

Abstract
Vascular endothelial growth factor (VEGF) is an angiogenic mitogen involved in promoting tumor angiogenesis inside the body. VEGF is a key protein required for progression of tumor from benign to malignant phenotype. In this study, we investigated the binding affinity of a previously selected 26-mer DNA aptamer sequence (SL2-B) against heparin binding domain (HBD) of VEGF165 protein. The SL2-B was first chemically modified by introduction of phosphorothioate linkages (PS-linkages). Subsequently, surface plasmon resonance (SPR) spectroscopy and circular dichroism (CD) were used to determine the binding affinity, specificity and to deduce the conformation of PS-modified SL2-B sequence. Finally, antiproliferative activity of the modified SL2-B sequence on Hep G2 cancer cells was investigated. Our results demonstrate a marked enhancement in the biostability of the SL2-B sequence after PS modification. The modified SL2-B sequence also exhibits enhanced antiproliferative activity against Hep G2 cancer cells in hypoxia conditions. In addition, modified SL2-B sequence inhibits the expression of Jagged-1 protein, which is one of the ligands to VEGF linked delta/jagged-notch signaling pathway.
Introduction
Cancer is one of the leading causes of death worldwide and accounted for 7.6 million deaths in 2008 [1], [2]. In the United States alone, approximately 1 in 4 people die due to cancer [3]. Currently, monoclonal antibodies are one of the most advanced therapeutic agents for cancer treatment in the market. Several FDA approved monoclonal antibody drugs, such as bevacizumab (trade name: Avastin) against vascular endothelial growth factor (VEGF) in colorectal, lung, and kidney cancer treatment, trastuzumab (trade name: Herceptin) against HER2/neu receptor in breast cancer treatment, and cetuximab (trade name: Erbitux) against epidermal growth factor receptor (EGFR) in metastatic colorectal, head and neck cancers, have been developed and are used either as a single agent or in combination with other drugs and radiation for cancer therapy [4]–[12].
In 1990, an in vitro selection process called systematic evolution of ligands by exponential enrichment (SELEX) was developed to screen single stranded nucleic acid molecules from random pool of library against the target ligand [13], [14]. These classes of single stranded molecules are referred as “aptamers”. They possess high binding affinity and specificity that are comparable to monoclonal antibodies. In addition, the small size, non-immunogenicity and ease of modification compared to conventional monoclonal antibody makes aptamers attractive for therapeutic application [15]. Based on the promising results in preclinical studies, two cancer targeting aptamers, ACT-GRO-777 (or AS1411) - a G-rich DNA aptamer targeting nucleolin for treatment of acute myeloid leukemia (AML) and NOX-A12 L-RNA aptamer targeting CXCL12 for treatment of multiple myeloma and lymphoma are already in clinical trials [16], [17].
One chief problem that arises in the therapeutic application of aptamers is their instability under in vitro and in vivo conditions [18]. They are susceptible to enzymatic nuclease attack in the cellular and serum fluids. To circumvent this problem, several chemical modification strategies have been employed to enhance their resistance against nucleases and to prolong their circulation half-life in the biological fluids. Such chemical modifications include incorporation of phosphorothioate linkages (PS-linkages) or locked nucleic acids (LNAs), addition of functional groups such as amino (-NH2), fluoro (-F), O-methyl (-OCH3) in 2′-position of ribose sugar, and conjugation to high molecular mass polyethylene glycol (PEG) or cholesterol [19]–[25]. Studies have demonstrated that, compared to the unmodified version, the chemically modified aptamers exhibit not only longer lifetime in the biological milieu but sometimes also better binding affinity and specificity to their targets [21], [26].
VEGF is a crucial angiogenic mitogen overexpressed in the tumor cells and induces their migration, excessive proliferation, invasion and metabolism inside the body. VEGF is considered to be the hallmark protein for tumor angiogenesis and has been associated with neoplastic transformation of cells inside the body [27]. It is generally thought to be secreted by endothelial cells to stimulate their proliferation and migration. Previous reports, however, indicate that different carcinoma and malignant mesothelioma cell lines also secrete this protein [28]–[31]. VEGF165 is the pre-dominant isoform of VEGF-A protein, one of the members of VEGF family, and primarily binds to its two tyrosine-kinase receptors VEGFR-1/Flt-1 and VEGFR-2/KDR/Flk-1 with very high affinity and to specific co-receptor neuropilins [27]. The mitogenic signaling and cell proliferation in tumor cells is induced by expression of VEGFR-2 [32], [33]. In contrast, activation of VEGFR-1 results in cell invasion and cell migration but not cell proliferation [34]–[36].
In our previous study, a 26-mer DNA aptamer against heparin binding domain (HBD) of VEGF165 protein (referred to as SL2-B) was obtained using stem-loop truncation strategy [37]. Compared to the original untruncated aptamer, the SL2-B aptamer exhibited more than 200-fold increase in the binding affinity to VEGF165 protein. Herein, we modified the SL2-B aptamer by incorporating phosphorothioate (PS) linkages, tested its binding affinity, specificity, biostability, secondary structure and the potential feasibility of the PS-modified SL2-B aptamer as antagonist on the proliferation activity of cancer cells. We demonstrated that, compared to unmodified SL2-B aptamer, the PS-modified SL2-B aptamer is an improved sequence in terms of serum stability and antiproliferative activity without sacrificing the binding affinity and specificity for VEGF165 protein.
Materials and Methods
Materials
The HPLC purified oligonucleotide (both unmodified and PS-modified) was purchased from Sigma-Aldrich. The recombinant human carrier free VEGF165 (molecular weight of 38 kDa, pI = 8.25) and VEGF121 (molecular weight of 28 kDa, pI = 6.4) proteins were purchased from R & D systems. CM5 sensor chips were purchased from GE Healthcare for protein immobilization. 1-ethyl-3- [3-dimethylaminopropyl] hydrochloride (EDC), N-hydroxysuccinimide (NHS), and ethanolamine-HCl were purchased from Sigma-Aldrich. Sodium acetate (anhydrous) was purchased from Fluka. Tween-20 was purchased from USB Corporation. Acrylamide/Bis-acrylamide (30%) and triton X-100 were purchased from BIO-RAD. Sodium dodecyl sulfate (SDS), phosphate buffer saline (PBS), and sodium hydroxide (NaOH) were purchased from 1st Base. Human hepatocellular carcinoma (Hep G2) cell line was a gift from Dr. Tong Yen Wah’s lab, which was purchased from ATCC. Human breast adenocarcinoma (MCF-7) cell line and human colorectal carcinoma cell line (HCT-116) were purchased from ATCC. The hypoxia chamber was purchased from Billups-Rothenberg. Dulbecco’s modified eagle’s media (DMEM) media, and fetal bovine serum (FBS) were purchased from Caisson laboratories. Trypsin-EDTA and 1% penicillin/streptomycin mixture were purchased from PAN biotech. Thiazolyl blue tetrazolium bromide (MTT, 97.5%) ammonium persulfate (APS), urea and N, N, N′, N′-methylene-bis-acrylamide (TEMED, 99%), nadeoxycholate and tris buffer were purchased from Sigma-Aldrich. Monoclonal anti-human Jagged-1 fluorescein antibody was purchased from R & D systems. Jagged-1 (28H8) rabbit monoclonal antibody was purchased from cell signaling. Purified mouse anti-calnexin antibody was purchased from BD transduction laboratories. The lysis and extraction buffer RIPA (Radio-Immunoprecipitation Assay) buffer for western blotting was prepared with the following reagents: RIPA Buffer (50 ml), 50 mM Tris (pH 7.8), 150 mM NaCl, 0.1% SDS (sodium dodecyl sulphate), 0.5% Nadeoxycholate, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF). One tablet of the protein inhibitor cocktail, complete mini tablet (Roche Applied Science, Switzerland) was dissolved in 10 ml of the buffer to complete the lysis buffer preparation. Polyvinyllidene difluoride (PVDF) membrane, wet pico chemiluminescence substrate and CL-exposure film were purchased from thermo scientific. The FITC annexin V apoptosis detection kit was purchased from BD Pharmingen, Germany. PMSF was purchased from CalBiochem.
Surface Plasmon Resonance (SPR) Spectroscopy
The binding affinity and specificity of modified aptamer sequence was investigated using surface plasmon resonance (SPR) spectroscopy, where VEGF165 and VEGF121 acted as ligands and were directly immobilized on the sensor chip. Briefly, the carboxylic group on the sensor chip was activated by standard amine coupling procedure using freshly prepared EDC/NHS. VEGF165 or VEGF121 (25 µg/ml) in acetate buffer (pH 6.0) were then injected into the sensor chip at flow rate 8 µl/min to reach ∼200 RU immobilization level. The deactivation was done by ethanolamine-HCl to block unreacted carboxyl groups. The binding analysis was carried out with modified aptamers at different concentrations (0.2 to 100 nM) using a BIAcore 2000 instrument (GE Healthcare). The running condition was set at 30 µl/min flow rate, 25°C, 3 min association time and 5 min dissociation time. PBS and tween-20 solution mixture was used as the running buffer, and 50 mM NaOH as the regeneration buffer. All the buffers were filtered and degassed prior to each experiment. Blank surfaces were used for background subtraction. Upon injection of the aptamers, sensorgrams recording the association/dissociation behavior of the VEGF-aptamer complex were collected. By varying the aptamer concentration, a series of sensorgrams (Figure 1) were obtained and subsequently analyzed using the 1∶1 Langmuir model provided in the BIAevaluation software (version 4.1) to calculate the equilibrium dissociation constant Kd. All SPR measurements were performed in triplicates.
Figure 1. Typical SPR sensorgrams demonstrating interaction of aptamer with immobilized VEGF165 protein at different concentration (bottom to top, 0.2 to 100.

nM). Point A to B corresponds to association phase and point B to C corresponds to the dissociation phase in all the sensorgrams. Shown here is PS-modified SL2-B aptamer (Kd = 0.56±0.44 nM).
Stability of SL2-B Aptamer Against Nucleases in Serum Containing Medium
To test the stability of the unmodified and PS-modified SL2-B aptamer against nucleases, 10 µM aptamer was incubated for different time intervals in DMEM media supplemented with 10% FBS at 37°C. 25 µl of sample was taken out at different time point (0, 12, 24, 48, and 72 hours) and immediately stored at −80°C to minimize unnecessary degradation. Samples were then subjected to 12% denaturing polyacrylamide gel electrophoresis (PAGE). The band density was quantitatively measured using gel densitometry and analyzed using gene tools software from Syngene.
Circular Dichroism (CD) Spectroscopy
To deduce the structure of PS-modified SL2-B aptamer, 10 µM of aptamer was dissolved in the PBS buffer for CD analysis. The CD spectrum was recorded in wavelength range of 200–320 nm at two different temperatures 25°C and 37°C and the data were the average of 10 scans. The CD spectrum analysis was performed using cuvette of 1-cm path length on a Jasco J-810 spectropolarimeter. The PBS buffer was used as blank for both the temperatures and the spectral data for SL2-B aptamer was blank corrected.
Antiproliferative Activity Assay
Hep G2 and MCF-7 cells were seeded at a density of 2000 cells/ml and HCT-116 cells were seeded at a density of 3000 cells/ml in 96-well plate at day 0 in DMEM media supplemented with 10% FBS and penicillin/streptomycin mixture. SL2-B aptamer (unmodified/PS-modified) and scrambled aptamer were incubated with cells at different concentrations and incubated for 3 days in hypoxia conditions (5% CO2, 1% O2, and 94% N2) inside the hypoxia chamber. The cell medium was not changed for 3 days. No cell transfecting or permeabilizing agent was added. The antiproliferative effect of aptamer on the cells was determined by measuring cell viability using colorimetric MTT assay. The optical density reading was recorded using microplate reader (Tecan, infinite M200) at 570 nm with background subtraction at 620 nm. The experiment was performed in triplicates.
Microscopy Imaging
The antiproliferative effect of PS-modified SL2-B aptamer on Hep G2 cells was assessed using optical microscopic imaging. Same conditions were maintained as for the antiproliferative activity assay and cells were imaged after 72 hours of aptamer treatment. Photomicrographs were taken on an Eclipse T5000 (Nikon, Japan) light microscope with Tame2u acquisition software.
Apoptosis Assay
Annexin V apoptosis assay was performed to investigate the cell death mechanism in Hep G2 cells according to manufacturer’s protocol. Cells were harvested by trypsinization and washed twice with cold PBS (1X) and subsequently stained with FITC Annexin V and propidium iodide. Analysis was performed on the Beckman-Couter CyAn™ ADP flow cytometer by counting 15000 events.
Flow Cytometry Analysis
Flow cytometry was used to study the effect of PS-modified SL2-B aptamer on Jagged-1 protein expression in Hep G2 cells. Hep G2 cells were seeded at a density of 80,000 cells/ml in 6-well plate at day 0 in DMEM media supplemented with 10% FBS and penicillin/streptomycin mixture. Following day after seeding, the cells were treated with modified SL2-B aptamer and scrambled aptamer sequence at 15 µM aptamer concentration. Same hypoxia conditions were maintained as for the antiproliferative activity assay. After 3 days of aptamer treatment, the cells were trypsinized, incubated with anti-human Jagged-1 fluorescein antibody for 1 hour, re-suspended in PBS buffer and analyzed immediately using a Beckman-Couter CyAn ADP flow cytometer by analyzing 15,000 events and relative fluorescence was determined using SUMMIT V 4.3.02 software.
Western Blot Analysis
The sequence specific effect of PS-modified SL2-B aptamer on Jagged-1 protein expression in Hep G2 cells was analyzed using western blotting. Same experimental conditions were maintained as for the flow cytometry. After 3 days of aptamer treatment, the cell medium was removed and the cells were washed once in cold 1×PBS. 500 µl of the complete lysis buffer was added to each 6-well and the cells were scrapped with a cell scrapper and collected into microcentrifuge tubes. The extracted proteins were resolved on an SDS-PAGE gel and transferred onto a PVDF membrane via wet transfer. Membranes were blocked in 5% non-fat milk and washed in tris-buffered saline with 1% tween. Subsequently, membranes were incubated with primary antibody (Jagged-1 rabbit monoclonal and purified mouse anti-calnexin antibody) and then with corresponding secondary antibody (goat anti-rabbit and anti-mouse IgG secondary antibody conjugated to horseradish peroxidase (HRP)) with 3 washing steps in between. The protein bands were developed with west pico chemiluminescence substrate and visualized on XPress CL blue ray film. Optical densities of bands were measured on a GS800 densitometer and band intensities were analyzed with Quantity One image analysis software (Biorad, USA).
Statistical Analysis
Data are presented as mean ± SD. A p-value <0.05 was considered statistically significant using student’s t-test.
Results and Discussion
Binding Analysis of PS-modified SL2-B Aptamer and VEGF Complex by Surface Plasmon Resonance (SPR)
As reported previously in our study, the unmodified SL2-B aptamer displayed a Kd = 0.5 nM to heparin binding domain (HBD) of VEGF165 protein determined via SPR technique (Table 1) [37]. The unmodified aptamer, however, exhibited low structural stability in the cellular conditions. This is due to the presence of exonucleases and endonucleases in biological fluids which degrade the aptamers by hydrolyzing the phosphate ester bond in the backbone [19]. To alleviate this problem, in this study, the SL2-B aptamer was chemically modified with phosphorothioate (PS) linkages at 5′ and 3′- terminus (Table 1) to protect the SL2-B aptamer from exonucleolytic digestion. The PS-modification involves the substitution of unbridged phosphoryl oxygen in phosphodiester linkage by sulfur atom. Since the excess incorporation of PS-linkages leads to non-specific binding and can perturb the aptamer conformation and its interaction with the target, the modification was introduced only at aptamer termini [38].
Table 1. Unmodified and PS-modified SL2-B aptamer sequences along with their equilibrium dissociation constant (Kd) values determined using surface plasmon resonance (SPR) spectroscopy.
| Sequences of original and PS-modified aptamer (5′–3′) | Kd |
| Unmodified SL2-B aptamer CAATTGGGCCCGTCCGTATGGTGGGT | 0.50±0.32 nM |
| PS-modified SL2-B aptamer C*AATTGGGCCCGTCCGTATGGTGGG*T | 0.56±0.44 nM |
“*” indicates the position of phosphorothioate (PS) modification.
The Kd value for PS-modified SL2-B aptamer was determined using SPR technique at different aptamer concentrations (Figure 1 and Table 1). The Kd value for the PS-modified SL2-B was found to be 0.56 nM, which is similar to the Kd for unmodified SL2-B. Introducing PS-modification does not appear to affect the binding affinity of the SL2-B aptamer. Moreover, the affinity of PS-modified SL2-B is similar to the FDA approved humanized anti-VEGF monoclonal antibody “bevacizumab” (Kd ∼ 0.5 nM) used for cancer treatment [4].
Specificity of PS-modified SL2-B Aptamer Sequence
VEGF165 as well as other VEGF isoforms, such as VEGF189 and VEGF206, are generated from splicing of a single VEGF gene that shares a carboxyl-terminal heparin-binding domain (HBD) of 50-residues and binds to heparin with different binding affinities [27], [39], [40]. HBD is responsible for enhancing the interaction of VEGF with its receptors (VEGFR-1/Flt-1 and VEGFR-2/KDR/Flk-1) and the specific co-receptor neuropilins to trigger the angiogenic response in malignant cells [41].
VEGF121, however, does not share the HBD as other VEGF isoforms and can be used as a control for HBD binding specificity study. The SPR sensorgram in Figure 2 shows that compared to VEGF165 protein at same aptamer concentration (80 nM), the response signal of PS-modified SL2-B binding to VEGF121 protein was weak and displayed a high Kd value of 17 µM. This indicates that PS modification does not reduce the binding specificity of SL2-B aptamer towards HBD significantly (Kd = 17 µM for PS-modified SL2-B towards VEGF121, Kd = 10 µM for unmodified SL2-B towards VEGF121). Compared to the “bevacizumab” monoclonal antibody that binds to all isoforms of VEGF, the PS-modified SL2-B is specific to HBD of VEGF165 protein [4]. Since VEGF-A is involved in normal physiological processes, such as formation of new blood vessels and wound healing process, the complete inhibition of VEGF protein can affect the maintenance of the normal vascular system inside the body [42], [43]. Therefore, inhibition of specific VEGF protein (for example, VEGF165 in this case) may be a better therapeutic approach.
Figure 2. SPR sensorgrams demonstrating interaction of PS-modified SL2-B aptamer with immobilized VEGF165 and VEGF121 protein at same concentration.

Point A to B corresponds to association phase and point B to C corresponds to the dissociation phase in both the sensorgrams. Shown here is PS-modified SL2-B aptamer binding with VEGF165 protein (Kd = 0.56±0.44 nM) and VEGF121 protein (Kd = 17±1.24 µM) at 80 nM aptamer concentration.
Stability of SL2-B Aptamer Against Nucleases in Serum Containing Medium
To test the biostability of the unmodified and PS-modified SL2-B aptamer against nucleases present in the biological fluids, both aptamers were incubated with 10% FBS for different time periods. Based on the results, the unmodified SL2-B degraded by 50% within 24 hours of incubation in serum (Figure 3). On the other hand, the PS-modified SL2-B displayed good stability, with more than 90% aptamer intact after 72 hours of incubation in the serum. The data demonstrates the importance of PS-linkages in the SL2-B sequence termini, which protects the aptamer sequence from exonuclease attack.
Figure 3. Nuclease-resistance stability of unmodified and modified SL2-B aptamer sequence in 10% FBS.

Aptamers were incubated with 10% FBS dissolved in DMEM media at 37°C for different time points and percentage of intact aptamer was determined by measuring the band density after running denaturing PAGE. Filled columns are PS-modified SL2-B, while open columns are unmodified SL2-B.
Structural Analysis by Circular Dichroism (CD) Spectroscopy
Structural studies have shown the impact of the conformation on the binding affinity and specificity of the aptamer for its target [44]. If the conformation changes with temperature, then the binding affinity results obtained from SPR spectroscopy (conducted at 25°C) may not be representative in in vitro assays (conducted at 37°C). Thus, the secondary conformation of the PS-modified SL2-B aptamer was investigated. Positive maxima peaks were observed at 260 nm and 220 nm as well as a negative minima peak at 240 nm and additional small shoulder peak at 290 nm (Figure 4). Based on the previous reports, such spectra reflect a typical hairpin stem-loop conformation [45]. Since no change in the spectra was observed between 25°C and 37°C, this confirms the preservation of the secondary conformation at the SPR conditions (25°C) where the Kd of the aptamer was determined and at physiological conditions (37°C). However, the CD spectroscopy does not provide the complete and validated information on the structure. Advanced techniques such as nuclear magnetic resonance (NMR) and X-ray crystallography are required for further in-depth structural analysis.
Figure 4. CD spectra of 10.

µM PS-modified SL2-B aptamer in phosphate buffer saline (PBS) buffer, pH-7.2. Spectra were measured at 25°C (solid line) and 37°C (dotted line).
Antiproliferative Activity Assay
The antiproliferative property of SL2-B aptamer was studied using Hep G2 cancer cells in hypoxia conditions. Previous studies have demonstrated that the expression of VEGF protein is potentiated in Hep G2 cells under hypoxia conditions [46]. Since no significant effect on cell proliferation was observed at 24 and 48 hours, both the unmodified and PS-modified SL2-B aptamers were tested for 72 hours duration. As shown in Figure 5, lower cell proliferation was observed at 15 µM modified SL2-B concentration after 72 hours of aptamer treatment (52±2.1%). However, no decrease in the cell proliferation was observed on further increasing aptamer concentration to 20 µM. A possible explanation for decrease in the cell proliferation could be that either the excess binding of modified SL2-B sequence to VEGF165 protein ultimately prevents the interaction of the protein to the VEGFR-2 (or KDR/Flk-1) receptor, which affects the cellular proliferation. Or aptamer after binding with VEGF protein binds with VEGFR-2, undergoes cellular internalization and interferes with the downstream VEGF linked intracellular signaling pathways. The result also indicates that VEGF protein may be involved in the proliferation of investigated Hep G2 cancer cells under hypoxia conditions. On the contrary, the unmodified SL2-B aptamer sequence did not exhibit significant inhibitory activity on the cellular proliferation. This could be due to the degradation of the unmodified sequence by nuclease enzymes in the media before pronouncing its effect on the cancer cells.
Figure 5. Relative % proliferation of Hep G2 cells (compared to control) after treating with unmodified and PS-modified SL2-B aptamers at different concentrations in hypoxia conditions.

The sequence specificity was determined using scrambled sequence for PS-modified SL2-B for each data point at same concentration to the modified SL2-B. Solid line is PS-modified SL2-B, dashed line is unmodified SL2-B, and dotted line is scrambled sequence.
To demonstrate that the antiproliferative effect of PS-modified SL2-B aptamer is sequence specific, a scrambled sequence was added to the Hep G2 cells at the same concentration as PS-modified SL2-B (Figure 5). The results showed minimal decrease on the cell proliferation with the scrambled sequence, confirming that the inhibitory effect on VEGF165 protein activity by PS-modified SL2-B was sequence specific in Hep G2 cells. The sequence specific inhibition was also confirmed by the cell count and morphological differences presented in photomicrographs (Figure 6). As shown in Figure 6A and 6B, the cells treated with modified sequence have noticeably fewer cells as compared with the scrambled sequence where there appears to be more cells per view and packed closely to one another. Furthermore, under the same magnification, the morphology of the cells treated with the modified sequence appears longer and thinner with many side projections as compared with the scrambled sequence, which are more angular and more defined in shape (Figure 6C and 6D). These findings indicate the potential of the PS-modified SL2-B aptamer sequence in inhibiting the Hep G2 cancer cells proliferation strongly and specifically.
Figure 6. Effect of PS-modified SL2–B aptamer sequence compared to the scrambled sequence on Hep G2 cells.

Low magnification view of (A) modified sequence treatment, (B) scrambled sequence treatment on Hep G2 cells after 72 hours under hypoxia condition. Scale bar = 200 µm. Close up views of (C) modified sequence treatment, (D) scrambled sequence treatment on Hep G2 cells after 72 hours under hypoxia condition. Cellular morphology differs upon the different treatments; modified sequence treatment produces cells which are thinner with more cellular projections while the scrambled sequence treatment shows cells which appear closer to the untreated Hep G2 cells. Scale bar = 50 µm.
To determine the cell death mechanism in Hep G2 cells, annexin V apoptosis assay was performed and analyzed using flow cytometry. In Figure 7A, the R9 and R11 quadrant cells in flow cytometry scatterplot were counted and expressed as percentage of cells in late and early apoptosis phase respectively. Early apoptotic cells include cell population that is annexin V positive only (R11), and late apoptotic cells include cell population that is both annexin V and PI positive (R9). The apoptosis assay showed increased percentage of cell death with modified sequence compared with the scrambled sequence treatment in late apoptosis phase (Figure 7B, p-value <0.05). However, the percentage of cells undergoing late apoptosis was not very high and no significant difference in cell count was observed between modified and scrambled sequence in early apoptosis phase (Figure 7C). This result indicates that besides apoptosis, other non-apoptotic cell death mechanism such as senescence may be involved in induction of cell death in the Hep G2 cells.
Figure 7. Annexin V assay of Hep G2 cells treated with modified sequence and scrambled sequence.

(A) The scatterplot depicting the distribution of cells with annexin V staining along the x-axis and those stained with propidium iodide (PI) along the y-axis. Region R10 denotes the viable population (double negative for annexin V and PI), R9 the non-viable cells (double positive for annexin V and PI), R11 shows the annexin V positive (PI negative) population while R8 are the damaged cells (PI positive but annexin-V negative). (B) % Histogram of the R9 quadrant data. The analysis of the triplicate samples for showed a significantly higher amount of dead cells (p-value <0.05) in the modified sequence treatment compared to the scrambled sequence control. (C) % Histogram of R11 quadrant data. The results show no significant difference for early apoptosis. Error bars = SEM.
To confirm the antiproliferative ability of the PS-modified SL2-B aptamer, we further investigated the effect with MCF-7 cells and HCT-116 cells since existing literature has shown that they also overexpress VEGF protein in hypoxia conditions [47], [48]. A 15 µM modified SL2-B concentration was used in this study but our results showed that both MCF-7 and HCT-116 cancer cells displayed only 23±3.2% and 9±1.8% decrease in cell proliferation was observed respectively. Based on these cell proliferation results, the effect of PS-modified SL2-B sequence on cell proliferation is believed to be cell type specific. Since antiproliferative effect on MCF-7 and HCT-116 cancer cells were not very substantial, they were not used for further studies below. Additional antiproliferative studies on various cancer cell types should be conducted to uncover the potential therapeutic targets and to identify the factors responsible for cell specific antiproliferative activity of this aptamer.
Flow Cytometry and Western Blot Analysis of Jagged-1 Protein Expression
Notch signaling is an evolutionary conserved signaling pathway affecting many cellular processes such as cell-fate determination, differentiation, proliferation, and survival. Five Notch ligands (Jagged-1, Jagged-2, Delta-1, Delta-3, and Delta-4) and four Notch receptors have been well established in mammals [49], [50]. Evidence indicates the biochemical linkage between VEGF and delta/jagged-notch pathways activation, and together both are involved in promoting tumor progression [51], [52]. In this linkage, VEGF pathway is essential for the initiation of tumor angiogenesis and acts as the upstream activating stimulus, whereas notch signaling which acts on downstream of the VEGF pathway, helps to respond to activating stimulus and shape the activation by making cell fate decisions [49]. Due to the crosstalk between VEGF and notch signaling pathways, the effect of PS-modified SL2-B aptamer was tested on Jagged-1, which is one of the notch ligands. Jagged-1 is overexpressed in various malignant tumors and has been associated with cancer recurrence [53]–[55]. Here, we examined the effect of PS-modified SL2-B aptamer on the expression of Jagged-1 protein in Hep G2 cells via flow cytometry technique. Compared to the untreated sample (only cells), modified SL2-B treatment exhibited decrease in the fluorescent signal (Figure 8). This shift in the peak indicates the downregulation of the Jagged-1 expression due to the addition of PS-modified SL2-B aptamer in Hep G2 cells (p-value <0.05).
Figure 8. Flow cytometry histogram of Jagged-1 protein expression in Hep G2 cells using anti-human Jagged-1 antibody and quantitative analysis of flow cytometry result.
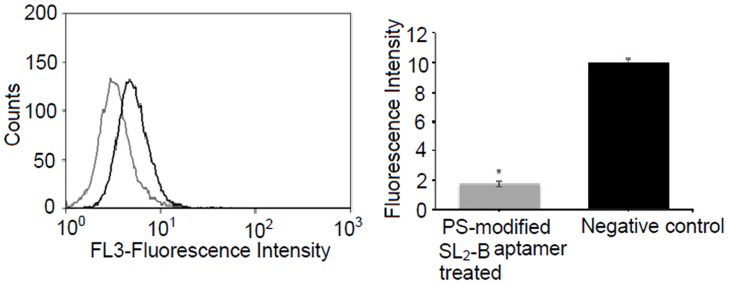
Each histogram curve represents the expression of Jagged-1 obtained with (gray line) and without (black line, negative control) treatment with PS-modified SL2-B aptamer at 15 µM concentration. *Significant difference from the negative control sample at p-value <0.05.
Besides flow cytometry, the effect of PS-modified SL2 aptamer on Jagged-1 protein expression in Hep G2 cells was analyzed using western blotting. The scrambled sequence of the modified aptamer was used as control. The modified aptamer appears to induce a lower expression of the Jagged-1 protein in Hep G2 cells as compared to the scrambled sequence (Figure 9). This confirms the sequence specific inhibition of the aptamer on Jagged-1 protein expression in Hep G2 cells. Based on both flow cytometry and western blotting results, it can be concluded that the binding of PS-modified SL2-B aptamer to VEGF protein exhibits its antiproliferative activity in Hep G2 cells not only by inhibiting VEGF pathway but also the interconnected delta/jagged-notch signaling pathway in Hep G2 cells. Further studies are warranted to determine the effect of the modified aptamer on different notch ligands and other VEGF linked signaling pathways.
Figure 9. Western blot of whole cell lysates from Hep G2 cells treated with the PS-modified SL2 aptamer and scrambled sequence (control).
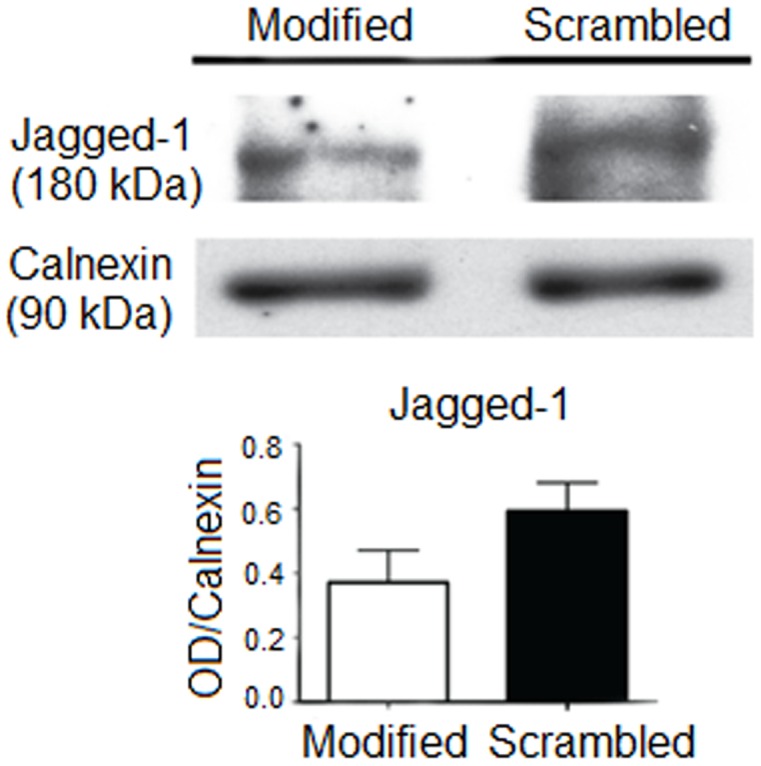
The expression of Jagged-1 protein in Hep G2 cells was assessed. Calnexin protein was used as a loading control. Error bar = SEM.
Conclusions
To summarize, this work attempted to study the antiproliferative potential of SL2-B aptamer in cancer cells. From the data, we conclude that post-modification, the PS-modified SL2-B aptamer retained its binding affinity and specificity for the heparin-binding domain (HBD) of VEGF165 protein. Furthermore, compared to the unmodified aptamer, the modified SL2-B demonstrated good biostability and exhibited its sequence specific antiproliferative activity on Hep G2 cancer cells in hypoxia conditions. Thus, based on the results of this work, it appears that chemical modification can be a useful approach in prolonging the half-life of the SL2-B aptamer in the in vitro conditions. This newly obtained SL2-B aptamer sequence can potentially be useful in oligomer-based cancer therapeutic applications, though further preclinical studies are required for better understanding of the SL2-B aptamer sequence and to evaluate its potential therapeutic value for cancer treatment.
Acknowledgments
The authors thank Dr Tong Yen Wah (Department of Chemical and Biomolecular engineering, National University of Singapore) for providing the Hep G2 cancer cells.
Funding Statement
This work was supported by research funding from the Singapore Ministry of Education Academic Research Fund Tier 2 grant MOE2008-T2-1-046 and Tier 1 grant R279000282112. The authors also appreciated the doctoral scholarship (for H.K.) from NUS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, et al. (2008) Estimates of worldwide burden of cancer in 2008: GLOBOCAN. Intl J Cancer 127: 2893–2917. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Bray F (2011) Center MM, Ferlay J, Ward E, et al (2011) Global cancer statistics. CA-Cancer J Clin 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 3. Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA-Cancer J Clin 60: 277–300. [DOI] [PubMed] [Google Scholar]
- 4. Ferrara N, Hillan KJ, Gerber HP, Novotny W (2004) Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3: 391–400. [DOI] [PubMed] [Google Scholar]
- 5. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, et al. (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New Eng J Med 350: 2335–2342. [DOI] [PubMed] [Google Scholar]
- 6. Los M, Roodhart JML, Voest EE (2007) Target practice: Lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist 12: 443–450. [DOI] [PubMed] [Google Scholar]
- 7. Leighl NB, Zatloukal P, Mezger J, Ramlau R, Moore N, et al. (2010) Efficacy and safety of bevacizumab-based therapy in elderly patients with advanced or recurrent nonsquamous non-small cell lung cancer in the phase III BO17704 study (AVAiL). J Thorac Oncol 5: 1970–1976. [DOI] [PubMed] [Google Scholar]
- 8. Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, et al. (2003) A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. New Eng J Med 349: 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Costa RB, Kurra G, Greenberg L, Geyer CE (2010) Efficacy and cardiac safety of adjuvant trastuzumab-based chemotherapy regimens for HER2-positive early breast cancer. Ann Oncol 21: 2153–2160. [DOI] [PubMed] [Google Scholar]
- 10. Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, et al. (2010) Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol 28: 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong SF (2005) Cetuximab: An epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin Ther 27: 684–694. [DOI] [PubMed] [Google Scholar]
- 12. Frampton JE (2010) Cetuximab: A review of its use in squamous cell carcinoma of the head and Neck. Drugs 70: 1987–2010. [DOI] [PubMed] [Google Scholar]
- 13. Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818–822. [DOI] [PubMed] [Google Scholar]
- 14. Tuerk C, Gold L (1990) Systemic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505–510. [DOI] [PubMed] [Google Scholar]
- 15. Jayasena SD (1999) Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin Chem 45: 1628–1650. [PubMed] [Google Scholar]
- 16. Bates PJ, Laber DA, Miller DM, Thomas SD, Trent JO (2009) Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp Mol Pathol 86: 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sayyed SG, Hägele H, Kulkarni OP, Endlich K, Segerer S, et al. (2009) Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 52: 2445–2454. [DOI] [PubMed] [Google Scholar]
- 18. Agrawal S, Temsamani J, Galbraith W, Tang J (1995) Pharmacokinetics of antisense oligonucleotides. Clin Pharmacokinet 28: 7–16. [DOI] [PubMed] [Google Scholar]
- 19. Kawasaki AM, Casper MD, Freier SM, Lesnik EA, Zounes MC, et al. (1993) Uniformly modified 2′-deoxy-2′-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J Med Chem 36: 831–841. [DOI] [PubMed] [Google Scholar]
- 20. Vester B, Wengel J (2004) LNA (Locked Nucleic Acid): High-affinity targeting of complementary RNA and DNA. Biochemistry 43: 13233–13241. [DOI] [PubMed] [Google Scholar]
- 21. Lin Y, Nieuwlandt D, Magallanez A, Feistner B, Jayasena SD (1996) High-affinity and specific recognition of human thyroid stimulating hormone (hTSH) by in vitro-selected 2′-amino-modified RNA. Nucleic Acids Res 24: 3407–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruckman J, Green LS, Beeson J, Waugh S, Gillette WL, et al. (1998) 2′-fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165): Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J Biol Chem 273: 20556–20567. [DOI] [PubMed] [Google Scholar]
- 23. Burmeister PE, Lewis SD, Silva RF, Preiss JR, Horwitz LR, et al. (2005) Direct in vitro selection of a 2′-O-methyl aptamer to VEGF. Chem Biol 12: 25–33. [DOI] [PubMed] [Google Scholar]
- 24. Boomer RM, Lewis SD, Healy JM, Kurz M, Wilson C, et al. (2005) Conjugation to polyethylene glycol polymer promotes aptamer biodistribution to healthy and inflamed tissues. Oligonucleotides 15: 183–195. [DOI] [PubMed] [Google Scholar]
- 25. de Smidt PC, Le Doan T, de Falco S, van Berkel TJC (1991) Association of antisense oligonucleotides with lipoproteins prolongs the plasma half-life and modifies the tissue distribution. Nucleic Acids Res 19: 4695–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang J, Lee MS, Copland JA III, Luxon BA, Gorenstein DG (2008) Combinatorial selection of a single stranded DNA thioaptamer targeting TGF-β1 protein. Bioorg Med Chem Lett 18: 1835–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13: 9–22. [PubMed] [Google Scholar]
- 28. Itakura J, Ishiwata T, Shen B, Kornmann M, Korc M (2000) Concomitant over-expression of vascular endothelial growth factor and its receptors in pancreatic cancer. Intl J Cancer 85: 27–34. [DOI] [PubMed] [Google Scholar]
- 29. Price DJ, Miralem T, Jiang S, Steinberg R, Avraham H (2001) Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ 12: 129–135. [PubMed] [Google Scholar]
- 30. Masood R, Cai J, Zheng T, Lynne Smith D, Hinton DR, et al. (2001) Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood 98: 1904–1913. [DOI] [PubMed] [Google Scholar]
- 31. Strizzi L, Catalano A, Vianale G, Orecchia S, Casalini A, et al. (2001) Vascular endothelial growth factor is an autocrine growth factor in human malignant mesothelioma. J Pathol 193: 468–475. [DOI] [PubMed] [Google Scholar]
- 32. Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NPH, et al. (1993) High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 72: 835–846. [DOI] [PubMed] [Google Scholar]
- 33. Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH (1994) Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem 269: 26988–26995. [PubMed] [Google Scholar]
- 34. Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, et al. (1996) Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 87: 3336–3343. [PubMed] [Google Scholar]
- 35. Fan F, Wey JS, McCarty MF, Belcheva A, Liu W, et al. (2005) Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene 24: 2647–2653. [DOI] [PubMed] [Google Scholar]
- 36. Wey JS, Fan F, Gray MJ, Bauer TW, McCarty MF, et al. (2005) Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer 104: 427–438. [DOI] [PubMed] [Google Scholar]
- 37. Kaur H, Yung L (2012) Probing High Affinity Sequences of DNA aptamer against VEGF165. PLoS ONE 7: e31196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thiviyanathan V, Somasunderam AD, Gorenstein DG (2007) Combinatorial selection and delivery of thioaptamers. Biochem Soc T 35: 50–52. [DOI] [PubMed] [Google Scholar]
- 39. Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N (1992) Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem 267: 26031–26037. [PubMed] [Google Scholar]
- 40. Robinson CJ, Stringer SE (2001) The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci 114: 853–865. [DOI] [PubMed] [Google Scholar]
- 41. Keyt BA, Berleau LT, Nguyen HV, Chen H, Heinsohn H, et al. (1996) The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. J Biol Chem 271: 7788–7795. [DOI] [PubMed] [Google Scholar]
- 42. Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA (2009) An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci USA 106: 18751–18756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, et al. (1992) Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med 176: 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eaton BE, Gold L, Zichi DA (1995) Let’s get specific: The relationship between specificity and affinity. Chem Biol 2: 633–638. [DOI] [PubMed] [Google Scholar]
- 45. Kypr J, Kejnovská I, Renčiuk D, Vorlíčková M (2009) Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 37: 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suzuki H, Seto K, Shinoda Y, Mori M, Ishimura Y, et al. (1999) Paracrine upregulation of VEGF receptor mRNA in endothelial cells by hypoxia-exposed Hep G2 cells. Am J Physiol Gastrointest Liver Physiol 276: G92–G97. [DOI] [PubMed] [Google Scholar]
- 47. Oswald J, Treite F, Haase C, Kampfrath T, Mading P, et al. (2007) Experimental hypoxia is a potent stimulus for radiotracer uptake in vitro: comparison of different tumor cells and primary endothelial cells. Cancer lett 254: 102–110. [DOI] [PubMed] [Google Scholar]
- 48. Calvani M, Trisciuoglio D, Bergamaschi C, Shoemaker RH, Melillo G (2008) Differential involvement of vascular endothelial growth factor in the survival of hypoxic colon cancer cells. Cancer Res 68: 285–291. [DOI] [PubMed] [Google Scholar]
- 49. Radtke F, Raj K (2003) The role of Notch in tumorigenesis: Oncogene or tumour suppressor. Nat Rev Cancer 3: 756–767. [DOI] [PubMed] [Google Scholar]
- 50. Wang Z, Li Y, Banerjee S, Sarkar FH (2008) Exploitation of the notch signaling pathway as a novel target for cancer therapy. Anticancer Res 28: 3621–3630. [PubMed] [Google Scholar]
- 51. Jiang X, Zhou JH, Deng ZH, Qu XH, Jiang HY, et al. (2007) Expression and significance of Notch1, Jagged1 and VEGF in human non-small cell lung cancer. J Cent South Univ 32: 1031–1036. [PubMed] [Google Scholar]
- 52. Thurston G, Kitajewski J (2008) VEGF and Delta-Notch: Interacting signalling pathways in tumour angiogenesis. Brit J Cancer 99: 1204–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, et al. (2004) JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res 64: 6854–6857. [DOI] [PubMed] [Google Scholar]
- 54. Gao J, Chen C, Hong L, Wang J, Du Y, et al. (2007) Expression of Jagged1 and its association with hepatitis B virus X protein in hepatocellular carcinoma. Biochem Biophys Res Co 356: 341–347. [DOI] [PubMed] [Google Scholar]
- 55. Wang Z, Li Y, Banerjee S, Kong D, Ahmad A, et al. (2010) Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-κB signaling pathways. J Cell Biochem 109: 726–736. [DOI] [PubMed] [Google Scholar]