

Abstract
Elevated concentrations of plasminogen activator inhibitor–1 (PAI-1) are associated with pleural injury, but its effects on pleural organization remain unclear. A method of adenovirus-mediated delivery of genes of interest (expressed under a cytomegalovirus promoter) to rabbit pleura was developed and used with lacZ and human (h) PAI-1. Histology, β-galactosidase staining, Western blotting, enzymatic and immunohistochemical analyses of pleural fluids (PFs), lavages, and pleural mesothelial cells were used to evaluate the efficiency and effects of transduction. Transduction was selective and limited to the pleural mesothelial monolayer. The intrapleural expression of both genes was transient, with their peak expression at 4 to 5 days. On Day 5, hPAI-1 (40–80 and 200–400 nM of active and total hPAI-1 in lavages, respectively) caused no overt pleural injury, effusions, or fibrosis. The adenovirus-mediated delivery of hPAI-1 with subsequent tetracycline-induced pleural injury resulted in a significant exacerbation of the pleural fibrosis observed on Day 5 (P = 0.029 and P = 0.021 versus vehicle and adenoviral control samples, respectively). Intrapleural fibrinolytic therapy (IPFT) with plasminogen activators was effective in both animals overexpressing hPAI-1 and control animals with tetracycline injury alone. An increase in intrapleural active PAI-1 (from 10–15 nM in control animals to 20–40 nM in hPAI-1–overexpressing animals) resulted in the increased formation of PAI-1/plasminogen activator complexes in vivo. The decrease in intrapleural plasminogen-activating activity observed at 10 to 40 minutes after IPFT correlates linearly with the initial concentration of active PAI-1. Therefore, active PAI-1 in PFs affects the outcome of IPFT, and may be both a biomarker of pleural injury and a molecular target for its treatment.
Keywords: pleural injury, plasminogen activator inhibitor–1, intrapleural fibrinolytic therapy
The incidence of complicated pleural infection and empyema, a serious infection of the pleural space often associated with pneumonia, is increasing in the United States (1) and other countries, in both adult and pediatric populations (2–5). The exact cause of this increase is unknown, although the increased prevalence of antibiotic-resistant bacteria, changes in empyema management, and changes in causative bacterial agents have been implicated (1, 3, 6). Pleural infections, empyema, or complicated parapneumonic effusions develop in approximately 80,000 patients in the United States and the United Kingdom annually (7). In the United Kingdom, a 20% mortality rate was reported for patients with empyema, and 20% of patients require surgical intervention after developing a pleural infection (7). When pleural effusions occur in association with high-grade inflammation, they can organize with the development of loculation, where an effusion becomes trapped behind partly fused visceral and parietal pleura, with pleural thickening (8–10). Persistent pleural loculation and fibrosis increase morbidity and mortality and can lead to irreversible scarring of the pleura, with permanent decrements in pulmonary function (10).
Intrapleural fibrinolytic therapy (IPFT) with urokinase and tissue-type plasminogen activators (uPA and tPA, respectively) has been used to treat pleural loculation since the 1940s (11), and remains an alternative to surgical drainage (12). The use of these agents is predicated on the intrapleural administration of quantities of fibrinolysin that overwhelm endogenous inhibitors. Although the efficacy of currently used fibrinolytics is a matter of ongoing debate (13, 14), the successful use of IPFT has been reported, particularly in pediatric settings (15–18).
Plasminogen activator inhibitor 1 (PAI-1) has been identified as the major endogenous intrapleural inhibitor of tPA and uPA (19). The concentration of PAI-1 is markedly increased in pleural loculation (20–22). Active PAI-1 spontaneously transitions to its inactive latent form (23), which is predominant under physiological conditions. Although high concentrations of PAI-1 antigen are likewise strongly correlated with the severity of human pleural injury (18, 20, 21, 24), the proportion of PAI-1 that remains active in pleural injury remains unclear, and its contribution to the severity of pleural injury has not, to our knowledge, been previously studied.
In this study, we used a rabbit model of tetracycline (TCN)–induced pleural injury, which offers several important advantages. This model recapitulates key aspects of the temporal progression of pleural injury in humans (20, 25, 26), and the intrapleural administration of fibrinolysins is well-tolerated. The pleural fluids (PFs) of these animals contain elevated concentrations of active PAI-1, which inhibits intrapleural plasminogen activation by uPA or tPA (27, 28). Fibrinolytic activity is therefore suppressed within the PFs of rabbits with TCN-induced pleural injury, which occurs with the concurrent expression of robust tissue factor–related procoagulant activity (28). These changes collectively promote intrapleural fibrin deposition, which rapidly organizes, with the infiltration of fibrinous intrapleural adhesion by collagen fibrils within 2 to 3 days of an intrapleural administration of TCN (25, 26). A recent report demonstrated the close similarity of rabbit fibrin structure to human fibrin, which provides another advantage for the use of rabbit models of fibrotic injury in preclinical testing (29).
In the present study, we induced the selective, transient overexpression of human PAI-1 (hPAI-1) and lacZ in the pleural mesothelium of rabbits, delivering the target gene using a recombinant adenovirus. By overexpressing hPAI-1, we sought to further our understanding of the role of active PAI-1 in pleural injury. We found that the overexpression of hPAI-1 in the rabbit pleural mesothelium did not induce acute pleural inflammation or the formation of effusions or adhesions for up to 5 days after transduction. However, the overexpression of hPAI-1 significantly exacerbated subsequent TCN-induced pleural injury in rabbits. Moreover, the level of active PAI-1 demonstrated a linear correlation with decreased intrapleural plasminogen-activating activity in hPAI-1–overexpressing animals with TCN-induced injury that were treated with single-chain (sc) uPA or sctPA.
Materials and Methods
Proteins and Reagents
Proteins and reagents were obtained as described in the online supplement.
Recombinant Adenoviruses
Three adenoviral constructs were used: (1) AdPAI-1, a construct with a gene coding for hPAI-1 under a cytomegalovirus (CMV) promoter; (2) AdlacZ, which expresses lacZ under a CMV promoter, producing β-galactosidase; and (3) AdEV, an empty vector. AdPAI-1 was cloned and expressed using the RAPAd CMV Adenoviral Expression System (CellBiolabs, Inc., San Diego, CA), according to the manufacturer’s protocol. AdlacZ and AdEV were purchased from CellBiolabs, Inc. Adenoviruses were amplified in HEK293AD cells (CellBiolabs, Inc.) and purified, and the functional titer was quantified using a ViraBind Adenovirus Purification Kit and QuickTiter Adenovirus Titer Immunoassay Kit, respectively (CellBiolabs, Inc.), according to the manufacturer’s protocol.
Animal Model
In total, 3 × 1010 plaque-forming units of the adenoviral vectors were administered intrapleurally, containing either a single vector (AdEV or AdlacZ), or a pair of vectors at a 3:1 ratio (AdEV/AdlacZ or AdPAI-1/AdlacZ). Details of the experimental protocol for the enhanced TCN injury model are provided in the online supplement. This work was performed under a protocol approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at Tyler.
Pleural Fluid
One-milliliter aliquots of PF were collected from the thoracic cavity over a course of time by placing a 1.25-inch, 18-gauge indwelling catheter into the rabbit’s chest at the time of intervention, or by using a 60-ml plastic syringe at the time of death, diluted 9:1 (volume/volume) with 3.8% sodium citrate, as described in the online supplement.
Primary Mesothelial Cells
Primary rabbit pleural mesothelial cells (RPMCs) were collected from uninjured rabbit pleural spaces postmortem, as previously described (28, 30).
Staining for β-Galactosidase Activity
This protocol was modified from that reported by Decologne and colleagues (9), as described in the online supplement.
Western Blotting
These immunoanalyses were performed as previously described (31).
PAI-1 Antigen and Activity Assay
Concentrations of active PAI-1 in the samples were determined by titrating the active inhibitor with solutions of uPA of a known concentration, as previously described (31). Total hPAI-1 antigen was assessed via an Imubind Tissue PAI-1 ELISA kit (American Diagnostica, Inc., Stamford, CT), using the manufacturer’s protocol.
Interactions between PAI-1 and Plasminogen Activators, Both Human and Rabbit
The association rate constants for reactions between human and rabbit PAI-1 (rPAI-1) and uPA were measured using stopped flow fluorimetry and were analyzed, and the stoichiometry of inhibition was calculated as described in the online supplement.
Fibrinolytic Activity
PF d-dimers were measured using an Imuclone d-Dimer ELISA kit (American Diagnostica, Inc.), following the manufacturer’s protocol. Enzymographic analyses were performed as previously described (32).
Plasminogen Activation Assay
These analyses were performed as previously described (33).
Immunohistochemistry
Tissue staining and assessments were performed as previously described (34).
Metrics of Pleural Injury
Gross loculation/adhesion scores were determined by direct assessments of intrapleural adhesions or webs and fibrin aggregates during necropsy, and were scored as reported previously (35–37), with minor modifications described in the online supplement.
Data Analysis and Statistics
Levels of statistical significance were determined using the Mann-Whitney rank sum test, as described previously (31, 37, 38).
Results
Transduction of Primary RPMCs with Recombinant Adenoviruses
Adenoviruses with a gene of interest expressed under a CMV promoter were used to transduce RPMCs, and included (1) AdPAI-1, used to deliver the gene encoding for hPAI-1; (2) AdlacZ, used to deliver the reporter gene lacZ, resulting in the overexpression of active β-galactosidase; and (3) AdEV, an empty vector. In preliminary experiments, we optimized the transduction strategy, after which RPMCs were transduced with a 3:1 mixture of AdEV/AdlacZ or AdPAI-1/AdlacZ. Negative control samples were treated with PBS (vehicle control). Cells from each group (PBS, AdEV/AdlacZ, and AdPAI-1/AdlacZ) were fixed and stained for β-galactosidase activity, as described in Materials and Methods. Cells successfully transduced with AdlacZ overexpressed β-galactosidase, turning a distinct blue color when treated with a staining solution containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) (data not shown). Because PAI-1 is a secreted protein, conditioned media as well as cellular lysates from each transduction experiment were collected to test for the presence of hPAI-1 by Western blotting (Figure E1 in the online supplement). Cells treated with AdEV/AdlacZ or AdPAI-1/AdlacZ expressed β-galactosidase (data not shown), but only cells transduced with AdPAI-1, as expected, expressed hPAI-1 (Figure E1). In both cell lysates and conditioned media from RPMCs treated with PBS or transduced with AdEV/AdlacZ or AdPAI-1/AdlacZ, the concentration of rPAI-1 antigen was below the limit of detection (data not shown).
Characterization of the Interactions of Human uPA and tPA with rPAI-1
Before our interventional experiments in rabbits with adenoviral transduction resulting in hPAI-1expression in the pleural space, the interactions between hPAI-1 and rabbit uPA and tPA were evaluated. The second-order rate constants for the inhibition of PAs with human and rabbit PAI-1 were determined using stopped flow fluorimetry. SDS-PAGE was used to compare the stoichiometry of inhibition (39) for the interactions between PAs and PAI-1 from both species, as previously described (23, 40). The rates of mechanism-based inhibition of both uPA (Figure E2) and tPA (data not shown) by PAI-1 were diffusion-limited (> 106 M−1s−1), and the values of the stoichiometry of inhibition were close to unity for any combination of rabbit and human proteins. Thus, both endogenous rPAI-1 and hPAI-1 effectively inhibited endogenous rabbit uPA and tPA, and both human fibrinolysins. Therefore, endogenous rabbit PAI-1 and overexpressed hPAI-1 can inhibit the human fibrinolysins used for IPFT in this study.
Adenoviral Transduction of the Rabbit Pleura Selectively Targeted the Mesothelium
To determine the efficacy of transduction in vivo, we transduced the rabbit pleural mesothelium. Rabbits received intrapleural PBS (negative control, n = 6), AdEV (viral control, n = 8), or AdlacZ (positive control, n = 18, and were killed between 2 and 10 days after intrapleural injection. The lungs and thoracic cavity were harvested and stained for β-galactosidase activity, to determine the tissue specificity of gene expression and level of expression after transduction with respect to time (Figure 1A). Although the rabbit mediastinum is occasionally communicating, gene transfer was typically limited to the right pleural space. The highest level of β-galactosidase expression was present 4–5 days after adenovirus injection, with a gradual decrement over 10 days (Figure 1A). These results recapitulated the time course observed for the adenoviral intrapleural delivery of transforming growth factor–β (TGF-β) in rats, with peak expression on Days 4–5 (9). As expected, the intrapleural administration of PBS or AdEV alone did not result in the expression of β-galactosidase (Figures 1B and 1C). Gene delivery was efficient, as indicated by the widespread overexpression of β-galactosidase on the visceral and parietal pleura, but was also selective, because it was limited to the pleural mesothelial monolayer (Figures 1D and 1E).
Figure 1.

Successful adenoviral transduction with adenoviral construct AdlacZ results in expression of active β-galactosidase in the rabbit pleural mesothelium, which is sustained for up to 10 days. The presence of active β-galactosidase is demonstrated by the distinct blue color of cells and tissues treated with a stain containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal). (A) Lungs harvested and stained 2, 4, 5, 8, and 10 days after intrapleural transduction with the lacZ gene, delivered using the AdlacZ adenovirus, demonstrated the blue color associated with the presence of active β-galactosidase. The peak of lacZ expression was observed on Days 4–5. (B and C) Lungs of control animals treated with PBS (B), empty vector (EV) (C), or not treated (NL; in the left lungs) (B and C), lacked a blue color after staining. (D and E) Transduction, and therefore lacZ expression, was selective to the mesothelial monolayer of the visceral (D) and parietal (E) pleura, as confirmed by staining for β-galactosidase activity.
Mesothelial Overexpression of hPAI-1 Did Not Induce Pleural Effusions, Organization, or Fibrosis
The transduction of the mesothelium with mixtures of AdPAI-1 and AdlacZ (n = 4) or AdEV and AdlacZ (n = 2) did not induce overt pleural injury, inflammation, effusions, adhesion formation, or fibrosis for up to 5 days after the intrapleural administration of the constructs. (Figure E3). β-galactosidase activity was present on both the visceral (Figure E4) and parietal (data not shown) pleural surfaces of rabbits receiving AdPAI-1/AdlacZ or AdEV/AdlacZ, providing proof of successful adenoviral delivery. To determine if hPAI-1 had been secreted into the pleural space by transduced RPMCs, the transduced pleural space was lavaged with Hanks’ balanced salt solution (HBSS; 10 ml) at the time of death, followed by tryptic lavage to collect RPMCs. Western blot analysis was used to visualize hPAI-1 expression in the pleural space and in collected cells (Figure 2A). hPAI-1 antigen was detected in HBSS lavages of AdPAI-1/AdlacZ transduced rabbits (Figure 2A, top, lavage, hPAI-1). As expected, no hPAI-1 antigen was detected in rabbits receiving PBS or AdEV/AdlacZ (Figure 2A). hPAI-1 was present in cell lysates of RPMCs harvested from rabbits receiving intrapleural AdPAI-1/AdlacZ (Figure 2A, middle, lysates, hPAI-1). No hPAI-1 was detected in RPMCs from rabbits injected with PBS or AdEV/AdlacZ (Figure 2A, middle, lysates, hPAI-1). The concentrations of rPAI-1 antigen were below the limit of detection of our assays in all three transduced groups.
Figure 2.

Human plasminogen activator inhibitor–1 (hPAI-1) in pleural lavages and rabbit pleural mesothelial cells (RPMCs) harvested from rabbits intrapleurally injected with AdPAI-1/AdlacZ. (A) hPAI-1 was detected in pleural lavages (top; Lavage, hPAI-1) and lysates (middle; lysates and hPAI-1) of RPMCs from rabbits injected with a 3:1 mixture of AdPAI-1/AdlacZ, but was not present in lavages or RPMCs of control (PBS or AdEV/AdlacZ) animals. β-actin was used as a loading control for lysates (bottom). (B) A significant fraction (20–25%) of hPAI-1 in pleural lavages was active. Pleural lavages from two animals (Lanes 1 and 2) were supplemented with exogenous human tc urokinase-type plasminogen activator (tcuPA) and incubated for 5 minutes at 4°C, and the reaction mixtures were subjected to SDS-PAGE followed by Western blot analysis of the hPAI-1 antigen. The upper band corresponds to the inhibitory PAI-1/uPA complex, which forms because of the mechanism-based inhibition of uPA by active PAI-1. The lower band corresponds to uncomplexed, latent PAI-1. (C) High levels of PAI-1 activity were detected in the pleural lavages of animals transduced with AdPAI-1/AdlacZ. Aliquots of pleural lavages were titrated with human tcuPA, as described in Materials and Methods. The concentration of active PAI-1 in each sample was calculated, assuming a stoichiometry of inhibition close to unity. A statistically significant difference (P > 0.05) between AdPAI-1/AdlacZ and the two control groups (P = 0.029 for both) is indicated by an asterisk.
The activity of hPAI-1 in rabbit pleural lavages was visualized by Western blotting (Figure 2B). An excess of exogenous human two-chain uPA (tcuPA) (100 nM) was added to the lavage, and the reaction mixture was subjected to SDS-PAGE, followed by Western blotting developed with anti-human PAI-1 monoclonal antibodies (Figure 2B). The inhibitory complex, which comprises acylated uPA (PAI-1/uPA), reflects the fraction of the active hPAI-1 in the lavage. The lower band (PAI-1), which represents inactive latent hPAI-1, was used to estimate the ratio of active/latent hPAI-1 in the lavages as 1:4–5 (Figure 2B). The concentration of active PAI-1 in the lavages was estimated by titrating with a known amount of human tcuPA. PAI-1 activity in the lavages of animals transduced with AdPAI-1 (40–80 nM) was significantly higher than that of AdEV/AdlacZ-treated animals or PBS control animals (around and less than 1 nM, respectively; P = 0.029 for both; Figure 2C). Based on the measurements of active hPAI-1, the total hPAI-1 in the lavages of AdPAI-1/AdlacZ-transduced rabbits was estimated as 200 to 400 nM. Thus, in contrast to the results of transduction with TGF-β, which caused pleural fibrosis in rats on Days 4 to 5 (9), the high concentrations of PAI-1 found at the peak of transgene expression in the rabbit pleural space did not induce inflammation, effusions, or fibrosis. Next, the effects of the intrapleural overexpression of hPAI-1 on TCN-induced pleural injury were studied.
The Development of TCN-Induced Pleural Injury on Pleural Transduction
The efficiency of the adenoviral delivery system in an injured pleural space was tested using AdlacZ. Rabbits received PBS or AdEV/AdlacZ (3:1) intrapleurally, with a TCN injection 2 days afterward (n = 5 and 8, respectively). The injection of TCN into the pleural space induced pleural effusions and the subsequent formation of adhesions during the next 3 days, as previously described (25, 26). In rabbits injected with AdEV/AdlacZ, β-galactosidase expression was visually detectable after staining, indicating successful transduction with lacZ (Figure E5A). β-galactosidase expression was limited to the pleural mesothelium. In contrast to animals without TCN-induced injury (Figure 1A), β-galactosidase expression in the visceral pleura, as determined by X-gal staining, decreased rapidly with time (Figure E5A). Pleural fibrosis progressed from discrete and easily quantifiable strands, present for up to 2 days, into sheets and webs complex and too numerous to count at 3 days and beyond. TCN-induced injury resulted in gaps in the mesothelial lining, suggesting that RPMCs had detached from the pleural surface (Figure E5B). Indeed, unlike cases of uninjured transduced mesothelium (Figures 1D and 1E), RPMCs found within fibrin strands after TCN injury stained positive for β-galactosidase activity (Figure E5B), providing novel evidence supporting the concept that mesothelial cells integrate within the forming intrapleural adhesions. These results demonstrate that the adenovirus-mediated transduction of the pleura leads to the overexpression of the proteins of interest, and this overexpression is successfully maintained in TCN-induced pleural injury.
Overexpression of hPAI-1 Exacerbates TCN-Induced Injury in Rabbits
To determine whether hPAI-1 overexpression affects the progression and severity of pleural injury, rabbits (n = 7) were first injected with AdPAI-1/AdlacZ (3:1) and then with TCN 3 days later, as described in Materials and Methods. This experimental design allowed us to assess the effects of PAI-1 at the peak of transgene expression (5 days), at a time point when we could quantify TCN injury (2 days) in control animals injected with AdEV/AdlacZ (3:1) or with PBS before TCN (Figures 3A and 3B). The severity of pleural injury in animals that successfully overexpressed hPAI-1 (Figure 3C) was clearly apparent versus control animals (Figures 3A and 3B). The extent of pleural injury was assessed by comparing gross loculation/adhesion injury scores, using a scoring system we previously described (38, 41), with minor modifications detailed in the online supplement. The gross loculation scores of pleural injury in rabbits injected with AdPAI-1/AdlacZ and overexpressing hPAI-1 were increased compared with animals intrapleurally injected with PBS (P = 0.029) or AdEV/AdlacZ (P = 0.021; Figure 3D). To verify intrapleural hPAI-1 expression, PFs (typically between 20 and 50 ml total volume) were analyzed for both rPAI-1(Figure 3E, top) and hPAI-1(Figure 3E, bottom) by Western blotting. As expected, the concentration of rPAI-1 antigen was similar for all three groups, but only the PF of AdPAI-1/AdlacZ rabbits contained significant amounts of hPAI-1 antigen. However, low-level cross-reactivity was observed between rPAI-1 antigen and the monoclonal antibody we used to detect hPAI-1, evident only when rPAI-1 was strongly overexpressed. Therefore, although the overexpression of hPAI-1 by itself caused no overt pleural inflammation, effusions, or fibrosis, it exacerbated TCN-induced pleural injury.
Figure 3.

Overexpression of hPAI-1 markedly increases the severity of tetracycline (TCN)–induced pleural fibrosis. (A–C) The typical level of injury 2 days after intrapleural injection of TCN and 5 days after intrapleural injection of PBS (A), AdEV/AdlacZ (B), or AdPAI-1/AdlacZ (C). (D) The severity of pleural injury in animals injected with PBS (n = 5), empty vector (n = 8), or AdhPAI-1 (n = 7) was evaluated as described briefly in Materials and Methods and detailed in the online supplement, and was expressed as a gross loculation injury for each animal. Data are shown as a box plot in which the 25% and 75% quartiles are indicated by the box and median values are shown as horizontal lines within the box, as previously described (31, 37). Individual scores for each animal are also shown as open circles, which overlap if more than one animal received an identical score. The fibrotic injury observed was uniformly worse (i.e., the gross loculation injury score was higher) in rabbits that received AdPAI-1/AdlacZ compared with PBS (P = 0.029) and AdEV/AdlacZ (P = 0.021). (C) This more complex pleural injury was characterized by the formation of fibrin webs and coalescent fibrinous sheets. (E) Western blot analysis of rabbit plasminogen activator inhibitor–1 (rPAI-1) (top) and hPAI-1 (bottom) antigens in pleural fluids (PFs) of animals with TCN-induced pleural injury. Whereas the concentration of rPAI-1 in the PF of all animals with TCN-induced injury was similar (top), the concentration of hPAI-1 (bottom) was markedly higher in the PF of rabbits transduced with AdPAI-1/lacZ. Monoclonal anti-human PAI-1 antibodies exhibit low-level cross-reactivity with rPAI-1 at higher concentrations.
The Effects of Mesothelial hPAI-1 Expression on Intrapleural Fibrinolytic Therapy with scuPA or sctPA
TCN-induced pleural fibrosis in rabbits was successfully treated with intrapleural fibrinolytic therapy, using effective doses of sctPA (0.145 mg/kg) or scuPA (0.5 mg/kg) (35, 37, 38). To test the effects of overexpressed hPAI-1 on IPFT, animals injected with AdPAI-1/AdlacZ and then with TCN 3 days after transduction (n = 6) were subjected to IPFT (on Day 5 after transduction) with effective doses of scuPA and sctPA, or with PBS (vehicle control) (n = 2 each). Five animals were used as controls for IPFT without adenoviral transduction, and were treated with the same doses of scuPA (n = 2) and sctPA (n = 3). PFs were collected before and after IPFT, using a 1.25-inch, 18-gauge indwelling catheter placed into the rabbit’s chest at the time of intervention. Aliquots of PF were withdrawn from animals immediately before the injection of fibrinolysin (time point 0), and then 10, 20, and 40 minutes after IPFT, as described in Materials and Methods, for further analysis. The overall results of IPFT were assessed by visual examination of the rabbit pleural spaces after killing animals 24 hours after IPFT (Figure 4). Whereas advanced pleural fibrosis was found in control animals receiving PBS in place of a fibrinolysin (gross injury score, 50; Figure 4A), the pleural spaces of animals overexpressing hPAI-1 (Figure 4B, C) and TCN-only control animals (data not shown) had fewer pleural adhesions (gross injury score, 0–5). Therefore, despite the overexpression of hPAI-1, both fibrinolysins were effective in the treatment of TCN-induced injury. IPFT with an effective dose of either fibrinolysin cleared most of the adhesions in all nine animals (gross loculation injury scores were in the single-digit range), indicating that the selected doses overcame the concentrations of active PAI-1 present in the pleural space.
Figure 4.
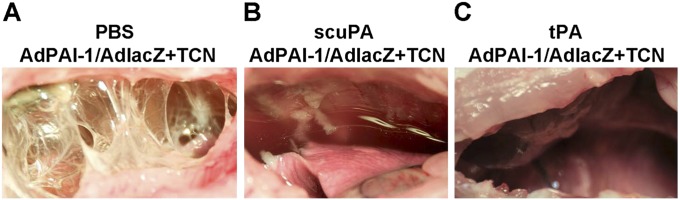
Intrapleural fibrinolytic therapy of pleural fibrosis in rabbits with overexpression of hPAI-1 and TCN-induced injury. Animals transduced with AdPAI-1/AdlacZ overexpressed hPAI-1 in the pleural space, and developed pleural effusions and adhesions 2 days after TCN was administered intrapleurally (see Figure 3C). When these animals were treated with single-chain uPA (scuPA) (0.5 mg per kg) (B) or tissue-type plasminogen activator (tPA) (0.145 mg per kg) (C), most of the pleural adhesions were effectively cleared 24 hours later. In contrast, control animals treated with PBS (A) demonstrated a severe, complex organization and adhesion formation.
Aliquots of PF were analyzed by SDS-PAGE, followed by enzymography (Figures 5A, 5B, 5D, and 5E) and Western blotting (Figures 5C and 5F), to visualize the PAI-1 antigen. Free uPA and tPA, as well as their inhibitory complexes with PAI-1, were detected in PF samples via enzymography (Figures 5A and 5B for uPA; Figures 5D and 5E for tPA). Whereas both free tPA and uPA were present in the pleural space, the concentration of PAI-1/enzyme complexes in PF (Figure 5, bands I and III) increased with time from 10 to 40 minutes (Figures 5A, 5B, 5D, and 5E). However, the difference in active PAI-1 concentrations (formation of PAI-1/uPA complexes, Figures 5A and 5B; formation of PAI-1/tPA complexes, Figures 5D and 5E) between hPAI-1 expressing (Figures 5B and 5E) and nonexpressing (Figures 5A and 5D) animals was visible, especially at 10 and 20 minutes after IPFT. Western blot analysis of hPAI-1 antigen (Figures 5C and 5F) showed high concentrations of latent (indicated as V) and cleaved (indicated as VI) forms of hPAI-1 accumulating in the PF of animals receiving AdPAI-1/AdlacZ. Notably, the cleaved PAI-1 was only detected after IPFT. That could indicate either a degradation of the PAI-1/enzyme complex, or else endogenous redirection of the reaction to the substrate branch, similar to that described for anti-PAI-1 monoclonal antibodies (23).
Figure 5.

Analysis of pleural fluids after fibrinolytic therapy. Enzymographic (A, B, D, and E) and Western blot (C and F) analyses of PF from animals with TCN-induced injury (A and D) and TCN-induced injury enhanced with the mesothelial expression of hPAI-1 (B, E, C, and F). PFs were sampled before (time point 0) as well as 10, 20, and 40 minutes after intrapleural treatment with an effective therapeutic dose of scuPA (A–C) or tPA (D–F). Arrows to the right indicate positions of uPA/PAI-1 inhibitory complexes (∼ 100 kD) (I), uPA (∼ 50 kD) (II), tPA/PAI-1 inhibitory complexes (∼ 110 kD) (III), tPA (∼ 63 kD) (IV), active and latent hPAI-1 (∼ 50 kD) (V), and cleaved hPAI-1 (VI). Endogenous plasminogen activators complexed with PAI-1 were detected before treatment, at 0 minutes (A and B, and D and E, respectively). However, these time point 0 enzymographic analyses required an incubation period of 2–3 hours or 2–4 hours (2- to -4-fold or 1.7- to 5-fold, respectively) longer for the bands representing these complexes to become fully visible. (G) Active rPAI-1 in animals with TCN injury (n = 5) and active and latent hPAI-1 in animals transduced with AdPAI-1/AdlacZ followed by TCN injury (n = 4). Active PAI-1 (rabbit or total rabbit plus human, respectively) in PF, withdrawn before intrapleural fibrinolytic therapy (IPFT), was measured by titration with known amounts of tcuPA, as briefly described in Materials and Methods, and as detailed in the online supplement. Concentrations of active rPAI-1 were similar between transduced and nontransduced animals (Figure 3E, top) and among animals with TCN-induced injury (G, rPAI-1 active). Thus, the concentrations of active hPAI-1 (G, hPAI-1 active) were calculated as [hPAI-1 active] = [total PAI-1 active] − [average rPAI-1 active]. The concentrations of latent hPAI-1 (G, hPAI-1 latent) were calculated as [total hPAI-1] (measured by ELISA) minus [hPAI-1 active]. Data are presented as a box plot in which the 25% and 75% quartiles are indicated in the box, and median values are shown as horizontal lines within the box, as previously described (31, 37).
The concentrations of active rPAI-1 and hPAI-1 in PF samples withdrawn before IPFT from nine animals were estimated as described previously (31). The adenoviral overexpression of hPAI-1 resulted in a 1.5-fold increase of PAI-1 activity in the PF of rabbits with TCN-induced injury. The concentrations of hPAI-1 antigen in the samples were also determined using ELISA, and the concentration of latent hPAI-1 in PF was calculated assuming that most of the intrapleural hPAI-1 at time point 0 was either in an active or latent form (Figures 5C and 5F). The observed distribution of hPAI-1 between active and latent forms in TCN injury (Figure 5G) was similar to that in animals subjected to adenoviral transduction without TCN injury (Figure 2B). Based on the similarity of the biochemical and biophysical properties of PAI-1 from humans and rabbits (Figure E2), we infer that the concentration of latent rPAI-1 in TCN-induced injury falls within the range of 25–50 nM.
The increase in PAI-1/PA complexes with time in both TCN-only animals (Figures 5A and 5D) and hPAI-1–overexpressing animals (Figures 5B and 5E) reflects the intrapleural neutralization of tPA and uPA, which could potentially affect intrapleural PA activity. To test the effects of intrapleural active PAI-1 on the PA activity of fibrinolysins during IPFT, the aliquots withdrawn at 10, 20, and 40 minutes were tested for their ability to activate human Glu-plasminogen, as previously described (33). The PA activity in PF at 10, 20, and 40 minutes was plotted versus the initial intrapleural concentration of active PAI-1 (Figure 6). Whereas PA activity decreased with time from 10 minutes (Figure 6, circles) to 40 minutes (Figure 6, triangles) in all samples (Figure 6), an increase in intrapleural PAI-1 activity was correlated with a decrease in intrapleural PA activity during IPFT at every time point for both fibrinolysins (Figure 6, solid lines). Therefore, intrapleural PA activity in hPAI-1–overexpressing animals was lower than that in the control group, and an increase in the intrapleural concentration of active PAI-1 resulted in a decrease of PA activity. These results support the hypothesis that active PAI-1 is not only a marker of pleural injury, but also a target for IPFT and a major determinant of the activity of intrapleurally administered fibrinolysins.
Figure 6.

Intrapleural plasminogen-activating (PA) activity during fibrinolytic therapy inversely correlates with the concentration of intrapleural active PAI-1. PFs of rabbits with TCN-induced pleural injury (n = 5) and TCN-induced injury enhanced by overexpressing hPAI-1 intrapleurally (n = 4) were treated with an effective dose of scuPA (open symbols) or sctPA (solid symbols) (0.5 and 0.145 mg/kg, respectively) (37). PAI-1 activity was measured in PF collected before IPFT, as described previously (33). PA activity was measured in PF collected 10 minutes (circles), 20 minutes (squares), and 40 minutes (triangles) after IPFT, as described elsewhere (33). The average of four measurements was plotted against the concentration of active PAI-1. Solid lines represent the best fit of a linear equation to the data for 10 minutes (circles; r2 = 0.77), 20 minutes (squares; r2 = 0.71), and 40 minutes (triangles; r2 = 0.71) after IPFT.
The concentration of d-dimers in PF samples withdrawn from animals treated with IPFT and controls was measured as described previously (37), to estimate levels of fibrinolytic activity (Figure E6). During IPFT, the concentration of d-dimers increased 2- to 3-fold (Figure E6, circles), and the levels of d-dimers in control (no IPFT) animal PF was unchanged (Figure E6, triangles). The observed similarity in d-dimer concentrations (fibrinolytic activity) between animals overexpressing hPAI-1 (Figure 6E, solid circles) and TCN-only control animals (Figure 6E, open circles) is in accordance with the similar efficacy of IPFT in both groups of animals (37).
Discussion
To the best of our knowledge, this is the first report of adenovirus-mediated gene delivery into the rabbit pleural space, and of the contribution of hPAI-1 overexpression to the development of pleural effusions and fibrosis. By delivering lacZ, we verified that the transduction was selective to the mesothelial monolayer of the rabbit visceral and parietal pleura, where β-galactosidase activity was exclusively localized. Target gene expression was transient, with lacZ expression peaking at 4 to 5 days after AdlacZ was administered intrapleurally, but was sustained for up to 10 days, as determined by staining for β-galactosidase activity. The injection of neither AdEV nor AdlacZ caused overt pleural injury, effusions, adhesions, or pleural fibrosis for up to 10 days.
By delivering AdPAI-1 intrapleurally, we found that the broadly distributed but selective transduction of the visceral and parietal mesothelial lining was achieved, similar to the transduction found with lacZ. The robust mesothelial overexpression of hPAI-1 was detected for up to 5 days, as documented by significant increments of hPAI-1 activity as well as antigen in pleural lavages. Moreover, significant amounts of hPAI-1 antigen were found in lysates of RPMCs harvested from treated animals. Despite the high levels of expression, hPAI-1 alone was insufficient to induce overt pleural injury in rabbits. Because we have shown that hPAI-1 efficiently inhibits both rabbit uPA and tPA, one could conclude that the development of acute pleural injury does not depend solely on the presence of high PAI-1 inhibitory activity. Recently, Decologne and colleagues reported that the adenovirus-mediated transduction of the rat pleura with TGF-β, and subsequent overexpression of it with a peak on Day 4, induced pleural thickening and mesomesenchymal transition (9). In a previous report, we showed that human PMCs and RPMCs are a source of PAI-1 in an injured pleural space, and that the mediators present within PFs, including TGF-β and TNF-α, can induce PAI-1 expression in these cells (42, 43). Our study demonstrates that PAI-1 overexpression, although known to be induced by TGF-β, was not by itself a cause of overt pleural injury, effusions, or fibrosis in the rabbit pleural space.
An increase of active rPAI-1 in the PF of animals with TCN-induced pleural injury, together with the suppression of fibrinolytic activity, indicates that endogenous PAI-1 could contribute to pleural fibrosis. To investigate the role of PAI-1 activity in the development of pleural effusions and fibrosis, an enhanced rabbit model, with the adenovirus-mediated delivery of a gene of interest to the pleural space followed by TCN-induced injury, was developed. First we showed that the adenovirus-mediated delivery of a control gene, lacZ, resulted in the robust transduction of the pleural mesothelium before and after TCN-induced injury, and exerted no significant effect on the development of pleural fibrosis. Evidence of successful gene retention was observed by detecting β-galactosidase activity within the mesothelial cells lining the surface of the TCN-injured lungs and chest wall. In contrast to uninjured pleural spaces, a number of transduced, β-galactosidase–expressing, mesothelial cells were found intercalated within fibrinous adhesions. This novel observation suggests that dehisced RPMCs could potentially promote adhesion formation by expressing tissue factor and PAI-1 or collagen (44, 45). The intrapleural overexpression of hPAI-1 intensified the severity of TCN-induced pleural injury, primarily associated with increased adhesion formation and density. The increment and complexity of adhesions were visually obvious in hPAI-1-transduced animals with superimposed TCN-induced pleural injury. However, pleural inflammation was not increased, nor was a clear increment in pleural thickening evident (Figure E7). The increments of hPAI-1 activity in rabbit PF from animals transduced with AdPAI-1 suggest that the enhanced inhibition of endogenous intrapleural plasminogen activators contributed to the worsening of pleural injury, and could potentially affect the outcome of IPFT. Although a considerable proportion of the intrapleural PAI-1 spontaneously reverted to an inactive, latent form, our data show that a significant increment of active PAI-1 was bioavailable in the rapidly harvested PFs.
Lastly, we compared the results of IPFT with single intrapleural doses of two fibrinolysins (scuPA and sctPA) (37) in rabbits with TCN-induced pleural injury previously transduced or not transduced with hPAI-1. At the doses used, both scuPA and sctPA cleared most of the pleural adhesions, consistent with our previous report of TCN-induced pleural injury without hPAI-1 transduction (37). However, the ability of either fibrinolysin to generate PA activity within PFs was strongly correlated with the level of intrapleural PAI-1 activity. Notably, such a direct correlation predicts an intrapleural concentration of active PAI-1 (> 50–70 nM) that would render the most common clinical dose of fibrinolysins ineffective. These results are of keen interest because the robust expression of PAI-1 antigen approximated at high concentrations (nM or μM) has been reported in human empyema (18, 20, 21). Thus, the overexpression of active PAI-1 in human pleural injury could limit the efficacy of IPFT, which may have contributed to the negative results reported for intrapleural sctPA used alone in the recent Second Multicentre Intrapleural Sepsis clinical trial (14).
In conclusion, we offer a new model of pleural transduction that is selective for the rabbit mesothelium, and we show that the mesothelial expression of hPAI-1 does not induce overt pleural inflammation, effusion formation, or fibrosis in rabbits. We show for the first time, to the best of our knowledge, that the expression of hPAI-1 in the rabbit mesothelium induces more severe intrapleural organization during TCN-induced pleural injury, which is primarily associated with gross evidence of increased adhesion formation. Lastly, we provide new information that in the setting of TCN-induced injury, high concentrations of active hPAI-1 can limit the PA activity of exogenous fibrinolysins. Based on our results, we speculate that the assessment of active PAI-1 in the pleural space in humans with empyema could be used to optimize the dosing of agents used for intrapleural fibrinolytic therapy.
Supplementary Material
Acknowledgments
The authors offer their profound thanks to the staff at the University of Texas Health Science Center at the Tyler Vivarium for their exemplary animal care and assistance with animal protocols.
Footnotes
This work was funded by National Institutes of Health grants PPG PO1 HL076406, and P50 HL107186 (S.I.), and by the Texas Lung Injury Institute.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0183OC on September 20, 2012
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Grijalva CG, Zhu Y, Nuorti JP, Griffin MR. Emergence of parapneumonic empyema in the USA. Thorax 2011;66:663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strachan RE, Cornelius A, Gilbert GL, Gulliver T, Martin A, McDonald T, Nixon GM, Roseby R, Ranganathan S, Selvadurai H, et al. Bacterial causes of empyema in children, Australia, 2007–2009. Emerg Infect Dis 2011;17:1839–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finley C, Clifton J, Fitzgerald JM, Yee J. Empyema: an increasing concern in Canada. Can Respir J 2008;15:85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obando I, Munoz-Almagro C, Arroyo LA, Tarrago D, Sanchez-Tatay D, Moreno-Perez D, Dhillon SS, Esteva C, Hernandez-Bou S, Garcia-Garcia JJ, et al. Pediatric parapneumonic empyema, Spain. Emerg Infect Dis 2008;14:1390–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roxburgh CS, Youngson GG, Townend JA, Turner SW. Trends in pneumonia and empyema in Scottish children in the past 25 years. Arch Dis Child 2008;93:316–318 [DOI] [PubMed] [Google Scholar]
- 6.Anthonisen NR. Empyema. Can Respir J 2008;15:69–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies HE, Davies RJ, Davies CW. Management of pleural infection in adults: British Thoracic Society pleural disease guideline 2010. Thorax 2010;65:ii41–ii53 [DOI] [PubMed] [Google Scholar]
- 8.Huggins JT, Sahn SA. Drug-induced pleural disease. Clin Chest Med 2004;25:141–153 [DOI] [PubMed] [Google Scholar]
- 9.Decologne N, Kolb M, Margetts PJ, Menetrier F, Artur Y, Garrido C, Gauldie J, Camus P, Bonniaud P. TGF-beta1 induces progressive pleural scarring and subpleural fibrosis. J Immunol 2007;179:6043–6051 [DOI] [PubMed] [Google Scholar]
- 10.Mutsaers SE, Prele CM, Brody AR, Idell S. Pathogenesis of pleural fibrosis. Respirology 2004;9:428–440 [DOI] [PubMed] [Google Scholar]
- 11.Tillett WS, Sherry S. doi: 10.1172/JCI102046. The effect in patients of streptococcal fibrinolysin (streptokinase) and streptococcal desoxyribonuclease on fibrinous, purulent and sanguinous pleural exudations J Clin Invest 1949;28:173–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Idell S. Update on the use of fibrinolysins in pleural disease, 12th edition. Philadelphia, PA: Lippincott, Williams and Wilkins; 2005. pp. 184–190. [Google Scholar]
- 13.Maskell NA, Davies CW, Nunn AJ, Hedley EL, Gleeson FV, Miller R, Gabe R, Rees GL, Peto TE, Woodhead MA, et al. UK controlled trial of intrapleural streptokinase for pleural infection. N Engl J Med 2005;352:865–874 [DOI] [PubMed] [Google Scholar]
- 14.Rahman NM, Maskell NA, West A, Teoh R, Arnold A, Mackinlay C, Peckham D, Davies CW, Ali N, Kinnear W, et al. Intrapleural use of tissue plasminogen activator and DNase in pleural infection. N Engl J Med 2011;365:518–526 [DOI] [PubMed] [Google Scholar]
- 15.Sonnappa S, Cohen G, Owens CM, van Doorn C, Cairns J, Stanojevic S, Elliott MJ, Jaffe A. Comparison of urokinase and video-assisted thoracoscopic surgery for treatment of childhood empyema. Am J Respir Crit Care Med 2006;174:221–227 [DOI] [PubMed] [Google Scholar]
- 16.Sonnappa S, Jaffe A. Treatment approaches for empyema in children. Paediatr Respir Rev 2007;8:164–170 [DOI] [PubMed] [Google Scholar]
- 17.Stefanutti G, Ghirardo V, Barbato A, Gamba P. Evaluation of a pediatric protocol of intrapleural urokinase for pleural empyema: a prospective study. Surgery 2010;148:589–594 [DOI] [PubMed] [Google Scholar]
- 18.Chung CL, Chen YC, Chang SC. Effect of repeated thoracenteses on fluid characteristics, cytokines, and fibrinolytic activity in malignant pleural effusion. Chest 2003;123:1188–1195 [DOI] [PubMed] [Google Scholar]
- 19.Dawson S, Henney A. The status of PAI-1 as a risk factor for arterial and thrombotic disease: a review. Atherosclerosis 1992;95:105–117 [DOI] [PubMed] [Google Scholar]
- 20.Idell S, Girard W, Koenig KB, McLarty J, Fair DS. Abnormalities of pathways of fibrin turnover in the human pleural space. Am Rev Respir Dis 1991;144:187–194 [DOI] [PubMed] [Google Scholar]
- 21.Philip-Joet F, Alessi MC, Philip-Joet C, Aillaud M, Barriere JR, Arnaud A, Juhan-Vague I. Fibrinolytic and inflammatory processes in pleural effusions. Eur Respir J 1995;8:1352–1356 [DOI] [PubMed] [Google Scholar]
- 22.Iglesias D, Alegre J, Aleman C, Ruiz E, Soriano T, Armadans LI, Segura RM, Angles A, Monasterio J, de Sevilla TF. Metalloproteinases and tissue inhibitors of metalloproteinases in exudative pleural effusions. Eur Respir J 2005;25:104–109 [DOI] [PubMed] [Google Scholar]
- 23.Komissarov AA, Zhou A, Declerck PJ. Modulation of serpin reaction through stabilization of transient intermediate by ligands bound to alpha-helix F. J Biol Chem 2007;282:26306–26315 [DOI] [PubMed] [Google Scholar]
- 24.Idell S, Zwieb C, Kumar A, Koenig KB, Johnson AR. Pathways of fibrin turnover of human pleural mesothelial cells in vitro. Am J Respir Cell Mol Biol 1992;7:414–426 [DOI] [PubMed] [Google Scholar]
- 25.Miller EJ, Kajikawa O, Pueblitz S, Light RW, Koenig KK, Idell S. Chemokine involvement in tetracycline-induced pleuritis. Eur Respir J 1999;14:1387–1393 [DOI] [PubMed] [Google Scholar]
- 26.Idell S, Pendurthi U, Pueblitz S, Koenig K, Williams T, Rao LV. Tissue factor pathway inhibitor in tetracycline-induced pleuritis in rabbits. Thromb Haemost 1998;79:649–655 [PubMed] [Google Scholar]
- 27.Strange C, Baumann MH, Sahn SA, Idell S. Effects of intrapleural heparin or urokinase on the extent of tetracycline-induced pleural disease. Am J Respir Crit Care Med 1995;151:508–515 [DOI] [PubMed] [Google Scholar]
- 28.Idell S, Mazar A, Cines D, Kuo A, Parry G, Gawlak S, Juarez J, Koenig K, Azghani A, Hadden W, et al. Single-chain urokinase alone or complexed to its receptor in tetracycline-induced pleuritis in rabbits. Am J Respir Crit Care Med 2002;166:920–926 [DOI] [PubMed] [Google Scholar]
- 29.Pretorius E, Humphries P, Ekpo OE, Smit E, van der Merwe CF. Comparative ultrastructural analyses of mouse, rabbit, and human platelets and fibrin networks. microsc res tech 2007;70:823–827 [DOI] [PubMed] [Google Scholar]
- 30.acencio mm, vargas fs, marchi e, carnevale gg, Teixeira LR, Antonangelo L, Broaddus VC. Pleural mesothelial cells mediate inflammatory and profibrotic responses in talc-induced pleurodesis. Lung 2007;185:343–348 [DOI] [PubMed] [Google Scholar]
- 31.Komissarov AA, Mazar AP, Koenig K, Kurdowska AK, Idell S. Regulation of intrapleural fibrinolysis by urokinase-alpha–macroglobulin complexes in tetracycline-induced pleural injury in rabbits. Am J Physiol Lung Cell Mol Physiol 2009;297:L568–L577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, McLarty J, Fair DS. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest 1989;84:695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komissarov AA, Florova G, Idell S. Effects of extracellular DNA on plasminogen activation and fibrinolysis. J Biol Chem 2011;286:41949–41962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Midde KK, Batchinsky AI, Cancio LC, Shetty S, Komissarov AA, Florova G, Walker KP, III, Koenig K, Chroneos ZC, Allen T, et al. Wood bark smoke induces lung and pleural plasminogen activator inhibitor 1 and stabilizes its MmRNA in porcine lung cells. Shock 2011;36:128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Idell S. Anticoagulants for acute respiratory distress syndrome: can they work? Am J Respir Crit Care Med 2001;164:517–520 [DOI] [PubMed] [Google Scholar]
- 36.Idell S, Mazar AP, Bitterman P, Mohla S, Harabin AL. Fibrin turnover in lung inflammation and neoplasia. Am J Respir Crit Care Med 2001;163:578–584 [DOI] [PubMed] [Google Scholar]
- 37.Idell S, Azghani A, Chen S, Koenig K, Mazar A, Kodandapani L, Bdeir K, Cines D, Kulikovskaya I, Allen T. Intrapleural low-molecular-weight urokinase or tissue plasminogen activator versus single-chain urokinase in tetracycline-induced pleural loculation in rabbits. Exp Lung Res 2007;33:419–440 [DOI] [PubMed] [Google Scholar]
- 38.Idell S, Allen T, Chen S, Koenig K, Mazar A, Azghani A. Intrapleural activation, processing, efficacy, and duration of protection of single-chain urokinase in evolving tetracycline-induced pleural injury in rabbits. Am J Physiol Lung Cell Mol Physiol 2007;292:L25–L32 [DOI] [PubMed] [Google Scholar]
- 39.Chaillan-Huntington CE, Gettins PGW, Huntington JA, Patston PA. The P6–P2 region of serpins is critical for proteinase inhibition and complex stability. Biochemistry 1997;36:9562–9570 [DOI] [PubMed] [Google Scholar]
- 40.Komissarov AA, Andreasen PA, Declerck PJ, Kamikubo Y, Zhou A, Gruber A. Redirection of the reaction between activated protein C and a serpin to the substrate pathway. Thromb Res 2008;122:397–404 [DOI] [PubMed] [Google Scholar]
- 41.Idell S, Jun NM, Liao H, Gazar AE, Drake W, Lane KB, Koenig K, Komissarov A, Tucker T, Light RW. Single-chain urokinase in empyema induced by Pasturella multocida. Exp Lung Res 2009;35:665–681 [DOI] [PubMed] [Google Scholar]
- 42.Kumar A, Koenig KB, Johnson AR, Idell S. Expression and assembly of procoagulant complexes by human pleural mesothelial cells. Thromb Haemost 1994;71:587–592 [PubMed] [Google Scholar]
- 43.Shetty S, Velusamy T, Shetty RS, Marudamuthu AS, Shetty SK, Florova G, Tucker T, Koenig K, Shetty P, Bhandary YP, et al. Post-transcriptional regulation of plasminogen activator inhibitor Type–1 expression in human pleural mesothelial cells. Am J Respir Cell Mol Biol 2010;43:358–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lucas A, Dai E, Liu L, Guan H, Nash P, McFadden G, Miller L. Transplant vasculopathy: viral anti-inflammatory serpin regulation of atherogenesis. J Heart Lung Transplant 2000;19:1029–1038 [DOI] [PubMed] [Google Scholar]
- 45.Tucker TA, Williams L, Koenig K, Kothari H, Komissarov AA, Florova G, Mazar AP, Allen TC, Bdeir K, Mohan Rao LV, et al. Lipoprotein receptor–related protein 1 regulates collagen 1 expression, proteolysis, and migration in human pleural mesothelial cells. Am J Respir Cell Mol Biol 2012;46:196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.