

Abstract
Previous work demonstrated that pre-exposure to ozone primes innate immunity and increases Toll-like receptor–4 (TLR4)–mediated responses to subsequent stimulation with LPS. To explore the pulmonary innate immune response to ozone exposure further, we investigated the effects of ozone in combination with Pam3CYS, a synthetic TLR2/TLR1 agonist. Whole-lung lavage (WLL) and lung tissue were harvested from C57BL/6 mice after exposure to ozone or filtered air, followed by saline or Pam3CYS 24 hours later. Cells and cytokines in the WLL, the surface expression of TLRs on macrophages, and lung RNA genomic expression profiles were examined. We demonstrated an increased WLL cell influx, increased IL-6 and chemokine KC (Cxcl1), and decreased macrophage inflammatory protein (MIP)-1α and TNF-α in response to Pam3CYS as a result of ozone pre-exposure. We also observed the increased cell surface expression of TLR4, TLR2, and TLR1 on macrophages as a result of ozone alone or in combination with Pam3CYS. Gene expression analysis of lung tissue revealed a significant increase in the expression of genes related to injury repair and the cell cycle as a result of ozone alone or in combination with Pam3CYS. Our results extend previous findings with ozone/LPS to other TLR ligands, and suggest that the ozone priming of innate immunity is a general mechanism. Gene expression profiling of lung tissue identified transcriptional networks and genes that contribute to the priming of innate immunity at the molecular level.
Keywords: ozone, Toll-like receptors, air pollution, gene expression profiles, macrophages
Air pollution accounts for substantial morbidity and mortality in children and adults (1). Air pollutants are known to exacerbate the symptoms of asthma, and may be involved in the initiation of this disease. In most urban areas, and increasingly in suburban areas, traffic-related emissions, including ozone precursors, comprise a major source of air pollution. Several studies reported increased emergency room visits by school-age children in response to ozone levels at or below the current standards (2–5). Recent evidence also suggests that ozone exposure may promote the development of asthma (6). Controlled ozone exposures in healthy volunteers have consistently demonstrated a decrease in forced expiratory volume in 1 second (FEV1), an increase in nonspecific airway hyperresponsiveness, and neutrophilic inflammation as early as 1 hours after exposure and persisting up to 24 hours (7, 8). Ozone exposure was also shown to cause an enhanced response to inhaled allergens, both in normal volunteers and in patients with allergic asthma (9–11). Animal models of allergic asthma have been used to study the complex interaction between ozone and allergen (12–14).
In addition to the role it plays in the development of allergic airway disease, ozone may also predispose individuals, particularly children who are more susceptible, to develop respiratory infections. A number of studies in animals suggested that pre-exposure to ozone exerts an influence on pulmonary host defense. Exposure to ozone was associated with the impaired clearance of several bacteria, including Klebsiella pneumoniae (15), Listeria monocytogenes (16), Mycobacterium tuberculosis (17), Staphylococcus aureus (18), Streptococcus pyogenes (19), and Streptococcus zooepidemicus (20). This impairment in antibacterial host defense is at least in part explained by the disruption of the epithelial barrier and the inefficiency of phagocytosis. Studies in humans have shown that exposure to ambient levels of ozone can impair selective epithelial permeability (21), and that this loss of epithelial integrity is parallel to but not necessarily coupled with increased inflammation in the lower respiratory tract (7, 22). Moreover, both human and murine alveolar macrophages in vitro demonstrated impaired phagocytosis and superoxide production in response to ozone exposure (23, 24).
We have hypothesized that the expression of Toll-like receptors (TLRs) in the lung are influenced by exposure to ozone, and that the dynamic expression of TLRs exerts profound effects on lung host defense. An earlier study from our laboratory demonstrated that ozone pre-exposure altered the innate immune response to inhaled LPS in mice. Pre-exposure to ozone resulted in enhanced airway hyperresponsiveness (AHR), increased concentrations of total protein and proinflammatory cytokines in whole-lung lavage (WLL), and reduced inflammatory cell recruitment to the lower airways (25). That study also showed how ozone exposure results in the increased surface expression of TLR4 and enhanced LPS-mediated signaling in lung tissue. To test whether priming of the innate immune system by ozone constitutes a more general mechanism, we investigated the effects of ozone in combination with Pam3CYS, a synthetic TLR2/TLR1 agonist. We demonstrated changes in the inflammatory response to Pam3CYS as a result of ozone pre-exposure that appears to be caused by the enhanced expression of cell-surface TLRs on macrophages. In addition, we identified molecular processes and transcriptional networks associated with these phenotypes by performing gene expression analyses of mRNA from lung tissue.
Materials and Methods
Animals
Male 7- to 8-week-old C57B/L6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). All experiments were performed according to National Institutes of Health guidelines, and were approved by the Institutional Animal Care and Use Committee at National Jewish Health.
Exposure Protocol
Mice were exposed to either 2 ppm ozone (n = 10) or filtered air (FA) (n = 10) for 3 hours, according to a published protocol (25) described in detail in the online supplement. Twenty-four hours after ozone exposure, mice from the ozone or FA groups were treated intratracheally with 100 μg of Pam3CYS in saline (n = 5) or saline alone (n = 5). Animals were killed 4 or 24 hours after Pam3CYS exposure. WLL, total and differential cell counts, ELISAs, and total protein assays were performed according to published protocols (25, 26), as described in detail in the online supplement.
Flow Cytometry
Fixed WLL cells were washed and resuspended in CD16/CD32 Mouse BD FC Block (BD Biosciences, San Jose, CA) for 15 minutes. Cells were then stained with several fluorochrome-labeled antibodies: F4/80:FITC (AbD Serotec, Oxford, United Kingdom), TLR-1:PE, TLR 2:Alexa Fluor647, and CD11c:APC-e780 (eBioscience, San Diego, CA). Flow cytometry was performed using a FacScan from BD Biosciences, and data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Statistical Analysis of Cell, Cytokine, and Flow Cytometry Data
Data were analyzed using GraphPad Prism statistical software (GraphPad, San Diego, CA), and are expressed as means ± SEMs (n = 10–15 from three independent experiments). Comparisons between groups were performed with the two-tailed Mann-Whitney U test.
Western Blots
Lung tissues were homogenized and lysed in ice-cold lysis buffer, as described in the online supplement. After centrifugation, tissue extracts were resolved by SDS-PAGE and analyzed by immunoblotting. The membranes were probed with antibodies to phospho-JUN N-terminal kinase (JNK), phospho-p44/42 mitogen-activated protein kinase (MAPK; Erk1/2), and p44/42 MAPK (Cell Signaling Technology, Danvers, MA). Blots were developed with SuperSignal West Dura (Thermo Scientific, Rockford, IL).
RNA Extractions, Microarray, and Quantitative RT-PCR
RNA from the right lung was extracted using an Ambion mirVana RNA extraction kit, according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA). Gene expression profiling was performed on Agilent Whole Mouse Genome arrays (Agilent Technologies, Santa Clara, CA), according to the manufacturer’s protocols, as briefly described in the online supplement. All primary data were deposited at the Gene Expression Omnibus database (accession number GSE38014). Quantitative RT-PCR with SYBRGreen (Life Technologies) was performed using standard protocols, as described in the online supplement, and primers were designed using Primer-BLAST (National Center for Biotechnology Information, Bethesda, MD) (Table E1 in the online supplement).
Statistical Analysis of Microarray and Quantitative RT-PCR Data
Expression data from 32 mRNA arrays (four exposure groups, two time points, and four animals/group) were analyzed using Partek software (Partek, Inc., St. Louis, MO). Intensity data were imported, log2-transformed, and quantile-normalized, using the robust multi-array average (RMA) algorithm (27). The differential expression of individual transcripts was identified using an ANOVA model incorporating exposure groups and time points. Differentially expressed genes with a false discovery rate (FDR) of 5% and 2-fold change between O3/saline versus FA/saline, O3/Pam3CYS versus O3/saline, and O3/Pam3CYS versus FA/Pam3CYS at two time points after Pam3CYS exposure were identified and analyzed for enriched pathways and transcriptional networks, using the Ingenuity Pathway Analysis (IPA) database (Ingenuity Systems, Redwood City, CA). Quantitative RT-PCR data were analyzed according to the ΔΔCt method (28).
Results
Ozone Pre-Exposure Alters the Inflammatory Response to Pam3CYS in WLL
To study the effects of ozone pre-exposure on the innate immune response to Pam3CYS in the lung, we first examined the inflammatory response in the lavage fluid of C57BL/6 mice that were exposed to 2 ppm O3 (2 ppm) or FA, followed by an intratracheal instillation of Pam3CYS or saline 24 hours after exposure to ozone. The WLL was examined either 4 hours or 24 hours after Pam3CYS instillation (Figure 1 and Figure E1 in the online supplement). The effect of ozone exposure (ozone/saline group) compared with FA (FA/saline group) was characterized by a significant decrease in total cell macrophages and neutrophils at 4 hours and neutrophils at 24 hours (Figure 1A). Not surprisingly, a significant increase of neutrophil influx was observed in response to Pam3CYS (mice exposed to FA/Pam3CYS or O3/Pam3CYS, compared with FA/saline and O3/saline). Importantly, ozone pre-exposure resulted in a significant increase of, neutrophil influx in response to Pam3CYS at both time points (O3/Pam3CYS versus FA/Pam3CYS) with a greater increase at 24 hours. Macrophage influx was also significantly increased in O3/Pam3CYS compared with O3/saline mice. These data demonstrate that O3 pre-exposure enhances the influx of both macrophages and neutrophils to Pam3CYS.
Figure 1.

Ozone alters the inflammatory response to the synthetic Toll-like receptor–2 (TLR2)/TLR1 agonist Pam3CYS in murine whole-lung lavage (WLL). (A) Cell counts and (B) selected cytokines and chemokines in the WLL of C57BL/6 mice in response to intratracheal instillation of saline or Pam3CYS 24 hours after ozone (2 ppm, 3 h) or filtered air (FA) exposure. Measurements were performed at two time points, 4 hours and 24 hours after Pam3CYS instillation (28 h and 48 h after ozone exposure, respectively). Data are expressed as mean ± SEM (n = 10–15 C57BL/6 mice from three independent experiments). Group comparisons included O3/saline versus FA/saline, O3/saline versus O3/Pam3CYS, FA/Pam3CYS versus FA/saline, and O3/Pam3CYS versus FA/Pam3CYS. Statistical significance was determined using the Mann-Whitney U test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P < 0.0001).
We next investigated the inflammatory cytokine and chemokine response in our model by measuring WLL total protein, IL-6, TNF-α, KC, MIP-1α, and MIP-2. As in previous studies (25, 29), O3/saline exposure increased total protein in WLL (compared with FA/saline) at both time points (Figure E1A). At 24 hours, O3/Pam3CYS exposure resulted in a significant increase of WLL protein compared with FA/Pam3CYS, and was higher than at 4 hours, reflecting an increase in lung permeability over time. As in previous reports, IL-6 in the WLL of O3 exposed mice was increased compared with FA at both 4 hours and 24 hours, but the difference did not reach statistical significance (Figure 1B). Pam3CYS exposure (FA/Pam3CYS and O3/Pam3CYS, compared with FA/saline and O3/saline) led to significant increases in IL-6 concentration. Similar to previous exposure studies with O3 and LPS (25), we observed a fivefold increase in IL-6 in the O3/Pam3CYS group compared with the FA/Pam3CYS group at 4 hours, suggesting an enhanced IL-6 response with O3 priming, followed by Pam3CYS. However, in contrast to our previous ozone/LPS studies (25), this response diminished over time. KC, a neutrophil chemokine, follows a similar pattern as IL-6, with the only difference involving a significant decrease in KC in the O3/Pam3CYS group compared with the FA/Pam3CYS group. A second well-characterized neutrophil chemokine, MIP-2, was increased in the WLL from animals exposed to FA/Pam3CYS and O3/Pam3CYS compared with their saline counterparts, but not significant compared with each other. Similar to IL-6 and KC, the 24-hour time point was significantly diminished compared with 4 hours, but with no differences between treatment groups (Figure E1B). In contrast to increased concentrations of IL-6, KC, and MIP-2, ozone pre-exposure before exposure to Pam3CYS did not affect concentrations of TNFα and MIP-1α. Taken together, these data demonstrate an enhanced, but not consistent, pulmonary inflammatory response to Pam3CYS as a result of ozone pre-exposure, providing a rationale for the further characterization of this response.
Ozone Exposure Leads to Increased Surface Expression of TLR1, TLR2, and TLR4 in Alveolar Macrophages
We next investigated the cell-surface expression of TLR1, TLR2, and TLR4 in alveolar macrophages. We hypothesized that the synergistic response of ozone and Pam3Cys in WLL cytokines may be at least partly attributable to an increase in cell-surface TLRs, in keeping with previous findings of an increase in TLR4 with O3 (25). We identified WLL macrophages from each treatment group by forward scatter and side scatter and by positivity for F4/80 and CD11b (Figure E2). Mice exposed to O3/saline demonstrated a significant increase in TLR1, TLR2, and TLR4 compared with FA/saline at 4 hours after saline. At 24 hours, a significant increase in TLR2 and TLR4 was still evident, but no difference in TLR1 was observed (Figure 2). Ozone pre-exposure resulted in a significant increase in the expression of TLR2, but not of TLR1 and TLR4, in response to Pam3CYS at 4 hours, with a significant increase in both TLR1 and TLR2, but not TLR4, at 24 hours (O3/Pam3CYS versus FA/Pam3CYS groups). Although no increase in TLR4 surface expression as a result of ozone pre-exposure was evident in response to Pam3CYS (O3/Pam3CYS versus FA/Pam3CYS), treatment with Pam3CYS (FA/Pam3CYS versus FA/saline, and O3/Pam3CYS versus O3/saline) surprisingly led to an increase in surface TLR4. These data confirm our earlier findings of an increase in surface TLR4 as a result of ozone exposure, and extend these findings to other TLRs, but also demonstrate the dynamic nature of TLR expression as a result of both ozone and TLR ligand exposure.
Figure 2.

Surface expression of TLR1, TLR2, and TLR4 on alveolar macrophages after ozone and Pam3CYS exposure (A–C) at 4 hours and (D–F) at 24 hours. Cell-surface expression of TLR1, TLR2, and TLR4 on alveolar macrophages collected from WLL after ozone (2 ppm, 3 h) or FA pre-exposure was followed by intratracheal instillation of saline or Pam3CYS. WLL macrophages were identified by forward scatter and side scatter and by positivity for F4/80 and CD11c, and were then analyzed for TLR expression. Group comparisons involved O3/saline versus FA/Saline, O3/saline versus O3/Pam3CYS, FA/Pam3CYS versus FA/Saline, and O3/Pam3CYS versus FA/Pam3CYS. Data are expressed as mean ± SEM (n = 10–15 C57 mice from three independent experiments). Statistical significance was determined using the Mann-Whitney U test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P < 0.0001).
We also investigated the expression of TLR1, TLR2, and TLR4 in lung tissue via quantitative RT-PCR (Figure E3). TLR1, TLR2, and TLR4 mRNA expression in whole lung exhibited a similar pattern to that identified by flow cytometry analysis on the cell surface of WLL macrophages. The most prominent change involved a greater than twofold increase in TLR2 mRNA expression in the O3/Pam3CYS group compared with FA/Pam3CYS at the 4-hour time point (Figure E3A). TLR expression in cell types other than alveolar macrophages and intracellular expression, at least in the case of TLR4, provides the likely explanation for any differences observed between surface expression measured by flow cytometry and expression in whole-lung tissue measured by quantitative RT-PCR.
Ozone Exposure Leads to Increased TLR Signaling in the Lung
To determine whether the ozone priming of innate immunity leads to enhanced downstream signaling in the TLR pathway, we investigated the effects of ozone alone or followed by Pam3CYS on downstream signaling in whole-lung homogenates. At 4 hours, ozone alone significantly increased the phosphorylation of p44/42 (Erk1/2) MAPK but not JNK, whereas Pam3CYS did not increase the phosphorylation of either kinase (Figure 3). Strikingly, ozone followed by Pam3CYS resulted in a large increase of phosphorylation for both kinases and a significant reduction in nonphosphorylated p44/42 MAPK. This enhanced signal associated with ozone/Pam3CYS treatment was not present at 24 hours (data not shown). These data, analogous to findings in our earlier study (25), demonstrate an enhancement in TLR signaling as a result of the ozone priming of innate immunity.
Figure 3.

Ozone pre-exposure resulted in significantly enhanced TLR signaling in response to Pam3CYS, as assessed by mitogen-activated protein kinase (MAPK) phosphorylation at 4 hours (28 h after ozone). Western blot analysis of lung homogenates was performed on animals exposed to FA/saline (FA), FA/Pam3CYS (P3C), ozone/saline (O3), and ozone/Pam3CYS (O3 + P3C).
Gene Expression Analysis in the Lung Reveals Novel Candidate Genes for the Ozone Priming of Innate Immunity
To characterize the priming effect of ozone on innate immunity at the molecular level in an unbiased manner, we performed gene expression profiling on lung tissue of mice from the four exposure groups. Differentially expressed genes were identified as those with an FDR of 5% in an ANOVA model and a two fold change in O3/saline versus FA/Saline (effect of ozone), O3/Pam3CYS versus O3/saline (effect of Pam3CYS), and O3/Pam3CYS versus FA/Pam3CYS (effect of ozone priming on Pam3CYS response) at two time points (4 h and 24 h). Venn diagrams for the three sets of genes (Figure 4A and Tables E2–E7) reveal that ozone exposure exerts the strongest effect on gene expression at the 4-hour time point, with the effect diminishing by the 24-hour time point. Pam3CYS alone or when preceded by ozone results in less pronounced transcriptional changes.
Figure 4.
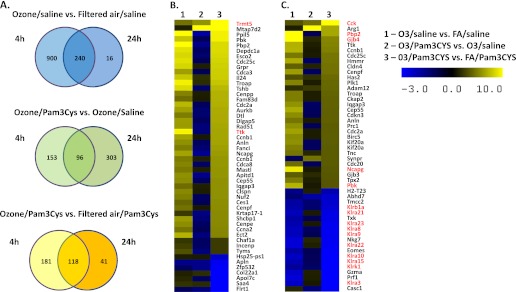
Gene expression in lung tissue after ozone pre-exposure and intratracheal instillation of Pam3CYS. (A) Venn diagrams depict overlaps in expression profiles at 4 hours (28 h after ozone) and 24 hours (48 h after ozone) after Pam3CYS instillation. Differentially expressed genes were defined as those with a false discovery rate (FDR) of 5% and 2-fold change between the comparison groups at two time points. (B and C) Heat maps of the most differentially expressed genes as a result of ozone pre-exposure, followed by Pam3CYS treatment at the (B) 4-hour time point and (C) 24-hour time point. The heat maps depict the 50 most up-regulated and down-regulated genes in the O3/Pam3CYS versus FA/Pam3CYS comparison.
We focused the remainder of our analyses on the effect of ozone priming on Pam3CYS response as the primary goal of a gene expression study, to identify molecular processes that mediate this effect. To examine the most prominent changes in gene expression, we focused on 50 genes at each time point with the highest mean fold change in the O3/Pam3CYS versus FA/Pam3CYS comparison (Figure 4B). Ozone pre-exposure before Pam3CYS treatment (O3/Pam3CYS versus FA/Pam3CYS) enhanced the induction of transfer RNA (tRNA) methyltransferase 5 (Trmt5) at 4 hours and cholecystokinin (Cck) at 24 hours. The expression of Ttk protein kinase (4-h time point), phosphatidylethanolamine binding protein 2 (Pbp2), gap junction protein, beta 4 (Gjb4), non-SMC condensin I complex, subunit G (Ncapg), and PDZ binding kinase (Pbk) (24-h time point) were also increased in O3/Pam3CYS versus FA/Pam3CYS, but to a significantly lesser extent than in the O3/saline versus FA/saline comparison. The most significant result among down-regulated genes involved the down-regulation at 24 hours of killer cell lectin-like receptors (Klra3, Klra8, Klra9, Klra10, Klra15, Klra21, Klra22, Klra23, Klrb1a, and Klrk1.
We also used ingenuity pathway analysis (IPA) to identify significantly overrepresented biological functions in the gene expression data. Biological function analysis revealed significant changes in cellular (Figure E4A) and immune (Figure E4B) function. Ozone exposure followed by exposure to Pam3Cys resulted in significant changes in cell cycle, DNA replication/recombination/repair, and cellular assembly and organization. These same gene categories were enhanced in response to ozone alone, but were different from Pam3CYS-induced genes, which were enhanced in cell signaling, immune cell trafficking, inflammatory response/disease, and infection. We also used IPA to identify transcriptional regulators whose binding sites are overrepresented in promoters of differentially expressed genes (Table E8). Among the most significant transcriptional regulators whose target gene expression changed as a result of ozone followed by Pam3CYS exposure were those involved in the control of the cell cycle (E2F and Rb families), cancer (Myc, Rb, and TP53), and chromatin structure (the SMARC family and HDAC1). These same transcription factors were overrepresented in response to ozone alone, but produced gene expression changes different from those induced by Pam3CYS, which are regulated by transcription factors involved in innate immunity and inflammation (the NF-κB, Sp1, AP1, HMGB1, CEBP, IRF, and STAT families).
Using quantitative PCR, we confirmed gene expression changes in six selected genes identified in the array analysis: Cck, Retnla (RELMα), Retnlb (RELMβ), Hhmr, Il24, and Tnf (Table E9).
Discussion
Our results extend previous findings in regard to ozone/LPS to other TLR ligands, and suggest that the ozone priming of innate immunity is likely a general mechanism. We demonstrate an increased inflammatory response to Pam3CYS as a result of ozone pre-exposure. We also observed an increased cell surface expression of TLRs in macrophages as a result of ozone alone or in combination with Pam3CYS. The gene expression profiling of lung tissue identified transcriptional networks and genes that may contribute to the priming of innate immunity at the molecular level.
Our study demonstrates that ozone priming of the innate immune system is likely a general mechanism. Components of air pollution such as ozone and endotoxin are well-documented to exacerbate the symptoms of asthma, and may also play a role in the initiation of this disease. Although our study used a high concentration of ozone, exposure to 2 ppm of ozone in a rodent was shown to be comparable to 0.2 ppm of ozone in a susceptible human subject (30). Ozone and LPS pre-exposures have been shown to alter the innate immune response to pathogens in mice, as well as in murine and human macrophages. However, the mechanisms underlying the host-defense response to the components of air pollution are poorly understood. Taken together, our previous and current work provides evidence for the role of ozone exposure in altering the basic innate immune signaling that renders the pulmonary environment more responsive to pathogens and possibly allergens.
Although the ozone priming of innate immunity appears to be a general mechanism and our findings are largely parallel to those in an earlier study by Hollingsworth and colleagues (25), a few differences between the two studies are evident. In regard to ozone alone exposure, Hollingsworth and colleagues (25) did not observe a difference in total protein in the BAL, whereas we observed a small but statistically significant increase in the group with ozone alone compared with the free air group. Given that the ozone exposure and the strain of mice in the two studies were the same, and that the timings of exposure were similar, the most likely explanation involves the larger number of animals included in each group in our study. In terms of the enhanced response to TLR stimulation, Hollingsworth and colleagues (25) observed a decrease in neutrophils as a result of the combined ozone/LPS exposure compared with LPS alone, whereas we observed a significant increase in neutrophils in the ozone/Pam3CYS group compared with the Pam3CYS-alone group. Moreover, a few subtle differences in cytokine production were detected in the ozone/Pam3CYS study compared with the earlier ozone/LPS study. These differences are most likely a result of differences in the TLR ligands used in these studies. In addition, the route of administration of the TLR ligand (intratracheal versus aerosolized) may also explain some of the observed differences.
The mechanisms by which ozone increases the expression of TLRs on the surface of alveolar macrophages are not understood. Previously, the increase in the response of TLR4 to ozone alone was determined to be a result of the ozone-induced release of endogenous ligands (hyaluronan and heat-shock proteins) that stimulate TLR4, resulting in the migration of TLR4 to the surface of the cell (31). A subsequent gene expression profiling study by another group showed that heat-shock protein–70 is an effector molecule downstream of TLR4, and is involved in the regulation of ozone-induced lung inflammation by triggering pathways similar to those of TLR4 (32). This is likely a general mechanism that acts on all TLRs and accounts for the increases of TLR1 and TLR2 in response to ozone alone in the present study. In addition, we observed a further enhancement in the expression of TLR2 and TLR4 as a result of Pam3CYS treatment after ozone pre-exposure. Potentially, endogenous ligands such as hyaluronan and heat shock proteins also contribute to this priming effect. Alternately, a recent study demonstrated that the recruitment of TLR4 to the cell membrane can be activated by mechanisms involving the generation of nicotinamide adenine dinucleotide phosphate–reduced oxidase–dependent reactive oxygen species (33). Both Pam3CYS and pulmonary ozone exposure can initiate a cascade of reactive oxygen species, thus establishing an environment conducive to TLR2 and TLR4 recruitment to the cell surface (34, 35).
Gene expression analysis identified novel molecular targets associated with the priming effect of ozone on the innate immune response. These include changes in cell cycle, DNA replication/recombination/repair, cellular assembly and organization, and chromatin structure. The expression of the Trmt5 and Cck genes was induced by ozone alone, and was further enhanced by subsequent Pam3Cys stimulation. Trmt5 is a DNA methyltransferase that methylates the N1 position of guanosine-37 in selected tRNAs. It is difficult to speculate how this gene may be involved in the priming of innate immunity, but recently, oxidative stress induced by ozone and other environmental toxins was postulated to regulate 5-methyl-cytosine and 5-hydroxymethyl-cytosine (36). On the other hand, the role of Cck in the priming of innate immunity may be more straightforward. Cck is a neuropeptide that was shown to inhibit inflammation by down-regulating the expression of proinflammatory cytokines during endotoxin shock (37), and it inhibits LPS-induced IL-1β production in pulmonary interstitial macrophages by modulating cAMP-dependent protein kinase (PKA), p38, and NF-κB activity (38).
Among genes highly induced by ozone alone, but whose expression was dampened in response to Pam3Cys after exposure to ozone, are Ttk, Pbp2, Gjb4, Ncapg, and Pbk. Three of these genes are related to kinase activity. Phosphatidylethanolamine-binding protein–2 is a member of the evolutionarily conserved family of proteins (i.e., RAF kinase inhibitory proteins) implicated in MAPK pathway regulation (39). Pbk is a member of the dual specific MAPKK family. Ttk, also known as Mps1, is a mitotic protein kinase with a well-established role in the cell cycle and in carcinogenesis (40). Finally, seven members of the killer cell lectin–like receptor family are down-regulated by ozone, and their expression is further dampened by Pam3CYS. Killer cell lectin–like receptors are preferentially expressed on natural killer T (NKT) cells, which are innate immune lymphocytes that play a major role in the host rejection of virally infected cells (41), and which have also been implicated in the pathogenesis of asthma (42). A previous study demonstrated that NKT cells are required for the development of both ozone-induced and allergen-induced AHR (43). Together with our findings regarding the potential role of NKT cells on the priming of innate immunity, these results strongly suggest that NKT cells mediate a unifying pathogenic mechanism for several distinct forms of airway inflammation, and may represent a therapeutic target.
Four published studies assessed the genomic transcription profiles of murine lung tissue after ozone exposure (26, 32, 44). Park and colleagues profiled lung tissue from C57BL/6 mice exposed to 2 ppm ozone for 3 hours, either 4 or 24 hours after exposure, but focused their analysis primarily on inflammatory markers (26). Gohil and colleagues used an early genomic array to identify 260 differentially expressed transcripts (out of 4,000 represented on the array) in C57BL/6 mice exposed to 1 ppm O3 for 3 consecutive nights (8 hours per night) (45). Among the differentially expressed transcripts were genes related to the immune response, DNA synthesis, cell-cycle progression, xenobiotic metabolism, and cytoskeletal functions. Williams and colleagues identified 400 differentially expressed genes (greater than twofold) in BALB/c mice 3 hours after ozone exposure, and also identified a number of markers of inflammation as the most affected genes (44). Bauer and colleagues profiled C3H/HeJ and C3H/HeOuJ mice to identify ozone candidate genes downstream of TLR4, and demonstrated a role for heat shock protein–70 (32). Our analysis of gene expression in the lung after ozone exposure differs from these previously published studies because it focused on the priming effect of ozone on innate immunity. As such, our study identified a number of novel targets for further exploration, both mechanistically and as therapeutic targets.
In conclusion, we have shown that the priming of innate immunity is a general mechanism, and we have identified novel molecular targets in this important process. Our study demonstrates that the expression of TLRs on macrophage surfaces is a dynamic process that is influenced by ozone, and that this process is associated with the differential expression of numerous previously unexplored genes. This dynamic nature of TLR expression is likely more general, and could be influenced by other components of air pollution and in cell types other than macrophages. This priming effect of air pollution and genes that are associated with the process may also lead to potential therapeutic targets for air pollutant exposure in the context of pulmonary infection or allergic airway inflammation.
Acknowledgments
The authors thank David MCKean for his assistance with the GEO submission.
Footnotes
This work was supported by National Institute of Environmental Health Sciences grant P01-ES18181 (J.L.O.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0187OC on September 20, 2012
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Ruckerl R, Schneider A, Breitner S, Cyrys J, Peters A. Health effects of particulate air pollution: a review of epidemiological evidence. Inhal Toxicol 2011;23:555–592 [DOI] [PubMed] [Google Scholar]
- 2.Gent JF, Triche EW, Holford TR, Belanger K, Bracken MB, Beckett WS, Leaderer BP. Association of low-level ozone and fine particles with respiratory symptoms in children with asthma. JAMA 2003;290:1859–1867 [DOI] [PubMed] [Google Scholar]
- 3.Romieu I, Meneses F, Ruiz S, Huerta J, Sienra JJ, White M, Etzel R, Hernandez M. Effects of intermittent ozone exposure on peak expiratory flow and respiratory symptoms among asthmatic children in Mexico City. Arch Environ Health 1997;52:368–376 [DOI] [PubMed] [Google Scholar]
- 4.Romieu I, Meneses F, Sienra-Monge JJ, Huerta J, Ruiz Velasco S, White MC, Etzel RA, Hernandez-Avila M. Effects of urban air pollutants on emergency visits for childhood asthma in Mexico City. Am J Epidemiol 1995;141:546–553 [DOI] [PubMed] [Google Scholar]
- 5.White MC, Etzel RA, Wilcox WD, Lloyd C. Exacerbations of childhood asthma and ozone pollution in Atlanta. Environ Res 1994;65:56–68 [DOI] [PubMed] [Google Scholar]
- 6.McConnell R, Berhane K, Gilliland F, London SJ, Islam T, Gauderman WJ, Avol E, Margolis HG, Peters JM. Asthma in exercising children exposed to ozone: a cohort study. Lancet 2002;359:386–391 [DOI] [PubMed] [Google Scholar]
- 7.Koren HS, Devlin RB, Graham DE, Mann R, McGee MP, Horstman DH, Kozumbo WJ, Becker S, House DE, McDonnell WF, et al. Ozone-induced inflammation in the lower airways of human subjects. Am Rev Respir Dis 1989;139:407–415 [DOI] [PubMed] [Google Scholar]
- 8.Foster WM, Brown RH, Macri K, Mitchell CS. Bronchial reactivity of healthy subjects: 18–20 hours postexposure to ozone. J Appl Physiol 2000;89:1804–1810 [DOI] [PubMed] [Google Scholar]
- 9.Peden DB. Pollutants and asthma: role of air toxics. Environ Health Perspect 2002;110:565–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kehrl HR, Peden DB, Ball B, Folinsbee LJ, Horstman D. Increased specific airway reactivity of persons with mild allergic asthma after 7.6 hours of exposure to 0.16 ppm ozone. J Allergy Clin Immunol 1999;104:1198–1204 [DOI] [PubMed] [Google Scholar]
- 11.Peden DB, Setzer RW, Jr, Devlin RB. Ozone exposure has both a priming effect on allergen-induced responses and an intrinsic inflammatory action in the nasal airways of perennially allergic asthmatics. Am J Respir Crit Care Med 1995;151:1336–1345 [DOI] [PubMed] [Google Scholar]
- 12.Depuydt PO, Lambrecht BN, Joos GF, Pauwels RA. Effect of ozone exposure on allergic sensitization and airway inflammation induced by dendritic cells. Clin Exp Allergy 2002;32:391–396 [DOI] [PubMed] [Google Scholar]
- 13.Last JA, Ward R, Temple L, Kenyon NJ. Ovalbumin-induced airway inflammation and fibrosis in mice also exposed to ozone. Inhal Toxicol 2004;16:33–43 [DOI] [PubMed] [Google Scholar]
- 14.Schelegle ES, Miller LA, Gershwin LJ, Fanucchi MV, Van Winkle LS, Gerriets JE, Walby WF, Mitchell V, Tarkington BK, Wong VJ, et al. Repeated episodes of ozone inhalation amplifies the effects of allergen sensitization and inhalation on airway immune and structural development in rhesus monkeys. Toxicol Appl Pharmacol 2003;191:74–85 [DOI] [PubMed] [Google Scholar]
- 15.Miller S, Ehrlich R. Susceptibility to respiratory infections of animals exposed to ozone: I. Susceptibility to Klebsiella pneumoniae. J Infect Dis 1958;103:145–149 [DOI] [PubMed] [Google Scholar]
- 16.Van Loveren H, Rombout PJ, Wagenaar SS, Walvoort HC, Vos JG. Effects of ozone on the defense to a respiratory Listeria monocytogenes infection in the rat: suppression of macrophage function and cellular immunity and aggravation of histopathology in lung and liver during infection. Toxicol Appl Pharmacol 1988;94:374–393 [DOI] [PubMed] [Google Scholar]
- 17.Thomas GB, Fenters JD, Ehrlich R, Gardner DE. Effects of exposure to ozone on susceptibility to experimental tuberculosis. Toxicol Lett 1981;9:11–17 [DOI] [PubMed] [Google Scholar]
- 18.Goldstein E, Tyler WS, Hoeprich PD, Eagle C. Ozone and the antibacterial defense mechanisms of the murine lung. Arch Intern Med 1971;127:1099–1102 [PubMed] [Google Scholar]
- 19.Aranyi C, Vana SC, Thomas PT, Bradof JN, Fenters JD, Graham JA, Miller FJ. Effects of subchronic exposure to a mixture of O3, SO2, and (NH4)2SO4 on host defenses of mice. J Toxicol Environ Health 1983;12:55–71 [DOI] [PubMed] [Google Scholar]
- 20.Gilmour MI, Park P, Doerfler D, Selgrade MK. Factors that influence the suppression of pulmonary antibacterial defenses in mice exposed to ozone. Exp Lung Res 1993;19:299–314 [DOI] [PubMed] [Google Scholar]
- 21.Foster WM, Stetkiewicz PT. Regional clearance of solute from the respiratory epithelia: 18–20 hours postexposure to ozone. J Appl Physiol 1996;81:1143–1149 [DOI] [PubMed] [Google Scholar]
- 22.Devlin RB, McDonnell WF, Mann R, Becker S, House DE, Schreinemachers D, Koren HS. Exposure of humans to ambient levels of ozone for 6.6 hours causes cellular and biochemical changes in the lung. Am J Respir Cell Mol Biol 1991;4:72–81 [DOI] [PubMed] [Google Scholar]
- 23.Becker S, Madden MC, Newman SL, Devlin RB, Koren HS. Modulation of human alveolar macrophage properties by ozone exposure in vitro. Toxicol Appl Pharmacol 1991;110:403–415 [DOI] [PubMed] [Google Scholar]
- 24.Valentine R. An in vitro system for exposure of lung cells to gases: effects of ozone on rat macrophages. J Toxicol Environ Health 1985;16:115–126 [DOI] [PubMed] [Google Scholar]
- 25.Hollingsworth JW, Maruoka S, Li Z, Potts EN, Brass DM, Garantziotis S, Fong A, Foster WM, Schwartz DA. Ambient ozone primes pulmonary innate immunity in mice. J Immunol 2007;179:4367–4375 [DOI] [PubMed] [Google Scholar]
- 26.Park JW, Taube C, Swasey C, Kodama T, Joetham A, Balhorn A, Takeda K, Miyahara N, Allen CB, Dakhama A, et al. Interleukin-1 receptor antagonist attenuates airway hyperresponsiveness following exposure to ozone. Am J Respir Cell Mol Biol 2004;30:830–836 [DOI] [PubMed] [Google Scholar]
- 27.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003;31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(t) method. Nat Protoc 2008;3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Potts EN, Piantadosi CA, Foster WM, Hollingsworth JW. Hyaluronan fragments contribute to the ozone-primed immune response to lipopolysaccharide. J Immunol 2010;185:6891–6898 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 30.Hatch GE, Slade R, Harris LP, McDonnell WF, Devlin RB, Koren HS, Costa DL, McKee J. Ozone dose and effect in humans and rats: a comparison using oxygen-18 labeling and bronchoalveolar lavage. Am J Respir Crit Care Med 1994;150:676–683 [DOI] [PubMed] [Google Scholar]
- 31.Garantziotis S, Li Z, Potts EN, Lindsey JY, Stober VP, Polosukhin VV, Blackwell TS, Schwartz DA, Foster WM, Hollingsworth JW. TLR4 is necessary for hyaluronan-mediated airway hyperresponsiveness after ozone inhalation. Am J Respir Crit Care Med 2010;181:666–675 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 32.Bauer AK, Rondini EA, Hummel KA, Degraff LM, Walker C, Jedlicka AE, Kleeberger SR. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: a role for heat-shock protein 70. Environ Health Perspect 2011;119:1091–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species–dependent manner. J Biol Chem 2009;284:27384–27392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hollingsworth JW, Kleeberger SR, Foster WM. Ozone and pulmonary innate immunity. Proc Am Thorac Soc 2007;4:240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med 2010;48:1121–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chia N, Wang L, Lu X, Senut MC, Brenner C, Ruden DM. Hypothesis: environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 2011;6:853–856 [DOI] [PubMed] [Google Scholar]
- 37.Ling YL, Meng AH, Zhao XY, Shan BE, Zhang JL, Zhang XP. Effect of cholecystokinin on cytokines during endotoxic shock in rats. World J Gastroenterol WJG 2001;7:667–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Ni Z, Cong B, Gao W, Xu S, Wang C, Yao Y, Ma C, Ling Y. Cck-8 inhibits LPS-induced IL-1beta production in pulmonary interstitial macrophages by modulating PKA, p38, and NF-kappaB pathway. Shock 2007;27:678–686 [DOI] [PubMed] [Google Scholar]
- 39.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, et al. Suppression of RAF-1 kinase activity and map kinase signalling by RKIP. Nature 1999;401:173–177 [DOI] [PubMed] [Google Scholar]
- 40.de Carcer G, Perez de Castro I, Malumbres M. Targeting cell cycle kinases for cancer therapy. Curr Med Chem 2007;14:969–985 [DOI] [PubMed] [Google Scholar]
- 41.Marcenaro E, Carlomagno S, Pesce S, Della Chiesa M, Parolini S, Moretta A, Sivori S. NK cells and their receptors during viral infections. Immunotherapy 2011;3:1075–1086 [DOI] [PubMed] [Google Scholar]
- 42.Umetsu DT, Dekruyff RH. Natural killer T cells are important in the pathogenesis of asthma: the many pathways to asthma. J Allergy Clin Immunol 2010;125:975–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, Zhu M, Iwakura Y, Savage PB, DeKruyff RH, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med 2008;205:385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams AS, Issa R, Leung SY, Nath P, Ferguson GD, Bennett BL, Adcock IM, Chung KF. Attenuation of ozone-induced airway inflammation and hyper-responsiveness by c-Jun NH2 terminal kinase inhibitor SP600125. J Pharmacol Exp Ther 2007;322:351–359 [DOI] [PubMed] [Google Scholar]
- 45.Gohil K, Cross CE, Last JA. Ozone-induced disruptions of lung transcriptomes. Biochem Biophys Res Commun 2003;305:719–728 [DOI] [PubMed] [Google Scholar]