole rapidly to regenerate 12. By contrast, hydroboration of 18 with catecholborane to form 26 is facile and gives an oxygen-substituted analog of our proposed deactivated catalyst that is free of N—H groups.
In situ preparation of 26 in a benzene/diglyme solution affords an opportunity to compare the rate and kinetic profile of 26-catalyzed ammonia borane dehydrogenation with those of the slow catalysis case (Scheme 7). The kinetic profiles each appear first order in AB, but the rate for dehydrogenation with catalyst precursor 26 is faster than slow catalysis by a factor of ca. three-fold. This faster rate could be a result of a more labile O—B bond between the catalyst’s hydroxycyclopentadiene and the corresponding boranes. A plot of ln (kobs) versus ln ([Ru]) gave a slope of 0.51(3) (Table 2), which is in agreement with the measured [Ru] dependence for slow catalysis. These data show us that the dimerization behavior that we see in the slow catalysis case is effectively recreated in borylated analog 26. Taken together these data provide good anecdotal evidence that the deactivated catalyst is an O-borylated form of the precatalyst.
Scheme 7.

Synthesis of a Mechanistic Analog for the Proposed Deactivated Catalyst Complex.
Table 2.
Ammonia Borane Dehydrogenation Catalyzed by Borylated Complex 26.
|
|
|
|---|---|---|
|
| ||
| 26 (mol %) | Rate (s−1) | |
| 5.0 | 1.26(3) × 10−4 | |
| 7.5 | 1.50(2) × 10−4 | |
| 10 | 1.82(2) × 10−4 | |
| 15 | 2.18(2) × 10−4 | |
Other Reaction Products and Intermediates in the Slow Catalysis Case
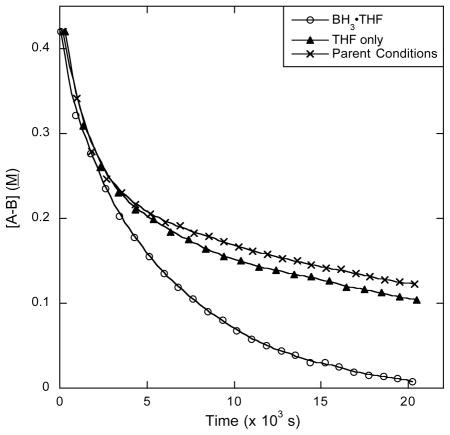
Ammonia borane dehydrogenation catalyzed by 12 generates multiple B, N intermediates throughout the reaction. In fast catalysis the intermediates detected by 11B NMR are the same as those observed for other catalysts that are known to liberate multiple equivalents of hydrogen from ammonia borane:9, 10,11 AB -> branched cyclotriborazane (2) -> borazine (3) (Figure 1A). In the slow catalysis case, however, a new species appears, aminodiborane 6. This species should be dehydrogenated to borazine, and in fact, in reactions in which this species is an intermediate, borazine remains the only product upon completion of the reaction. We believe that 6 is an adduct formed from BH3, dissociated from ammonia borane, and NH2BH2, generated transiently after the first dehydrogenation of ammonia borane.16, 24 We propose that formation of 6 is reversible, and this is only a mechanistic cul-de-sac, rather than an in-line intermediate in the dehydrogenation sequence. To test this hypothesis, we added (a) 0.5 eq 1 M BH3-THF and (b) a comparable volume of THF to two otherwise identical runs of ammonia borane dehydrogenation with 12 (Table 3). A strong signal for 6 was observed by 11B NMR in tube (a) in the beginning of the reaction, much earlier than the first emergence of 6’s peak in the THF control experiment (tube b). Both reactions proceeded through fast catalysis at about the same rate (Figure 3). Furthermore, the rates of AB consumption in slow catalysis case are similar, ca. 25% difference, which shows that although there is a large excess of 6 in tube (a) compared to tube (b), this has a disproportionately small effect on the rate of AB consumption. We therefore know that 6 goes on to dehydrogenate to borazine and does not significantly interfere with the rates of the steps in slow catalysis as it forms and disappears. It further appears that BH3 does not hydroborate and deactivate the catalyst in the same way as borazine or catecholborane.
Table 3.
Rate of Slow Catalysis in the Presence and Absence of BH3.a
| |
|---|---|
| Conditions | Slow Catalysis kobs (s−1) |
| BH3·THF | 1.57(7) × 10−4 |
| THF only | 7.0(13) × 10−5 |
| Parent Conditionsb | 6.3(6) × 10−5 |
Data calculated from 11B NMR kinetic studies at 70 °C. Smoothed curves are empirical fits; kobs values shown are for slow catalysis, not the entire curve. See Supporting Information.
A parallel run under parent conditions has kobs in statistical agreement with others reported herein.
If free BH3 from the dissociation of ammonia borane is impacting the course of the reaction in the slow catalysis case, then free NH3 must also be present, and NH3 is known to modulate the reactivity of the Shvo system.23 Thus, we propose a second mechanism of catalyst deactivation, which is reversible formation of 23 by NH3 ligation to the catalyst.
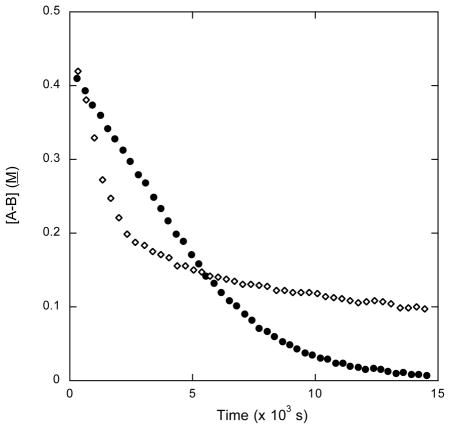
To interrogate directly the reactivity of ammonia adduct 23, we prepared it independently through the addition of ammonia gas to 18 (Scheme 9). Analogous to 12, the 11B NMR kinetic profile of AB dehydrogenation with 23 appears to have 2 distinct kinetic cases, one linear case, with a reaction rate of 5.12 × 10−5 M s−1, and one exponential decay case, with a rate constant of 4.22 × 10−4 s−1 (Table 4). This is similar to the second and third cases (fast and slow catalysis) of dehydrogenation with 12, but since 23 is monomeric, it stands to reason that there should not be an initiation delay analogous to the one observed in reactions featuring 12. We account for this behavior by proposing that NH3 reversibly can ligate 17 as previously documented23 and thereby temporarily sequester it from its catalytic roles. Thus, NH3 ligation provides a second mechanism for catalyst deactivation, although this one appears to be less deleterious than hydroboration of 17.
Scheme 9.
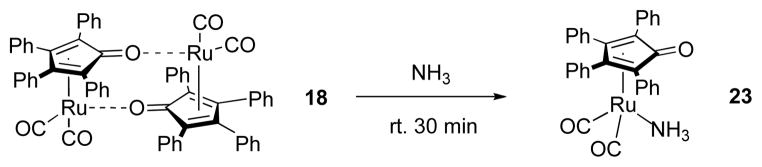
Synthesis of Ammonia Adduct 23.
Table 4.
[AB] Dehydrogenation with 12 and 23.a
| |||
|---|---|---|---|
| Fast Catalysis | Slow Catalysis | ||
|
| |||
| Catalyst | Rate (M s−1) | Catalyst | kobs (s−1) |
| 12 | 1.47(9) × 10−4 | 12 | 3.06(39) × 10−4 |
| 23 | 5.12(3) × 10−5 | 23 | 4.23(33) × 10−4 |
Data calculated from 11B NMR kinetic studies. 0.25 mol AB and 0.035 mol [Ruatom] are added to 0.6 mL diglyme/benzene-d6. Both reactions were run at 70 °C. Black circles and diamonds are kinetic profiles of reactions catalyzed by 23 and 12 respectively.
Homogeneous versus Heterogeneous Catalysis
We propose that this reaction is homogeneous throughout its duration on the basis on four observations. First, the reactor maintains its homogeneous appearance through the duration of the reactions. No metallic residue is observed. Second, the rate of catalysis is not impacted by the addition of Hg(0). By contrast, a mercury drop does inhibit the catalytic hydrogenation of benzene based on the [Ru3(μ2-H)3(η6-C6H6)(η6-C6Me6)2(μ3-O)]+ catalyst precursor, which is part of the evidence for heterogeneous reduction in that system.25 This result is germane to the present discussion because it shows a documented system wherein the mercury drop experiment was effective with ruthenium. Still, the best evidence we have (point three) for homogeneous catalysis remains the foregoing kinetics data in which the data remain pseudo-first order in the slow catalysis case through > 90% conversion (> 3 half-lives). A forth piece of evidence favoring homogeneous catalysis comes from a quantitative poisoning experiment wherein the reaction is run in the presence of a small portion of 1,10-phenanthroline.
Quantitative poisoning is an experiment in which less than one molar equivalent (relative to the proposed monomeric catalyst) is introduced into the reaction, and one monitors the rate to see if it is affected proportionally to the concentration of the poison. If the drop in rate upon poisoning is disproportionally large, this can be evidence for heterogeneous catalysis, because < 100% metal atoms (and often ≤ 50%)26 are on the surface of a nanoparticle and thus ≤ 50% are available to be poisoned.
In this present case, two quantitative poisoning experiments were conducted wherein 0.1 and 0.5 equivalents of 1,10-phenanthroline (phen) relative to [Ruatom] were added to two otherwise standard ammonia borane dehydrogenation runs with catalyst 12 (0.25 mol AB, 70 °C, diglyme/benzene-d6). Although these are not first order reactions, generally we see that phen accelerates the reaction, apparently by prolonging fast catalysis portion of the reaction (Figure 4). By contrast, 0.5 equivalents of phen (relative to ruthenium atoms) completely quenches catalytic heterogeneous hydrogenation of benzene based on the [Ru3(μ2-H)3(η6-C6H6)(η6-C6Me6)2(μ3-O)]+ catalyst precursor.25 Thus, we take our evidence to argue against the formation of a ruthenium nanoparticle. The origin of the acceleration behavior seems as if the phenanthroline “poison” is protecting the catalyst from borazine-mediated deactivation. This concept is further discussed in Supporting Information.
Figure 4.

[AB] Dehydrogenation with 12 in 1,10-phenanthroline. Data calculated from 11B NMR kinetic studies. 0.25 mol AB and 5 mol% 12 are added to 0.6 mL diglyme/benzene-d6 at 70 °C.
A tetranuclear catalyst (e.g. Ru4Ln) is unlikely, but cannot be rigorously eliminated. Such a hypothesis is disfavored because an analogous Rh4Cp*2.4Cl4Hc cluster has been shown to be the likely active catalyst in the hydrogen of benzene with [Cp*RhCl2]2, and this Rh4 cluster is deactivated by Hg(0).27 Further, that Rh4-based system is deactivated by 4 equivalents of 1,10-phenanthroline (i.e., 1:1 phen: Rhatom), while ours is not.
A Second-Generation Catalyst
To avoid the problem of catalyst O-borylation that is intrinsic to the deactivation of the Shvo catalyst, we devised the second-generation system highlighted in Scheme 10 below.10, 17 One design feature of this motif is that if the catalyst is borylated by analogy to 21, the resulting borylated oxygen atom can be lost from the catalyst, so that the catalytic species is not deactivated. Although we do not know the mechanism of ammonia borane dehydrogenation with 13, we observe that it can liberate > 2 equiv. of H2 from ammonia borane, even in repeated uses and with exposure to the atmosphere, and shows first order consumption of ammonia borane throughout conversion.
Scheme 10.

A Second-Generation Catalyst.
Conclusion
In conclusion, we were able to propose a full picture of the catalyst deactivation mechanism (Scheme 11). A full scheme showing the origin of the slow catalysis case is illustrated in a diagram in Supporting Information. The upper-right half of Scheme 11 is the fast catalysis mechanism, which is sketched in Scheme 3. We propose that catalyst deactivation is caused by the introduction of borazine, the product of selective dehydrogenation, into solution. When the rate of reduction of oxidized catalyst 17 by ammonia borane becomes competitive with its hydroboration, AB consumption ceases to be linear. When hydroboration of 17 becomes fast relative to reduction of 17, then 21 becomes a resting state of [Ru], and the catalysis “dies”, as sketched in the lower-left of the cycle in Scheme 11. In this phase of the reaction, conversion has first order dependence on [AB], apparently because ammonia borane is needed to convert borazine species 21 back into an active species.
Scheme 11.

Mechanitic Proposal of Slow Catalysis.
Reversible ammonia ligation of 17 is almost certainly happening throughout the reaction. We know, however, from the rates of (a) fast catalysis and (b) slow catalysis with ammonia complex 23 that 23 is a minor contributor to the total [Ru atoms] in fast catalysis because (i) the addition of one equivalent of NH3 relative to ruthenium slows fast catalysis, and (ii) the rate of AB consumption is constant throughout the fast catalysis case. We also know that NH3 ligation to ruthenium does not significantly alter the rate of slow catalysis.
In summary, the mechanism of AB dehydrogenation catalyzed by Shvo catalyst 12 was investigated. This reaction initiates with dissociation of the dimeric precatalyst 12, then goes through a fast dehydrogenation reaction wherein the H—H bond formation is the turnover-limiting step. As the concentration of borazine increases, it adds to the reactive form of the catalyst to give ruthenium species, which are not as reactive as their mechanistic predecessor in ammonia borane dehydrogenation. Presumably, these deactivated ruthenium species are reactivated by ammonia borane itself and proceed to further ammonia borane dehydrogenation. This observation gives us insight into the higher reactivity of our second-generation catalyst, 13.
Experimental Section
I. General Procedures
All air and water sensitive procedures were carried out either in a Vacuum Atmosphere glove box under nitrogen (0.5–10 ppm O2 for all manipulations) or using standard Schlenk techniques under nitrogen. Deuterated NMR solvents were purchased from Cambridge Isotopes Laboratories. Benzene-d6 and diethylene glycol dimethyl ether (diglyme, J. T. Baker) were dried over sodium benzophenone ketyl and distilled prior to use. Shvo’s catalyst was purchased from Strem Chemicals. Ammonia borane (NH3BH3, AB) was purchased from Sigma Aldrich. Borazine was synthesized and purified by the method used by Wideman and Sneddon.28 1H and 11B NMR spectra were obtained on a Varian 600 spectrometer (600 MHz in 1H, 192 MHz in 11B) with chemical shifts reported in units of ppm. All 1H chemical shifts are referenced to the residual 1H solvent (relative to TMS). All 11B chemical shifts are referenced to a BF3-OEt2 in diglyme in a co-axial external standard (0 ppm). NMR spectra were taken in 8″ J-Young tubes (Wilmad) with Teflon valve plugs. The NMR tubes were shaken vigorously for several minutes with chlorotrimethylsilane then dried in vacuo on a Schlenk line prior to use.
Safety Note. Extreme caution should be used when carrying out these reactions as the release of hydrogen can lead to sudden pressurization of reaction vessels.
II. Mechanistic Studies Utilizing 11B and 1H NMR Spectroscopy
In a typical reaction, 7.7 mg AB was combined with Shvo’s catalyst (12, 13.6 mg, 5 mol%) in a 2 mL J-Young NMR tube while in a glovebox under nitrogen. The AB concentration and catalyst concentrations may be varied. Diglyme (0.4 mL) and benzene-d6 (0.2 mL) were added to the tube as was the BF3 insert. The sample tube was immediately inserted into a preheated NMR (70 °C) and the kinetic monitoring commenced after quickly locking and shimming. Disappearance of AB in the solution was monitored by the relative integration of its characteristic peak in the 11B spectrum (−22 ppm) and the BF3-OEt2 standard. All spectra were processed using VNMRJ (v. 2.3). The acquisition involved a 1.67 sec pulse sequence in which 4,096 complex points were recorded, followed by 1 sec relaxation delay. To eliminate B-O peaks from the borosilicate NMR tube and probe, the 11B FIDs were processed with back linear prediction, ca. 5–15 points.
A. Determination of Catalyst Order in Conversion of AB in Case 3 (Slow Catalysis)
The rate values for the slow catalysis case were determined using 11B NMR in sealed NMR tubes, as described above. Data treatments are shown in the Supporting Information. The amount of AB was 7.7 mg (0.25 mmol) and catalyst concentrations were varied (6.8 mg, 10.2 mg, 13.6 mg, 20.3 mg 12, (2.5, 3.75, 5.0, 7.5 mol%)). The results were plotted as a ln/ln relationship to determine order in catalyst (Table 1, right).
B. Determination of Order in AB in Case 3 (Slow Catalysis)
The rate values for the slow catalysis case were again determined using 11B NMR in sealed NMR tubes. Data treatments are shown in the Supporting Information. The amount of 12 was 13.6 mg (0.013 mmol) and AB concentrations were varied (7.7 mg, 8.7 mg, 13.5 mg, 17.4 mg AB (0.42, 0.53, 0.73, 0.94 M)). The results were plotted as a ln/ln relationship to determine order in catalyst (Table 1, left).
C. Kinetic Isotope Effects in Case 3 (Slow Catalysis)
To determine the kinetic isotope effects on the reaction rate, 8.5 mg ND3BH3, 8.5 NH3BD3 or 9.2 mg ND3BD3 (0.25 mmol) were added to a J Young NMR tube. To each was added 13.6 mg 12 (5 mol%), diglyme (0.4 mL), and benzene-d6 (0.2 mL). Again, we analyzed [AB] vs. time for the third case of the reaction conditions. Data treatments are shown in the Supporting Information. KIEs were determined from the quotient of protic AB kobs (5.99(12) × 10−5 s−1) divided by deuterated AB kobs. HD was found signal in the 1H NMR resulted from H-D exchange is also shown is Figure S5.
D. Kinetics for AB Dehydrogenation in the Presence of Added BH3-THF
To examine the role of aminodiborane (6) in the mechanism of AB dehydrogenation, we manipulated its concentration by adding (tube A) 0.125 mL 1 M BH3-THF (0.125 mmol, 0.5 equiv to AB) or (tube B) 0.125 mL THF to otherwise typical AB dehydrogenation reactions (7.7 mg (0.25 mmol) AB, 13.6 mg, (5 mol%) 12, 0.6 mL 2:1 diglyme/benzene-d6). 11B NMR showed 6 (−26.7 ppm) in tube A in the beginning of the reaction (see Supporting Information). The kobs values for these runs were determined using 11B NMR; data treatment is shown in Supporting Information.
E. Kinetics for AB dehydrogenation by NH3-ligated species 23
Rate values for 23-catalyzed dehydrogenation run at 70 °C were determined using 11B NMR as shown in the Supporting Information. In this reaction 7.7 mg AB (0.25 mmol) was combined with (tube A) 20.7 mg 23 (35 μmol, 14 mol%), (tube B) 38.0 mg 12 (35 μmol, 14 mol%). Data are shown in Table 4.
F. 1,10-Phenanthroline Poisoning Experiments
Fractional poisoning experiments employing separately 0.5, 0.1, and 0 molar equivalents of 1,10-phenanthroline relative to [Ruatom] were done in conditions otherwise identical to our standard conditions for ammonia borane dehydrogenation by 12.
1,10-Phenanthroline (2.2 mg, 12 μmol, 50 mol% versus [Ruatom]) was added to a solution of 7.7 mg ammonia borane (0.25 mmol) and 13.6 mg 12 (25 μmol, 5 mol% versus AB) in 0.6 mL 2:1 diglyme/benzene-d6. The rates for both the fast and slow catalysis cases at 70 °C were determined using 11B NMR as shown in the Supporting Information.
1,10-Phenanthroline (2.2 mg) was dissolved in 0.5 mL diglyme to make a 1,10-phenanthroline stock solution. A portion of this solution (0.1 mL, 12 μmol, 10 mol% 1,10-phenanthroline versus [Ruatom]) was added to a solution of 7.7 mg ammonia borane (0.25 mmol) and 13.6 mg 12 (25 μmol, 5 mol% versus AB) in 0.5 mL 2:1 diglyme/benzene-d6. The rates for both the fast and slow catalysis cases at 70 °C were determined using 11B NMR as shown in the Supporting Information.
A further fractional poisoning regarding the role of 1,10-phenanthroline in the 26-catalyzed dehydrogenation of ammonia borane is also conducted. These data show little change in reactivity upon addition of the poison (see Supporting Information).
III. Preparative and Spectroscopic Details
Adduct 23
Ru-NH3 adduct 23 was prepared by delivering ammonia gas to a benzene (5 mL) solution of 18 (50 mg, 0.046 mmol). The reaction was stirred at room temperature for 15 minutes. A black precipitate was filtered out and successively washed with deionized water, acetone, benzene and hexanes. The solid was then dried under vacuum to give a pale gray power in 59% yield (30 mg).
1H NMR (pyridine-d5, 600 MHz): δ 8.05 (d, JHH = 7.1 Hz, 4H, Ph), 7.47 (d, JHH = 7.1 Hz, 4H, Ph), 7.18 (t, JHH = 7.1 Hz, 4H, Ph), 7.13 (m, 6H, Ph), 7.07 (t, JHH = 7.1 Hz, 4H, Ph), 4.25 (br. s, 3H, NH3). 13C{1H} NMR (pyridine-d5, 150 MHz): δ 202.5 (CO), 165.5 (C1 of Cp), 134.9 (Ph), 133.4 (Ph), 133.2 (Ph), 131.3 (Ph), 128.5 (Ph), 128.4 (Ph), 128.3 (Ph), 126.9 (Ph), 104.3 (C2,5 of Cp), 83.0 (C3,4 of Cp). Data are consistent with a known compound.23
Supplementary Material
Scheme 8.
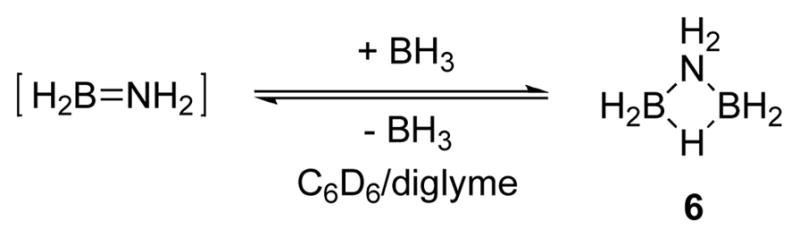
Formation of 6.
Acknowledgments
We thank Richard Finke and Ercan Bayram (Colorado State) for insightful discussions. This work was sponsored by the National Science Foundation (CHE-1054910) and the Hydrocarbon Research Foundation. We are grateful to the National Science Foundation (DBI-0821671, CHE-0840366), the National Institutes of Health (1 S10 RR25432), and the University of Southern California for their sponsorship of NMR spectrometers at USC.
Literature Cited
- 1.(a) Imarisio G. Int J Hydrogen Energy. 1981;6:153–158. [Google Scholar]; (b) Zoulias E, Varkaraki E, Lymberopoulos N, Christodoulou CN, Karagiorgis GN. TCJST. 2004;4:41–71. [Google Scholar]; (c) Liu R-S, Zhang L, Sun X, Liu H, Zhang J. Water Electrolysis for Hydrogen Generation. In: Millet P, editor. Electrochemical Technologies for Energy Storage and Conversion. 1, 2 Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2011. [Google Scholar]
- 2.United States Dept. of Energy. Multi-Year Research Development and Demonstration Plan, Sect 3.3. GPO; Washington: 2009. Hydrogen Fuel Cells and Infrastructure Technology Program. [Google Scholar]
- 3.Satyapal S, Petrovic J, Read C, Thomas G, Ordaz G. Catal Today. 2007;120:246–256. [Google Scholar]
- 4.For a review, see Staubitz A, Robertson APM, Manners I. Chem Rev. 2010;110:4079–4124. doi: 10.1021/cr100088b.For thermolysis of ammonia borane see Baitalow F, Wolf G, Jaeicke-Rossler K, Leitner G. Thermochim Acta. 2006;445:121–125.Baitalow F, Baumann J, Wolf G, Jaenicke-Rossler K, Leitner G. Thermochim Acta. 2002;391:159–168.Wolf G, Baumann J, Baitalow F, Hoffmann FP. Thermochim Acta. 2000;343:19–25.Wang JS, Geanangel RA. Inorg Chim Acta. 1988;148:185–190.Bluhm ME, Bradley MG, III, RB, Kusari U, Sneddon LG. J Am Chem Soc. 2006;128:7748–7749. doi: 10.1021/ja062085v.Rassat SD, Aardahl CL, Autrey T, Smith RS. Energy Fuels. 2010;24:2596–2606.Nylen J, Sato T, Soignard E, Yarger JL, Stoyanov E, Haussermann U. J Chem Phys. 2009;131:104506.Heldebrant DJ, Karkamakar A, Hess NJ, Bowden M, Rassat S, Zheng F, Rappe K, Autrey T. Chem Master. 2008;20:5332–5336.For hydrolysis: Marder TB. Angew Chem Int Ed. 2007;46:8116–8118. doi: 10.1002/anie.200703150.Yan JM, Zhang XB, Akita T, Haruta M, Xu Q. J Am Chem Soc. 2010;132:5326–5327. doi: 10.1021/ja910513h.Jiang HL, Umegaki T, Akita T, Zhang XB, Haruta M, Xu Q. Chem-Eur J. 2010;16:3132–3137. doi: 10.1002/chem.200902829.Ramachandran PV, Gagare PD. Inorg Chem. 2007;46:7810–7817. doi: 10.1021/ic700772a.
- 5.(a) Stephens FH, Pons V, Baker RT. Dalton Trans. 2007:2613–2626. doi: 10.1039/b703053c. [DOI] [PubMed] [Google Scholar]; (b) Smythe NC, Gordon JC. Eur J Inorg Chem. 2010:509–521. [Google Scholar]; (c) Hamilton CW, Baker RT, Staubitz A, Manners I. Chem Soc Rev. 2009;38:279–293. doi: 10.1039/b800312m. [DOI] [PubMed] [Google Scholar]; (d) Sutton AD, Burrell AK, Dixon DA, EBG, Gordon JC, Nakagawa T, Ott KC, Robinson JP, Vasiliu M. Science. 2011;331:1426–1429. doi: 10.1126/science.1199003. [DOI] [PubMed] [Google Scholar]
- 6.Jaska CA, Templ K, Lough AJ, Manners I. J Am Chem Soc. 2003;125:9424–9434. doi: 10.1021/ja030160l.Jaska CA, Manners I. J Am Chem Soc. 2004;126:1334–1335. doi: 10.1021/ja039162w.Sgrestha RP, Diyabalanage HVK, Semelsberger TA, Ott KC, Burrell AK. Int J Hydrogen Energy. 2009;34:2616–2621.Regarding borane-Rh coordination, see Douglas TM, Chaplin AB, Weller AS. J Am Chem Soc. 2008;130:14432–14433. doi: 10.1021/ja806582n.Alcaraz G, Sabo-Etienne S. Angew Chem Int Ed. 2010;49:7170–7179. doi: 10.1002/anie.201000898.
- 7.Denny MC, Pons V, Hebdon TJ, Heinekey M, Goldberg KI. J Am Chem Soc. 2006;128:12048–12049. doi: 10.1021/ja062419g. [DOI] [PubMed] [Google Scholar]
- 8.Blaquiere N, Diallo-Garcia S, Gorelsky I, Black A, Fagnou K. J Am Chem Soc. 2008;130:14034–14035. doi: 10.1021/ja804235t. [DOI] [PubMed] [Google Scholar]; (f) Käβ M, Fridrich A, Drees M, Schneider S. Angew Chem Int Ed. 2009;48:905–907. doi: 10.1002/anie.200805108. [DOI] [PubMed] [Google Scholar]
- 9.Conley BL, Williams TJ. Chem Commun. 2010;46:4815–4817. doi: 10.1039/c003157g. [DOI] [PubMed] [Google Scholar]
- 10.Conley BL, Guess D, Williams TJ. J Am Chem Soc. 2011;133:14212–14215. doi: 10.1021/ja2058154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keaton RJ, Blacquiere JM, Baker RT. J Am Chem Soc. 2007;129:1844–1845. doi: 10.1021/ja066860i. [DOI] [PubMed] [Google Scholar]
- 12.Kim S-K, Han W-S, Kim T-J, Kim T-Y, Nam SW, Mitoraj M, Piecoś Ł, Michalak A, Hwang S-J, Kang SO. J Am Chem Soc. 2010;132:9954–9955. doi: 10.1021/ja101685u. [DOI] [PubMed] [Google Scholar]
- 13.(a) Vance JR, Robertson APM, Lee K, Manners I. Chem Eur J. 2011;17:4099–4103. doi: 10.1002/chem.201003397. [DOI] [PubMed] [Google Scholar]; (b) Baker RT, Gordon JC, Hamilton CW, Henson NJ, Lin PH, Maguire S, Murugesu M, Scott BL, Smythe NC. J Am Chem Soc. 2012;134:5598–5609. doi: 10.1021/ja210542r. [DOI] [PubMed] [Google Scholar]
- 14.Chapman AM, Haddow MF, Wass DF. J Am Chem Soc. 2011;133:8826–8829. doi: 10.1021/ja201989c. [DOI] [PubMed] [Google Scholar]
- 15.Conley BL, Pennington-Boggio MK, Boz E, Williams TJ. Chem Rev. 2010;110:2294–2312. doi: 10.1021/cr9003133. [DOI] [PubMed] [Google Scholar]
- 16.(a) Wright WRH, Berkeley ER, Alden LR, Baker RT, Sneddon LG. Chem Commun. 2011;47:3177–3179. doi: 10.1039/c0cc05408a. [DOI] [PubMed] [Google Scholar]; (b) Alcaraz G, Vendier L, Clot E, Sabo-Etienne S. Angew Chem Int Ed. 2010;49:918–920. doi: 10.1002/anie.200905970. [DOI] [PubMed] [Google Scholar]
- 17.Conley BL, Williams TJ. J Am Chem Soc. 2010;132:1764–1765. doi: 10.1021/ja909858a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Casey CP, Singer SW, Powell DR, Hayashi RK, Kavana M. J Am Chem Soc. 2001;123:1090–1100. doi: 10.1021/ja002177z. [DOI] [PubMed] [Google Scholar]; (b) Casey CP, Beetner SE, Johnson JB. J Am Chem Soc. 2008;130:2285–2295. doi: 10.1021/ja077525c. [DOI] [PubMed] [Google Scholar]; (c) Casey CP, Johnson JB, Singer SW, Cui Q. J Am Chem Soc. 2005;127:3100–3109. doi: 10.1021/ja043460r. [DOI] [PubMed] [Google Scholar]
- 19.Johnson JB, Bäckvall J-E. J Org Chem. 2003;68:7681–7684. doi: 10.1021/jo034634a. [DOI] [PubMed] [Google Scholar]
- 20.Thorson MK, Klinkel KL, Wang J, Williams TJ. Eur J Inorg Chem. 2009:295–302. [Google Scholar]
- 21.Hu MG, Van Paasschen JM, Geanangel RA. J Inorg Nucl Chem. 1997;39:1247–1250. [Google Scholar]
- 22.Koren-Selfridge L, Query IP, Hanson JA, Isley NA, Guzei IA, Clark TB. Organometallics. 2010;29:3896–3900. doi: 10.1021/om1005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollmann D, Jiao H, Spannenberg A, Bähn S, Tillack A, Parton R, Altink R, Beller M. Organometallics. 2009;28:473–479. [Google Scholar]
- 24.Stephens FH, Pons V, Baker RT. Dalton Trans. 2007:2613–2626. doi: 10.1039/b703053c. [DOI] [PubMed] [Google Scholar]
- 25.Hagen CM, Vieille-Petit L, Laurenczy G, Süss-Fink G, Finke RG. Organometallics. 2005;24:1819–1831. [Google Scholar]
- 26.(a) Widegren JA, Finkie RG. J Mol Catal. 2003;198:317–341. [Google Scholar]; (b) Kovács G, Nádasdi L, Joó F, Laurenczy G. C R Acad Sci Paris, Chim. 2000;3:601. [Google Scholar]
- 27.Bayram E, Linehan JC, Fulton JL, Roberts JAS, Szymczak NK, Smurthwaite TD, Özkar S, Balasubramanian M, Finke RG. J Am Chem Soc. 2011;133:18889–18902. doi: 10.1021/ja2073438. [DOI] [PubMed] [Google Scholar]
- 28.Wildeman T, Sneddon LG. Inorg Chem. 1995;34:1002–1003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.